

ABSTRACT
Recent reports have made important revelations, uncovering direct regulation of DNA damage response (DDR)-associated proteins and chromatin ubiquitination (Ubn) by macroautophagy/autophagy. Here, we report a previously unexplored connection between autophagy and DDR, via a deubiquitnase (DUB), USP14. Loss of autophagy in prostate cancer cells led to unrepaired DNA double-strand breaks (DSBs) as indicated by persistent ionizing radiation (IR)-induced foci (IRIF) formation for γH2AFX, and decreased protein levels and IRIF formation for RNF168, an E3-ubiquitin ligase essential for chromatin Ubn and recruitment of critical DDR effector proteins in response to DSBs, including TP53BP1. Consistently, RNF168-associated Ubn signaling and TP53BP1 IRIF formation were reduced in autophagy-deficient cells. An activity assay identified several DUBs, including USP14, which showed higher activity in autophagy-deficient cells. Importantly, inhibiting USP14 could overcome DDR defects in autophagy-deficient cells. USP14 IRIF formation and protein stability were increased in autophagy-deficient cells. Co-immunoprecipitation and colocalization of USP14 with MAP1LC3B and the UBA-domain of SQSTM1 identified USP14 as a substrate of autophagy and SQSTM1. Additionally, USP14 directly interacted with RNF168, which depended on the MIU1 domain of RNF168. These findings identify USP14 as a novel substrate of autophagy and regulation of RNF168-dependent Ubn and TP53BP1 recruitment by USP14 as a critical link between DDR and autophagy. Given the role of Ubn signaling in non-homologous end joining (NHEJ), the major pathway for repair of IR-induced DNA damage, these findings provide unique insights into the link between autophagy, DDR-associated Ubn signaling and NHEJ DNA repair.
Abbreviations: ATG7: autophagy related 7; CQ: chloroquine; DDR: DNA damage response; DUB: deubiquitinase; HR: homologous recombination; IR: ionizing radiation; IRIF: ionizing radiation-induced foci; LAMP2: lysosomal associated membrane protein 2; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MIU1: motif interacting with ubiquitin; NHEJ: non homologous end-joining; PCa: prostate cancer; TP53BP1/53BP1: tumor protein p53 binding protein 1; RNF168: ring finger protein 168; SQSTM1/p62 sequestosome 1; γH2AFX/γH2AX: H2A histone family member X: phosphorylated, UBA: ubiquitin-associated; Ub: ubiquitin; Ubn: ubiquitination; USP14: ubiquitin specific peptidase 14.
KEYWORDS: Autophagy, deubiquitinase, DNA damage response, RNF168, USP14
Introduction
Ionizing radiation (IR) is an effective treatment modality in solid tumors, including prostate cancer (PCa). However, often tumors are, or become, resistant to treatment with IR. Therefore, research focus in the field has been to determine effective chemo- and radiotherapy combinations which are more effective in tumors with low cytotoxicity to normal tissue [1]. IR is a prototypical DNA-damaging agent and IR-induced DNA double-strand breaks (DSBs) are recognized by the DNA damage response (DDR) network of cellular pathways that sense, signal, and repair DNA lesions. DSB induction recruits the kinase ATM (ATM serine/threonine kinase), which phosphorylates Ser139 on histone H2AFX/H2AX (known as γH2AFX). γH2AFX, via MDC1 (mediator of DNA damage checkpoint 1), recruits the E3 ubiquitin ligases RNF8 (ring finger protein 8) and RNF168, which initiate a ubiquitination (Ubn) signaling cascade involving Ubn of H2A and γH2AFX. This Ubn signaling plays a crucial role in regulating the recruitment of downstream effectors of the DDR pathway, such as TP53BP1/53BP1 (tumor protein p53 binding protein 1) and the BRCA1 complex, and hence determine the cellular response to DNA damage through non-homologous end-joining (NHEJ) or homologous recombination (HR), respectively [2–4]. NHEJ is the major DSB repair pathway activated in response to IR for which TP53BP1 is critical as it inhibits BRCA1-RBBP8/CTIP (RB binding protein 8, endonuclease) complex-dependent DSB end-resection, and thus promotes the NHEJ repair pathway [5,6]. The process of ubiquitination is a very dynamic process and is counterbalanced by deubiquitinating (DUB) enzymes [7]. Several DUBs, including USP3 [8,9], BAP1 [10], USP26, USP37 [11], USP44 [8], USP34 [12], USP7 [13], OTUB1 [14], OTUB2 [15] and the BRISC complex-BRCC3/BRCC36 [16] have been reported to be involved in deubiquitination in the context of the DDR; when overexpressed, each of these DUBs can reverse various aspects of ubiquitin (Ub)-dependent protein assembly at DSB sites [17].
Autophagy is a cellular quality control as well as a stress adaptive pathway activated in response to most, if not all, chemotherapeutics as well as IR [18–21]. In many tumors, there is altered expression and/or subcellular localization of autophagy-regulatory proteins [22–26]. As apoptosis contributes to only 20% or less of radiation-induced cell death, there is a great deal of interest in understanding the role of autophagy in regulating IR-induced cell death [27,28]. However, those studies have yielded conflicting results. Activation of autophagy by various means such as MTOR complex 1 (MTORC1) inhibition [29,30], mitogen-activated protein kinase (MAPK) inhibition [31], or endoplasmic reticulum (ER) stress [32] have enhanced IR-induced cell death. By contrast in other cellular contexts, inhibition of autophagy enhances radiosenstivity of tumors [33,34]. Thus, a better understanding of the inter-relationship between autophagy and molecular pathways associated with radiosensitivity, such as DDR, is needed to identify molecular determinants of whether activation or inhibition of autophagy is the most effective treatment strategy in a given context.
The importance of autophagy in the DDR process has recently emerged. Lack of functional autophagy in tumor cells results in genomic instability, manifested by increased DNA damage, gene amplification, and aneuploidy [35–37]. However, molecular mechanisms that lead to direct cross-talk between the 2 pathways have been largely unclear. Recent studies have shown that autophagy directly regulates the levels of nuclear components [38–40], such as levels of the critical DDR-associated protein Sae2, the yeast homolog of RBBP8 [41], CHEK1/CHK1 (checkpoint kinase 1) [42], and CBX/HP1 [43]. In addition, SQSTM1/p62 (sequestome 1), a cargo receptor for degradation of ubiquitinated substrates by autophagy, mediates proteasomal degradation of FLNA (filamin A) and RAD51, critical regulators of HR [44]. Alternatively, SQSTM1 could directly bind and inhibit RNF168, and thus impair downstream DDR signaling [45]. These reports point to a potential role of autophagy in DNA repair by HR; however, there are no reports on how autophagy has an impact on NHEJ, the major DDR pathway for IR-induced DSBs.
Given the importance of the ubiquitination-deubiquitination equilibrium in maintaining effective DDR, and the well-established role of autophagy in regulating the clearance of ubiquitinated substrates, an important question that still remains unaddressed is whether deubiquitination is involved in autophagy-regulated DDR. In this study, we have examined the role of autophagy in regulation of the DDR in response to IR using PCa cell lines as a model system. We show that IR-induced autophagy facilitated DNA repair signaling. Inhibiting autophagy signaling led to inhibition of the DSB repair due to impaired DDR-associated Ub signaling. We identified USP14 as a highly active DUB in autophagy-deficient cells. USP14 underwent IRIF formation in response to DNA damage in the nucleus. It also colocalized and interacted with both MAP1LC3B/LC3B and SQSTM1 in the cytosol. USP14 protein half-life and IRIF formation were increased in autophagy-deficient cells, suggesting that USP14 is a substrate of autophagy that involves SQSTM1. USP14, in turn, negatively regulated levels and Ubn of RNF168 and suppressed RNF168-dependent Ub signaling. As a result, TP53BP1 recruitment to the sites of DNA DSBs was prevented in autophagy-deficient cells, leading to diminished DNA repair and increased cell death in response to IR. Our study identifies USP14 as a substrate of autophagy, and SQSTM1 as a nuclear protein critical for a novel molecular mechanism involving protein deubiquitination that connects 2 important cellular and genomic quality control pathways, autophagy and the DDR.
Results
Inhibition of autophagy leads to impaired DNA damage response in prostate cancer cells
IR treatment activates autophagy in many tumors, including PCa, as a cytoprotective mechanism in response to lethal DNA DSBs [46]. To study the crosstalk between autophagy and DDR signaling, we first examined induction of autophagy in PCa cell lines following IR. Autophagy was determined by MAP1LC3B–phosphatidylethanolamine conjugate (MAP1LC3B-II) puncta formation, considered as the gold standard marker for the induction of autophagy [47]. To compare steady state as well as flux inhibited levels of MAP1LC3B-II, the autophagy inhibitor chloroquine (CQ) was used to inhibit the autophagic flux [47]. To directly determine autophagic flux, GFP-mCherry-MAP1LC3B fusion protein-expressing C4-2 and PC3 stable cell lines were generated and MAP1LC3B-II puncta were visualized by confocal imaging. Autophagic flux was determined by counting autophagosomal (yellow) MAP1LC3B and autolysosomal (red) MAP1LC3B [47]. Autophagosomal MAP1LC3B levels were increased following IR in both cell lines followed by an increase in autolysosomal MAP1LC3B, suggesting that autophagy was induced as well as completed (Figure 1(a–d)). Upon addition of CQ, autophagosomal MAP1LC3B accumulated, suggesting a block in autolysosomal formation (Figure 1(a–d)). Moreover, immunostaining and confocal microscopy analysis of endogeneous MAP1LC3B showed that MAP1LC3B-II puncta significantly increased in both PC3 and C4-2 cells following IR (Fig. S1A, B).
Figure 1.
