

ABSTRACT
The macroautophagy/autophagy-inducing disaccharide, trehalose, has been proposed to be a promising therapeutic agent against neurodegenerative and cardiometabolic diseases. We recently showed that trehalose attenuates hepatic steatosis in part by blocking hepatocyte glucose transport to induce hepatocyte autophagic flux. However, although every major demonstration of trehalose action invokes activating autophagic flux as its primary function, the mechanism of action of trehalose in whole-body energy metabolism remains poorly defined. Here, we demonstrate that trehalose induces hepatocyte TFEB (transcription factor EB)-dependent thermogenesis in vivo, concomitant with upregulation of hepatic and white adipose expression of UCP1 (uncoupling protein 1 [mitochondrial, protein carrier]). Mechanistically, we provide evidence that hepatocyte fasting transcriptional and metabolic responses depend upon PPARGC1A (peroxisome proliferative activated receptor, gamma, coactivator 1 alpha), TFEB, and FGF21 (fibroblast growth factor 21) signaling. Strikingly, hepatocyte-selective TFEB knockdown abrogated trehalose induction of thermogenesis and white adipose tissue UCP1 upregulation in vivo. In contrast, we found that trehalose action on thermogenesis was independent of LEP (leptin) and the autophagy pathway, as there was robust thermogenic induction in trehalose-treated ob/ob, Becn1, Atg16l1, and Epg5 mutant mice. We conclude that trehalose induces metabolically favorable effects on whole-body thermogenesis in part via hepatocyte-centered fasting-like mechanisms that appear to be independent of autophagic flux. Our findings elucidate a novel mechanism by which trehalose acts as a metabolic therapeutic agent by activating hepatic fasting responses. More broadly, the hepatic glucose fasting response may be of clinical utility against overnutrition-driven disease, such as obesity and type 2 diabetes mellitus.
KEYWORDS: Beclin 1, ectopic P-granules autophagy protein, fibroblast growth factor, glucose transport, solute carrier 2A, transcription factor EB
Introduction
The adaptive fasting response is a promising metabolic pathway that may be leveraged to mitigate or reverse metabolic disease [1]. Physiological fasting activates a coordinated response that includes mobilization of fatty acids from peripheral fat stores in order to provide fuel for fatty acid β-oxidation in the hepatocyte. The hepatocyte occupies a unique node between the gut, where macronutrients are first encountered, sensed, and absorbed from the portal circulation, and the systemic circulation. Thus, hepatocytes play a critical role in nutrient status sensing to coordinate whole-body energy metabolism [2]. The signal transduction pathways initiating the hepatic fasting response largely involve blocking MTOR (mechanistic target of rapamycin kinase) [3,4] and activating AMPK (AMP-activated protein kinase), which is turned on in response to acute rises in AMP:ATP ratios [5–7]. Both of these proximal starvation signals feed into key transcriptional regulators to consummate the broader hepatocyte fasting response program. AMPK activates PPARGC1A (peroxisome proliferative activated receptor, gamma, coactivator 1 alpha), a key transcriptional regulator that prepares the hepatocyte for mitochondrial biogenesis and fat metabolism [8,9]. In parallel with AMPK sensing of hepatic fasting, MTOR blockade facilitates activation and translocation of the fasting response regulator TFEB (transcription factor EB) [10,11]. TFEB and PPARGC1A coordinate transcription of FGF21 (fibroblast growth factor 21), a circulating hepatokine that coordinates extrahepatic energy homeostasis and induces thermogenesis in response to fasting [9,12–18]. Specific manipulation each of these key adaptive fasted-state factors remains under investigation as promising means to ameliorate metabolic disease, including non-alcoholic fatty liver disease, obesity, and type 2 diabetes mellitus. On the basis that physiological fasting broadly and coordinately activates the pathways described, pharmacologically inducing the hepatic glucose fasting response itself was recently proposed [2,19–21].
