

ABSTRACT
Macroautophagy/autophagy is a survival mechanism that facilitates protein turnover in post-mitotic cells in a lysosomal-dependent process. Mitophagy is a selective form of autophagy, which arbitrates the selective recognition and targeting of aberrant mitochondria for degradation. Mitochondrial content in cells is the net balance of mitochondrial catabolism via mitophagy, and organelle biogenesis. Although the latter process has been well described, mitophagy in skeletal muscle is less understood, and it is currently unknown how these two opposing mechanisms converge during contractile activity. Here we show that chronic contractile activity (CCA) in muscle cells induced mitochondrial biogenesis and coordinately enhanced the expression of TFEB (transcription factor EB) and PPARGC1A/PGC-1α, master regulators of lysosome and mitochondrial biogenesis, respectively. CCA also enhanced the expression of PINK1 and the lysosomal protease CTSD (cathepsin D). Autophagy blockade with bafilomycin A1 (BafA) reduced mitochondrial state 3 and 4 respiration, increased ROS production and enhanced the accumulation of MAP1LC3B-II/LC3-II and SQSTM1/p62. CCA ameliorated this mitochondrial dysfunction during defective autophagy, increased PPARGC1A, normalized LC3-II levels and reversed mitochondrially-localized SQSTM1 toward control levels. NAC emulated the LC3-II reductions induced by contractile activity, signifying that a decrease in oxidative stress could represent a mechanism of autophagy normalization brought about by CCA. CCA enhances mitochondrial biogenesis and lysosomal activity, and normalizes autophagy flux during autophagy suppression, partly via ROS-dependent mechanisms. Thus, contractile activity represents a potential therapeutic intervention for diseases in which autophagy is inhibited, such as vacuolar myopathies in skeletal muscle, by establishing a healthy equilibrium of anabolic and catabolic pathways.
Abbreviations: AMPK: AMP-activated protein kinase; BafA: bafilomycin A1; BNIP3L: BCL2/adenovirus E1B interacting protein 3-like; CCA: chronic contractile activity; COX4I1: cytochrome c oxidase subunit 4I1; DMEM: Dulbecco’s modified Eagle’s medium; GFP: green fluorescent protein; LSD: lysosomal storage diseases; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MTORC1: mechanistic target of rapamycin kinase complex 1; NAC: N-acetylcysteine; PPARGC1A: peroxisome proliferative activated receptor, gamma, coactivator 1 alpha; PINK1: PTEN induced putative kinase 1; ROS: reactive oxygen species; SOD2: superoxide dismutase 2, mitochondrial; SQSTM1/p62: sequestosome 1; TFEB: transcription factor EB
KEYWORDS: Autophagy, bafilomycin, C2C12 muscle cells, contractile activity, mitochondrial biogenesis, mitophagy
Introduction
Endurance exercise has long been known to evoke favorable molecular adaptations, including an increased capacity for aerobic metabolism in skeletal muscle [1], and this enhancement conveys a potential therapeutic intervention against muscle wasting [2,3]. To study the cellular mechanisms underlying the health benefits of exercise, in vivo [4,5] and in vitro [6–9] models of chronic contractile activity (CCA) can be employed. CCA of skeletal muscle activates a transcriptional program of oxidative genes, some of which are upregulated via the transcriptional regulator PPARGC1A [7], leading to mitochondrial biogenesis and resistance to fatigue [10]. However, it is now recognized that an understanding of the mitochondrial adaptations to exercise requires consideration of organelle turnover, involving not only biogenesis, but also the selective autophagic degradation of mitochondria, termed mitophagy [11,12].
The mitophagy pathway is a pro-survival mechanism that operates through the degradation and recycling of mitochondrial components. Dysfunctional organelles are tagged and sequestered in transient compartments, phagophores, that mature into double-membrane vesicles termed autophagosomes that subsequently fuse with the lysosome for proteolytic degradation. The dysregulation of autophagy/mitophagy has sparked considerable interest for its role in the pathophysiology of Parkinson disease [13], cardiomyopathies [14], and cancer [15–17]. Beyond their bioenergetic role in determining the rate of oxidative phosphorylation (OXPHOS) in eukaryotic cells, mitochondria can also radically modulate cellular homeostasis. Aberrant mitochondria participate in cell fate decisions by initiating apoptotic pathways [18], facilitating proinflammatory signaling [19] and disrupting cellular redox status by elevating reactive oxygen species (ROS) production [20–22]. Stimuli that can trigger mitochondria for mitophagic degradation include electron transport chain abnormalities [23], mitochondrial DNA damage [24], hypoxia [25], and dissipation of the membrane potential (ΔΨm) [26]. Thus, proper functioning of the mitophagy pathway mitigates apoptotic signaling [27] and tempers cellular stress responses by invoking indispensable quality control.
PINK1 is a conserved serine/threonine kinase, possessing an N-terminal mitochondrial targeting sequence [28], with a dense expressional profile in brain, muscle and testes [29]. When mitochondria are healthy, PINK1 is rapidly imported and degraded by resident proteases. However, upon the loss of mitochondrial membrane potential, PINK1 is unable to be imported and it stabilizes on the outer mitochondrial membrane. From there, PINK1 recruits the E3-ubiquitin (Ub) ligase PARK2 through phosphorylation of PARK2 and ubiquitin [30,31]. PARK2-mediated ubiquitination of proteins provides a binding scaffold for the autophagy receptor SQSTM1/p62 [32], and primes mitochondria for degradation independently of the canonical ubiquitin-proteasome system. LC3B undergoes post-translational modifications to produce 2 isoforms, LC3-I and LC3-II [33]. LC3-I is found in the cytosol, but when covalently bound to phosphotidylethanolamine (PE) by ATG7- and ATG3-dependent conjugation events, converting it to LC3-II, this permits LC3-II to participate in the maturation of the phagophore and/or autophagosome [34]. Indeed, LC3-II is regarded as a bona fide marker of autophagosomes [35]. Targeted substrates, such as mitochondria, are engulfed in phagophores through the binding of receptor proteins such as SQSTM1 [36] to LC3-II.
In skeletal muscle, autophagy can be induced by mitochondrially-generated ROS [37] and by acute exercise [38], likely via AMPK activation [39]. Several studies have demonstrated an upregulation in autophagy concomitant with the physiological adaptations to exercise [40–46] in muscle. The pool of healthy mitochondria available to skeletal muscle is a function of the balance between organelle synthesis and mitophagy. Whereas biogenesis has been investigated for many years, the study of mitophagy remains in its infancy. Some evidence in humans suggests that mitophagy is downregulated with ageing and inactivity [47]. Indeed, a decrement in the pathway of mitophagy between the dysfunctional organelle and the lysosome may underlie functional deficits of muscle function with age [48]. However, little is known about the implications of autophagy suppression on mitochondrial function, and the potential response to contractile activity. In this study, we sought to investigate whether chronic contractile activity could offset the mitochondrial dysfunction induced by the inhibition of autophagy/mitophagy. Our results indicate that chronic contractile activity enhances mitochondrial biogenesis as well as lysosomal capacity to improve mitochondrial turnover and augment organelle function.
