

ABSTRACT
Adaptation to endoplasmic reticulum (ER) stress depends on the activation of the sensor inositol-requiring enzyme 1α (IRE1), an endoribonuclease that splices the mRNA of the transcription factor XBP1 (X-box-binding protein 1). To better understand the protein network that regulates the activity of the IRE1 pathway, we systematically screened the proteins that interact with IRE1 and identified a ribonuclease inhibitor called ribonuclease/angiogenin inhibitor 1 (RNH1). RNH1 is a leucine-rich repeat domains-containing protein that binds to and inhibits ribonucleases. Immunoprecipitation experiments confirmed this interaction. Docking experiments indicated that RNH1 physically interacts with IRE1 through its cytosolic RNase domain. Upon ER stress, the interaction of RNH1 with IRE1 in the ER increased at the expense of the nuclear pool of RNH1. Inhibition of RNH1 expression using siRNA mediated RNA interference upon ER stress led to an increased splicing activity of XBP1. Modulation of IRE1 RNase activity by RNH1 was recapitulated in a cell-free system, suggesting direct regulation of IRE1 by RNH. We conclude that RNH1 attenuates the activity of IRE1 by interacting with its ribonuclease domain. These findings have implications for understanding the molecular mechanism by which IRE1 signaling is attenuated upon ER stress.
KEYWORDS: Endoplasmic reticulum stress, IRE1, RNH1, XBP1, Unfolded Protein Response
Introduction
A wide range of cellular conditions can disrupt the efficiency of protein folding in the endoplasmic reticulum (ER) and lead to the accumulation of misfolded proteins within this organelle, a state known as ‘‘ER stress’’. Adaptation to ER stress is mediated through the Unfolded Protein Response (UPR), an integrated signaling pathway transduced by the three ER stress sensors ATF6 (Activated Transcription Factor 6), IRE1α (Inositol Requiring Enzyme 1α) and PERK (Protein Kinase RNA (PKR)-like ER kinase). Of particular interest in the UPR is the IRE1α-XBP1 axis. IRE1α (hereafter referred to as IRE1) signals by assembling a dynamic protein platform referred to as the UPRosome, in which different modulator and adaptor proteins assemble to regulate the kinetics and amplitude of UPR effector responses [1,2]. IRE1 is a type I transmembrane protein that contains a serine/threonine kinase and endoribonuclease (RNase) domain on its cytosolic face. In response to unfolded/misfolded protein accumulation in the ER, IRE1 oligomerizes and trans-autophosphorylates, thereby inducing a conformational change that activates its RNase domain to catalyze the excision of a 26-nt intron within the XBP1 mRNA [3,4]. Together with the concerted action of the tRNA ligase RtcB [5], this leads to the unconventional splicing of XBP1 mRNA that shifts its open reading frame and is translated into an active transcription factor, known as Spliced XBP1 (sXBP1). sXBP1 controls the expression of genes that encode factors that modulate protein folding, secretion, ER-associated degradation (ERAD), protein translocation into the ER, and lipid synthesis [6]. In addition, active IRE1 RNase is involved in RNA degradation through Regulated IRE1 Dependent Decay (RIDD), a process critical for ER homeostasis maintenance [7]. Thus, the ribonuclease activity of IRE1 is critical for cell adaptation capacity under stress conditions.
To investigate the regulation of IRE1, several protein-protein interactions approaches were conducted with IRE1 leading to the concept of the UPRosome, of which some IRE1 partners regulate the amplitude and duration of IRE1 signals [1,8]. Several examples suggest that the association of these regulators with IRE1 potentiate its activity. Indeed, the amplitude of IRE1 signaling is controlled at the ER membrane by the formation of a protein complex between bcl-2-like protein 4- Bcl-2 homologous antagonist/killer (BAX–BAK) and the cytosolic domains of IRE1 [9]. Similarly, the Abelson (ABL) kinase scaffolds and hyperactivates IRE1 [10]. IRE1 signaling is also induced by a physical interaction with pro-apoptotic ASK1-interacting protein 1 (AIP1) which facilitates IRE1 oligomerization [11]. Heat shock proteins HSP72 and HSP90 were identified as IRE1 interactors that potentiate its signaling in maintaining its stability [12,13], and nonmuscle myosin-IIB (NMIIB) and the actin cytoskeleton contribute to stabilizing IRE1 clusters for optimal activation [14]. In contrast, the mechanisms by which IRE1 gets inactivated remain elusive. Dephosphorylation mechanisms were first identified through the action of diverse phosphatases to control its phosphorylation/activation status [15–18]. The control of IRE1 expression levels is also involved in IRE1 signaling either through the RIDD of its own mRNA [19], or its degradation mediated by Protein Disulfide Isomerase (PDI) A6 [20]. The formation of stable interaction with Bax inhibitor 1 (BI-1) or Fortilin with IRE1 inhibits its activity upon sustained ER stress [21,22]. However, all these mechanisms do not provide a clear mechanism for tight and direct regulation of the activity of IRE1 RNase.
Here, in an attempt to identify new IRE1 regulator proteins, we performed a proteomic study and identified an association between the ribonuclease inhibitor RNH1 and IRE1. We demonstrated that RNH1 directly interacts with IRE1 and inhibits IRE1 ribonuclease activity upon late ER stress, thereby suggesting that RNH1 might represent the first direct regulator of IRE1 RNase activity.
Results
RNH1 interacts with IRE1 at the surface of the ER
To search for new proteins that might associate with and interact with IRE1, we performed endogenous IRE1 immunoprecipitation (IP) followed by tandem mass spectrometry (MS/MS). This approach led to the identification of proteins such as Filamin A, Tubulin α and β, Vimentin, glucose regulated protein (GRP) 78, HSP72 and pyruvate kinase, which are known to interact with IRE1 or to belong to the UPRosome [2,23] (Figure 1(A)). In addition, we identified ribonuclease/angiogenin inhibitor 1 (RNH1) (also known as placental ribonuclease inhibitor, PRI) in IRE1-IP (Figure 1(A)). Using in the same extracts than those used for MS/MS, we verified the presence of endogenous RNH1/IRE1 complexes when IRE1 was immunoprecipitated followed by immunoblotting (Figure 1(B)). Notably, Tunicamycin (an ER stress inducer) increased the ratio of RNH1 to immunoprecipitated IRE1, indicating that IRE1 may be enriched with RNH1 upon ER stress (the amount of IRE1 in IPs under ER stress was consistently lower compared with control conditions in our hands).
Figure 1.