Trehalose is an α,α-1,1 glycoside-linked disaccharide that activates autophagy to ameliorate pathological protein and fat aggregation in multiple disease models, including abnormal protein aggregation in models of amyotrophic lateral sclerosis, prion disease, and Huntington disease as well as atherosclerotic plaque formation and adipocyte hypertrophy [19,20,22–28]. We recently reported that trehalose induces hepatic autophagy in part by blocking energy substrate uptake (e.g. of glucose and fructose) via the SLC2A/GLUT (solute carrier family 2 [facilitated glucose transporter]) family of facilitative carbohydrate transporters [20,25]. Blocking energy substrate transport results in a pharmacological starvation-like state, which includes activation of hepatocyte AMPK and blockade of MTOR signaling. Indeed, trehalose treatment in vitro and in vivo blocks fructose-induced hepatic steatosis and dyslipidemia, in part in an ATG16L1 (autophagy related 16-like 1 [S. cerevisiae])-dependent manner [19,20].
Abundant prior data suggest that trehalose has broad, extrahepatic metabolic effects, particularly in the neuron [26] and vasculature [27], yet the effects of trehalose on global energy metabolism and the mechanisms by which trehalose alters global energy homeostasis have not been fully explored. Moreover, despite the beneficial neural, vascular, and hepatic effects of oral trehalose treatment, the previously-reported low peripheral bioavailability [20,27] of trehalose suggested that trehalose may render at least part of its metabolic effects from within the enterohepatic circulation.
Here, we tested the hypothesis that trehalose induces a hepatocyte fasting-like response to modulate hepatic and extrahepatic energy metabolism. We provide evidence that trehalose, an SLC2A inhibitor, induces a canonical adaptive hepatic fasting response cascade that includes TFEB- and PPARGC1A-dependent induction of FGF21 and UCP1 (uncoupling protein 1 [mitochondrial, protein carrier]) in vitro and in vivo. This signaling response was associated with enhanced heat generation and oxygen consumption in trehalose-fed mice. Furthermore, hepatocyte-directed TFEB and FGF21 knockdown attenuated trehalose-induced heat generation in vivo. In contrast, a number of autophagy pathway proteins, ATG16L1, EPG5 (ectopic P-granules autophagy protein 5 homolog [C. elegans]), BECN1 (beclin 1, autophagy related), and LEP (leptin), were dispensable for trehalose action on thermogenesis. Trehalose thus exerts an adaptive, TFEB-dependent, pro-thermogenic hepatic fasting response program that is independent of leptinergic and autophagy machinery.
Results
Oral trehalose induces whole-body heat generation
Treatment with oral trehalose for 10 days reduced high-fructose diet-induced hepatic steatosis in vivo and in vitro in association with hepatic AMPK activation, MTOR inhibition, and autophagy induction [19,20]. Prior studies indicated that AMPK activation and MTOR blockade are metabolically favorable, in particular with regard to thermogenic induction as a means of excess energy dissipation [29]. Here, we evaluated the effects of oral trehalose treatment on whole-organism heat production and O2/CO2 exchange by indirect calorimetry. Treatment with 3% trehalose in water fed ad libitum for 5 days significantly increased light- and dark-cycle thermogenesis (Figure 1(a and b)), oxygen consumption (Figure 1(c)), and CO2 production (Figure 1(d)) associated with light-cycle-restricted increases in respiratory exchange ratio (RER, Figure 1(e)). No locomotor changes were observed in response to trehalose feeding (not shown).
Figure 1.

Enhanced thermogenesis in response to trehalose feeding is leptin-independent. (A) Heat production over time in mice (n = 8 mice per group) fed regular chow diet with either sterile water (black line) or 3% trehalose in water ad libitum (5 total days, purple line) and placed in indirect calorimeters for at least 24 h prior to death. (B) Quantification of dark cycle (left) and light cycle (right) thermogenesis in mice fed sterile water or 3% trehalose in water. (C-E) Oxygen consumption (C), CO2 production (D), and respiratory exchange ratio (RER) (E) in same mice presented in (A). (F-I) Quantification of oxygen consumption (F), CO2 production (G), heat generation (H), and RER (I) by indirect calorimetry in 8-wk-old leptin-deficient ob/ob mice fed with or without 3% trehalose water ad libitum (5 days); n = 5 per group. * and **** P < 0.05 and < 0.0001 respectively versus control by Student’s T-test.