Results
Mitochondrial dysfunction with autophagy inhibition is ameliorated by chronic contractile activity
CCA successfully induced mitochondrial biogenesis, resulting in a 1.6-fold increase in cytochrome c oxidase (COX) activity (Figure 1(a)). This was corroborated by a significant 2.8-fold increase in COX4I1 (cytochrome c oxidase subunit 4I1) protein levels (Figure 1(b) and (c)). This effect of contractile activity was not observed when autophagy was inhibited by treatment with BafA. However, sustained autophagy inhibition invoked the accretion of mitochondria that presumably escape degradation, as demonstrated by an increase in mitochondrial enzyme activity. Significant elevations of both COX activity and COX4I1 protein were observed with BafA treatment alone (P < 0.05). The combination of BafA treatment with CCA did not lead to an additive response in the quantity of mitochondria.
Figure 1.
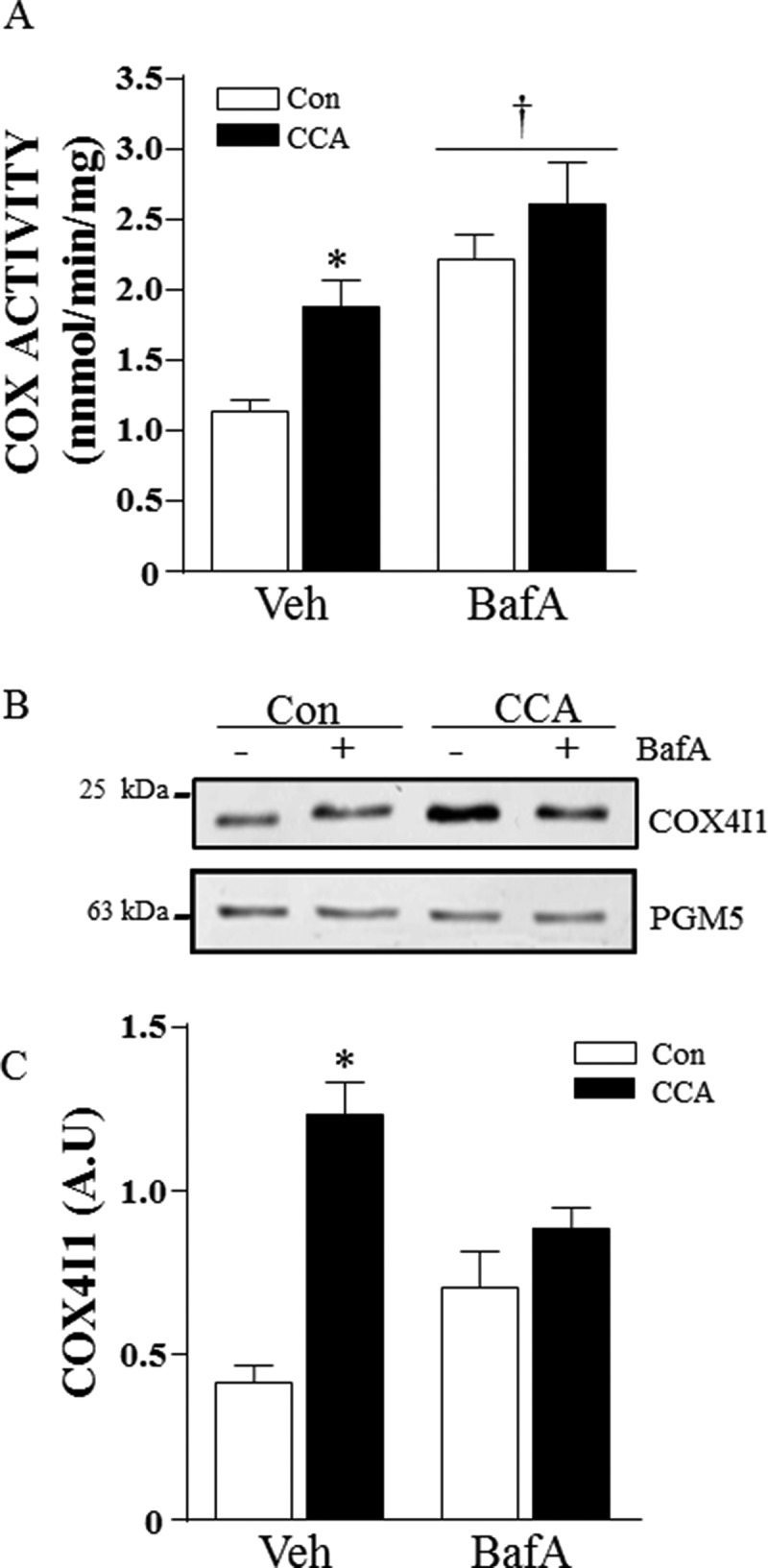
CCA-induced mitochondrial biogenesis. Differentiated myotubes were chronically stimulated to induce mitochondrial biogenesis in the presence of vehicle or bafilomycin A1 (BafA). (A) Cytochrome oxidase (COX) activity (*P < 0.05, vs. vehicle control; †P < 0.05, main effect of BafA vs. vehicle; n = 6). (B) Representative western blot of whole cell extracts probed for COX4I1 and PGM5 protein expression. (C) Graphical densitometric quantification, COX4I1 normalized to PGM5. (*P < 0.001 vs. vehicle Con; n = 8.) A.U., arbitrary units.
To assess changes in organelle function, we quantified oxygen consumption in isolated mitochondrial fractions. As expected, state 3 respiration was 3–4-fold greater than state 4 respiration in mitochondria from control cells. BafA treatment produced mitochondria with severe 2.5–3-fold depressions in both state 4 and state 3 respiration (Figure 2(a)). CCA did not alter the rates of oxygen consumption observed under control conditions, but successfully reversed the respiratory dysfunction induced by BafA, resulting in a 2–3-fold improvement over the BafA-treated cells, back toward control conditions. Mitochondrial ROS emission was elevated in response to autophagic disruption in state 3 (2.3-fold) and state 4 (1.6-fold) respiration (Figure 2(b)). CCA had no effect on ROS production in control, vehicle-treated cells, but ameliorated BafA-induced ROS emission and reversed the increase in ROS emission by 55% to levels that matched untreated controls. These results indicate that CCA can positively regulate mitochondrial function under conditions of cellular stress, and attenuate the increase in ROS production resulting from autophagic defects. The effects of CCA on ROS levels in isolated mitochondria were independent of changes in SOD2/manganese superoxide dismutase, because levels of the protein were unaffected by CCA, but increased by BafA (Figure 2(c), 2(d)).
Figure 2.

Mitochondrial respiration and ROS production. Mitochondria were isolated from myotube tissue culture by differential centrifugation. State 4 and state 3 respiration were measured and normalized to protein concentration. ROS production was determined on isolated mitochondria and normalized to respiration. (A) State 4 and state 3 respiration (*P < 0.05, vs. Con state 4; †P < 0.001, vs. Con state 3; ‡P < 0.05, BafA+ CCA vs. BafA control; n = 4). (B) Mitochondrial ROS production (*P < 0.05, vs. Con state 4; †P < 0.001, vs. con State 3; ‡P < 0.05, BafA+ CCA vs. BafA control; n = 4). (C) Representative western blot of SOD2 and TUBA/α-TUBULIN protein expression. (D) Graphical quantification, SOD2 normalized to TUBA († P < 0.001, main effect of BafA vs. vehicle; n = 6). A.U., arbitrary units. natoms, nanoatoms.
