

ABSTRACT
Macroautophagy/autophagy abnormality has been recently associated with metabolic disorders, such as type 2 diabetes (T2D). However, the effect of autophagy activation in systemic energy metabolism was poorly understood. In our recent study, we demonstrated that autophagy plays different roles in distinct metabolic tissues, using an autophagy-hyperactive mouse model. In insulin-producing β cells, excess autophagy degrades insulin-containing vesicles (a process termed vesicophagy), resulting in decreased insulin contents and systemic glucose intolerance; whereas in insulin-responsive cells, activating autophagy decreases endoplasmic reticulum (ER) stress and improves insulin sensitivity.
KEYWORDS: β cell, BECN1, ER stress, insulin granules, insulin sensitivity, type 2 diabetes, vesicophagy
Type 2 diabetes (T2D) is characterized by hyperglycemia resulting from an inability of pancreatic β cells to secrete enough insulin to compensate for insulin resistance in insulin-responsive organs, including liver, muscle, and adipose tissue. In the past decade, a growing body of evidence indicates that dysfunction of autophagy is linked to metabolic disruption and the etiology of T2D. For example, β cell-specific knockout (KO) of Atg7 (autophagy related 7) in mice results in decreased β cell mass, aberrant islet morphology, and glucose intolerance under either regular diet (RD) or high-fat diet (HFD) feeding conditions, suggesting that basal autophagy is required for the maintenance of β cell function. However, the results are largely based on the loss of function of basal autophagic activity, and metabolic effects of constitutive autophagy activation were still unknown. Furthermore, tissue-specific requirement of autophagy activity in metabolic regulation among different cell types, such as insulin-secreting β cells versus insulin-responsive cells, was poorly understood. To address these questions, we sought to utilize a genetic model of hyperactive autophagy [1], because currently known pharmacological autophagy inducers also regulate other pathways and are not specific to autophagy. The model we chose is the BECN1F121A knockin mouse line we recently generated, carrying a single amino acid substitution of Phe by Ala in the BH3 domain of the essential autophagy protein BECN1. This BECN1F121A point mutation leads to dissociation of BECN1 from its inhibitor, BCL2, resulting in constitutive activation of autophagy systemically, as evidenced by the GFP-LC3 puncta number, LC3-II levels, and SQSTM1/p62 levels.
Because autophagy targets and degrades a variety of nutrient storage molecules, including glycogen, lipid droplets, ribosomes, and proteins, we initially hypothesized that autophagy activation is beneficial for nutrient mobilization and metabolic maintenance. Unexpectedly, we found that after HFD feeding, the autophagy-hyperactive BECN1F121A mice behave similarly to the autophagy-deficient Becn1+/− KO mice in glucose tolerance tests: both of them show exacerbated glucose intolerance compared to WT mice. Yet, different from Becn1+/− KO mice, glucose intolerance in BECN1F121A mice is not caused by insulin resistance in peripheral organs, but rather by dysfunction of pancreatic β cells, because when we inject exogenous insulin in insulin tolerance tests, BECN1F121A mice show improved insulin sensitivity and insulin-AKT signaling in insulin-responsive tissues. Although we are still investigating the mechanisms underlying the improved insulin sensitivity, we found that a reduction in HFD-induced endoplasmic reticulum (ER) stress partially contributes, because tunicamycin, an ER stress inducer, can partially abolish insulin sensitization in BECN1F121A mice.
The idea of autophagy hyperactivation leading to reduced insulin secretion and storage was confirmed by glucose-stimulated insulin secretion assays in vivo (hyperglycemic clamp), ex vivo (isolated islets), and in vitro (BECN1F121A-expressing β cell line). We were able to restore not only insulin storage, but also glucose tolerance, in HFD-fed BECN1F121A mice, by short-term administration of an autophagy inhibitor, SBI-0206965. Importantly, the reduction of insulin storage in autophagy-hyperactive mice is not due to islet degeneration. In fact, in comparison to β cell-specific deletion of Atg7, which causes diminished β cell mass, we found that hyperactivation of autophagy increases β cell mass. It is yet unclear how autophagy drives the compensatory β cell hyperplasia under HFD treatment. In the future it will be intriguing to investigate whether high autophagy promotes islet expansion and β cell proliferation, using MKI67/Ki-67 or BrdU labeling.
Electron microscopy (EM) of β cells revealed that the number of mature insulin granules is decreased in HFD-treated BECN1F121A mice. By both EM and immunoisolation of islet autophagosomes, we discovered that in WT mice under fed conditions, insulin granules are not normally an autophagy cargo; however, in autophagy-hyperactive BECN1F121A mice, insulin granules are sequestered within autophagosomes or autolysosomes. We termed this process vesicophagy, referring to the autophagic degradation of secretory vesicles. Therefore, we propose a model in which different metabolic tissues require different levels of autophagy for optimal function: in insulin-responsive cells, constitutive autophagy activation by the BECN1F121A mutant is beneficial to maintain insulin sensitivity under HFD-treated conditions, whereas in insulin-secreting β cells, excess autophagy activation leads to degradation of insulin granules by vesicophagy, and decreased insulin secretion (Figure 1). Notably, among the metabolic tissues we have analyzed, including islets, liver, and muscle, islet β cells show the highest level of autophagy at both basal conditions and upon fasting induction. These data support the above model that in β cells autophagy is high at baseline levels, and it should be more tightly regulated.