LEP is a pro-thermogenic hormone that is required for heat generation during the cold response [30]. To genetically dissect mechanisms by which trehalose induces thermogenesis in a disease of obesity model in vivo, we quantified heat generation in obese, LEP-deficient (ob/ob) mice treated with or without oral trehalose (5 days, ad libitum). Trehalose induced robust light- and dark-cycle heat production (Figure 1(f)) and O2/CO2 exchange with trends toward increased RER independent of germline LEP deletion (Figure 1(g-i)).
Several reports documenting the therapeutic effects of trehalose suggest that trehalose-induced autophagy is central to its mechanism of action [20,22–24,27,28,31–34]. Accordingly, the therapeutic effects of trehalose on fructose-induced fat accumulation were attenuated in murine hepatocytes harboring homozygous hypomorphic mutant alleles of the autophagy regulatory gene, Atg16l1 (Atg16l1HM mice [20]). This demonstrated that wild-type Atg16l1 is required for the anti-steatotic metabolic actions of trehalose. We therefore quantified the thermogenic response of Atg16l1HM mice to 5-day, 3% trehalose water treatment ad libitum by indirect calorimetry. Surprisingly, trehalose induced robust thermogenesis in Atg16l1HM mice during both light- and dark-cycles (Figure 2(a)) in wild-type and Atg16l1HM mice. Light- and dark-cycle oxygen consumption (Figure 2(b)) and CO2 production (Figure 2(c)) were also induced in both WT and Atg16l1HM mice. Trehalose did not significantly change dark-or light-cycle RER, however, we observed a modest increase in dark cycle RER in Atg16l1HM mice (Figure 2(d)).
Figure 2.
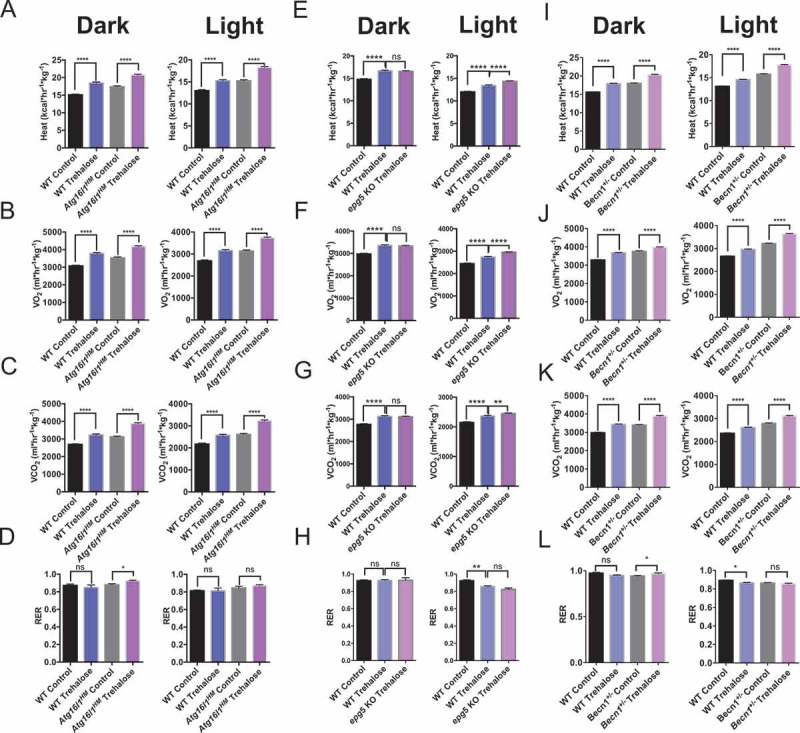
Trehalose induces thermogenesis in autophagy-deficient Atg16l1HM mice. (A) Dark and light cycle heat generation in wild-type or Atg16l1HM mice fed regular chow diet with either sterile water or 3% trehalose in water ad libitum. (B-D) Oxygen consumption (B), CO2 production (C), and respiratory exchange ratio (RER) (D) in same mice presented in (A); n = 5 mice per group. (E-H) Heat production (E), oxygen consumption (F), CO2 production (G), and RER (H) in wild-type and epg5 null mice treated with or without oral trehalose during dark and light cycles; n = 3–4 mice per group. (I-L) Heat production (I), oxygen consumption (J), CO2 production (K), and RER (L) in wild-type and Becn1+/- mice treated with or without oral trehalose during dark and light cycles; n = 4 mice per group. *P < 0.05, **P < 0.01 and ****P < 0.0001 versus control by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests.