CCA and autophagy suppression induce mitophagy gene expression
In order to explain some of the molecular mechanisms regulating these contractile activity-induced adaptations, relative changes in gene expression of Pink1, Park2, Bnip3l/NIX, Map1lc3b, Tfeb, Sqstm1 and Ppargc1a were quantified. CCA led to 1.5–2-fold increases in Ppargc1a, Pink1 and Sqstm1 mRNA levels (Figure 3), but had no effect on the mRNA encoding the ubiquitin E3 ligase, Park2, or the mitophagy receptor, Bnip3l/NIX. Autophagy suppression with BafA also led to a large increase in Ppargc1a, Pink1 and Sqstm1 gene expression, as well as more modest changes in PARK2 and BNIP3L/NIX (P < 0.05). Map1lc3b gene-expression was enhanced 2-fold following CCA in the BafA condition, while both Ppargc1a and Sqstm1 mRNA were upregulated in an additive fashion, by 2–4-fold, when the 2 treatments (BafA and CCA) were combined, compared to vehicle control.
Figure 3.
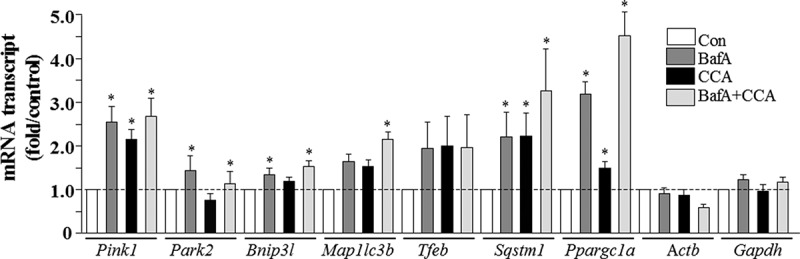
Real-time PCR analysis of mitophagy mRNA expression in control and CCA myotubes, treated with vehicle (Con) or bafilomycin A1 (BafA). Transcript levels were normalized to both Actb and Gapdh (*P < 0.05, vs. control levels of the same transcript).
CCA normalizes autophagy flux and enhances lysosomal activity
We next assessed autophagic flux by examining the levels of autophagy proteins LC3-I and LC3-II, as well as the autophagy substrate SQSTM1 using immunobloting. Treatment with BafA resulted in a large 24-fold accumulation of LC3-II (Figure 4(a), 4(b)). The difference in LC3-II between the vehicle-treated and BafA-treated cells illustrates the autophagy flux present in normal, control C2C12 myotubes. This was paralleled by the 15-fold increase in the LC3-II:LC3-I ratio (Figure 4(c)). These changes corresponded to the large increase in autophagosomal GFP-LC3 puncta evident with immunofluorescence microscopy (Con-BafA vs. Con; Figure 4(d)), as well as a more modest upregulation of the colocalization of mitochondria with autophagosomes, as evident from the enhanced yellow-orange fluorescence visible in the merged image. CCA had no effect on the very low levels of LC3-II, or on the LC3-II:LC3-I ratio in vehicle-treated cells. However, CCA produced a 50% reduction in LC3-II levels (P < 0.001) with BafA treatment, with a similar significant downregulation in the LC3-II:LC3-I ratio (Figure 4(b), 4(c)). Importantly, these data corresponded with a marked decrease in visible autophagosome puncta (Figure 4(d)), indicating that the myotubes had adapted to a lower level of autophagy flux in response to CCA. CCA also induced a greater mitochondrial content as indicated by mitochondrial Ds-Red fluorescence, as predicted by the changes in COX activity observed (Figure 1(a)). These changes in autophagy were not observed after only 1 day of CCA treatment, suggesting that they represent an adaptation to repeated bouts of contractile activity.
Figure 4.

Expression of autophagy proteins in response to CCA in C2C12 myotubes, treated with bafilomycin A1 (BafA). (A) Representative western blot. (B) Graphical densitometric quantification; LC3-II normalized to TUBA/α-tubulin protein (*P < 0.001, BafA CCA vs. BafA control; †P < 0.001, main effect of BafA vs. vehicle n = 8). (C) LC3-II normalized to LC3-I protein (*P < 0.001, BafA CCA vs. BafA control; †P < 0.001, main effect of BafA vs. vehicle n = 8). A.U., arbitrary units. (D) Fully differentiated myotubes were chronically stimulated to induce mitochondrial biogenesis in the presence of vehicle or BafA. Fluorescence microscopy of C2C12 myotubes co-expressing GFP-LC3 and mitoDs-Red at 60x magnification; scale bar: 10 µm.
Whole cell SQSTM1 levels were also significantly enhanced following BafA treatment (Figure 5(a),5(b)). CCA had no significant impact on SQSTM1 levels in whole cell lysates under vehicle-treated conditions, but attenuated the increase of SQSTM1 in the presence of BafA (Figure 5(b)). The difference in SQSTM1 levels between vehicle- and BafA-treated cells was reduced (p < 0.05) with CCA treatment, indicating a CCA-induced reduction in autophagy flux. BafA treatment also led to a dramatic 30-fold increase in SQSTM1 levels associated with the isolated mitochondria (Figure 5(c), 5(d)). CCA produced a modest, but statistically significant decline in the localization of SQSTM1 to mitochondria, indicating a lower drive for mitophagy in the presence of enhanced mitochondrial quality.
Figure 5.

Expression of autophagy proteins in response to CCA in C2C12 myotubes treated with bafilomycin A1 (BafA). Representative western blots. (A) Whole cell extracts were probed for SQSTM1 and GAPDH, and (C) mitochondrial fractions were probed for SQSTM1 and VDAC. Graphical densitometric quantification. (B) Total SQSTM1 normalized to GAPDH (†P < 0.01, main effect of BafA vs. vehicle; * P < 0.05, vehicle BafA vs. BafA + CCA; #, P < 0.05 interaction effect between CCA and BafA treatment, n = 5). (D) Mitochondrial SQSTM1 (†P < 0.001, main effect of BafA vs. vehicle; *P < 0.01, BafA control vs. BafA + CCA; n = 3). A.U., arbitrary units.
Chronic contractile activity resulted in the selective induction of lysosomal markers, such as mature CTSD protein (34 kDa), which was significantly increased by 2.5-fold (Figure 6(a), 6(b)). CCA also resulted in an increase in TFEB protein, parallel to the change in PPARGC1A, the master regulator of mitochondrial biogenesis (Figure 6(a), (d), and (e)). Interestingly, BafA treatment significantly reduced TFEB levels independently of MTORC1 signaling, as suggested by the lack of change in phosphorylated (p)-RPS6KB/p706SK kinase phosphorylation (Figure 6(a)). This effect was not rescued by CCA. BafA treatment also led to a large increase in Lamp2 levels (Figure 6(c)), indicative of an accumulation of lysosomal membranes. This effect was attenuated by CCA. It should be noted that the antibody used for LAMP2 detection cross-reacts with all LAMP2 isoforms.
Figure 6.

Expression of lysosomal proteins in response to CCA in C2C12 myotubes treated with bafilomycin A1 (BafA). (A) Representative western blots of whole cell extracts were probed for CTSD, LAMP2, TFEB, p-RPS6KB, PPARGC1A, GAPDH and PGM5. Graphical densitometric quantification: (B) CTSD normalized to PGM5 (*P < 0.05, CCa vs. Con, n = 6); (C) LAMP2 normalized to PGM5 (*P < 0.05, BafA control vs. vehicle control, n = 6); (D) TFEB normalized to GAPDH (*P < 0.05, CCA vs. vehicle control, n = 5). (E) PPARGC1A normalized to GAPDH (*P < 0.05 CCA vs. vehicle control, n = 4).