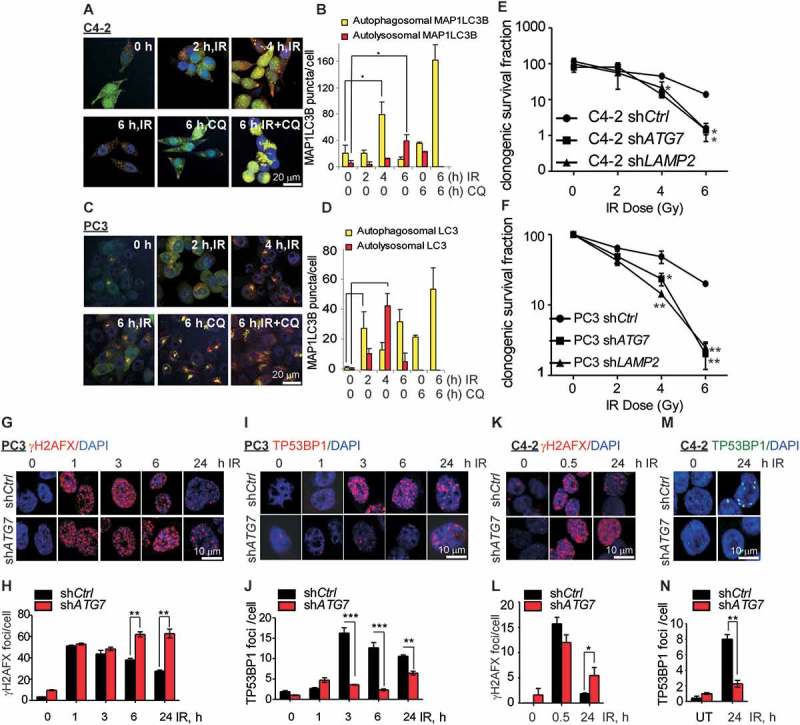
Inhibition of autophagy leads to an impaired DNA damage response in prostate cancer cells. (a and c) Representative confocal images of C4-2 and PC3 cells, respectively, stably expressing GFP-mCherry-MAP1LC3B at the indicated time following ionizing radiation (IR) ±chloroquine (CQ). (b and d) Quantification of autophagosomal (yellow) and autolysosomal (red) MAP1LC3B from the confocal images of C4-2 and PC3 cells stably expressing GFP-mCherry-MAP1LC3B at the indicated time following IR+/− CQ. (e and f). Clonogenic survival of C4-2 and PC3 cells stably expressing pLKO.1 vector control (shCtrl) or short hairpin RNA for ATG7 (shATG7), or LAMP2 (shLAMP2) following the indicated doses of IR. Representative confocal images and quantification of γH2AFX (g and h) and TP53BP1 (i and j) in PC3 cells stably expressing shCtrl or shATG7 at the indicated times following IR treatment. Nuclei were stained with DAPI. Representative confocal images and quantification of γH2AFX (k and l) and TP53BP1 (m and n) in C4-2 cells stably expressing shCtrl or shATG7 at the indicated times following IR treatment. Nuclei were stained with DAPI. Data shown are the means ± SEM (n = 3) P < 0.05 *. P < 0.01 **. P < 0.001***.
The effect of autophagy inhibition on cell survival in response to IR in PCa cell lines was investigated by CQ treatment and short-hairpin RNA (shRNA)-mediated genetic knockdown of ATG7 (autophagy related 7), an E1 Ub ligase-like protein involved in the MAP1LC3B lipidation step that is critical for facilitating autophagosome formation [48], or LAMP2 (lysosomal associated membrane protein 2), which mediates the fusion of autophagosomes with lysosomes to facilitate the autophagic flux [49]. Long-term survival in response to IR treatment, as measured by clonogenic survival, was reduced in C4-2 (Figure 1(e)) and PC3 (Figure 1(f)) cells expressing shATG7 compared to shCtrl cells. Furthermore, inhibition of autophagy by expression of shRNA against LAMP2 in C4-2 and PC3 cells, resulted in a similar decrease in long-term survival in response to IR (Figure 1(e, f)). Immunoblot analyses indicate efficient knockdown of ATG7 and LAMP2 in C4-2 and PC3 cells (Fig. S1E, F). Accumulation of SQSTM1 in shATG7-expressing C4-2 and PC3 (Fig. S1E), and shLAMP2-expressing PC3 cells (Fig. S1F) further indicated inhibition of autophagy in these cells. In addition, inhibition of autophagy using CQ in C4-2 (Fig. S1C, p < 0.01) and PC3 (Fig. S1D, p < 0.01) cells, and shATG7 expression in C4-2 cells (Fig. S1G, p < 0.05), resulted in significantly increased cell death in response to IR as measured by IncuCyte. These findings suggest that IR induces autophagy as a cytoprotective mechanism against IR-induced cell death.
Autophagy plays an important role in the maintenance of genomic stability by regulating the outcome of treatment with DNA damaging agents in cancer cells [20,50]. Therefore, we investigated the effect of autophagy inhibition on the DNA damage response (DDR). DDR signaling was determined by the formation of IR-induced foci (IRIF) for γH2AFX, a well-established DNA DSB marker [51], using immunostaining and confocal microscopy. The initial γH2AFX IRIF formation, i.e. at 1 h following IR treatment, was similar in shATG7 and shCtrl in both PC3 (Figure 1(g, h)) and C4-2 cells (Figure 1(k, l)), indicating that a similar extent of DNA damage was induced. Analyzing γH2AFX IRIF formation at various time points over a 24-h time period suggested that the early recruitment, i.e. at 1 and 3 h, of γH2AFX (Figure 1(g, h)) to the DSBs was similar among shCtrl- and shATG7-expressing PC3 cells. However, a significantly higher number of γH2AFX foci was unresolved at 6 h (p < 0.001) and 24 h (p < 0.001) following IR treatment in shATG7- compared to shCtrl-expressing PC3 cells (Figure 1(g, h)), suggesting an increase in unrepaired DNA DSBs in autophagy-deficient cells.
To study whether autophagy regulates the NHEJ repair pathway, we compared TP53BP1 IRIF formation in the presence and absence of functional autophagy. TP53BP1 was recruited as early as 3 h and was retained through the late time points, i.e. 6 and 24 h following IR treatment in shCtrl-expressing cells. However, in shATG7-expressing PC3 cells TP53BP1 IRIF formation was significantly reduced compared to shCtrl-expressing cells at all the time points examined (Figure 1(i, j), p < 0.01). These data suggest that the NHEJ DNA damage response is impaired upon inhibition of autophagy. Similarly, inhibition of autophagy by shATG7-expression or CQ pretreatment in C4-2 cells resulted in persistent γH2AFX (Figure 1(k, l) and S1H, I, respectively), and reduced TP53BP1 IRIF formation following IR (Figure 1(m, n) and S1J, K, respectively), suggesting that the DNA damage repair is impaired upon inhibition of autophagy. Together, these data suggest that the autophagy-proficient cells can efficiently repair the DNA damage. In contrast, autophagy-deficient cells are not able to repair the DNA damage induced by IR and hence are sensitized to IR-induced cell death.
USP14 disrupts DDR signaling in autophagy-deficient cells
Ubiquitination of histones H2A and H2AFX/H2AX in response to DNA DSBs is a critical post-translational modification that mediates downstream signaling leading to recruitment of DNA repair factors, including TP53BP1 [52]. Thus, ubiquitination and deubiquitination signaling dynamically regulates effective DDR [17]. A role of autophagy in regulating Ub signaling in DDR was recently shown by direct inhibition of the E3 ligase activity of RNF168 by SQSTM1 [45]. We have asked, therefore, whether a deubiquitinase (DUB) might be involved in regulating DDR in autophagy-deficient cells. To test this possibility, active DUBs were labeled with Ub vinyl sulfone in cellular extracts. This analysis identified several DUBs, including USP14 and USP5, that were more active in ATG7-deficient C4-2 cells (data not shown). Using a small-molecule DUB inhibitor, WP-1130, that inhibits both USP5 and USP14, we examined their possible involvement in DNA repair. WP-1130 pretreatment prevented persistent γH2AFX formation and restored TP53BP1 IRIF formation in ATG7-deficient cells (Fig. S2A, B). Gene-specific shRNA-mediated knockdown of USP5 and USP14 revealed the critical role of USP14 but not USP5 in regulating DDR. USP14 knockdown derivatives of ATG7-deficient cells resolved γH2AFX IRIFs (Figure 2(a, b)) and restored TP53BP1 (Figure 2(a, c)) IRIF formation by 24 h to a level similar to that of parental cells, indicative of decreased DNA damage. Immunoblot analyses indicated efficient knockdown of ATG7 and USP14 in C4-2 cells (Fig. S2C).
Figure 2.