The process of autophagy consists of multiple steps, which include initiation, phagophore elongation, autophagosome maturation, and degradation [35]. ATG16L1 is a key component of a complex that mediates elongation [35–38], and this was dispensable for trehalose-induced thermogenesis. To better understand the contributions of autophagosome initiation and maturation to the thermogenic effects of trehalose, we examined trehalose-induced thermogenesis in Becn1 haploinsufficient mice and in homozygous epg5 null mice [36,39–42]. Trehalose induced heat generation and O2 and CO2 exchange in wild-type mice, and again, trehalose-induced heat generation was not attenuated in mice lacking either Epg5 (Figure 2(e-g)) or Becn1 (Figure 2(i-k)). No differences in RER were observed during dark or light cycle when comparing trehalose-treated epg5 null mice with their trehalose-treated wild-type controls (Figure 2(h)). In Becn1 haploinsufficient mice, trehalose induced only minor changes in RER during dark cycle, but not during light cycle when compared with untreated Becn1 haploinsufficient mice (Figure 2(l)). Together, these data suggested that trehalose induces thermogenesis independent of autophagy and suggest that additional pathways mediate the therapeutic activity of trehalose.
Trehalose induces hepatocyte starvation by blocking cellular respiration and glycolytic flux
We next determined the metabolic consequences of hepatocyte trehalose treatment at the cellular level. To accomplish this, we utilized the AML12 murine hepatocyte model system and analyzed oxygen flux using the Seahorse XF96 Mito Stress Kit analysis [42]. We chose the AML12 hepatocyte model because it was resistant to the extensive washing process involved in performing Seahorse analysis. Trehalose-treated AML12 hepatocytes exhibited time-dependent decreases in basal oxygen consumption rate (OCR) and maximal (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone [FCCP]-stimulated) respiration after the 72-h trehalose stimulation time course (Figure 3(a-c)). However, cellular oxygen consumption devoted to ATP synthesis trended lower after 24- to 72-h trehalose incubation, but none of these differences reached statistical significance (Figure 3(d)). We previously demonstrated lower cellular ATP concentrations in response to trehalose treatment in hepatocytes [20]. In light of the current data demonstrating modest alterations in oxygen consumption devoted to ATP production, we directly tested the hypothesis that trehalose blocks glycolytic flux [19,20] by quantifying the extracellular acidification rate (ECAR) in trehalose-treated cultures. We showed that trehalose blocked ECAR 24, 48, and 72 h post-treatment (Figure 3(e and f)).
Figure 3.

Trehalose inhibits hepatocyte respiration and glycolysis to induce the fasting response. (A) Seahorse (XF96) analysis by mito stress test kit assay. Shown is the oxygen consumption rate (OCR) over time (n = 6–8 cell cultures per group), with injection of oligomycin (‘oligo’), FCCP, and rotenone and antimycin (‘Rot/Ant’) at the indicated time points. (B) Quantification of basal OCR in cells treated with or without trehalose (24–72 h). (C) Quantification of maximal (FCCP-stimulated) respiration in cells treated with or without trehalose. (D) Quantification of oxygen consumption that is devoted to ATP production, as determined in hepatocytes after oligomycin treatment. (E) Extracellular acidification rate (ECAR) quantified in cultures analyzed in (A-D) over time. (F) Quantification of basal ECAR, measured immediately prior to oligomycin injection. * P < 0.05, ** P < 0.01, *** P < 0.001 by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests.