N-acetyl-l-cysteine treatment
In view of the fact that CCA attenuated mitochondrial defects as well as autophagy flux, and that this was accompanied by a reversal in ROS production in BafA-treated groups, we sought to investigate whether these alterations could be mediated by the reduced ROS production. Thus, we treated myotubes with N-acetyl-L-cysteine (NAC), a thiol antioxidant, for either 24 or 72 h, to assess the role of ROS in modulating autophagy flux with BafA treatment. As expected, the presence of BafA led to a marked increase in LC3-II accumulation at both 24 and 72 h, revealing a significant level of autophagy flux in vehicle-treated C2C12 myotubes (black bars in Figure 7(b)). The presence of NAC significantly attenuated LC3-II autophagic flux at both 24 and 72 h of BafA treatment, by 50–66% (shaded bars in Figure 7(b)), similar to our observations with CCA (Figure 4). In our previous work we have shown that NAC completely and effectively neutralizes ROS effects in C2C12 cells [49]. The reductions to LC3-II flux observed with NAC treatment were independent of alterations in SQSTM1 (Figure 7(a)), indicating that antioxidant treatment did not affect the clearance of this protein. These results implicate ROS in the modulation of autophagy flux in muscle cells.
Figure 7.
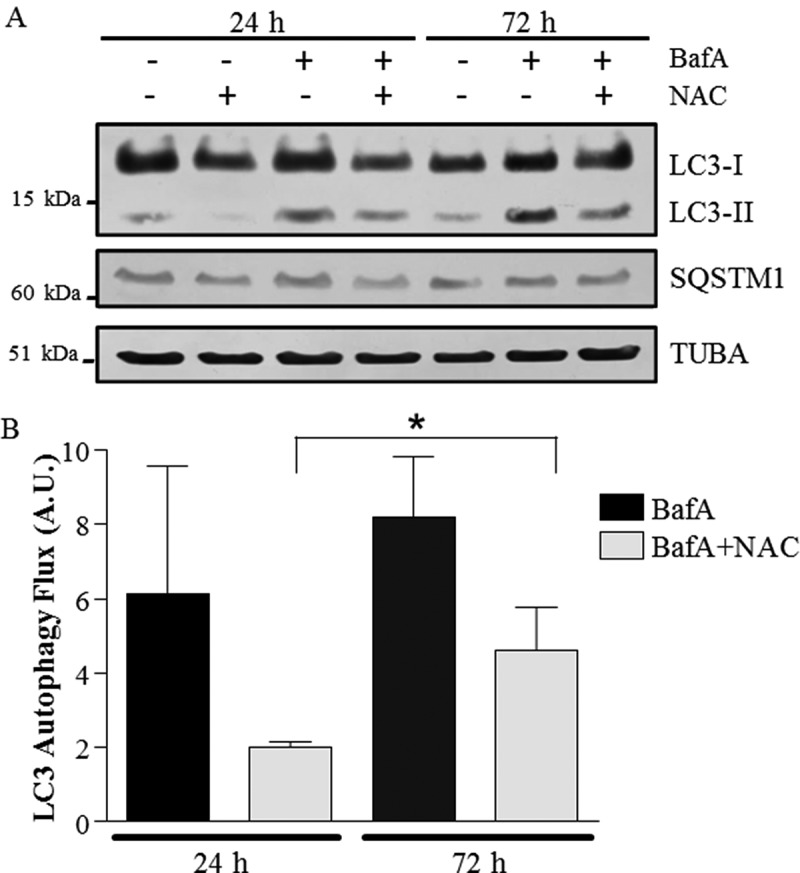
Autophagy flux with co-incubation of NAC (N-acetylcysteine) and BafA in muscle cells. (A) Representative western blots of whole cell extracts probed with antibodies to LC3, SQSTM1 and TUBA/α-TUBULIN, used as a loading control. (B) Graphical representation of autophagy flux. (*P < 0.05, overall effect of NAC vs. vehicle; n = 3.) A.U., arbitrary units.
Discussion
The literature reveals a growing appreciation for the role of autophagy in maintaining skeletal muscle health and cellular homeostasis [12,50]. A failure of autophagy has been linked to congenital muscular dystrophies, and the reinstatement of normal autophagy flux ameliorates the dystrophic phenotype and myofibrillar degeneration [51]. Mice with muscle-specific autophagy knockouts, atg7−/-, display a loss of fiber-integrity and muscle mass, and the accumulation of abnormal mitochondria [52]. Autophagy disruption has been noted in a variety of myopathologies, including sporadic inclusion body myositis, and polymyositis with mitochondrial pathology [53], characterized by protein aggregation and progressive muscle weakness. Thus, the inefficient clearance of toxic protein aggregates likely facilitates decrements in muscle function, leading to pathology. In healthy cells, autophagy is a tightly regulated process, and it has consistently been shown to be transiently upregulated in response to acute exercise in skeletal muscle [37,42,54,55]. This is likely because contractile activity induces a host of signaling, transcriptional and metabolic alterations that can promote an enhanced drive for autophagy. Moreover, the process has been suggested to necessitate the adaptation to a more oxidative phenotype in muscle [44].
Mitochondria are both key mediators of cellular bioenergetics and robust initiators of cellular apoptotic signaling [56]; thus, mitochondrial quality control is both integral to energy production and cytoprotection [55]. Steady-state organelle content within cells reflects a dynamic relationship between catabolism (i.e., mitophagy), and the anabolic process of biogenesis in response to enhanced metabolic demands. CCA has been established as an effective model of mitochondrial biogenesis [7], which activates a transcriptional profile resembling the response to chronic treadmill training in mice [57]. Acute bouts of muscle contraction, which ultimately lead to an adaptive change in mitochondrial biogenesis, result in bursts of ROS [58], as well as 5ʹAMPK activation, and can mediate the cross-talk between acute exercise and autophagy through a convergence on MTOR signaling. AMPK activation targets include the autophagy-initiator kinase ULK1 [59], along with PPARGC1A, which positively regulates TFEB [60], a transcription factor important for lysosomal biogenesis. However, the mechanisms by which exercise modulates selective autophagy are far from clear cut, and the effects of autophagy suppression on mitochondrial function and content have yet to be investigated. Thus, the purpose of our study was to examine the role of autophagy in the mitochondrial adaptations to contractile activity, and the implications of autophagy suppression on mitochondrial quality and content in muscle cells. Our expectation was that a block in autophagy would inhibit mitochondrial turnover via impaired mitophagy, leading to the accumulation of aberrant organelles, and that CCA would counterbalance mitochondrial dysfunction by providing nascent, functional organelles.