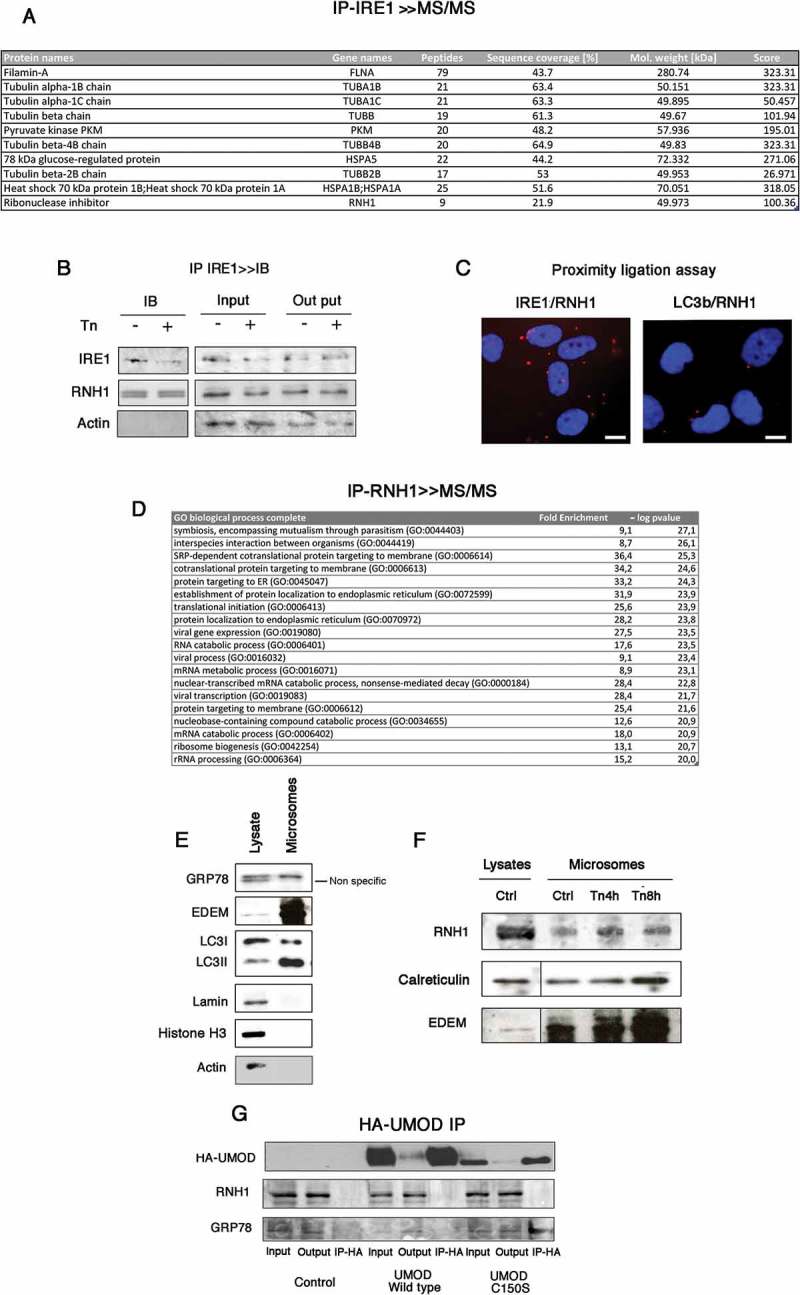
RNH1 interacts with IRE1 at the surface of the ER. A. Endogenous IRE1 was immunoprecipitated (IP) followed by tandem mass spectrometry. The IPs presented were processed as described in the Materials and Methods and analyzed by tandem mass spectrometry. Peptides identified for RNH1 and proteins known to interact with IRE1 in the analysis are indicated. B. Endogenous interaction between RNH1 and IRE1 were analyzed by immunoblotting after the IP of IRE1. Cells were incubated or not with 2.5 μg/l tunicamycin for 4 h. IRE1 was immunoprecipitated and analyzed by western blot to detect RNH1 and IRE1 expression. Data are representative of 3 independent experiments. Input refers to the lysate before IP, and output to the lysate after IP. C. Proximity ligation assay was performed using antibodies directed against IRE1, LC3b and RNH1. Red dots indicate proximity between IRE1 and RNH1 of less than 40 nm. Bar represents 10 μm D. Endogenous RNH1 was immunoprecipitated (IP), followed by tandem mass spectrometry. Proteins were annotated using the Gene Ontology Biological process, and enrichments analyses were performed using the PANTHER Overrepresentation Test on a list of 144 proteins identified in 3 independent experiments. The GO terms in red are associated with a p value < 10−20 after Bonferroni correction for multiple testing. E. Western-blot analysis of microsomes preparations and whole cell lysate for the expression of ER markers, nucleus marker and cytoplasmic markers. F. Western-blot analysis of microsome preparations and whole cell lysate for the expression of ER markers, nucleus marker and cytoplasmic markers. G. Cells were incubated or not with 2.5 μg/l tunicamycin for up to 8 h and then microsomes and protein extracts were prepared. RNH1, Calreticulin and EDEM expression in cell lysates and microsomes was analyzed by western blotting. Data are representative of 3 independent experiments. H. Cells were transfected with an expression vector for UMOD wild type-HA or mutated UMOD C150S or empty vector. Western blots for GRP78 and RNH1 were performed on IP-HA. Data are representative of 3 independent experiments.
To further demonstrate the interaction of endogenous RNH1 with IRE1, we used a Proximal Ligation Assay that identifies individual interactions between two proteins in their native form [24]. Consistent with the results of the IP, red dots indicative of the proximity between RNH1 and IRE1 were found throughout the cytoplasm whereas microtubule-associated proteins 1A/1B light chain 3B (LC3b), a cytoplasmic protein did not interact with RNH1 (Figure 1(C)), confirming that IRE1 physically interacts with RNH1 with some specificity.
RNH1 is known as a nuclear and cytoplasmic protein (www.proteinatlas.org). We reasoned that the corollary of the interaction of IRE1 with RNH1 is that RNH1 should be located at the ER. To do this, we characterized the RNH1 interactome after endogenous RNH1 immunoprecipitation followed by tandem mass spectrometry. Consistent with this hypothesis, the most significantly enriched (higher size effect and lower p values) Gene Ontology Biological Functions in the RNH1-IP were related to ER-related functions, including translation (Figure 1(D)), which is in line with a recently published RNH1 interactome [25]. To directly demonstrate that RNH1 is localized to the ER, we isolated microsomes by ultracentrifugation. Compared with whole cell lysates, microsome preparations were enriched in ER resident proteins, such as ER degradation-enhancing α-mannosidase-like protein (EDEM), GRP78, and phosphatidylethanolamine conjugated LC3 [26], and was completely devoid of nuclear and cytoplasmic components, reflecting the purity of the fraction (Figure 1(E)). RNH1 was present in these microsomes preparation and, as expected, at lower levels compared with total lysate RNH1, and its presence increased upon ER stress, as did the ER-resident proteins calreticulin and EDEM, which expression is increased during ER stress (Figure 1(F)). These results indicate that RNH1 is localized at the vicinity of the ER. To provide further information about the localization of RNH1 on the ER (cytoplasmic or luminal), we reasoned that if RNH1, which carries a hydrophobic leucine rich repeat domain, was present within the ER lumen, it could interact with misfolded proteins, which exposes hydrophobic residues. To test this hypothesis, we overexpressed C150S UMOD, a misfolded protein that accumulates within the ER lumen and promotes ER stress [27–29]. C150S UMOD, and not wild-type UMOD, interacted with the chaperone GRP78, as expected, but not with RNH1, indicating that RNH1 likely does not interact with misfolded proteins within the ER lumen (Figure 1(G)). These results are consistent with the fact that RNH1 lacks a signal peptide, and therefore should not be present within the ER lumen. Together, these results indicate that RNH1 forms a dynamic protein complex with IRE1 at the surface of the ER.
RNH1 physically interacts with IRE1
RNH1 is composed of seven leucine-rich repeats and a conserved structure domain that is often involved in protein–protein interactions [30]. The cytoplasmic region of IRE1 encompasses a protein kinase domain (n-lobe and c-lobe) followed by a C-terminal kinase-extension nuclease (KEN) domain, and dimerization of the kinase domain activates the ribonuclease function of the protein [31]. To provide insights into how IRE1 and RNH1 interact with each other, we modeled their interaction based on the published structures of the 3P23 crystal structure of the human kinase and RNase domain of IRE1 (residues 547 to 966) [31] and the 2Q4G crystal structure of RNH1 [32]. The docking studies demonstrated that the most probable interaction pattern was with the KEN domain (residues 835 to 963) (Figure 2(A)). The candidate residues of RNH1 interacting with IRE1 appear to lie on each loop, as it passes from a α-helix to a β-strand. The contacts with the IRE1-KEN domain are distributed over the domain 6 helices (Figure 2(B)). This conformation is reminiscent of the interaction of RNH1 with angiogenin (RNASE5) [33] (Figure 2(C)). The vast majority of the residues of the RNH1-IRE1-KEN interface are charged, and the minority appears to be hydrophobic (Figure 2(D,E)), suggesting that the interaction is largely governed by electrostatic interactions. We tested the nature of the IRE1-RNH1 interaction by incubating RNH1-IP in different solvents, followed by IRE1 IB. Disrupting weak hydrophobic interactions by adding 1,6 hexandiol was not sufficient to reduce the IRE1-RNH1 stability. However, incubation with 2 M urea significantly reduced the quantity of IRE1 bound to RNH1-IP, indicating that the stability of the interaction is due to electrostatic interactions (hydrogen bonds) (Figure 2(F)). In summary, the docking analysis of IRE1 with RNH1 indicates that the interaction is stabilized by hydrogen bonds between polar residues located at the line of the β–α loops of RNH1 and α-helix of the “KEN” RNase domain of IRE1.