Figure 1.
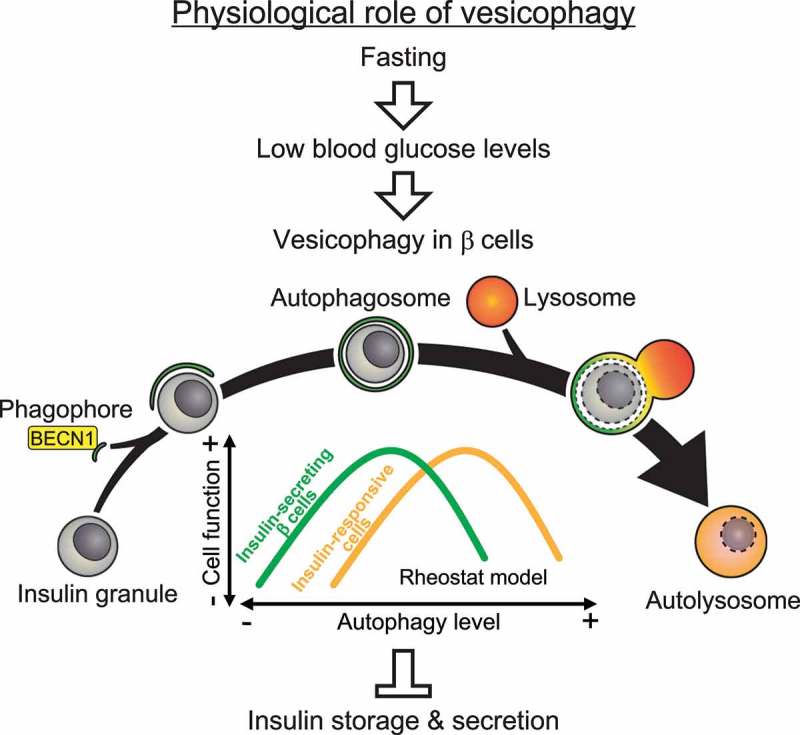
Proposed model and physiological function of autophagic degradation of insulin granules (vesicophagy). In pancreatic β cells, fasting (> 24 h) induces autophagy, which selectively engulfs and degrades unnecessary insulin granules under the hypoglycemia condition and prevents accidental insulin release. Conversely, in insulin-responsive tissues, activation of autophagy increases insulin sensitivity by reducing ER stress.
Nonetheless, we do not observe the above glucose metabolism phenotypes (exacerbated glucose intolerance) in BECN1F121A mice fed with RD; it is possible that under RD-feeding conditions, the systemic requirement for insulin is not as high, and insulin storage can be replenished in a timely manner. Conversely, although we have not performed islet transplantation studies, we do not expect that insulin sensitization in insulin-responsive tissues of BECN1F121A mice is a compensatory response to lowered insulin secretion, as under HFD-feeding conditions, pharmacologically reducing the insulin supply does not improve insulin sensitivity in animal models. Similarly in humans, type 1 diabetes (T1D) patients, who have decreased insulin secretion, are not naturally protected from T2D. In fact, many T1D patients are also insulin resistant (‘double diabetic’).
We propose that the vesicophagy process has physiological importance: fasting leads to hypoglycemia, where additional insulin release from islets would be detrimental; thus, autophagy simultaneously induced by fasting in β cells can reduce insulin load and leakage risks (Figure 1). It remains to be studied how vesciophagy is regulated. Our in vitro data suggest that ATG5 and ATG7, but not VMP1, may be involved. Our future work will focus on the molecular mechanisms governing vesicophagy in vitro and in vivo. Translationally, based on our results in BECN1F121A mice, we are testing whether periodic (such as 2-weeks on/2-weeks off) activation of autophagy is more metabolically beneficial than chronic activation in T2D mouse models, and whether it represents a new direction for T2D cure development. Finally, besides diabetes, we expect that the autophagy-hyperactive BECN1F121A mice will be a useful tool to study the role of autophagy in many different diseases and research directions.
Funding Statement
This work was supported by funding to C.H. from NIH/NIDDK (R01 DK113170), American Federation for Aging Research (AFAR), and Northwestern Cognitive Neurology and Alzheimer’s Disease Center (NIA AG13854).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Yamamoto S, Kuramoto K, Wang N, et al. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 2018. June 12;23(11):3286–3299. PMID: 29898399. [DOI] [PMC free article] [PubMed] [Google Scholar]