To evaluate the effects of autophagy inhibition on the molecular adaptations to CCA, we treated differentiated C2C12 myotubes with a specific inhibitor of the lysosomal vacuolar-type H+-translocating ATPase, BafA, for the duration of the CCA protocol to monitor the effects of sustained autophagy deficit on the function and quality of the mitochondria, in order to model the prolonged defects characteristic of pathological states. As previously reported [6,7], CCA effectively increased mitochondrial content, as observed by approximately 2-fold increases in COX4I1 protein expression and COX activity, as well as by 1.5-fold increases in PPARGC1A levels. In the absence of CCA, BafA led to severe deficits in mitochondrial respiration, illustrating that mitophagy is an integral mechanism that maintains organelle fitness and function. This effect was completely reversed following CCA, indicating that contractile activity led to the adaptive production of nascent, functional organelles, and that contractile activity can rescue the oxidative respiration insult induced by defective mitochondrial turnover. BafA treatment also upregulated the mitophagy-specific genes Pink1, Park2 and Bnip3l, suggesting that defective organelle clearance induces the expression of genes responsible for mitophagy signaling. The fact that CCA alone resulted in the enhanced expression of both Pink1 and Ppargc1a indicates a heightened reliance on mitochondrial quality-control pathways during contractile activity-induced mitochondrial biogenesis. These findings are in agreement with our recent work which demonstrated a greater mitophagy drive post-exercise in skeletal muscle [38,61].
Reactive oxygen species are a normal by-product of oxidative phosphorylation, escaping complexes I and III in the process of oxidative phosphorylation. Mitochondrial matrix antioxidants such as SOD2/MnSOD efficiently quench superoxide anion radicals; however, with mitochondrial dysfunction, ROS can out-live internal defense systems, leading to an exacerbation of ROS emission. Excessive ROS levels trigger apoptotic signaling and potentiate muscle wasting stimuli [62]. Our results indicate that ROS were upregulated with autophagic inhibition, despite the marked elevation in SOD2 protein with BafA treatment. CCA reversed this increase in ROS production, thereby restoring levels back toward control values, in concert with overturning the dysfunctional phenotype induced by aberrant organelle elimination. These reductions in mitochondrial ROS produced by CCA occur as a result of improved organelle integrity, and may be due, in part, to the ROS detoxification program initiated by the enhanced activation of PPARGC1A [63].
To investigate the effects of CCA on autophagy, we compared cells treated with either BafA or vehicle to assess flux, and we measured LC3-II and SQSTM1 accumulation, as well as the LC3-II:LC3-I ratio. BafA drastically disrupted autophagic degradation, resulting in a 25-fold increase in LC3-II accretion, accompanied by a large increase in autophagosome puncta. CCA alone had little impact on SQSTM1 or LC3-II levels, but resulted in a marked increase in mitochondrial content within contracting myotubes. In the presence of BafA, CCA effectively normalized autophagic degradation, and reduced the elevated LC3-II:LC3-I ratio, as well as the number of visible autophagosomes. This cellular adaptation is likely a reflection of 2 processes brought about by CCA. First, CCA induced the biogenesis of higher quality mitochondria, reflected by higher levels of PPARGC1A as well as improved rates of respiration and reduced ROS emission, thereby decreasing the requirement for mitophagy. Consistent with this is the modest reduction in mitochondrial-associated SQSTM1 with CCA. A similar phenomenon was recently illustrated by Jiang and colleagues, who found a reduction of flux with exercise following chloroquine treatment [41]. Second, the normalization of autophagic flux by CCA is a result of enhanced cargo clearance via an increase in lysosomal capacity. CCA led to a 2.5-fold increase in CTSD and a 1.5-fold increase in the regulatory protein TFEB, indicating an effect of contractile activity on the transcriptional regulation of the autophagy-lysosomal pathway, as we have shown recently [9]. In support of this, we have also recently shown that CCA in vivo leads to an early enhancement of TFEB expression along with lysosomal capacity within muscle, prior to the occurrence of mitochondrial biogenesis [5]. Indeed, Mansueto et al [64] have also recently shown strong evidence for the role of TFEB in regulating mitochondrial biogenesis in muscle. The fact that changes in TFEB and PPARGC1A expression occur concomitantly in response to CCA both in myotube cultures as well as in vivo supports the idea that TFEB and PPARGC1A are coordinately regulated to mediate a cross-talk between mitochondrial biogenesis and autophagy [60].
These CCA-induced changes in lysosomal proteins are important for the maintenance of muscle health. For example, CTSD knockdown during development conveys a congenital myopathy in zebrafish, suggestive of a critical role for this protease in muscle homeostasis [65]. Two prominent genetically inherited lysosomal storage diseases (LSDs) include Pompe disease and Danon disease, characterized by muscle weakness and poor muscle tone, owing to a block in autophagy and an inability of lysosomes to break down glycogen [66,67]. A recent report on Danon disease patients revealed the accumulation of dysfunctional mitochondria as an early feature of cardiac contractile failure in these patients with autophagy blockade [68]. Deficiencies of lysosomal enzymes as evident in these LSDs causes the intra-lysosomal accumulation of undegraded substrates and an expansion of the autophagic compartment within cells [69], often accompanied by the obvious accumulation of the ‘aging pigment’ lipofuscin in electron micrographs of muscle. Lipofuscin is a non-degradable aggregate contained within lysosomes, which indicates deficiencies in the terminal execution of autophagy at the level of the lysosome. Although therapies for LSDs is multi-factorial [69], our data support the use of contractile activity (i.e., exercise) to overcome autophagy blockade in muscle. Consistent with these findings, we view the large increase in the expression of the lysosomal membrane protein LAMP2 in the presence of BafA as an expansion of the autophagic compartment with autophagy suppression. The fact that CCA was able to reverse this pathological increase suggests that the normalized autophagy flux produced by CCA is also associated with improved lysosomal clearance.
We postulated that the observed CCA-associated decrement in ROS could mediate the ability of CCA to re-establish autophagic flux with BafA treatment. To examine this possibility, we co-treated differentiated myotubes with the nonspecific antioxidant NAC, along with BafA. Our results indicate that NAC reduced LC3-II accretion in BafA-treated cells to an equivalent extent as CCA, by approximately 50%. The ability of NAC to normalize autophagy in this model of autophagy suppression suggests that CCA modulates ROS production to restore autophagy flux. This is supported by the in vivo data of Lo Verso et al. [70] who found that the chronic treatment of mice with an anti-oxidant suppresses basal autophagy in skeletal muscle. Thus, we speculate that in models of suppressed autophagy, a major fraction of the reduced autophagy flux can be attributed to disruptions in cellular redox status.
In conclusion, our results indicate that CCA enhances functional autophagy, promotes lysosomal clearance and flux normalization, and mediates a reversal of mitochondrial deficits induced by aberrant autophagy. A limitation of our study is that these investigations were performed using contracting myotubes in culture, and experiments are currently underway to verify our data using animal models. Our results lead us to believe that contractile activity, or exercise in an in vivo setting, merits consideration as a potential therapeutic intervention against mitochondrial dysfunction in conditions of autophagy dysregulation, such as vacuolar myopathies. Mitophagy mediates an indispensable mitochondrial quality control pathway in muscle cells, and chronic contractile activity can blunt autophagy defects to promote muscle health.
Materials and methods
Cell culture
C2C12 murine myoblasts (ATCC, CRL 1772) were proliferated on 6-well and 10-cm culture dishes coated with 0.1% gelatin (Sigma-Aldrich, G1890) in Dulbecco’s modified Eagle’s medium (DMEM; Wisent, 319–015-CL) supplemented with 10% fetal bovine serum (GIBCO, 12,483–020) and 1% penicillin-streptomycin (P-S; Wisent, 450–201-EL). At 90–95% confluency, differentiation into myotubes was induced by replacing growth medium with DMEM supplemented with differentiation media (DM) containing 5% heat-inactivated horse serum (GIBCO, 16,050–114) and 1% P-S. DM was replenished daily for 4 days until fully differentiated myotubes were observed.