Figure 2.
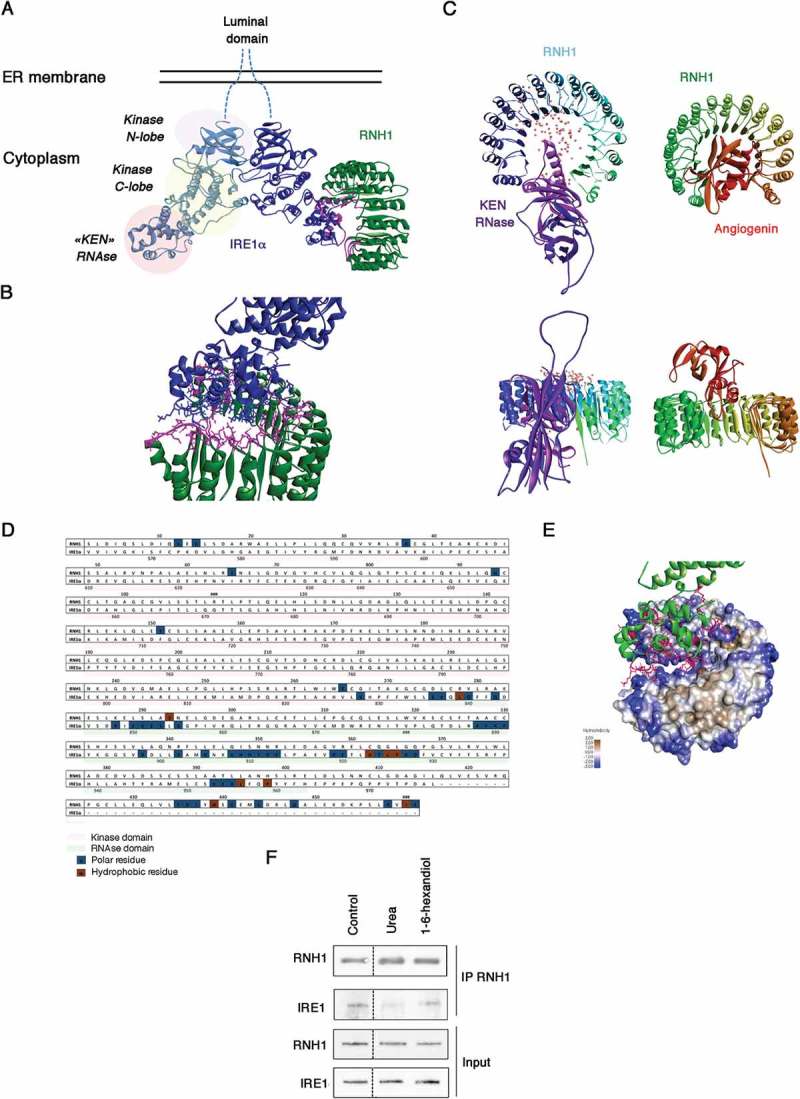
Physical interaction between RNH1 and IRE1. A. Docking studies on the interaction of the cytoplasmic domains of IRE1 in dimeric form, with RNH1. The 3D structure of the human kinase and KEN RNase domains are shown in blue, and the 3D structure of RNH1 is shown in green. The most probable interaction interfaces between the alpha helices of the IRE1-KEN domain and α-to-α or α-to-β loops of RNH1 are shown in turquoise. B. Enlargement of the putative interface between IRE1 and RNH1. The Cα atoms plus side chains of the contact residues involved in this interaction are shown. C. Overall view of the RNH1-KEN-domain (left) and RNH1-ANG dimer (right). Upper panel, parallel to the plane of the horseshoe, and lower panel, perpendicular to the plane of the horseshoe). The probability of the interaction is color-coded and increases with the intensity of the color of the dots. D. Amino acid sequence alignment of the RNH1 and IRE1 cytoplasmic domains. The contact residues are shown in boxes: polar resides are shown in blue, and hydrophobic residues are shown in brown. E. Molecular surface of RNH1 and the interface with the KEN domain of IRE1. Polarity is color-coded: from blue for the less hydrophobic to brown for the most hydrophobic resides. F. RNH1 was immunoprecipitated and then incubated with urea or 1–6-hexanediol to destabilize electrostatic or hydrophobic interactions. Western blotting was performed for the detection of RNH1 and IRE1.
ER stress increases the RNH1-ire1α interaction
To characterize the dynamic aspect of the IRE-RNH1 interaction, we performed an IP of endogenous RNH1 under basal and ER stress conditions (up to 4 hours with 2.5 μg/ml tunicamycin). We then validated the interaction of IRE1 with endogenous RNH1 protein and found that tunicamycin enhanced the association of RNH1 with IRE1 (Figure 3(A)). Consistent with the results of the IP, the number of red dots (indicative of the proximity between RNH1 and IRE1) provided by Proximity Ligation Assay increased and became statistically significant after 4 hours of exposure to tunicamycin (Figure 3(B)). These results indicate that ER stress facilitates the interaction of RNH1 and IRE1. We next examined the possible contribution of different cellular pools of RNH1 in the interaction with IRE1 and their relation to ER stress. Tunicamycin-induced ER stress did not impact whole cell RNH1 transcripts (Figure 3(C)) and protein (Figure 3(D)) expression levels. As a control, we included expression of the ER chaperon GRP78, which as expected, was clearly increased upon ER stress. In line with the enhanced interaction of RNH1 with the ER resident protein IRE1, the microsomal pool of RNH1 was enriched (Figure 3(E)). Since RNH1 is classically distributed in the cytoplasm and nucleus [34], we tested whether enrichment of ER in RNH1 could be obtained at the expense of the cytosolic or nucleus pool. IB performed in nucleus extracts of cells incubated with tunicamycin revealed a drastic reduction of RNH1 contents after 4 hours (Figure 3(F)), whereas virtually no changes were observed in cytosolic preparations (Figure 3(G)). These results support a process during which ER stress promotes shuttling of RNH1 from the nucleus to the ER and increased its interaction with IRE1.
Figure 3.
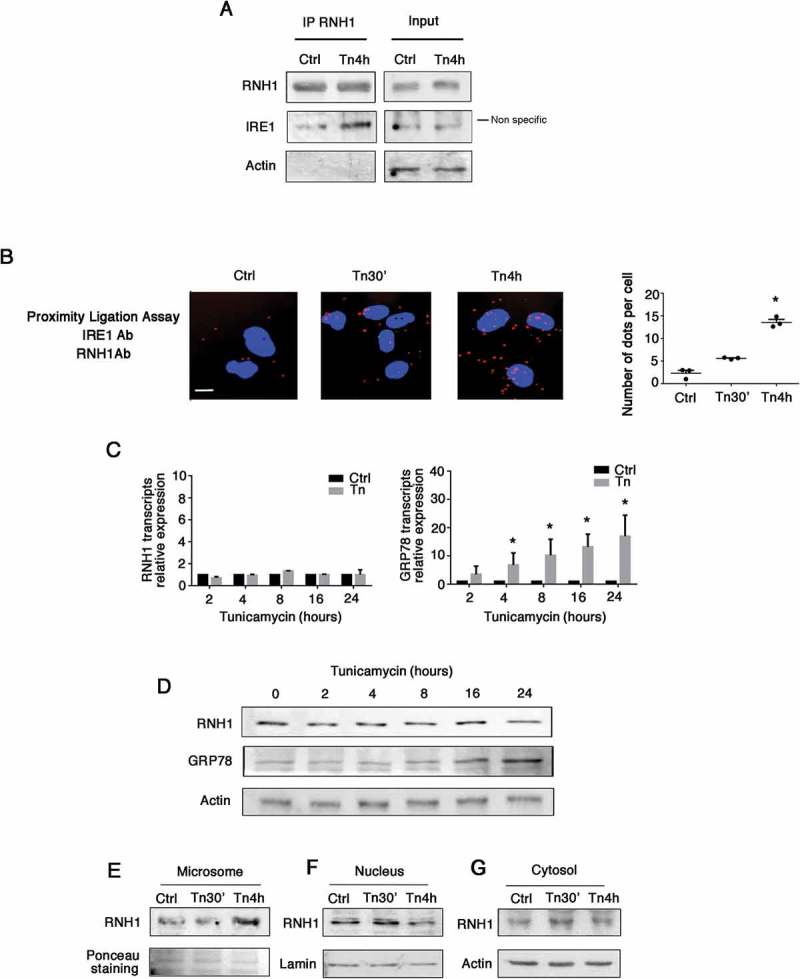
ER stress increases RNH1-IRE1 interaction. A. Cells were incubated with 2.5 μg/ml tunicamycin for 4 hours. RNH1-IP was performed and then subjected to western blotting for the detection of RNH1, IRE1, actin and histone H3. B. Cells were incubated with 2.5 μg/ml tunicamycin for up to 4 hours, and a proximity ligation assay was performed using antibodies directed against IRE1 and RNH1. Red dots indicate proximity between IRE1 and RNH1 of less than 40 nm. Dots were manually counted in 3 power fields per experiment for 3 experiments. *, p < 0.05, T test, compared with control condition. Bar represents 10 μm. C. Cells were incubated with 2.5 μg/ml tunicamycin for up to 24 hours, and the relative expression levels of RNH1 and GRP78 transcripts were measured by real-time quantitative PCR, using non-treated cells as a control and RPL13A as the reference gene. D. Cells were incubated with 2.5 μg/ml tunicamycin for up to 24 hours, and expression of RNH1 and GRP78 in whole cell lysates was measured by western blotting. *, p < 0.05, T test after Bonferroni correction for multiple testing, compared with control condition E,F,G. Cells were incubated with 2.5 μg/ml tunicamycin for up to 4 hours and the microsomes (E), nuclei (F) and cytosol (G) were separated. Expression of RNH1, lamin and actin was measured by western blotting.