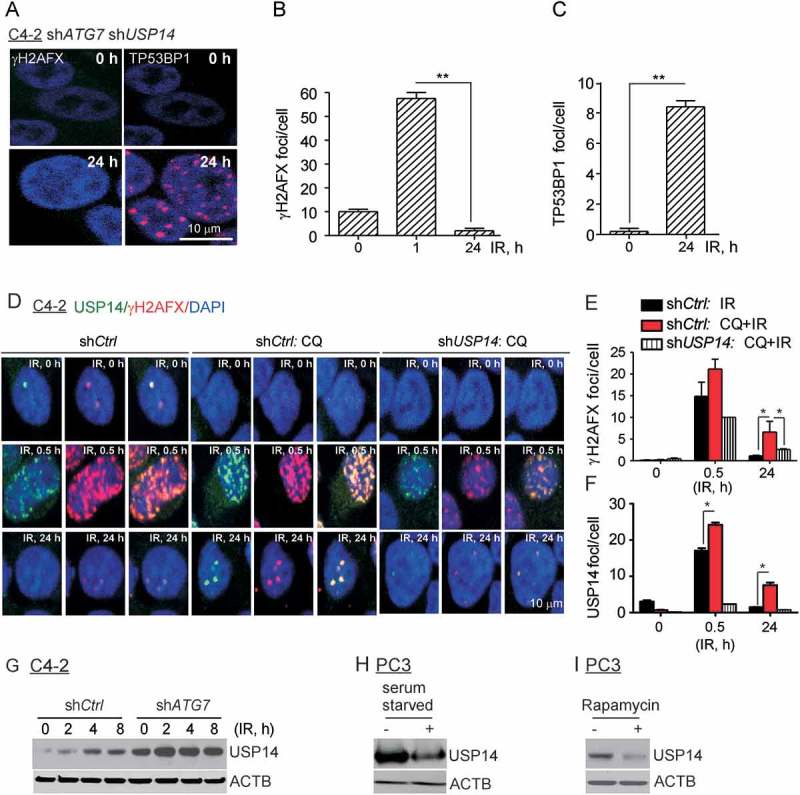
USP14 disrupts DDR signaling in autophagy-deficient cells. (a–c) Confocal immunostaining and graphical representation of γH2AFX and TP53BP1 foci following IR treatment in C4-2 cells co-expressing shATG7 and shUSP14. Nuclei were stained with DAPI. (d–f) Confocal immunostaining and graphical representation of γH2AFX and USP14 foci following IR+/− CQ treatment in C4-2 cells expressing shCtrl and shUSP14. Nuclei were stained with DAPI. Data shown are the means ± SEM (n = 2); P < 0.05 *, P < 0.01 **. (g) Western blot analysis for USP14 in shCtrl-expressing vs shATG7-expressing C4-2 cells following IR treatment for the indicated time. ACTB/β-actin was used as a loading control. Western blot analysis for USP14 in PC3 cells treated with (h) serum starvation and (i) rapamycin. ACTB/β-actin was used as a loading control.
Consistent with our findings in Figure 1, the γH2AFX foci in response to IR appeared as early as 1 h and were resolved by 24 h (Fig. S2D, E) in PC3 cells. In addition, TP53BP1 foci, essential for the NHEJ DDR pathway, were generated efficiently (Fig. S2F, G). By contrast, inhibition of autophagy using CQ pretreatment in PC3 cells led to impaired DDR, as indicated by persistent γH2AFX foci at 24 h (Fig. S2D, E), as well as the absence of TP53BP1 foci formation in these cells (Fig. S2F, G). Strikingly, inhibition of USP14 using a specific pharmacological inhibitor, IU1 [53], in PC3 cells pretreated with CQ resolved γH2AFX (Fig. S2D,E) and restored TP53BP1 IRIF to the level observed in autophagy-proficient cells (Fig. S2F, G). Overall, these findings indicate that USP14 negatively regulates DNA DSB repair in response to IR in autophagy-deficient cells.
Given the potential role of USP14 in regulating DDR signaling, we next investigated whether USP14 was recruited to DNA DSB sites by using co-immunostaining and confocal microscopy of γH2AFX and USP14 nuclear foci. Consistent with our findings (Figure 1), γH2AFX foci formation was significantly increased at 24 h following IR treatment in shCtrl-expressing C4-2 cells following pretreatment with CQ (p < 0.05), which were resolved in shUSP14-expressing cells (Figure 2(d, e)). Similar to γH2AFX (Figure 2(d, e)), USP14 foci formation was greatly increased at 0.5 h following IR treatment and was resolved by 24 h in shCtrl-expressing C4-2 cells (Figure 2(d, f)). Colocalization of USP14 with γH2AFX foci indicated recruitment of USP14 to DSB sites (Figure 2(d)). Importantly, USP14 IRIF formation was significantly increased in shCtrl-expressing C4-2 cells following pretreatment with CQ (Figure 2(d, f), p < 0.05). The signal for USP14 was specific because it was greatly reduced in shUSP14-expressing cells (Figure 2(d, f)).
We next investigated whether IR and/or autophagy regulated the total protein level of USP14. Interestingly, USP14 levels were increased in response to IR, and further augmented upon inhibition of autophagy by shATG7 expression (Figure 2(g)). In addition, serum starvation (Figure 2(h)) and rapamycin treatment (Figure 2(i)) decreased USP14 levels, further indicating that autophagy regulates USP14 levels. Overall, these findings identify USP14 as a novel negative regulator of DDR signaling in response to DSBs that is suppressed in autophagy-proficient cells. Inhibition of autophagy upregulates USP14, which, in turn, impairs DSB repair.
SQSTM1 directly interacts with and regulates the levels of USP14
Inhibition of autophagy increased USP14 IRIF formation and protein levels in response to IR. Therefore, we next tested the possibility of whether USP14 is a substrate of autophagy by examining whether it directly interacts with key components of the autophagic machinery. Initially, we assessed whether USP14 interacts with the Ub-associated protein SQSTM1. Immunoprecipitation of USP14 using anti-flag antibody from 293T cells co-expressing Flag-HA-USP14 and EGFP-SQSTM1 indicated a direct interaction between SQSTM1 and USP14 (Figure 3(a, b)). To define the role of SQSTM1 in regulating USP14, we further investigated the interaction between USP14 and various mutant forms of SQSTM1 (Figure 3(a, b)). Interestingly, deletion or mutation in the UBA domain of SQSTM1 completely inhibited interaction between USP14 and SQSTM1 (Figure 3(b)). Further, colocalization analysis using immunofluorecence staining of endogenous proteins showed that SQSTM1 and USP14 colocalized in both C4-2 (Figure 3(c, d)) and PC3 cells (Fig. S3C, D). As has been reported for other DDR signaling proteins, including γH2AFX and TP53BP1, while a fraction of USP14 foci contained SQSTM1 [44], the majority of colocalization was observed in the cytoplasm (Figure 3(a) and S3C, D).
Figure 3.
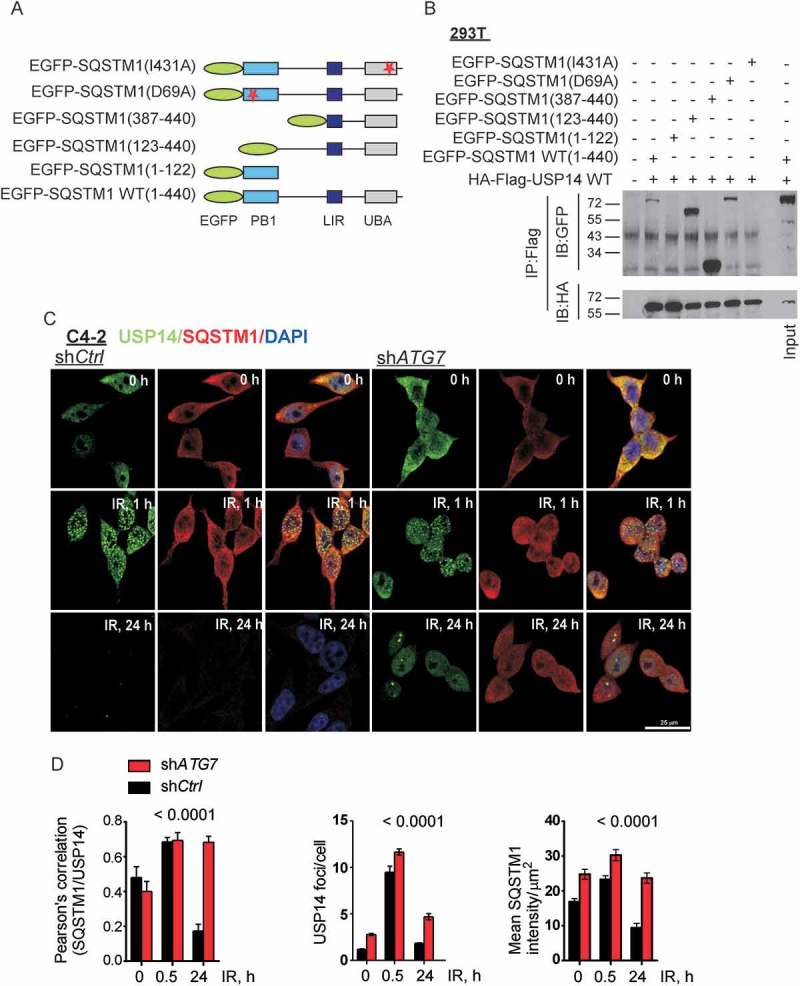
SQSTM1/p62 directly interacts with and regulates the levels of USP14. (a) Schematic of SQSTM1 mutant constructs. (b) 293T cells were transfected with the indicated constructs, followed by immunoprecipitation as indicated and immunoblotting using the anti-GFP and -HA antibodies. The corresponding whole cell lysates (WCL) were used as input controls and probed with the indicated antibodies. (c and d) Representative confocal images and quantification of SQSTM1 and USP14 levels and colocalization in C4-2 cells stably expressing shCtrl or shATG7 at the indicated time following IR. Nuclei were stained with DAPI. Data shown are the means ± SEM (n = 2); P < 0.0001****.
Interestingly, in response to IR treatment, the USP14-SQSTM1 colocalization decreased in a time-dependent manner in autophagy proficient shCtrl-expressing C4-2 and PC3 cells. However, in autophagy-deficient cells, i.e. shATG7-expressing C4-2 cells and shLAMP2-expressing PC3 cells colocalization was maintained and was significantly higher than in autophagy-proficient cells (Figure 3(c, d), p < 0.0001; S3C, D, p < 0.05, respectively). These data suggest that SQSTM1 interacts with and regulates USP14 degradation.
Because nuclear SQSTM1 has been shown to directly interact with RNF168 and inhibit DDR in autophagy-deficient cells [45], we investigated whether accumulation of SQSTM1 in the nucleus upon inhibition of autophagy led to an increase in RNF168-SQSTM1 interaction by immunoprecipitating endogenous RNF168, followed by immunoblotting for SQSTM1 (Fig. S3A). We did not observe interaction between SQSTM1 and RNF168 in our autophagy-deficient PCa cells. Moreover, while SQSTM1 was abundant in the cytoplasm, it was barely detectable in the nuclear fraction of C4-2 cells under all conditions tested (Fig. S3B). Thus, USP14-dependent regulation of RNF168 may be the major mechanism to keep RNF168 under check, in order to prevent genomic imbalance resulting from mutagenic NHEJ.