Trehalose-induced hepatocyte starvation increases hepatic FGF21 expression
Trehalose at the cellular level blocks glycolysis and respiratory potential, whereas at the organism level, trehalose induces whole-body thermogenesis via an ATG16L1-independent mechanism. We consequently evaluated ATG16L1- and LEP-independent mechanisms that may govern trehalose-induced thermogenesis. Trehalose is a hepatocyte SLC2A inhibitor [20,34] that induces cellular metabolic signaling responses consistent with glucose starvation, including MTOR inhibition and PRKAA1 activation. Prior work indicated that hepatic starvation signaling via MTOR regulates whole body heat generation partly via FGF21, a pro-thermogenic hepatokine [43,44,45] that is released in response to hepatocyte macronutrient deprivation [46]. We therefore hypothesized that trehalose induces hepatocyte FGF21 production. In vivo, trehalose (24 h) induced hepatic Fgf21 mRNA and protein expression (Figure 4(a and b)) and greater circulating FGF21 peptide within 48 h treatment (Figure 4(c)). FGF21 regulation by trehalose was then modeled in vitro to evaluate whether trehalose exerts hepatocyte-autonomous effects on FGF21. Indeed, in both primary hepatocytes and AML12 murine hepatocytes (Figure 4(d)), Fgf21 mRNA was significantly increased 24 h after trehalose treatment (100 mM). To quantify whether Fgf21 gene expression changes were paralleled with FGF21 protein accumulation, primary hepatocytes were incubated with or without trehalose (8–24 h) in the presence or absence of trehalose in growth media. Trehalose treatment in primary hepatocytes enhanced FGF21 protein accumulation at both 8 h and 24 h trehalose treatment (Figure 4(e)).
Figure 4.
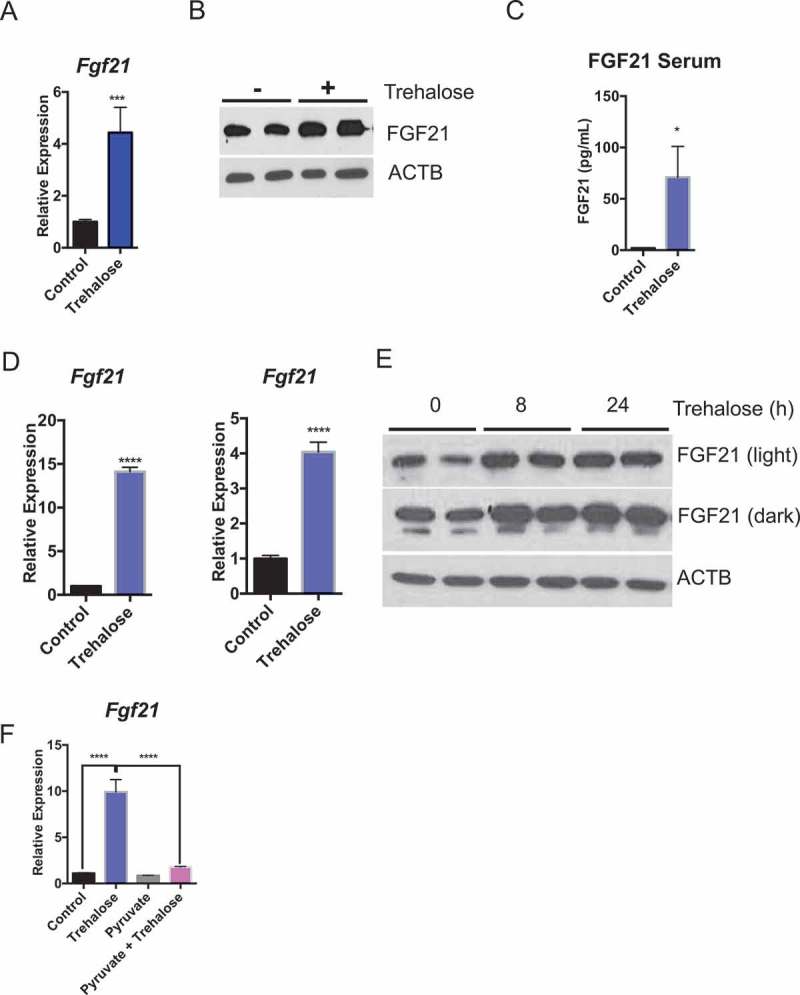
Trehalose induces hepatocyte FGF21 expression. (A) Fgf21 mRNA accumulation in mice treated with trehalose for 24 h with 3% trehalose water ad libitum (n = 5 mice per group). (B) Hepatic FGF21 protein in livers of mice treated with or without 3% trehalose water ad libitum (n = 5 mice per group). ACTB is used as a loading control. (C) FGF21 ELISA showing circulating FGF21 levels in wild-type mice in response to sterile water or 3% trehalose water. (D) Fgf21 mRNA accumulation in primary hepatocytes (left) and AML12 mouse hepatocytes (right) treated with or without trehalose for 24 h. (E) FGF21 immunoblot showing increased FGF21 peptide in primary hepatocytes treated with or without trehalose from 8–24 h. (F) Fgf21 mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of 5 mM pyruvate. * P < 0.05, *** P < 0.001, **** P < 0.0001 versus control by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests.