RNH1 reduces IRE1 ribonuclease activity
To explore the putative role of RNH1 in the IRE1 activation profile, we modulated the expression of RNH1. Transcient overexpression of RNH1 did not impact IRE1 activity (not shown). We next inhibited expression of RNH1 using siRNA-mediated RNA interference (Figure 4(A)). Depletion of RNH1 upon ER stress did not impact the phosphorylation levels of IRE1 (using a Phoshotag (Figure 4(B)). To gain insight into the impact of RNH1 depletion on the IRE1 splicing activity, we incubated cells with 2.5 μg/ml tunicamycin, and XBP1 mRNA splicing was assessed by polymerase chain reaction (PCR). To discriminate the two forms of XBP1 mRNA, we developed a highly sensitive method for measuring spliced and unspliced sXBP1 mRNA levels. Because the XBP1 mRNA is 26 nucleotides longer than sXBP1 (Figure 4(C)), we used a performed a fragment analysis to detect changes in the length of the XBP1 mRNA before and after splicing using PCR with fluorescent dye-labeled primers specific for the XBP1 coding sequence. The PCR amplicons were separated according to their sizes and analyzed on a capillary electrophoresis-based DNA sequencing instrument. The fragment (amplicon) corresponding to the unspliced XBP1 mRNA was predicted to be 166 nucleotides, whereas the fragment corresponding to spliced XBP mRNA as predicted to contain 140 nucleotides. In untreated cells, sXBP1 was not detected (Figure 4(C)), and the intensity of the fluorescent peak increased with tunicamycin, whereas the intensity of the unspliced XBP1 peak decreased (Figure 4(C)). Compared with cells transfected with scramble siRNA and incubated with tunicamycin for up to 16 hours, cells transfected with siRNA targeting RNH1 presented an enhancement in the levels of sXBP1 mRNA (Figure 4(D)) and protein (Figure 4(E)), indicating that RNH1 might interfere with IRE1-mediated XBP1 splicing.
Figure 4.
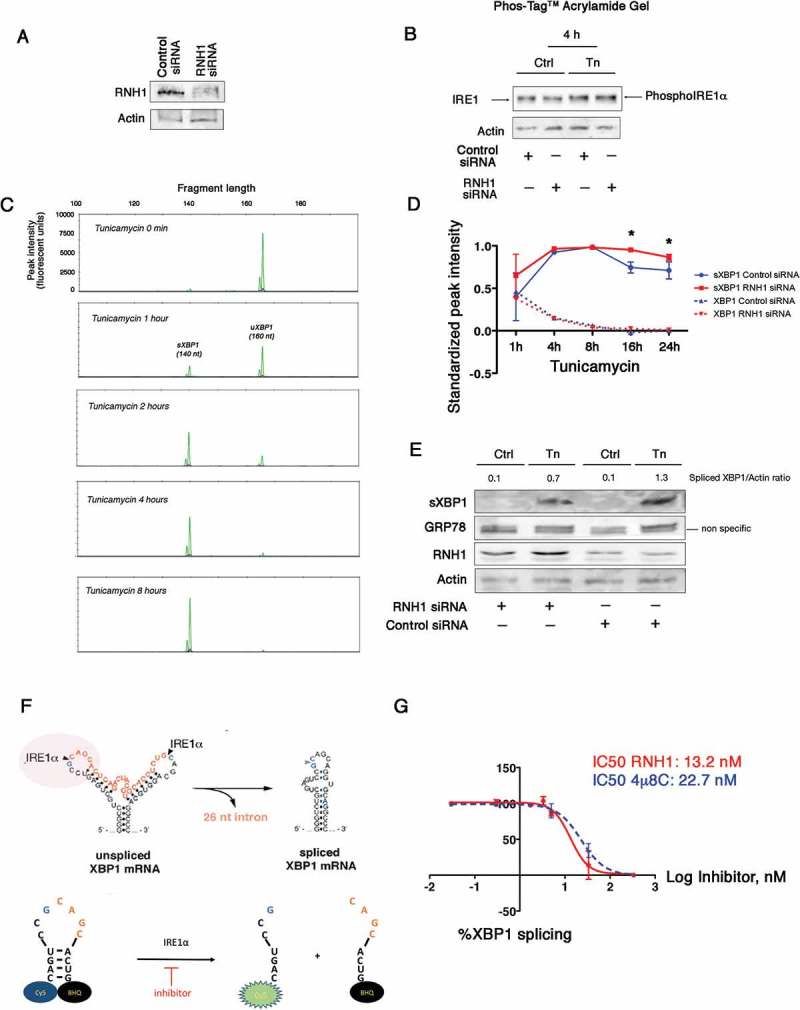
RNH1 reduces IRE1 activity. A. Cells were transfected with siRNA targeting RNH1 or with a scrambled sequence without a known target. RNH1 expression was analyzed by western blotting. B. Cells were incubated with 2.5 μg/ml tunicamycin for up to 4 hours. Cell lysates were separated on a SuperSep™ PhosTag™ gel, which separates proteins according to their phosphorylation status. Expression of IRE1 (phosphorylated or not) was measured by western blotting using antibodies directed toward IRE1. C. Total mRNAs were extracted from cells that had been incubated with 2.5 μg/ml tunicamycin for various periods of time to induce ER stress. sXBP1 and XBP1 amplicons were detected and quantified by fragment analysis after PCR. The pattern of peaks resulting from the fragment analysis using capillary electrophoresis was analyzed with GeneMapper©. Green peaks denote XBP1 (166 nucleotides) and sXBP1 (140 nucleotides) amplicons. Red peaks denote the size standards. The height of each peak corresponds to its relative fluorescence intensity. The x-axis denotes the fragment size. D. Graph representing the variation in XBP1 and sXBP1 amplicons at each time of ER stress induced by tunicamycin according to the RNH1 status of the cell (control/scramble siRNA or RNH1 siRNA). E. Cells were transfected with siRNA targeting RNH1 or with a scrambled sequence without a known target for 24 hours. Whole cell lysates were extracted from cells that had been incubated with 2.5 μg/ml tunicamycin for 8 hours to induce ER stress. sXBP1, RNH1, GRP78 and actin expression were characterized by immunoblotting. F. An in vitro IRE1 RNase activity assay was performed by incubating human recombinant IRE1 with human recombinant RNH1 and the substrate tagged XBP1 RNA stem loop, the cleavage of which would allow the cyanine 5 to fluoresce. G. IRE1 was incubated with increasing concentrations of 4μ8c or RNH1 and the XBP1 RNA stem loop. The % of XBP1 splicing was calculated by dividing the fluorescence for a given concentration of 4μ8C or RNH1 by that of the vehicle control, from which the half maximal inhibitory concentration (IC50) was calculated.