USP14 is an autophagy substrate
Next, we investigated whether USP14 is a direct substrate of autophagy. Colocalization analysis using immunofluorescence staining showed that MAP1LC3B and USP14 colocalized in punctate structures in the cytoplasm of both C4-2 (Figure 4(a, c)) and PC3 (Fig. S4A, B) cells following IR treatment. Importantly, the MAP1LC3B-USP14 colocalization was significantly higher in autophagy-deficient, CQ-pretreated C4-2 (Figure 4(a, c), p < 0.01) and shLAMP2-expressing PC3 (Fig. S4A, B, p < 0.05) cells than in autophagy-proficient cells. SQSTM1 knockdown using siRNA significantly decreased USP14-MAP1LC3B colocalization following IR treatment in C4-2 cells pretreated with CQ (Figure 4(a, c), p < 0.001) and IR-treated shLAMP2-expressing PC3 cells (Fig. S4A, B, p < 0.05). In addition, the levels of USP14 were reduced following CQ and IR treatment in siSQSTM1-expressing compared to siCtrl-expressing C4-2 cells (Figure 4(a, b), p < 0.001). These data further substantiate the role of SQSTM1 and autophagy in the degradation of USP14.
Figure 4.
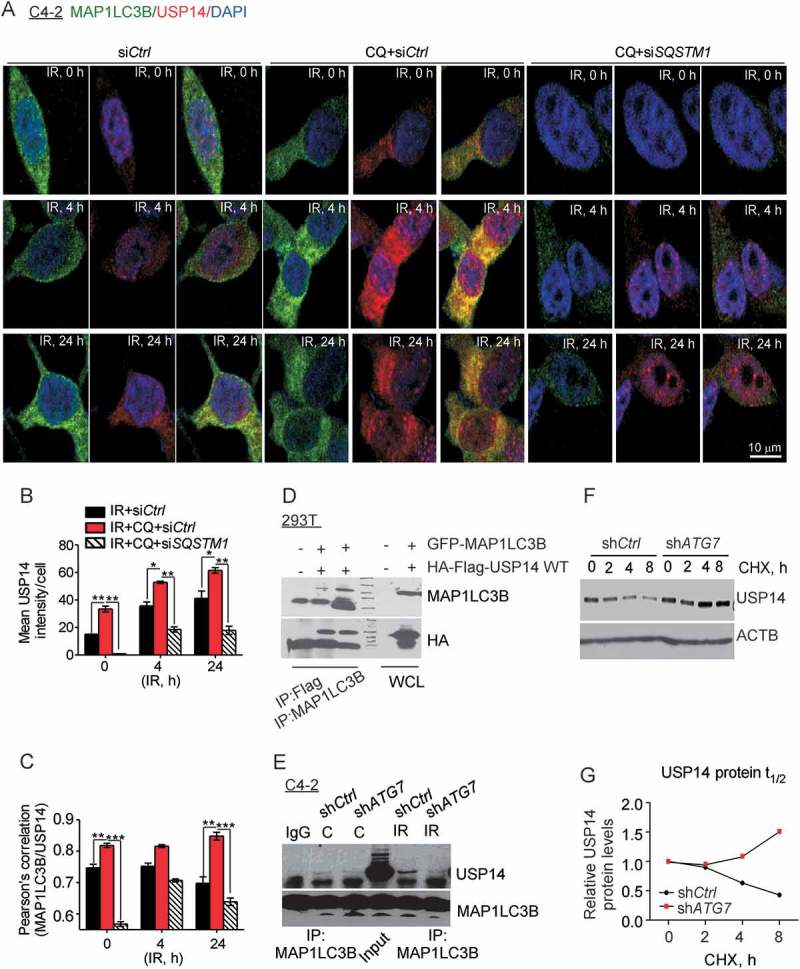
USP14 is a substrate of autophagy. (a–c) Representative confocal images and quantification of MAP1LC3B and USP14 levels and colocalization in C4-2 cells treated as indicated. Nuclei were stained with DAPI. (d) 293T cells were transfected with the indicated constructs, followed by immunoprecipitation as indicated and immunoblotting using the anti-MAP1LC3B and -HA antibodies. The corresponding WCLs were used as input controls and probed with the indicated antibodies. (e) MAP1LC3B was immunoprecipitated from C4-2 cells stably expressing shCtrl or shATG7, treated as indicated, followed by western blotting using anti-USP14 and -MAP1LC3B antibodies. (f) Western blot analysis and (g) relative protein quantification for USP14 in shCtrl vs shATG7-expressing C4-2 cells following cyclohexamide (CHX) treatment for the indicated time. ACTB/β-actin was used as a loading control. Data shown are the means ± SEM (n = 2); P < 0.05 *, P < 0.01 **, P < 0.001***.
Immunoprecipitation of USP14 or MAP1LC3B from 293T cells co-expressing Flag-HA-USP14 and GFP-MAP1LC3B indicated direct interaction between MAP1LC3B and USP14 (Figure 4(d)). Consistently, we found a direct interaction between endogenous USP14 and MAP1LC3B-II following IR treatment in shCtrl- but not shATG7-expressing C4-2 cells (Figure 4(e)). Immunoblot analysis, following a time course of cyclohexamide chase treatment to block protein translation, revealed that the half-life of USP14 protein was increased considerably, in shATG7-expressing C4-2 cells compared to that in shCtrl-expressing cells (Figure 4(f, g)). These data establish the role of autophagy in regulating cellular levels of USP14.
USP14 negatively regulates RNF168-dependent ubiquitination signaling
Next, we investigated the mechanism by which USP14 inhibited TP53BP1 IRIF formation in autophagy-deficient cells. The recruitment of the E3 Ub-ligases RNF8 and RNF168 to the DSBs to catalyze ubiquitination of histones H2A and H2AFX/H2AX at K63-linked chains on K13 and K15 is an essential upstream event for the recruitment of DNA repair factors, including TP53BP1 [2,3,54]. Thus, we examined how inhibition of autophagy affects RNF168 recruitment in response to IR. Similar to TP53BP1, there was a significant decrease in the number of RNF168 foci at various time points following IR treatment in shATG7-expressing compared to shCtrl-expressing PC3 (Figure 5(a, b), p < 0.01) and C4-2 cells (Figure 5(c, d), p < 0.001). However, we did not observe any difference in IR-induced RNF8 foci formation in shATG7-expressing compared to shCtrl-expressing C4-2 cells (Fig. S5A, B). USP14 knockdown in ATG7-deficient cells restored RNF168 IRIF formation (Figure 5(c, d)). Importantly, diminished RNF168 IRIF formation in PC3 cells upon autophagy inhibition by pretreatment with CQ could be restored using IU1 treatment (Fig. S5C, D).
Figure 5.
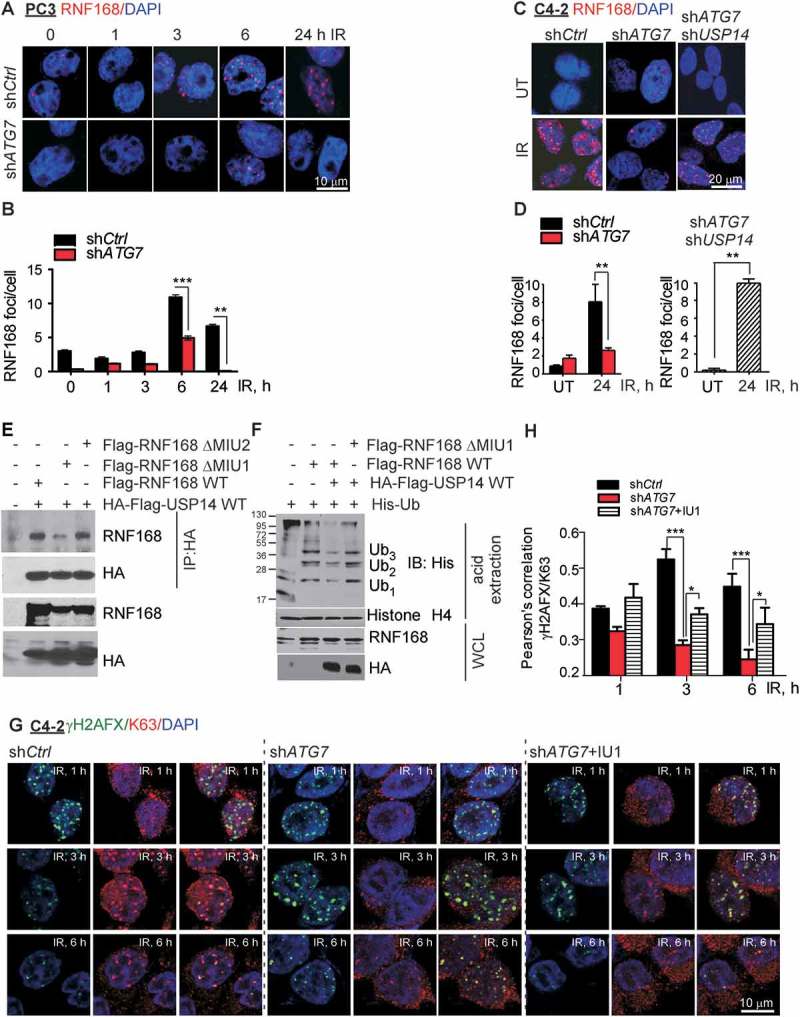
USP14 negatively regulates RNF168-dependent ubiquitination signaling. (a and b) Representative confocal images and quantification of RNF168 foci in PC3 cells stably expressing shCtrl or shATG7 at the indicated times following IR treatment. Nuclei were stained with DAPI. (c and d) Confocal immunostaining and graphical representation of RNF168 foci following IR treatment in C4-2 cells expressing shCtrl and shATG7, and co-expressing shATG7 and shUSP14. Nuclei were stained with DAPI. (e) 293T cells were transfected with the indicated constructs, followed by immunoprecipitation using anti-HA antibody and immunoblotting using the anti-RNF168 and -HA antibodies. The corresponding WCLs were used as input controls and probed with the indicated antibodies. (f) 293T cells were transfected with the indicated constructs, followed by acid extraction of histones and immunoblotting using the indicated antibodies. The corresponding WCLs were used as input controls and probed with the indicated antibodies. (g and h) Representative confocal images and quantification of γH2AFX/K63 colocalization in C4-2 cells expressing shCtrl- or shATG7 or shATG7 +/− IU1 treatment at the indicated times following IR treatment. Nuclei were stained with DAPI. Data shown are the means ± SEM (n = 2); P < 0.05 *, P < 0.01 **, P < 0.001***.