Stimulation of muscle cells to induce chronic contractile activity, drug treatment and autophagy flux calculations
Lids from plastic 6-well and 10-cm dishes were fitted with 2 platinum wire electrodes such that lengths ran parallel to each other at opposite ends, and when placed in the dish become submerged in media. Fully differentiated myotubes were subjected to electrical stimulation-induced contractile activity in a parallel circuit attached to a stimulator unit at a frequency of 5 Hz and an intensity of 9 V, chronically for 3 h/day over 4 successive days beginning on day 4 of differentiation. This stimulation protocol has been previously described in detail [7,9,71,72]. Differentiation medium was replenished 1 h prior to stimulation, containing vehicle (anhydrous ethanol) or 3 nM bafilomycin A1 (BafA; Invivogen, tlrl-baf1). Stimulation was performed at 37̊°C and 5% CO2, and the media was replenished following stimulation across all conditions. Autophagy flux was calculated from the difference in LC3-II or SQSTM1 levels, corrected for protein loading, between Vehicle- and BafA-treated cells.
NAC treatment
C2C12 myoblasts were differentiated as described above. On day 4 of differentiation, media was supplemented with 1 of 4 conditions: vehicle, 10 mM N-acetyl-L-cysteine (NAC; Sigma-Aldrich, A8199), 3 nM BafA, or 10 mM NAC plus 3 nM BafA. Media containing the indicated concentrations was replenished daily. Following 24 and 72 h of treatment, cells were lysed as described and the supernatant fraction was used for immunoblotting analysis.
Mitochondrial isolation
Mitochondria were isolated from myotubes in tissue culture using an adapted protocol [73] via differential centrifugation. Briefly, myotubes grown in 10-cm plates were washed 2x in ice-cold phosphate-buffered saline (PBS;Wisent, 311–425-C1) and scraped on ice using rubber policemen in mitochondrial isolation buffer (MIB; 10% 0.1 M Tris-MOPS [Bioship, 1132–61–2], 1% EGTA-Tris [Sigma-Aldrich, E4378], 20% 1 M sucrose [CALEDON, 8720–1], pH 7.4). Cells were pelleted at a centrifugation speed of 600 g (Beckman JA25.5) for 10 min at 4ºC and pellets were resuspended in 3 ml of MIB on ice. Suspensions were transferred to chilled 15-ml glass potters and subjected to homogenization with an Elvehjem PTFE Tissue Grinder (Wheaton, 358,009) at 800 rpm for 35 strokes. Homogenates were transferred to fresh isolation tubes and recentrifuged at 600 g for 10 min. The supernatant fractions containing mitochondria and cytosol were collected and passed through a 40-μm filter (BD Falcon, 352,340) and the pellet discarded. Filtered homogenates were centrifuged at 9,000 g for 10 min and the resulting mitochondrial fractions were resuspended in 500 μl of MIB and transferred to 1.5 ml microcentrifuge tubes. The mitochondrial pellets were subject to a final wash-spin in a microcentrifuge at 9,000 g for 10 min, the supernatant fractions were discarded and the pellets were resuspended in a final volume of 120 μl of MIB. Fresh mitochondria were used immediately for respiration and ROS assays.
Mitochondrial oxygen consumption
Fresh mitochondria (100 μl) were incubated with 200 μl of VO2 buffer (250 mM sucrose, 50 mM KCl, 25 mM Tris base, 10 mM K2HPO4, pH 7.4), and oxygen consumption was measured at 30°C with continuous stirring in a Clark electrode respiratory chamber (Strathkelvin Instruments, North Lanarkshire, Scotland) in the presence of 10 mM glutamate (Sigma Aldrich, G1505) to assess state 4 respiration, followed by glutamate plus 0.44 mM ADP (Sigma Aldrich, 2754) to elicit state 3 respiration. NADH (Sigma Aldrich, N0505) addition during state 3 respiration was used to evaluate the integrity of the inner mitochondrial membrane.
ROS detection
Mitochondrial levels of ROS were quantified using 2ʹ,7ʹ dichlorodihydrofluorescein diacetate (D2DCFDA; Invitrogen, D399). When oxidized, the compound is converted to the highly fluorescent DCF, with an emission spectrum between 480–520 nm, proportional to levels of ROS. Mitochondria (75 μg) were isolated as described and incubated in VO2 buffer and D2DCFDA for 45 min at 37°C, with glutamate (state 4) or glutamate and ADP (state 3). Fluorescence was assessed in a microplate reader using KC4 software and corrected for oxygen consumption for the corresponding state of respiration.
RNA isolation and mrna expression analysis
Total RNA was isolated from cultured C2C12 myotubes using TRIzol reagent (Ambien, 15,596,018) according to the manufacturer’s instructions. RNA concentration and quality was assessed using spectrophotometry (Ultrospec 2100; Biochrom, UK) and further verified with RNA gels. The mRNA expression was quantified using the StepONE Plus PCR System (Applied Biosystems, CA, USA) and SYBR® Green Supermix (Quanta Biociences, 95,056–100). First-strand cDNA synthesis from 2 μg of total RNA was performed with primers using Superscript III reverse transcriptase and Oligo(dt)20 (Invitrogen, 18,418,020) according to the manufacturer’s instructions. Forward and reverse primers were optimized to verify primer efficiency and dissociation melt curves were analyzed for primer specificity. All samples were run in duplicate, simultaneously with negative controls that contained no cDNA. Primers used are described in Table 1. Transcript levels were normalized to 2 housekeeping genes, Gapdh and Actb, and analyzed using the 2−ΔΔCt method. Statistical significance was calculated with a two-way ANOVA followed by Bonferroni post-hoc tests on the unlogged ΔCt values.
Table 1.
List of primer oligonucleotide sequences used in real-time qPCR analysis for Mus musculus.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Map1lc3b | 5ʹ-GCTTGCAGCTCAATGCTAAC-3’ | 5ʹ-CCTGCGAGGCATAAACCATGT-3’ |
| Pink1 | 5ʹ-GCTTGCCAATCCCTTCTATG-3’ | 5ʹ-CTCTCGCTGGAGCAGTGAC-3’ |
| Park2 | 5ʹ-TGTGACCTGGAACAACAGAGTA-3’ | 5ʹ-TCAGGTCCACTCGTGTCAA-3’ |
| Bnip3l | 5ʹ-TGAGTGACAGACAGGAAACAGA-3’ | 5ʹ-GGCCTGAAACATTCCTTACAA-3’ |
| Sqstm1 | 5ʹ- TGTGGTGGGAACTCGCTATAA-3ʹ, | 5ʹ-CAGCGGCTATGAGAGAAGCTAT-3’ |
| Ppargc1a | 5ʹ-TTCCACCAAGAGCAAGTAT-3’ | 5ʹ-CGCTGTCCCATGAGGTATT-3’ |
| Tfeb | 5ʹ-AGCTCCAACCCGAGAAAGAGTTTG-3’ | 5ʹ- CGTTCAGGTGGCTGCTAGAC-3’ |
| Gapdh | 5ʹ- AACACTGAGCATCTCCCTCA-3’ | 5ʹ-GTGGGTGCAGCGAACTTTAT-3’ |
| Actb | 5ʹ-TGTGACGTTGACATCCGTAA-3’ | 5ʹ-GCTAGGAGCCAGAGCAGTAA-3’ |
Fluorescence microscopy
C2C12 myoblasts were plated in 6-well dishes on a circular glass cover slip coated with 0.1% gelatin. At approximately 80% confluence, myoblasts were co-transfected with pBABE GFP-LC3 (Cell Biolabs, CBA-401) and pDs-Red2-Mito (Clontech, PT3633–5) using Lipofectamine 2000 (Invitrogen, 11,668–019). GFP-LC3 allows for visualization of autophagosomes upon maturation (LC3-II) while Ds-Red localizes to mitochondria through the COX8 mitochondrial-targeting sequence. Following transfection, myoblasts were cultured in differentiation medium (DMEM, 5% heat-inactivated horse serum [GIBCO, 16,050–114], 1% P-S) to form myotubes. Myotubes were subjected to 4 days of CCA (3 h/day). Following the last 21-h recovery, cells were washed twice with warm PBS then fixed in 4% paraformaldehyde solution for 15 min. Myoblasts were washed twice for 5 min with PBS then cover slips were lifted from the 6-well dish and allowed to dry while protected from light. Cover slips were mounted on microscope slides with ProLong Diamond Antifade Mountant (ThermoFisher, p36961) and sealed with nail polish. Myotube fluorescence was visualized using a laser scanning Nikon C2 Confocal with a 60x oil-immersion objective. Laser intensity was constant across all conditions and fluorescence of green and red was captured in channel series to prevent color bleedthrough. Representative images are shown of n = 3 experiments with ~ 5 myotubes visualized per condition.