To provide direct evidence that IRE1 activity is inhibited by RNH1, we performed in vitro studies using recombinant human IRE1 incubated with increasing amounts of recombinant human RNH1. We evaluated the activity of recombinant IRE1 using a synthetic mRNA stem loop corresponding to the XBP1 substrate sequence. This stem loop incorporates a Cy5 fluorophore on its 5ʹ end and the black hole quencher (BHQ) on its 3ʹ end, resulting in fluorescence only upon site-specific cleavage by IRE1 (Figure 4(F)). As a positive control, we assayed 4μ8c, which inhibits XBP1 mRNA splicing by IRE1 [35]. The IC50 value was calculated by the fluorescence readout, which was inversely correlated with the capability of a compound to inhibit IRE1 from cleaving the stem-loop. We measured an IC50 for 4μ8c of 22.7 nM, which is in the range of values previously reported in the literature [36]. We then evaluated the potency of recombinant RNH1 to inhibit XBP1 splicing and calculated an IC50 of 13.2 nM (Figure 4(G)). Altogether, these results suggest that RNH1 regulates IRE1 possibly through a direct modulation of its RNase activity.
Discussion
Although the biology of IRE1 and its role in health and disease has been broadly studied, the mechanisms mediating its regulation remain to be comprehensively characterized. However, understanding how IRE1 activity resolves upon late ER stress is of critical importance because ER stress mediates multiple pathological conditions, including cancer, inflammation, neurodegeneration, or metabolic disorders, and the intensity and the duration of the ER stress response dictate the fate of the cell and the status of the disease. Here we have uncovered that RNH1 physically interacts with IRE1 and allows titration of IRE1 activity (Figure 5). As such, RNH1 is the first endogenous regulator that directly modulates the ribonuclease activity of IRE1. RNH1 is one of the most abundant cellular proteins, accounting for approximately 0.1% of the total cytosolic protein content [37]. RNH1 binds to ANG (RNASE5, which cleaves tRNA into fragments called tiRNA) with a Kd of <1 fM and regulates its ribonucleolytic activity in different subcellular locations and under different growth conditions [33,34]. RNH1 also forms high affinity dimers with components of the pancreatic ribonuclease family, including RNASE1 (a secreted endonuclease that degrades single and double strand-RNAs) [38], RNASE2 (a non-secreted ribonuclease that cleaves viral RNA) [39], and RNASE7 (a ribonulcease with antibacterial activity) [40]. In addition, RNH1 controls cell growth and survival, with equivocal consequences since its reported antioxidant effects (RNH1 is a cysteine-rich protein) promote cell survival [41,42], whereas it could also protect against cancer progression and metastasis [43,44].
Figure 5.
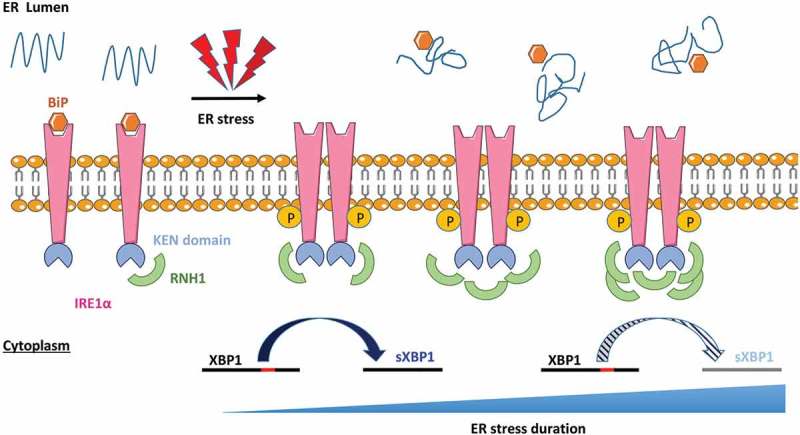
Working model. RNH1 interacts with IRE1 in basal conditions, but without interfering with its RNAse activity. Upon ER stress, the amount of RNH1 interacting with the KEN domain of IRE1 increases, which progressively interfere with and reduces its RNase activity. As a consequence, the splicing of XBP1 is reduced during late ER stress.
The data presented here indicate that RNH1 dynamically interacts with the ER and the UPR transducer IRE1 to limit its catalytic activity. We propose that export of RNH1 out of the nucleus is driven by ER stress, the mechanisms of which remain to be determined, allowing for its interaction with the KEN domain of IRE1 on the surface of the ER. In turn, RNH1 reduces the amount of IRE1-mediated splicing of XBP1 mRNA at the late phase of the ER stress response. Whereas our in vitro studies using recombinant IRE1 indicate that RNH1 is a potent inhibitor, we observed a limited (albeit highly reproducible) effect in vivo (i.e. in cellulo). Numerous factors may explain this observation, including the residual expression of RNH1, the intensity and nature of the stress, the cellular model, the redox status of the cell and possible proteins that interact with and modulate IRE1 activity. The expanding number of proteins that interact with IRE1α might also counteract the inhibitory effects of RNH1 by limiting its access to the molecular platform that organizes around IRE1. Nevertheless, under conditions of prolonged ER stress, the attenuation of XBP1 mRNA splicing by RNH1 could participate in the termination of the UPR.
Our results support the concept of nucleus shuttling of RNH1 during stressful conditions: RNH1 accumulates in the ER during ER stress at the expense of the nuclear pool. RNH1 is considered to be a cytoplasmic protein [37] and has also been detected in the nucleus (and in the mitochondria) [45], especially when cells are stressed [34]. The mechanisms that regulate the shuttling of RNH1 during ER stress are not known. The inner nuclear membrane is surrounded by the outer nuclear membrane, which is continuous with the membrane of the ER. Nuclear import/export receptors are structurally related, and the import and export transport systems work in similar ways, but in opposite directions. The GTPase Ran is found in both the cytosol and the nucleus and is critically required for both the nuclear import and export systems [46]. Interestingly, our RNH1-IP interactome experiments provided evidence that Ran was consistently associated with RNH1. Still based on the RNH1 interactome, the protein interacting with RNH1 that was the most significantly impacted during ER stress was nucleophosmin, a phosphoprotein that moves between the nucleus and cytoplasm as well as among numerous cellular functions and regulates nuclear shuttling of ribosomes [47].
As a stress rheostat, the UPRosome involves multiple proteins that determine the amplitude and kinetics of the UPR and hence the cell’s ability to adapt to ER stress. Unlike activation of IRE1, the “deactivation” process is not well known. Experimental evidence suggests that deactivation of IRE1 to inhibit its RNase activity and attenuate the UPR signal is a critical event that prevents cells from undergoing chronic deleterious ER stress [48]. Indeed, deactivation of IRE1 does not seem to simply follow the decrease in the amount of misfolded proteins in the ER lumen [49] but to occur independently of the phosphorylation state of IRE1, suggesting that other mechanisms are involved in attenuation of the UPR signal [50]. Our results support a model by which the ribonuclease inhibitor RNH1 is fully part of the negative control of IRE1 signaling by directly exerting its activity on the KEN cytosolic domain.
Overall, the current study has uncovered a previously unanticipated biological function of RNH1, contributing to our understanding of how IRE1 signaling is directly titrated. Because RNH1 is an abundant ribonuclease inhibitor that is ubiquitously expressed, a corollary of our discovery is that it may have broad physiological and therapeutic implications. Indeed, pharmacological manipulation of RNH1 might lead to a new, unifying therapeutic avenue for mitigating the toxic effects of excess activation of IRE1 to treat human diseases associated with ER stress.