To study the mechanism by which USP14 regulates DDR in autophagy-deficient cells, we hypothesized that USP14 directly regulates Ubn of one or more DDR signaling proteins. We asked whether RNF168 is a direct substrate for deubiquitination by USP14. We examined the interaction between HA-Flag-USP14 and Flag-WT-RNF168 following transient transfection in 293T cells. As shown in Figure 5(e), RNF168 directly interacted with USP14. Further, RNF168 recognizes RNF8-dependent ubiquitination and other protein partners through its MIU motifs [2,45]. Therefore, to investigate whether USP14 interacted with RNF168 through one of the MIU motifs, we co-expressed HA-Flag-USP14 with Flag-RNF168ΔMIU1, or Flag-RNF168ΔMIU2 in 293T cells, followed by immunoprecipitation of HA-Flag-USP14 and immunoblotting for RNF168. The MIU1 domain mutant construct of RNF168 showed greatly decreased interaction with USP14 (Figure 5(e)), suggesting that the MIU1 domain is required for the binding of RNF168 to USP14. As RNF168 catalyzes DNA-damage induced Ubn of chromatin [2,45,52], we investigated whether USP14 regulates RNF168-dependent Ub-signaling by measuring Ubn of acid-extracted histones. Expression of RNF168 resulted in chromatin Ubn (Figure 5(f)). However, co-expression of Flag-HA-USP14 and Flag-RNF168 resulted in reduced ubiquitination of acid-extracted histones (Figure 5(f)) compared to that in cells overexpressing RNF168 alone. Importantly, co-expression of USP14 with mutant MIU1-RNF168, which failed to interact with USP14, prevented USP14 from inhibiting histone Ubn (Figure 5(f)). Consistently, we observed significantly reduced γH2AFX-Ub in shATG7-expressing compared to shCtrl- expressing C4-2 cells (Figure 5(g, h), p < 0.001), as determined by colocalization analysis of γH2AFX and K63 by immunostaining and confocal microscopy. Importantly, IU1 treatment significantly restored γH2AFX-Ub in shATG7-expressing cells (Figure 5G, H, p < 0.05). These data indicate that USP14 interacts directly with RNF168 and inhibits RNF168-dependent Ub signaling.
USP14 regulates the levels of RNF168 by modulating RNF168 ubiquitination
To elucidate the mechanism by which USP14 regulates RNF168 function, we investigated Ubn of RNF168 by immunoprecipitation of RNF168 from lysates of 293T cells co-transfected with plasmids expressing Flag-RNF168 WT and His-Ub, with or without Flag-HA-USP14, followed by western blotting using anti-His antibody. We observed that Ubn of RNF168 was decreased in the presence of USP14 (Figure 6(a, b)). Similarly, we found that the IR-induced Ubn of endogenous RNF168, as determined by immunoprecipitation of RNF168 and western blotting for FK2, which identifies mono and poly-Ub [54], was reduced upon inhibition of autophagy using CQ pretreatment in both PC3 (Fig. S5A) and C4-2 (Figure 6(c)) cells. Consistently, co-immunostaining and colocalization analysis of RNF168 and FK2 nuclear foci, to study Ubn of RNF168, gave similar results. In shCtrl-C4-2 cells, pretreatment with CQ significantly reduced IR-induced RNF168-containing FK2 foci (Fig. S5F, G, p < 0.01). Importantly, Ub of RNF168 in autophagy-deficient conditions was significantly restored in shUSP14-expressing C4-2 cells (Figure 6(c) and S5F, G, p < 0.05).
Figure 6.
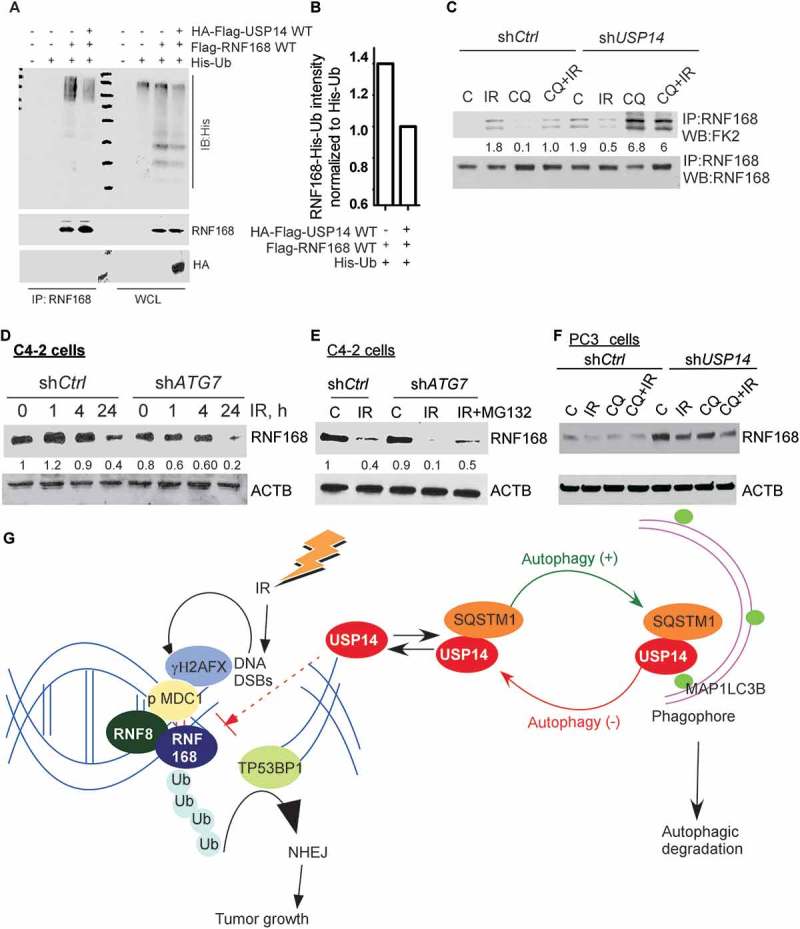
USP14 regulates the levels of RNF168 by modulating RNF168 ubiquitination. (a) 293T cells were transfected with the indicated constructs, followed by immunoprecipitation of RNF168 and immunoblotting using the indicated antibodies. The corresponding WCL were used as input controls. (b) Quantitative representation of RNF168-Ub determined in A, in the presence or absence of USP14, normalized to His-Ub levels. (c) Ub of RNF168 determined by immunoprecipitation of RNF168 in shCtrl or shUSP14 expressing C4-2 and following IR +/− CQ followed by western blotting using anti-FK2 antibody. The numbers below the blots correspond to band signal intensities compared to the untreated control in shCtrl cells. (d and e) Western blot analysis for RNF168 following the indicated treatments in C4-2 cells stably expressing shCtrl or shATG7, and (f) PC3 cells stably expressing shCtrl or shUSP14. ACTB/β-actin was used as the loading control. (g) Model for role and regulation of USP14 in autophagy-dependent DNA damage response. IR induced DNA DSBs are marked by γH2AFX followed by MDC1, which in turn recruits the RNF8-RNF168 E3 ligases to ubiquitinate (Ub) histones, leading to TP53BP1 recruitment and hence the NHEJ repair pathway that promotes tumor growth. USP14 antagonizes Ub-signaling in DDR by deubiquitinating RNF168 and inhibiting TP53BP1 recruitment. In addition, USP14 directly interacts with SQSTM1 and is targeted for autophagic degradation under normal conditions. Thus, USP14 levels are kept under check by autophagy. When autophagy is inhibited, USP14 is sequestered in SQSTM1 aggregates, and hence there is an increase in USP14 total protein levels and its recruitment to DSB sites. This, in turn, impairs DDR and, therefore, autophagy inhibition sensitizes PCa cells to IR by inhibiting DNA repair that is rescued by USP14 inhibition or depletion.
Deubiquitination regulates the fate and function of Ub-conjugated proteins. To determine the functional consequence of the modified Ubn state of RNF168, we investigated RNF168 protein turnover in response to IR and/or disruption of autophagy and/or USP14. Interestingly, in autophagy-proficient C4-2 cells, RNF168 levels first increased at 1 h, and then decreased below basal levels at 24 h following IR treatment (Figure 6(d)). However, in shATG7-expressing C4-2 cells no increase in RNF168 levels following IR treatment was observed (Figure 6(d)). In addition, the levels of RNF168 following IR were reduced in shATG7-expressing compared to shCtrl-expressing cells at any time point (Figure 6(d)). The levels of RNF168 following IR in shATG7-expressing cells could be restored up to those in shCtrl-expressing cells by MG132 treatment, suggesting an increased proteasomal turnover of RNF168 in shATG7-expressing cells following IR treatment (Figure 6(e)). Importantly, shUSP14-expressing PC3 cells had greatly increased RNF168 levels compared to shCtrl-expressing cells where RNF168 was barely detectable (Figure 6(f)).
Overall, these findings establish USP14 as an autophagy substrate. Inhibition of autophagy led to increased levels and DSB recruitment of USP14. USP14 antagonized RNF168-dependent Ub signaling and downstream TP53BP1 recruitment (Figure 6(g)). Thus, autophagy inhibition results in inhibition of DNA damage repair signaling following IR treatment and sensitizes PCa cells to IR by inhibiting DNA repair, which was effectively rescued by USP14 inhibition or depletion.