Cytochrome c oxidase (COX) activity assay
Cells were harvested 21 h after the last stimulation period and measurement of COX activity was performed as previously described [7]. Briefly, cells were resuspended in 120 μl of enzyme extraction buffer (71.4 mM Na2HPO4, 28.6 mM KH2PO4-phosphate, 2 mM EDTA, pH 7.2), sonicated 3 × 3 sec on ice, and subjected to repeated freeze-thaw cycles in liquid N2. The supernatant fraction containing the enzyme extracts was removed and used to measure COX enzyme activity. COX activity was measured over time as a reduction in absorbance at 550 nm of reduced equine CYCS/cytochrome c, and activity was quantified using a 96-well plate in a microplate reader (Bio-Tek Synergy HT, Winooski, VT, USA) at 30°C. The data were compiled using KC4 software and corrected for protein concentration to determine COX activity.
Immunoblotting and protein extraction
Cultured myotubes were washed 2x with ice-cold PBS and scraped in Passive Lysis Buffer (Promega, E194A) supplemented with Complete Protease Inhibitor Cocktail Tablets (Roche, 11,697,498,001). Following extractions, protein was quantified using the Bradford method (ethanol, phosphoric acid, Brilliant Blue G [Sigma-Aldrich, B0770]). Whole cell and mitochondrial fraction extracts (20–40 μg of protein) were separated using 8–15% SDS-PAGE and then transferred to nitrocellulose membranes. Membranes were blocked in 5% skim milk in TBST buffer (100 mM TRIS, 100 mM NaCl, 0.1% Tween 20 [Sigma, P1379]) for 1 h, and then incubated overnight at 4°C with primary antibodies directed against COX4I1 (1:1000; Abcam, ab14744), SOD2 (1:000; Upstate Biotechnology, 06–984), BNIP3L/NIX (1:500; Abcam, ab109414), LC3B (1:1000; Cell Signaling Technology, 2775), SQSTM1/p62 (1:40,0000; Sigma-Aldrich, P0067), PGM5/ACICULIN (1:500; in house), VDAC (1:5000; Abcam, ab1473), PPARGC1A/PGC1α (1:1000; Millipore Corporation, ab3243), p-RPS6KB (1:1000, Cell Signaling Technology, 9202), pan-LAMP2 (abcam, ab13524; 1:1000), CTSD (1:1000; Santa Cruz Biotechnology, SC6486), Tubulin (1:90,000, Calbiochem, CP06), ACTB (1:1000, Santa Cruz Biotechnology, 47,778) and TFEB (1:1000; MyBioSource, mbs120432). Secondary antibodies were used as per the manufacturer’s suggestions (Santa Cruz Biotechnology, SC2031; SC2054).
Statistical analysis
The means and standard errors were calculated for all measured values, and statistical significance between groups was determined by ANOVA with Boneferroni post-hoc tests (Graph Pad, La Jolla, CA, USA). Results were considered statistically significant when P ≤ 0.05. Graph bars represent the means and SEM.
Funding Statement
This work was supported by the Government of Canada | Natural Sciences and Engineering Research Council of Canada (NSERC) [38462].
Acknowledgments
D.A. Hood is the holder of a Canada Research Chair in Cell Physiology. This research was supported by funding from NSERC of Canada.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hood DA. Invited review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001;90:1137–1157. [DOI] [PubMed] [Google Scholar]
- [2].Gielen S, Sandri M, Kozarez I, et al. Exercise training attenuates MuRF-1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized leipzig exercise intervention in chronic heart failure and aging catabolism study. Circulation. 2012;125:2716–2727. [DOI] [PubMed] [Google Scholar]
- [3].Zinna EM, Yarasheski KE. Exercise treatment to counteract protein wasting of chronic diseases. Curr Opin Clin Nutr Metab Care. 2003;6:87–93. [DOI] [PubMed] [Google Scholar]
- [4].Memme JM, Oliveira AN, Hood DA. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am J Physiol Cell Physiol. 2016;310:C1024–C1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim Y, Hood DA. Regulation of the autophagy system during chronic contractile activity‐induced muscle adaptations. Physiol Rep. 2017;5:e13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carter HN, Hood DA. Contractile activity-induced mitochondrial biogenesis and mTORC1. Am J Physiol Cell Physiol. 2012;303:C540–547. [DOI] [PubMed] [Google Scholar]
- [7].Uguccioni G, Hood DA. The importance of PGC-1α in contractile activity-induced mitochondrial adaptations. Am J Physiol Endocrinol Metab. 2011;300:E361–71. [DOI] [PubMed] [Google Scholar]
- [8].Zhang Y, Uguccioni G, Ljubicic V, et al. Multiple signaling pathways regulate contractile activity-mediated PGC-1 gene expression and activity in skeletal muscle cells. Physiol Rep. 2014;2:e12008–e12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Erlich AT, Brownlee DM, Beyfuss K, et al. Exercise induces TFEB expression and activity in skeletal muscle in a PGC-1$α$-dependent manner. Am J Physiol Cell Physiol. 2017; 314, C62–C72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ljubicic V, Joseph A-M, Adhihetty PJ, et al. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany NY). 2009;1:818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yan Z, Lira VA, Greene NP. Exercise training-induced regulation of mitochondrial quality. Exerc Sport Sci Rev. 2013; 40, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vainshtein A, Hood DA. The regulation of autophagy during exercise in skeletal muscle. J Appl Physiol. 2016;120:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang Q, Mao Z. Dysregulation of autophagy and Parkinson’s disease: the MEF2D link. Apoptosis. 2010;15:1410–1414. [DOI] [PubMed] [Google Scholar]
- [14].Albin R, Dowell RT, Zak R, et al. Synthesis and degradation of mitochondrial components in hypertrophied rat heart. Biochem J. 1973;136:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–4465. [DOI] [PubMed] [Google Scholar]
- [16].Jiang X, Li X, Huang H, et al. Elevated levels of mitochondrion-associated autophagy inhibitor LRPPRC are associated with poor prognosis in patients with prostate cancer. Cancer. 2014;120:1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lehman JJ, Barger PM, Kovacs A, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Petrosillo G, Ruggiero FM, Paradies G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 2003;17:2202–2208. [DOI] [PubMed] [Google Scholar]
- [19].Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ishikawa K, Kimura S, Kobayashi A, et al. Increased reactive oxygen species and anti-oxidative response in mitochondrial cardiomyopathy. Circ J. 2005;69:617–620. [DOI] [PubMed] [Google Scholar]
- [21].Murphy MP. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 2013;18:145–146. [DOI] [PubMed] [Google Scholar]
- [22].Kumari U, Ya Jun W, Huat Bay B, et al. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene. 2014;33:165–172. [DOI] [PubMed] [Google Scholar]
- [23].Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shokolenko IN, Wilson GL, Alexeyev MF. Persistent damage induces mitochondrial DNA degradation. DNA Repair (Amst). 2013;12:488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. [DOI] [PubMed] [Google Scholar]
- [26].Wang Y, Nartiss Y, Steipe B, et al. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–1476. [DOI] [PubMed] [Google Scholar]