Materials and methods
Cell culture
All experiments except the UMOD experiments (see below) were performed on the HK-2 (human kidney-2) cell line, immortalized with the HPF 16 E6/E7 genes (Cat. No. CSC-C9430L), which was obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle’s medium (Gibco, ref 41,965–039) supplemented with 1% fetal bovine serum (PAA Laboratories), hydrocortisone 0,5 µg/mL (Sigma-Aldrich), Insulin Transferrin Selenium 1X (Sigma-Aldrich), Epithelial Growth Factor 10 ng/mL (Sigma-Aldrich), triiodothyronine 6,5 ng/mL (Sigma-Aldrich), and penicillin-streptomycin 25 UI/mL and 25 µg/mL (Gibco) at 37°C in an atmosphere containing 5% CO2. Tunicamycin and 4µ8C were purchased from Sigma-Aldrich. For UMOD experiments, mTAL (mouse Thick Ascending Limb) cells were a generous gift from Prof. S. Bachmann (Charité University Medicine, Berlin, Germany). The cells were derived by microdissection of the nephron thick ascending limb (TAL) of the Henle’s loop segments of the immortomouse® (Charles River, Wilmington, MA) [51] and grown as previously described [52].
Immunoprecipitation
Immunoprecipitation of RNH1 and IRE1 were performed with 2 mg of a protein lysate of HK-2 cells. First, protein G-agarose (Roche) was washed 3 times in lysis buffer. Then, the lysate was pre-cleared with 50 µL of protein G-agarose to remove nonspecific interactions. Immunoprecipitations were performed with 40 µL of washed protein G-agarose and 3 µg of primary antibodies rose against the targets of interest (listed in Supplementary Table 1) overnight at 4°C. After 3 washes with lysis buffer, immunoblots were performed. For the modification of interaction assay, 1,6-Hexandiol 5% w/v and Urea at 2M purshased from Sigma-Aldrich were incubated with the immunoprecipitate before immunoblotting. For UMOD IP, mTAL cells were grown to confluence in 10 cm dishes and lysed in 1 mL of octylglucoside lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 60 mM octyl β-D-glucopyranoside, 10 mM NaF, 0.5 mM sodium orthovanadate, 1 mM glycerophosphate and protease inhibitor cocktail (Sigma)) for 1 h at 4°C under rotation followed by centrifugation 10 min at 17,000 g. Cell lysates (1.5 mg) were incubated under rotation for 4 h at 4°C with 20 µl of protein G-sepharose beads for pre-clearing (GE Healthcare). Pre-cleared lysates were then incubated under rotation for 16 hours at 4 °C with 20 µl of Anti-HA.11 Epitope Tag Affinity Matrix was from Biolegend (Cat# 900,801). The beads were washed 3 times in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl and IGEPAL 0.5%. The immuno-precipitated material was eluted by incubation for 10 min at 98°C in 50 µl of 100 mM Tris-HCl pH 8, 4% SDS.
SDS-PAGE and tandem mass spectrometry
After immunoprecipitation of the target (IRE1α or RHN1) eluted in 50 µL of Laemmli buffer 4X (Biorad), peptides were prepared by Filter Aided Separation method (FASP) essentially as described [53]. Briefly, proteins were reduced and alkyled simultaneously in 50 mM Tris/HCl, pH 8.5, 20 mM TCEP, 50 mM chloroacetamide and heated for 5 min at 95°C. After cooling to room temperature, extracts were diluted with 300 µL Tris Urea buffer (Urea 8 M, 50 mM Tris/HCl (pH 8.5) and transferred onto 30 kDa centrifuged filters and prepared for digestion. Proteins were digested overnight at 37°C with 1 µg trypsin (Promega) and peptides were desalted on C18 StageTips [54]. Mass spectrometry (MS) analyses were performed on a Dionex U3000 RSLC nano-LC system coupled to a Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific). After drying, peptides were solubilized in 10 µL of 0.1% trifluoroacetic acid (TFA) containing 10% acetonitrile (ACN). One µL was loaded, concentrated and washed for 3 min on a C18 reverse phase precolumn (3 µm particle size, 100 Å pore size, 75 µm inner diameter, 2 cm length, Thermo Fisher Scientific). Peptides were separated on a C18 reverse phase resin (2 µm particle size, 100Å pore size, 75 µm inner diameter, 25 cm length from Thermo Fisher Scientific) with a 1 hour gradient starting from 99% of solvent A containing 0.1% formic acid in H2O and ending in 40% of solvent B containing 80% acetonitrile, 0.085% formic acid in H2O. The mass spectrometer acquired data throughout the elution process and operated in a data-dependent scheme with full MS scans acquired with the Orbitrap, followed by MS/MS HCD fragmentations acquired with the Ion Trap on the most abundant ions detected in top speed mode for 3 seconds. Resolution was set to 60,000 for full scans at AGC target 2.0e5 within 60 ms maximum injection ion time (MIIT). The MS scans spanned from 350 to 1500 m/z. Precursor selection window was set at 1.6 m/z, and MS/MS scan resolution was set with AGC target 2.0e4 within 100 ms MIIT. HCD Collision Energy was set at 30%. Dynamic exclusion was set to 30 s duration. The mass spectrometry data were analyzed using Maxquant version 1.5.6.0 [55]. The database used was a concatenation of human sequences from the Uniprot-Swissprot database (Uniprot, release 2017–05) and a list of contaminant sequences from MaxQuant. The enzyme specificity was trypsin. The precursor mass tolerance was set to 4 ppm and the fragment mass tolerance to 0.56 Da for Fusion data. Carbamidomethylation of cysteins was set as constant modification and acetylation of protein N-terminus and oxidation of methionines were set as variable modification. For analysis, LFQ results from MaxQuant were imported into the Perseus software (version 1.5.3.30). Reverse and contaminant proteins were excluded from analysis. Annotations for RNH1 and IRE1-IPs were obtained from the Gene Ontology Project using the DAVID Gene-Enrichment and Functional Annotation (https://david.ncifcrf.gov) or by using the PANTHER Classification System (http://www.pantherdb.org). Only a subset of GO terms (GO slim) was used, and any parent terms were removed. To identify the biological processes that were statistically overrepresented in our protein list, binomial statistics tools were used to compare the classifications of annotated terms in our list with a reference list. Only enriched terms with a p-value of < 0.05 were used.
Protein extraction and western blot analysis
HK-2 cells were lysed in mPER lysis buffer (Thermo Fisher Scientific) with protease (HaltTM Protease Inhibitor Cocktail 100X, Thermo Fisher Scientific) and phosphatase inhibitors (HaltTM Phosphatase Inhibitor Cocktail 100X, Thermo Fisher Scientific). The concentrations were determined by using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and Tecan Safire® plate reader, and then, 25 µg of each sample was separated by SDS-PAGE under denaturing conditions and transferred to PVDF membranes (GE Healthcare). The primary antibodies (listed in Supplementary Table 1) were visualized using horseradish peroxidase-conjugated polyclonal secondary antibodies and detected by ECL reagent (GE Healthcare).
Sirna transfection
For transient knockdown of RNH1, two different siRNA duplexes were used (Hs_RNH1_4 and Hs_RNH1_5, Qiagen) targeting, respectively, the following sequences 5ʹ-CAGGTTTCTCCTGGAGCTACA-3ʹ and 5ʹ-CTGGGCGATGTCGGCGTGCA-3ʹ of RNH1 mRNA. In these experiments, siRNA was transfected at 20 nM/50,000 cells with HiPerfect (Qiagen) for 24 h to obtain an inhibition of >80% of RNH1 transcripts. Scrambled siRNA as a negative control siRNA (Qiagen) was used at 40 nM. After 24 h of transfection, cells were treated with tunicamycin 2,5 µg/mL.
UMOD experiments
Human uromodulin expression was induced in mTAL cells by transduction with lentiviral particles [56]. HA-tagged wild-type uromodulin was cloned into a transfer vector under the hPGK synthetic bidirectional promoter. The transfer vector for the mutant uromodulin C150S isoform was obtained by mutagenesis using a Quikchange Lightning mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer’s instructions. The primers were designed using the software QuikChange® Primer Design Program (hUMOD C150S forward 5ʹ-GATGGCACTGTGAGTCCTCCCCGGGCTCCTG-3ʹ; reverse 5ʹ-CAGGAGCCCGGGGAGGACTCACAGTGCCATC-3ʹ). Lentiviral particles were produced in HEK293T cells by transient four-plasmid (transfer, packaging [pMDLg/p.RRE and pILV001], and envelope [pMD2.VSV-g]) transfection by calcium phosphate precipitation and concentrated by ultracentrifugation. All experiments were performed using mixed populations that were obtained after transduction with lentiviral constructs expressing the indicated uromodulin isoforms.