Discussion
Here, we identify USP14 as a novel autophagy substrate, and USP14-dependent regulation of RNF168 as a critical link between autophagy and DDR. We found that USP14 interacts directly with both MAP1LC3B and SQSTM1. We show that loss of autophagy results in increased protein levels of and IRIF formation by USP14 in response to IR. In addition, USP14 negatively regulates RNF168-dependent Ub signaling. USP14 directly interacts with RNF168 and regulates its protein levels, which leads to decreased levels and IRIF formation by RNF168. As a result, DDR-associated Ub-signaling is decreased, and TP53BP1 cannot be recruited to the sites of DNA DSBs in autophagy-deficient cells. Autophagy inhibition increases USP14 levels which, in turn, leads to diminished DSB repair and increased radiosensitization. USP14 inhibition or knockdown restored RNF168 levels and downstream DDR signaling in autophagy-deficient cells.
Substantial evidence exists to support the importance of autophagy in maintaining genomic stability either through regulation of ROS [35–37] or degradation of nuclear components [38–40]. More recently, autophagy deficiency was shown to directly impair DDR signaling [41,42,44,45]. Consistently, our data suggest that functional autophagy promotes cell survival and efficient repair of DSBs in response to IR. Inhibition of autophagy led to: (i) persistent γH2AFX foci, suggesting unrepaired DSBs, and (ii) inhibition of TP53BP1 foci, indicating impaired NHEJ DDR, due to (iii) defective recruitment of RNF168 in response to IR. Homologous recombination (HR) and NHEJ are the 2 major DSB repair pathways, known to compensate for each other when one is dysfunctional [4]. The increased cell death, diminished clonogenic survival, and persistent DSBs upon inhibition of autophagy in response to IR, suggest a lack of compensatory HR. Moreover, impaired RNF168 recruitment inhibits both NHEJ and HR [2,52,55]. Thus, we hypothesize that perhaps both pathways are impaired in this case, which will be investigated in our future studies. While the previous studies have shown that inhibition of autophagy affects HR [42,44,45], ours is the first report to show inhibition of NHEJ in the context of autophagy deficiency.
Our data identify USP14 as a critical negative regulator of DSB DDR signaling upon loss of autophagy. We show that inhibition of USP14 was necessary and sufficient to overcome suppression of DDR signaling in autophagy-deficient PCa cells. Ub conjugation events promoted by the E3 ligases RNF8 and RNF168 play a crucial role in recruiting downstream effectors of the DDR pathway [2–4]. By virtue of their activity as Ub-isopeptidases, DUB enzymes are prime candidates for negative regulation of DSB-induced ubiquitination [17]. Thus, our study uncovers an important missing link, i.e. a DUB in autophagy-regulated DDR-associated chromatin Ub signaling.
USP14 colocalized and interacted with MAP1LC3B and SQSTM1. Inhibition of autophagy flux by CQ or shLAMP2 expression led to an increase in USP14 protein levels, IRIF formation, and colocalization with MAP1LC3B and SQSTM1. These data clearly indicate that USP14 is a substrate of autophagy. Further, we found that USP14 interacted with the UBA domain of SQSTM1. SQSTM1 interacts with Ub substrates through its UBA domain and delivers them to either MAP1LC3B for autophagic degradation through interaction between the LC3-interacting region domain of SQSTM1 and MAP1LC3B, and/or sequesters them into SQSTM1-aggregates due to its ability to oligomerize using its PB1 domain [56,57]. In addition, knockdown of SQSTM1 in autophagy-deficient cells decreased USP14 levels. Thus, our data suggest that USP14 is an autophagy and SQSTM1 substrate. An important question that emerges now is the physiological relevance of USP14 regulation by autophagy. USP14 is a major proteasome-associated DUB that plays an important role in the maintenance of Ub homeostasis, and regulation of proteasomal activity [7,58–61]. Cellular Ub stress upregulates overall, and proteasomal-associated USP14 levels in mammals and yeast [62]. At the same time, proteasome function is essential to maintain free Ub pools to assure a functional response to DDR and other cellular stresses [63,64]. Moreover, accumulation of SQSTM1 upon inhibition of autophagy impairs Ub-proteasome system activity due to its ability to oligomerize and sequester ubiquitinated proteins, leading to reduced delivery of ubiquitinated substrates to the proteasome [65]. Thus, one possibility points to a broader role of autophagy in maintaining protein quality control and Ub homeostasis by regulating proteasomal activity. In autophagy-proficient cells, SQSTM1 and USP14 are kept under check so that proteasomes function optimally. Inhibition of autophagy will reduce proteasomal activity by multiple mechanisms, including hampered delivery of Ub substrates due to: (i) sequestration in SQSTM1-Ub aggregates, and (ii) USP14 upregulation, leading to cellular Ub stress. How autophagy regulates proteasomal function and Ub-homeostasis in a USP14-dependent manner will be an interesting question for future studies. Recently, USP14 was found to directly interact with and inhibit the critical autophagy protein BECN1 in a proteasomal-independent manner [66]. Thus, whether regulation of USP14 by autophagy affects proteasomal-independent functions of USP14, and how USP14 and autophagy negatively regulate each other will be of interest. It may be noted that IR treatment simultaneously induces both autophagic-flux (Figure 1) and upregulates USP14 in autophagy-proficient cells (Figure 2(d, f, g)). This suggests that there may be multiple mechanisms, in addition to autophagy, of regulating USP14 protein levels in response to IR. Nevertheless, our studies identify autophagy as a critical regulator of USP14 protein levels.
We found that USP14 directly targets RNF168-dependent Ub signaling and regulates RNF168 protein expression and IRIF formation. Consistently, levels of USP14 were upregulated and those of RNF168 were downregulated upon inhibition of autophagy. We propose that the decreased levels of RNF168 in autophagy-deficient cells in response IR are due to an increase in its proteasomal degradation. Indeed, in IR-treated autophagy-deficient cells, the levels of RNF168 were restored to that of IR-treated autophagy-proficient cells by treatment with the proteasomal inhibitor MG132. In addition to maintenance of Ub homeostasis, USP14 is a major regulator of protein degradation by the proteasome, and has the unusual capacity to both activate and inhibit multiple steps in substrate degradation [61]. Thus, USP14 depletion both increases and decreases substrate protein levels [59,60,67]. USP14 directly interacted with RNF168, and protein levels of RNF168 and its Ubn were increased in shUSP14-expressing cells. Whereas USP14 is a critical regulator of RNF168, we cannot rule out the possibility that USP14 and RNF168 are part of a larger complex that involves yet other unidentified protein partners, which, in turn, can regulate Ubn and chromatin recruitment of RNF168 and/or TP53BP1.
Autophagy deficiency inhibits RNF168-mediated chromatin Ub signaling through accumulation of nuclear SQSTM1 [45]. This may be explained due to different cellular contexts in the 2 studies. Indeed, we and others have reported that SQSTM1 is cytoplasmic in more advanced PCa [22,23]. Indeed, whereas SQSTM1 was abundant in the cytoplasm, under all conditions we tested it was barely detectable in the nuclear fraction of C4-2 cells, which are derivatives of metastatic PCa. Thus, in cancer cells that lack nuclear SQSTM1, USP14- dependent regulation of RNF168 may be the major mechanism to keep RNF168 under check, in order to prevent genomic imbalance resulting from mutagenic NHEJ. It will be interesting to investigate whether nuclear SQSTM1-dependent RNF168 regulation is an alternative mechanism in cells lacking nuclear USP14.
In summary, our studies identify a previously unexplored connection between USP14 and 2 critical pathways of tumor cell response to radiotherapy, DDR, and autophagy. Increased expression of USP14 negatively regulated DDR signaling and promoted radiosensitization in autophagy-deficient cells. Conversely, disruption of USP14 in autophagy-deficient cells restored DDR signaling and radiation resistance. These findings have important therapeutic implications. First, since autophagy signaling is elevated in high-grade PCa [22,26], autophagy inhibitors can serve as potential therapeutic agents in sensitizing cancer cells to radiotherapy. Second, it is tempting to propose that USP14 expression levels can predict radiosensitivity in the clinic. USP14 has been reported to be a potential oncogene; however, its role in the DDR has not been reported. Although the success of autophagy inhibitors in clinical trials has been so far limited, understanding the cross-talk between autophagy and key signaling pathways that regulate radiosensitization, such as DDR signaling, holds the promise to reveal druggable targets and potential biomarkers.
Materials and methods
Reagents and plasmids
The following reagents were purchased: puromycin (Sigma-Aldrich, P7255), polybrene (Santa Cruz Biotechnology, sc-134220), Lipofectamine 2000 (Thermo Fisher Scientific, 11668-019), Fugene 6 (Promega, E2691), vectashield with DAPI (Vector laboratories, H-1500), chloroquine diphosphate (Sigma-Aldrich, C6628), rapamycin (Cell Signaling Technology, 9904), protease inhibitor cocktail tablets (Millipore Sigma, 11836153001), phosphatase inhibitor cocktail 2 (Millipore Sigma, P5726), phosphatase inhibitor cocktail 3 (Millipore Sigma, P0044), Bio-Rad Protein Assay Dye Reagent (Bio-Rad, 5000006), protein G dynabeads (Thermo Fisher Scientific, 10004D), ON-TARGET plus SQSTM1 siRNA (Dharmacon, L-010230-00-0005), ON-TARGET plus Non-targeting Control siRNA (Dharmacon, D-001810-01-05), USP14 inhibitor IU1 (Millipore Sigma, 662210), MG132 (Millipore Sigma, 474791), cyclohexamide (Millipore Sigma, 239765), N-ethylmaleimide/NEM (Sigma-Aldrich, 128-53-0), normal rabbit IgG (Santa Cruz Biotechnology, sc-2027), normal mouse IgG (Santa Cruz Biotechnology, sc-2025), Sytox Green (Thermo Fisher Scientific, S7020) and Syto 59 Red dyes (Thermo Fisher Scientific, S11341).