- [27].Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou C, Huang Y, Shao Y, et al. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA. 2008;105:12022–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taymans J-M, Van Den Haute C, Baekelandt V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem. 2006;98:951–961. [DOI] [PubMed] [Google Scholar]
- [30].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. [DOI] [PubMed] [Google Scholar]
- [31].Panicker N, Dawson VL, Dawson TM. Activation mechanisms of the E3 ubiquitin ligase parkin. Biochem J. 2017;474:3075–3086. [DOI] [PubMed] [Google Scholar]
- [32].Narendra D, Tanaka A, Suen D, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. [DOI] [PubMed] [Google Scholar]
- [34].Otomo C, Metlagel Z, Takaesu G, et al. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. [DOI] [PubMed] [Google Scholar]
- [36].Lim J, Kim H-W, Youdim MBH, et al. Binding preference of p62 towards LC3-ll during dopaminergic neurotoxin-induced impairment of autophagic flux. Autophagy. 2011;7:51–60. [DOI] [PubMed] [Google Scholar]
- [37].Rahman M, Mofarrahi M, Kristof AS, et al. Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxidants {&} Redox Signal. 2013. g20:443–459. [DOI] [PubMed] [Google Scholar]
- [38].Vainshtein A, Tryon LD, Pauly M, et al. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308:C710–C719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ferraro E, Giammarioli AM, Chiandotto S, et al. Exercise-induced skeletal muscle remodeling and metabolic adaptation: redox signaling and role of autophagy. Antioxid Redox Signal. 2014;21:154–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jiang D, Chen K, Lu X, et al. Exercise ameliorates the detrimental effect of chloroquine on skeletal muscles in mice via restoring autophagy flux. Acta Pharmacol Sin. 2014;35:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nair U, Klionsky DJ. Activation of autophagy is required for muscle homeostasis during physical exercise. Autophagy. 2011;7:1405–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Grumati P, Coletto L, Schiavinato A, et al. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy. 2011;7:1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lira VA, Okutsu M, Zhang M, et al. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013;27:4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ju JS, Jeon SI, Park JY, et al. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J Physiol Sci. 2016;66:417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Drummond MJ, Addison O, Brunker L, Hopkins PN, McClain DA, LaStayo PC & Marcus RL (2014). Downregulation of e3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J Gerontol A Biol Sci Med Sci 69, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging, Eur. J Biochem. 2002;269:1996–2002. [DOI] [PubMed] [Google Scholar]
- [49].Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am J Physiol Cell Physiol. 2009;296:C116–C123. [DOI] [PubMed] [Google Scholar]
- [50].Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol. 2016;6:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Grumati P, Coletto L, Sabatelli P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–1320. [DOI] [PubMed] [Google Scholar]
- [52].Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. [DOI] [PubMed] [Google Scholar]
- [53].Hiniker A, Daniels BH, Lee HS, et al. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun. 2013;1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jamart C, Naslain D, Gilson H, et al. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state, Am. J Physiol Endocrinol Metab. 2013;305:964–974. [DOI] [PubMed] [Google Scholar]
- [55].Lokireddy S, Wijesoma IW, Teng S, et al. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012;16:613–624. [DOI] [PubMed] [Google Scholar]
- [56].Yang Y, Xing D, Zhou F, et al. Mitochondrial autophagy protects against heat shock-induced apoptosis through reducing cytosolic cytochrome c release and downstream caspase-3 activation. Biochem Biophys Res Commun. 2010;395:190–195. [DOI] [PubMed] [Google Scholar]
- [57].Burch N, Arnold A-S, Item F, et al. Electric pulse stimulation of cultured murine muscle cells reproduces gene expression changes of trained mouse muscle. PLoS One. 2010;5:e10970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Heinzel FR, Luo Y, Dodoni G, et al. Formation of reactive oxygen species at increased contraction frequency in rat cardiomyocytes. Cardiovasc Res. 2006;71:374–382. [DOI] [PubMed] [Google Scholar]
- [59].Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tsunemi T, Ashe TD, Morrison BE, et al. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Saleem A, Carter HN, Hood DA. p53 is necessary for the adaptive changes in the cellular milieu subsequent to an acute bout of endurance exercise. Am J Physiol Cell Physiol. 2014;306:C241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Aucello M, Dobrowolny G, Musarò A. Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy. 2009;5:527–529. [DOI] [PubMed] [Google Scholar]
- [63].Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. [DOI] [PubMed] [Google Scholar]
- [64].Mansueto G, Armani A, Viscomi C, et al. Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 2017;25:182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Follo C, Ozzano M, Montalenti C, et al. Knockdown of cathepsin D in zebrafish fertilized eggs determines congenital myopathy. Biosci Rep. 2013;33:e00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nascimbeni AC, Fanin M, Angelini C, et al. Autophagy dysregulation in Danon disease. Cell Death Dis. 2017;8:e2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tarnopolsky M, Katzberg H, Petrof BJ, et al. Pompe disease: diagnosis and management. Evidence-based guidelines from a Canadian expert panel. Can J Neurol Sci/J Can Des Sci Neurol. 2016;43:472–485. [DOI] [PubMed] [Google Scholar]
- [68].Hashem SI, Murphy AN, Divakaruni AS, et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J Mol Cell Cardiol. 2017;108:86–94. [DOI] [PubMed] [Google Scholar]
- [69].Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy, Annu. Rev Med. 2015;66:471–486. [DOI] [PubMed] [Google Scholar]
- [70].Lo Verso F, Carnio S, Vainshtein A, et al. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy. 2014;10:1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Connor MK, Irrcher I, Hood DA. Contractile activity-induced transcriptional activation of cytochrome c involves Sp1 and is proportional to mitochondrial ATP synthesis in C2C12 muscle cells, J. Biol Chem. 2001;276:15898–15904. [DOI] [PubMed] [Google Scholar]
- [72].Mesbah Moosavi ZS, Hood DA. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am J Physiol Cell Physiol. 2017;312:C583–C594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Maitra PK, Estabrook RW. Studies of baker’s yeast metabolism. II. The role of adenine nucleotides and inorganic phosphate in the control of respiration during alcohol oxidation. Arch Biochem Biophys. 1967;121:129–139. [DOI] [PubMed] [Google Scholar]