Microsome extraction
To obtain cellular pellets, 2 75 cm2 flasks of HK-2 cells were used. They were then lysed by sonication in 1 mL of STE Buffer (Sucrose 0,25 M, Tris 10 mM, EDTA 1 mM) with a protease inhibitor added. Then, the lysate was centrifuged at 9000 g, 20 min at 4°C. The supernatant was ultracentrifuged at 100 000 g, 4°C for 1 h. The pellet was resuspended in 100 µL of microsome buffer (100 mM NaPO4, 10 mM MgCl2, 20% glycerol (v/v) pH 7,4).
In-silico modelization and protein-protein docking
Human IRE1 (PDB code: 3P23) and human RNH1 (PDB code 2Q4G) were used for docking experiments. The structures were cleaned and prepared using the prepared protocol of Biovia Discovery studio software. Loops were refined using MODELER. The final selected structures were used for docking. The protein-protein docking experiment was performed using protein docking (ZDOCK) module (DS Modeling 4.5). Docking was performed without constraints or specific residue selection for the IRE1 or RNH1 structures. The obtained poses (2000 per run) were refined using the RDOCK module (DS Modeling 4.5). All poses were regrouped into several clusters (energy/number/position based), and the most significant clusters were selected for interaction studies. Figures and sequence alignments were performed using DS 4.5.
Proximity ligation assay
Proximity Ligation Assay (PLA) probe Duolink assay (Sigma-Aldrich) was performed following the manufacturer’s instructions with IRE1 and RNH1 primary antibodies (reported in Supplementary Table 1). Briefly, primary antibodies bind their target, and secondary antibodies with oligonucleotides (PLA probes) are added to the reaction. Ligation and amplification of the fluorescently labeled oligonucleotides of the two PLA probes can only occur when the targets of the primary antibodies are close enough (<40 nm). All slides were observed under a LSM 510 confocal microscope (Zeiss). Identical acquisition parameters were used to observe the different slides.
RNA extraction and real-time qPCR analysis
Total RNA from HK-2 cells was extracted using a RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. The RNA sample concentrations were determined by using Nanodrop (Thermo Fisher Scientific). For each sample, 1 µg was reverse-transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Expression of target genes was analyzed by SYBR Green (Applied Biosystems) RT-qPCR was performed on a ABI PRISM 7900 sequence detector system (Applied Biosystems). The primers were designed using Amplify 3X® (Bill Engels, 2015, University of Wisconsin) (a list of all of the primers used in this study is reported in Supplementary Table 2). Expression of the genes of interest was normalized to RPL13A. The relative mRNA expression was calculated following the 2−ΔΔCt method [57].
Nucleus and cytosol preparations
Cell lysis and extraction of separate protein fractions were performed using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (ThermoScientific) according to the manufacturer’s protocol.
Phos-tag™ gel
IRE1 phosphorylation status was evaluated using SuperSep™ PhosTag™ Acrylamide Gel (Wako), which contains molecules that binds specifically phosphorylated ions, and allows the specific separation of proteins according to their phosphorylation levels.
Fragment analysis
RNA was extracted from the pellets using a RNeasy Mini Kit® (Qiagen) and reverse-transcribed into cDNAs using TaqMan® Reverse Transcription Reagents (Applied Biosystems). Basically, the fragment analysis involved the following 3 steps: (1) Amplification of the sXBP1 and XBP1 cDNAs by PCR using fluorogenic oligonucleotide primers. Several differently colored fluorescent dyes are detectable in one sample. We designed the following fluorogenic oligonucleotide primers (tagged with hexachloro-fluorescein, HEX, green) for fluorometric detection of the sXBP1 and XBP1 mRNA levels: forward primer: (5ʹ-HEX)GGAGTTAAGACAGCGCTTGG-3ʹ and reverse primer: 5ʹ-GAGATGTTCTGGAGGGGTGA-3ʹ. We performed PCR using HotStart Taq® DNA polymerase on a thermal cycler with the following program: 95°C for 10 min; 40 cycles of 94°C for 30 sec, 59°C for 30 sec, and 72°C for 30 sec; and a final step of 72°C for 10 min. (2) Labeled fragments (amplicons) were separated by size using capillary electrophoresis, and the fluorescence intensity was measured using the Applied Biosystems™ 3730xl DNA Analyzer. One of the dye colors (GENESCAN® ROX 400 HD size standard, Applied Biosystems™, red) was used to detect a labeled size standard in each sample. Fragments and ROX 400 HD were mixed with HiDi™ Formamide (Applied Biosystems) prior to capillary electrophoresis. (3) The data were analyzed using GeneMapper® Software to determine the relative size of each dye‐labeled fragment in the sample by comparing fragments with the standard curve for that specific sample.
In vitro IRE1 rnase activity assay
RNase activity of IRE1 in the presence of RNH1 or 4µ8C was assayed in vitro using a fluorescence method as described previously [22,58]. Briefly, 40 µL of recombinant human IRE1 (20 µg, OriGene) was pre-incubated for 30 min with 40 µL of various concentrations of recombinant human RNH1 (20 µg, OriGene) or 4µ8C (0,1 nM–1 µM) in RNase Assay Buffer (50 mM HEPES, 100 mM KOAc, 5 mM DTT, 5% glycerol, 0,005% Triton X-100, pH 7,2). Then, 40 µL of 150 nM fluorescently tagged XBP1 RNA stem loop (5ʹ-FAM-CAGUCCGCAGCACUG-Iowa-Black-FQ-3ʹ) was added to the solution and incubated for 2 h and protected from light at room temperature. Fluorescence was read with excitation at 495 nm and emission at 520 nm using a Tecan Safire® plate reader.
Statistical analysis
All data are expressed as the means ±SD of at least three independent experiments, unless otherwise specified. Biological and histological data were compared using Student’s t-test were performed using Prism 4 (Prism-GraphPad Software, Inc.). Any p-values less than 0.05 were considered significant.