The GFP-mCherry-MAP1LC3B and pCL10 plasmids were a kind gift from Dr. Jayanta Debnath (University of California, San Francisco, USA). Flag-WT-RNF168, Flag-A179G-RNF168 (Flag-RNF168ΔMIU1), or Flag-A450G-RNF168 (Flag-RNF168ΔMIU2) plasmids were a kind gift from Dr. Daniel Durocher (Lunenfeld-Tanenbaum Research Institute, Toronto, Canada). EGFP-WT-SQSTM1, EGFP-1-122-SQSTM1, EGFP-123-387-SQSTM1, EGFP-387-440-SQSTM1, EGFP-D69A-SQSTM1, EGFP-I431A-SQSTM1, plasmids were a kind gift from Dr. Terje Johansen (Institute of Medical Biology, University of Tromsø, Norway), Flag-HA-USP14 was from Addgene (22569, deposited by Wade Harper). The following shRNA-expressing lentiviral plasmids were made in pLKO.1-puro and purchased from Sigma-Aldrich, with the clone numbers indicated: ATG7 (TRCN0000092164), LAMP2 (TRCN0000029262), USP14 (TRCN0000007426). The lentiviral packaging plasmids pCMV-VSV-G (Sigma-Aldrich, pMISSIONvsvg) and pCMV-GAP-POL (Sigma-Aldrich, pMISSIONgagpol) were from Invitrogen.
The primary antibodies used in this study were: γ-H2AFX (Millipore Sigma, 05-636), TP53BP1/53BP1 (Novus Biologicals, NBP2-54753), USP14 for immunostaining (Proteintech, 14517-1-AP), USP14 for immublotting (Cell Signaling Technology, 11931), ACTB/β-actin (Sigma Aldrich, A2228), FLAG clone M2 (Sigma Aldrich, F1804), HA clone 7 (Sigma Aldrich, H3663), GFP clone B2 (Santa Cruz Biotechnology, sc-9996), SQSTM1/p62 clone A6 (Santa Cruz Biotechnology, sc-48402), SQSTM1/p62 (Fitzgerald Industries International, 20R-PP001), MAP1LC3B for co-immunostaining with USP14 (MBL International, M152-3), MAP1LC3B for immunoprecipitation and immunoblotting (Cell Signaling Technology, 3868), RNF168 for immunostaining (Millipore Sigma, ABE-367 or Novus Biologicals, H00165918-M01), RNF168 for immunoblotting (Millipore Sigma, ABE-367), RNF168 for immunoprecipitation (Santa Cruz Biotechnology, sc-101125), His (Thermo Fisher Scientific, MA1-135), histone H4 (Cell Signaling Technology, 13919), ubiquitin, Lys63-specific, clone Apu3 (Millipore Sigma, 05-1308), mono- and polyubiquitinated conjugates, clone FK2 (Enzo, BML-PW8810-0100), ATG7 (Cell Signaling Technology, 8558), LAMP2 (Santa Cruz Biotechnology, sc-18822), and RNF8 (Santa Cruz Biotechnology, sc- 271462). Secondary antibodies were against mouse HRP (Jackson Immuno Research Laboratories, Inc., 115-035-174) and rabbit HRP (Jackson ImmunoResearch Laboratories, Inc., 211-032-171), guinea pig HRP (Santa Cruz Biotechnology, sc- 2903), Alexa Fluor 488 rabbit (Thermo Fisher Scientific, A-11034), Alexa Fluor 488 mouse (Thermo Fisher Scientific, A-32723), Alexa Fluor 568 mouse (Thermo Fisher Scientific, A-11004), Alexa Fluor 568 rabbit (Thermo Fisher Scientific, A-11036), and Alexa Fluor 594 mouse (Thermo Fisher Scientific, A-11032).
Cell culture and treatments
Prostate cancer (PCa) cell lines PC3 cells (Cleveland Clinic Research Institute Cell Culture Core) and LNCaP-derived C4-2 cells were a kind gift from Dr. Warren D.W. Heston (Lerner Research Institute, the Cleveland Clinic Foundation, Cleveland, OH, USA) were maintained in RPMI medium containing L-glutamine (Cleveland Clinic Research Institute Media Preparation Core, 10-500), and 293T cells (Cleveland Clinic Research Institute Cell Culture Core) were maintained in DMEM (Lerner Research Institute, 11-500)containing L-glutamine supplemented with 10% fetal bovine serum (Invitrogen, FBS-500HI), and 100× antibiotic-antimycotic (Thermo Fisher Scientific, 15240062). Cells were grown in a humidified incubator at 37°C and 5% CO2.
Cells were irradiated with 10 Gy, unless otherwise mentioned, at 25°C, using a Mark I Irradiator (J. C. Shepherd & Associates, Irvine, CA, USA) with a 137Cs source emitting at a fixed dose-rate of 2.0 Gy/min, as described previously [68]. Cells were treated with 10 µM CQ (Sigma-Aldrich, C6628).
Lentiviral transduction of shrna
293T cells were transfected with shRNAs together with the VSVG and gag-pol plasmids in a 3:1:1 ratio using the Fugene transfection reagent (Promega, E2311). Media containing viral particles was then collected 48 h after transfection, passed through a 0.22-micron filter, and added to PC3 and C4-2 cells along with polybrene (10 µg/ml; Santa Cruz Biotechnology, sc-134220). After overnight incubation at 37°C, the media was replenished and cells were selected for puromycin resistance (2 µg/ml) for 3 days, after which knockdown was further validated. As controls pLKO.1 vector (Sigma-Aldrich, SHC001) and non-target shRNA-expressing stable cell lines were generated similarly.
Cell death and proliferation analyses
Cell Viability was determined using Incucyte (Essen BioScience). Briefly, 105 cells per well were seeded in a 6-well plate. The next day, immediately following 4-Gy irradiation treatment, Sytox Green and Syto Red dyes were simultaneously added to the cells and the cell plate was scanned by the IncuCyte Live-Cell Analysis System, every 4 h, until the end of the experiment. Cell death was determined as % (green object count per image)x100/(red object count per image).
For clonogenic cell survival assay, cells were counted and plated in 6-well plates in triplicate. Following drug treatments, cells were allowed to grow for 14 days, fixed and stained in methanol:acetic acid (75:25, v:v) containing 0.5% crystal violet (w:v) to visualize colonies of at least 50 cells.
Immunoblotting and immunoprecipitation
Cells were lysed following the respective treatments in cell lysis buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.2% NP-40 (Sigma-Aldrich, 9016459), supplemented with the phosphatase inhibitor cocktails and complete protease inhibitor tablet. Protein concentration was determined using Bradford’s assay. Equal amount of protein in each sample was subjected to gel electrophoresis, followed by immunoblotting. Western blot detection was done by x-ray film-based enzyme chemiluminescence. Band signal intensity was quantified using the Image Studio Lite Version 5.2 (LI-COR), which were then normalized to the ACTB/β-actin controls. The differences in the band signal intensities were compared with the untreated control and were plotted as fold change. To verify linearity of signal intensity for USP14 and RNF168 westerns, serial dilutions of cell lysate starting at 100 µg protein were run on gels, and quantified, and linear trend lines with R2 > 0.95 were used as the coefficient of determination. Accordingly, ~10 µg protein that were loaded for USP14, and ~ 50 µg for RNF168 were found to be within the linear range. ACTB at these concentrations was also found to be within the linear range. For immunoprecipitation, cell lysates were prepared in 1x phosphate-buffered saline (PBS; 123-1000) with 1–2% CHAPS (Sigma-Aldrich, 75621-03-3) supplemented with the phosphatase inhibitor cocktail II, III and complete protease inhibitor tablet. Immunoprecipitation for detecting ubiquitination was performed under denaturing conditions using 1% SDS. Acid extraction of histones was done using a histone extraction kit from Abcam (ab221031) using the manufacturer’s protocol.
Confocal immunostaining
Cells were plated at 2 × 105 cells/cm2 on 22 × 22-mm coverslips in 35-mm culture dishes. Immunostaining was performed as previously described [21]. Briefly, following the respective treatments, cells were fixed with 2.0% paraformaldehyde in PBS for 15 min at room temperature, washed 3× for 10 min each, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich 9002-93-1) in PBS for 10 min, and blocked in 10% fetal bovine serum in PBS for 1 h. The coverslips were then immunostained using the primary antibodies diluted in blocking buffer, followed by fluorescently-conjugated secondary antibody, washed 3× for 10 min each, and mounted using Vectashield containing DAPI (Vector Laboratories, H-1200). Images were collected using an HCX Plan Apo 63X/1.4N.A. oil immersion objective lens on a Leica TCS-SP2 confocal microscope (Leica Microsystems AG). Quantification was based on data observed from at least 50 cells.
Mass spectrometry identification of active DUBs
HA-ubiquitin vinyl sulfone (Boston Biochem, u212) was used to identify Ub-binding proteins in C4-2 and derivative cells stably expressing shATG7. Cells were lysed in 50 mM Tris-HCl, pH 7.5, 0.1% NP-40, 5 mM MgCl2, 250 mM sucrose (Sigma-Aldrich, 57501), 1 mM DTT, 0.1 mM ATP (Sigma-Aldrich, 34369-07-8). The lysate was incubated with HA-ubiquitin vinyl sulfone for 30 min at 37°C. HA immune-precipitated proteins were fractionated on an SDS-PAGE gel and 5 areas were cut from the gel lane. The protein samples were subjected to in-gel digestion in which the bands were washed in 50% ethanol, 5% acetic acid. The gel pieces were then dehydrated in acetonitrile, dried in a Speed-vac, and digested overnight at room temperature by adding 50 ng of trypsin (Sigma-Aldrich, EMS0004) in 50 mM ammonium bicarbonate. The peptides were extracted and brought up in a final volume of ~30 µL for LC-MS analysis. The LC-MS system was a LTQ-Obitrap Elite hybrid mass spectrometer system coupled to a Dionex 3000 ultimate nano LC (Thermo Scientific). The HPLC column was a Dionex 15-cm x 75-µm id Acclaim Pepmap C18, 2 µm, 100 Å reversed-phase capillary chromatography column. Five µL volumes of the extract were injected and the peptides were eluted from the column in an acetonitrile, 0.1% formic acid gradient at a flow rate of 0.25 µL/min. The digest was analyzed in a data-dependent manner in which full mass scans at a resolution of 60,000 were obtained followed by MS/MS scans on the most abundant peptides. The LC-MS/MS data were searched with the program Mascot against the full human reference sequence database. The parameters used in this search include a peptide mass accuracy of 10 ppm, fragment ion mass accuracy of 0.6 Da, carbamidomethylated cysteines as a constant modification, and oxidized methionine and ubiquitination of K as a dynamic modification. Positive protein identification requires at least 2 peptides with a Mascot ion score greater than 40. All positively identified proteins were manually validated.