We next tested the hypothesis that hepatocyte FGF21 induction by trehalose depends upon a cellular energy substrate deficit. To that end, primary hepatocytes were stimulated with trehalose in the presence or absence of 5 mM pyruvate as an energy substrate that bypasses the SLC2A transporter. Again, trehalose significantly induced Fgf21 expression in primary hepatocytes, but pyruvate pre-treatment abrogated Fgf21 mRNA accumulation (Figure 4(f)).
Trehalose induces PPARGC1A expression upstream of FGF21
Fgf21 is canonically regulated by upstream fasting-induced transcription factors TFEB and PPARGC1A [47]. Both TFEB and PPARGC1A are key determinants of the fasting-state gene expression program in liver and in other tissues [8,11,48]. We first tested whether hepatic Ppargc1a was induced in trehalose-treated mice. Oral trehalose feeding ad libitum robustly induced hepatic Ppargc1a mRNA and protein accumulation within 48 h of initial exposure (Figure 5(a and b)). We again modeled trehalose induction of Ppargc1a mRNA and protein in primary hepatocytes and in AML12 hepatocytes treated with 100 mM trehalose (24 h, Figure 5(c and d)). We then evaluated Ppargc1a expression in primary hepatocytes treated with or without trehalose in the presence or absence of 5 mM pyruvate to quantify the dependence of Fgf21 induction by trehalose on energetic deficits. Trehalose stimulated Ppargc1a mRNA and PPARGC1A protein accumulation in pyruvate-naïve hepatocytes, but pyruvate supplementation blocked trehalose-induced Ppargc1a mRNA accumulation (Figure 5(e)). We then examined the role of PPARGC1A in trehalose-stimulated FGF21 accumulation by treating primary hepatocytes with scrambled antisense oligonucleotide (ASO) or with Fgf21-directed ASOs prior to trehalose treatment. Trehalose enhanced Ppargc1a expression in scrambled ASO-treated hepatocyte controls, whereas Fgf21 ASO-treated hepatocytes exhibited modestly decreased Ppargc1a mRNA abundance (Figure 5(f)). Conversely, Ppargc1a ASO-treated cells were unable to upregulate Fgf21 mRNA expression and protein accumulation in response to trehalose when compared with cultures treated with scrambled ASO (Figure 5(g and h)). Together, the data suggested that PPARGC1A activation is energy deficit-dependent, and that it lies primarily upstream of FGF21 in the trehalose signal transduction pathway.
Figure 5.