Funding Statement
This work was supported by the Agence Nationale de la Recherche;Agence de la Biomédecine;
Acknowledgments
This work was funded by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM), l’Agence de la Biomédecine et l’Agence Nationale de la Recherche.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Woehlbier U, Hetz C.. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36:329–337. [DOI] [PubMed] [Google Scholar]
- [2].Hetz C, Papa FR.. The Unfolded Protein Response and cell fate control. Mol Cell. 2018;69:169–181. [DOI] [PubMed] [Google Scholar]
- [3].Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS.. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, Martinez J.. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014;33:2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee AH, Iwakoshi NN, Glimcher LH.. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Maurel M, Chevet E, Tavernier J, Gerlo S.. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. 2014;39:245–254. [DOI] [PubMed] [Google Scholar]
- [8].Hetz C, Glimcher LH.. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. [DOI] [PubMed] [Google Scholar]
- [10].Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, Mehdizadeh M, Ghosh R, Wang L, Colon-Negron K, et al. Targeting ABL-IRE1alpha signaling spares ER-stressed pancreatic beta cells to reverse autoimmune diabetes. Cell Metab. 2017;25:883–97 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Luo D, He Y, Zhang H, Yu L, Chen H, Xu Z, Tang S, Urano F, Min W.. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J Biol Chem. 2008;283:11905–11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A.. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8:e1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L.. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol. 2002;22:8506–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].He Y, Beatty A, Han X, Ji Y, Ma X, Adelstein RS, Yates JR 3rd, Kemphues K, Qi L.. Nonmuscle myosin IIB links cytoskeleton to IRE1alpha signaling during ER stress. Dev Cell. 2012;23:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guo J, Polymenis M.. Dcr2 targets Ire1 and downregulates the unfolded protein response in Saccharomyces cerevisiae. EMBO Rep. 2006;7:1124–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lu G, Ota A, Ren S, Franklin S, Cd Rau, Ping P, Tf Lane, Zh Zhou, Reue K, Aj Lusis, et al. PPM1l encodes an inositol requiring-protein 1 (IRE1) specific phosphatase that regulates the functional outcome of the ER stress response. Mol Metab. 2013;2:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ren S, Lu G, Ota A, Zhou ZH, Vondriska TM, Lane TF, Wang Y.. IRE1 phosphatase PP2Ce regulates adaptive ER stress response in the postpartum mammary gland. PLoS One. 2014;9:e111606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Welihinda AA, Tirasophon W, Green SR, Kaufman RJ.. Protein serine/threonine phosphatase Ptc2p negatively regulates the unfolded-protein response by dephosphorylating Ire1p kinase. Mol Cell Biol. 1998;18:1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ.. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000;14:2725–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Eletto D, Eletto D, Dersh D, Gidalevitz T, Argon Y.. Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide-dependent association. Mol Cell. 2014;53:562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, JH Lin, Walter P, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell. 2009;33:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pinkaew D, Chattopadhyay A, King MD, Chunhacha P, Liu Z, HL Stevenson, Chen Y, Sinthujaroen P, OM McDougal, Fujise K.. Fortilin binds IRE1alpha and prevents ER stress from signaling apoptotic cell death. Nat Commun. 2017;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sepulveda D, Rojas-Rivera D, DA Rodriguez, Groenendyk J, Kohler A, Lebeaupin C, Ito S, Urra H, Carreras-Sureda A, Hazari Y, et al. Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1alpha. Mol Cell. 2018;69:238–52 e7. [DOI] [PubMed] [Google Scholar]
- [24].Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U.. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. [DOI] [PubMed] [Google Scholar]
- [25].Chennupati V, Veiga DF, Maslowski KM, Andina N, Tardivel A, Yu EC, Stilinovic M, Simillion C, Duchosal MA, Quadroni M, et al. Ribonuclease inhibitor 1 regulates erythropoiesis by controlling GATA1 mRNA translation. J Clin Invest. 2018;128:1597–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Karim MR, Kanazawa T, Daigaku Y, Fujimura S, Miotto G, Kadowaki M.. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy. 2007;3:553–560. [DOI] [PubMed] [Google Scholar]
- [27].Devuyst O, Olinger E, Rampoldi L.. Uromodulin: from physiology to rare and complex kidney disorders. Nat Rev Nephrol. 2017;13:525–544. [DOI] [PubMed] [Google Scholar]
- [28].Trudu M, Schaeffer C, Riba M, Ikehata M, Brambilla P, Messa P, Martinelli-Boneschi F, Rastaldi MP, Rampoldi L.. Early involvement of cellular stress and inflammatory signals in the pathogenesis of tubulointerstitial kidney disease due to UMOD mutations. Sci Rep. 2017;7:7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bernascone I, Vavassori S, Di Pentima A, Santambrogio S, Lamorte G, Amoroso A, Scolari F, Ghiggeri GM, Casari G, Polishchuk R, et al. Defective intracellular trafficking of uromodulin mutant isoforms. Traffic. 2006;7:1567–1579. [DOI] [PubMed] [Google Scholar]
- [30].Kobe B, Deisenhofer J.. Proteins with leucine-rich repeats. Curr Opin Struct Biol. 1995;5:409–416. [DOI] [PubMed] [Google Scholar]
- [31].Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011;30:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johnson RJ, McCoy JG, Bingman CA, Phillips GN Jr., Raines RT.. Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein. J Mol Biol. 2007;368:434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Papageorgiou AC, Shapiro R, Acharya KR.. Molecular recognition of human angiogenin by placental ribonuclease inhibitor–an X-ray crystallographic study at 2.0 A resolution. EMBO J. 1997;16:5162–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pizzo E, Sarcinelli C, Sheng J, Fusco S, Formiggini F, Netti P, Yu W, D’Alessio G, Hu GF.. Ribonuclease/angiogenin inhibitor 1 regulates stress-induced subcellular localization of angiogenin to control growth and survival. J Cell Sci. 2013;126:4308–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Maly DJ, Papa FR.. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014;10:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Haigis MC, Kurten EL, Raines RT.. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003;31:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Arnold U, Leich F, Neumann P, Lilie H, Ulbrich-Hofmann R.. Crystal structure of RNase A tandem enzymes and their interaction with the cytosolic ribonuclease inhibitor. FEBS J. 2011;278:331–340. [DOI] [PubMed] [Google Scholar]
- [39].Teufel DP, Kao RY, Acharya KR, Shapiro R.. Mutational analysis of the complex of human RNase inhibitor and human eosinophil-derived neurotoxin (RNase 2). Biochemistry. 2003;42:1451–1459. [DOI] [PubMed] [Google Scholar]
- [40].Spencer JD, Schwaderer AL, Eichler T, Wang H, Kline J, Justice SS, Cohen DM, Hains DS.. An endogenous ribonuclease inhibitor regulates the antimicrobial activity of ribonuclease 7 in the human urinary tract. Kidney Int. 2014;85:1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cui XY, Fu PF, Pan DN, Zhao Y, Zhao J, Zhao BC.. The antioxidant effects of ribonuclease inhibitor. Free Radic Res. 2003;37:1079–1085. [DOI] [PubMed] [Google Scholar]
- [42].Medugno L, Costanzo P, Lupo A, Monti M, Florio F, Pucci P, Izzo P.. A novel zinc finger transcriptional repressor, ZNF224, interacts with the negative regulatory element (AldA-NRE) and inhibits gene expression. FEBS Lett. 2003;534:93–100. [DOI] [PubMed] [Google Scholar]
- [43].Xiong D, Liou Y, Shu J, Li D, Zhang L, Chen J.. Down-regulating ribonuclease inhibitor enhances metastasis of bladder cancer cells through regulating epithelial-mesenchymal transition and ILK signaling pathway. Exp Mol Pathol. 2014;96:411–421. [DOI] [PubMed] [Google Scholar]
- [44].Chen J, Ou-Yang X, Gao J, Zhu J, He X, Rong J.. Knockdown of ribonuclease inhibitor expression with siRNA in non-invasive bladder cancer cell line BIU-87 promotes growth and metastasis potentials. Mol Cell Biochem. 2011;349:83–95. [DOI] [PubMed] [Google Scholar]
- [45].Furia A, Moscato M, Cali G, Pizzo E, Confalone E, Amoroso MR, Esposito F, Nitsch L, D’Alessio G.. The ribonuclease/angiogenin inhibitor is also present in mitochondria and nuclei. FEBS Lett. 2011;585:613–617. [DOI] [PubMed] [Google Scholar]
- [46].Matsuura Y. Mechanistic insights from structural analyses of Ran-GTPase-Driven nuclear export of proteins and RNAs. J Mol Biol. 2016;428:2025–2039. [DOI] [PubMed] [Google Scholar]
- [47].Colombo E, Alcalay M, Pelicci PG.. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011;30:2595–2609. [DOI] [PubMed] [Google Scholar]
- [48].Chawla A, Chakrabarti S, Ghosh G, Niwa M.. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol. 2011;193:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sanchez-Alvarez M, Del Pozo MA, Bakal C.. AKT-mTOR signaling modulates the dynamics of IRE1 RNAse activity by regulating ER-mitochondria contacts. Sci Rep. 2017;7:16497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Papa FR, Zhang C, Shokat K, Walter P.. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. [DOI] [PubMed] [Google Scholar]
- [51].Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D.. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88:5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schaeffer C, Merella S, Pasqualetto E, Lazarevic D, Rampoldi L.. Mutant uromodulin expression leads to altered homeostasis of the endoplasmic reticulum and activates the unfolded protein response. PLoS One. 2017;12:e0175970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wisniewski JR, Zougman A, Nagaraj N, Mann M.. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. [DOI] [PubMed] [Google Scholar]
- [54].Rappsilber J, Mann M, Ishihama Y.. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. [DOI] [PubMed] [Google Scholar]
- [55].Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M.. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 2009;4:698–705. [DOI] [PubMed] [Google Scholar]
- [56].Schaeffer C, Santambrogio S, Perucca S, Casari G, Rampoldi L.. Analysis of uromodulin polymerization provides new insights into the mechanisms regulating ZP domain-mediated protein assembly. Mol Biol Cell. 2009;20:589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Livak KJ, Schmittgen TD.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [58].Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del Valle JR, Hu CC.. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. 2014;124:2585–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.