Statistical analyses
Statistical comparisons between 2 groups were conducted by using the Student’s t test and between multiple groups using 2-way ANOVA using the Prism software.
Funding Statement
This work was supported by the HHS | NIH | National Institute of General Medical Sciences (NIGMS) [GM112895]; HHS | NIH | National Cancer Institute (NCI) [CA184137]; HHS | NIH | National Cancer Institute (NCI) [P30CA043703].
Acknowledgments
The authors acknowledge the assistance of the Cleveland Clinic Lerner Research Institute Proteomics and Metabolomics as well as Imaging Core, partially supported by P30CA043703. Confocal “Artemis” utilized the Leica SP8 confocal microscope that was purchased with funding from NIH SIG grant 1S10OD019972-01. Confocal “Titan” utilized the Leica SP5 confocal/multi-photon microscope that was purchased with partial funding from NIH SIG grant 1S10RR026820-01. The Orbitrap Elite instrument was purchased via an NIH shared instrument grant, 1S10RR031537-01. This work was supported by the National Institutes of Health (NCI) grant 5R01CA184137 to AA and 5R01GM112895 to MS.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Material
Supplemental data for this article can be accessed here.
References
- [1].Begg AC, Stewart FA, Vens C.. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. [DOI] [PubMed] [Google Scholar]
- [2].Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. [DOI] [PubMed] [Google Scholar]
- [3].Huen MS, Grant R, Manke I, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. [DOI] [PubMed] [Google Scholar]
- [5].Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. [DOI] [PubMed] [Google Scholar]
- [6].Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. [DOI] [PubMed] [Google Scholar]
- [7].Sah DP, Tian G, Finley D. Meddling with fate: the proteasomal deubiquitinating enzymes. J Mol Biol. 2017;429:3525–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mosbech A, Lukas C, Bekker-Jensen S, et al. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem. 2013;288:16579–16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sharma N, Zhu Q, Wani G, et al. USP3 counteracts RNF168 via deubiquitinating H2A and gammaH2AX at lysine 13 and 15. Cell cycle. 2014;13:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yu H, Pak H, Hammond-Martel I, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A. 2014;111:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Typas D, Luijsterburg MS, Wiegant WW, et al. The de-ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Res. 2015;43:6919–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sy SM, Jiang J, O WS, et al. The ubiquitin specific protease USP34 promotes ubiquitin signaling at DNA double-strand breaks. Nucleic Acids Res. 2013;41:8572–8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhu Q, Sharma N, He J, et al. USP7 deubiquitinase promotes ubiquitin-dependent DNA damage signaling by stabilizing RNF168. Cell cycle. 2015;14:1413–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nakada S, Tai I, Panier S, et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature. 2010;466:941–946. [DOI] [PubMed] [Google Scholar]
- [15].Kato K, Nakajima K, Ui A, et al. Fine-tuning of DNA damage-dependent ubiquitination by OTUB2 supports the DNA repair pathway choice. Mol Cell. 2014;53:617–630. [DOI] [PubMed] [Google Scholar]
- [16].Shao G, Lilli DR, Patterson-Fortin J, et al. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci U S A. 2009;106:3166–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kee Y, Huang TT. Role of deubiquitinating enzymes in DNA repair. Mol Cell Biol. 2015;36:524–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sharma A, Singh K, Mazumder S, et al. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013;4:e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Singh K, Matsuyama S, JA Drazba, et al. Autophagy-dependent senescence in response to DNA damage and chronic apoptotic stress. Autophagy. 2012;8:236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Singh K, Sharma A, Mir MC, et al. Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer. 2014;13:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kitamura H, Torigoe T, Asanuma H, et al. Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology. 2006;48:157–161. [DOI] [PubMed] [Google Scholar]
- [24].Saleem A, Dvorzhinski D, Santanam U, et al. Effect of dual inhibition of apoptosis and autophagy in prostate cancer. Prostate. 2012;72:1374–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ladoire S, Chaba K, Martins I, et al. Immunohistochemical detection of cytoplasmic LC3 puncta in human cancer specimens. Autophagy. 2012;8:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Burdelski C, Reiswich V, Hube-Magg C, et al. Cytoplasmic accumulation of sequestosome 1 (p62) is a predictor of biochemical recurrence, rapid tumor cell proliferation, and genomic instability in prostate cancer. Clin Cancer Res. 2015;21:3471–3479. [DOI] [PubMed] [Google Scholar]
- [27].Schleicher SM, Moretti L, Varki V, et al. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resist Updat. 2010;13:79–86. [DOI] [PubMed] [Google Scholar]
- [28].Tam SY, Wu VW, Law HK. Influence of autophagy on the efficacy of radiotherapy. Radiat Oncol. 2017;12:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kim KW, Moretti L, Mitchell LR, et al. Combined Bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin Cancer Res. 2009;15:6096–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cao C, Subhawong T, Albert JM, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66:10040–10047. [DOI] [PubMed] [Google Scholar]
- [31].Yuk JM, Shin DM, Song KS, et al. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy. 2010;6:46–60. [DOI] [PubMed] [Google Scholar]
- [32].Chiu HW, Lin SW, Lin LC, et al. Synergistic antitumor effects of radiation and proteasome inhibitor treatment in pancreatic cancer through the induction of autophagy and the downregulation of TRAF6. Cancer Lett. 2015;365:229–239. [DOI] [PubMed] [Google Scholar]
- [33].Rouschop KM, Van Den Beucken T, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Koukourakis MI, Kalamida D, Mitrakas A, et al. Intensified autophagy compromises the efficacy of radiotherapy against prostate cancer. Biochem Biophys Res Commun. 2015;461:268–274. [DOI] [PubMed] [Google Scholar]
- [35].Karantza-Wadsworth V, Patel S, Kravchuk O, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mortensen M, Soilleux EJ, Djordjevic G, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Park Y-E, Hayashi YK, Bonne G, et al. Autophagic degradation of nuclear components in mammalian cells. Autophagy. 2009;5:795–804. [DOI] [PubMed] [Google Scholar]
- [39].Ivanov A, Pawlikowski J, Manoharan I, et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol. 2013;202:129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Robert T, Vanoli F, Chiolo I, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu EY, Xu N, O’Prey J, et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci USA. 2015;112:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen S, Wang C, Sun L, et al. RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol Cell Biol. 2015;35:406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hewitt G, Carroll B, Sarallah R, et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy. 2016;12:1917–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang Y, Zhang N, Zhang L, et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol Cell. 2016;63:34–48. [DOI] [PubMed] [Google Scholar]
- [46].Zois CE, Koukourakis MI. Radiation-induced autophagy in normal and cancer cells: towards novel cytoprotection and radio-sensitization policies? Autophagy. 2009;5:442–450. [DOI] [PubMed] [Google Scholar]
- [47].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edn). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tanida I, Sou YS, Minematsu-Ikeguchi N, et al. Atg8L/Apg8L is the fourth mammalian modifier of mammalian Atg8 conjugation mediated by human Atg4B, Atg7 and Atg3. FEBS J. 2006;273:2553–2562. [DOI] [PubMed] [Google Scholar]
- [49].Fortunato F, Burgers H, Bergmann F, et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350–60,60.e1-5. [DOI] [PubMed] [Google Scholar]
- [50].Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage In: Bjergbæk L, ed. DNA Repair Protocols. Totowa, NJ: Humana Press; 2012. p. 613–626. [DOI] [PubMed] [Google Scholar]
- [52].Stewart GS, Panier S, Townsend K, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. [DOI] [PubMed] [Google Scholar]
- [53].Lee B-H, Lee MJ, Park S, et al. Enhancement of Proteasome Activity by a Small-Molecule Inhibitor of Usp14. Nature. 2010;467:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Thorslund T, Ripplinger A, Hoffmann S, et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;527:389–393. [DOI] [PubMed] [Google Scholar]
- [55].Luijsterburg MS, Typas D, Caron MC, et al. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife. 2017;6:e20922. doi: 10.7554/eLife.20922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu WJ, Ye L, Huang WF, et al. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Borodovsky A, Kessler BM, Casagrande R, et al. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Anderson C, Crimmins S, Wilson JA, et al. Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J Neurochem. 2005;95:724–731. [DOI] [PubMed] [Google Scholar]
- [60].Lee BH, Lu Y, Prado MA, et al. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature. 2016;532:398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kim HT, Goldberg AL. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J Biol Chem. 2017;292:9830–9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Park CW, Ryu KY. Cellular ubiquitin pool dynamics and homeostasis. BMB Rep. 2014;47:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007;67:7395–7405. [DOI] [PubMed] [Google Scholar]
- [64].Butler LR, Densham RM, Jia J, et al. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012;31:3918–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Korolchuk VI, Mansilla A, Menzies FM, et al. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Xu D, Shan B, Sun H, et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016;30:1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mialki RK, Zhao J, Wei J, et al. Overexpression of USP14 protease reduces I-kappaB protein levels and increases cytokine release in lung epithelial cells. J Biol Chem. 2013;288:15437–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen Q, Gong B, Almasan A. Distinct stages of cytochrome c release from mitochondria: evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000;7:227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.