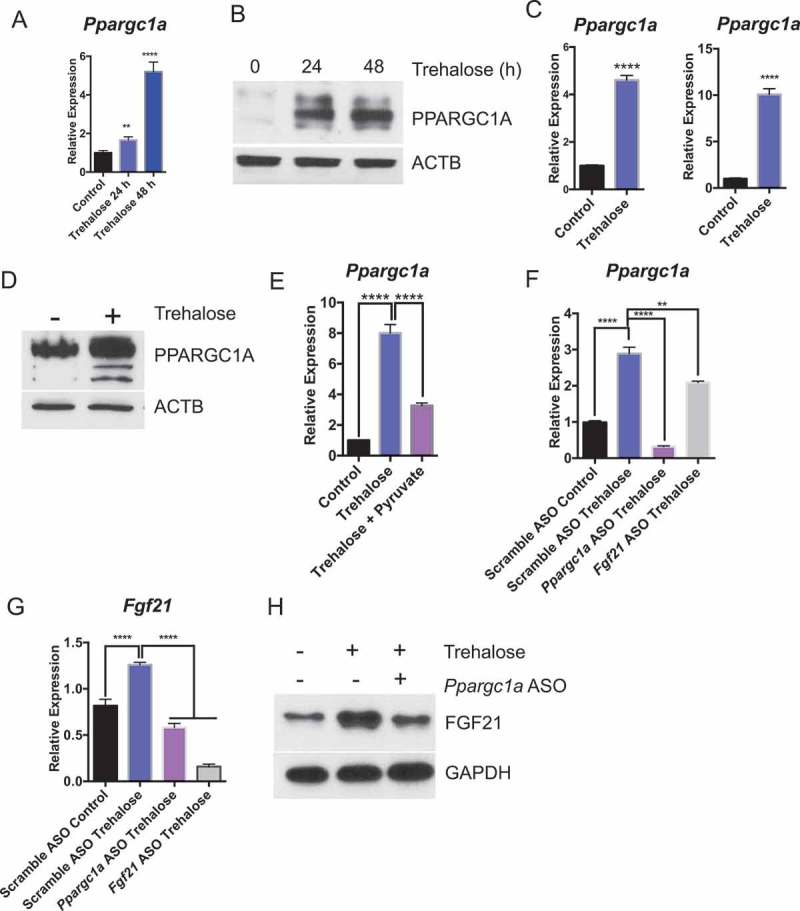
Trehalose activates hepatocyte PPARGC1A upstream of FGF21. (A) Ppargc1a mRNA accumulation in mice treated with trehalose 0–48 h with 3% trehalose water ad libitum (n = 4 mice per group). (B) PPARGC1A immunoblot showing hepatocyte PPARGC1A in mouse livers after 96 h treatment with sterile water (group labeled ‘0 h trehalose’) or 24–48 h treatment with 3% trehalose water. (C) Ppargc1a mRNA accumulation in primary hepatocytes treated with or without trehalose for 24 h. (D) PPARGC1A immunoblot showing hepatocyte PPARGC1A after 24 h trehalose exposure in regular growth media. (E) Ppargc1a mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of 5 mM pyruvate. (F) Ppargc1a mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of scrambled, Ppargc1a-, or Fgf21-directed antisense oligonucleotides (ASO). (G) Fgf21 mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of scrambled or Ppargc1a- or Fgf21-directed ASOs. (H) FGF21 protein accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of scrambled or Ppargc1a-directed ASO. GAPDH is used as a loading control. *** P < 0.001, **** P < 0.0001 versus control by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests.
TFEB and PPARGC1A mediate trehalose induction of hepatic UCP1 expression
PPARGC1A is a transcriptional regulator of Ucp1 in brown adipose tissue [49]. Moreover, inducing hepatic heat dissipation via UCP1 has been proposed as a therapeutic pathway to manipulate obesity and type 2 diabetes pathogenesis [50–52]. To examine whether this analogous PPARGC1A-FGF21-UCP1 pathway also exists in hepatocytes, we tested the hypothesis that trehalose increases hepatic UCP1 levels. We first tested whether trehalose induced Ucp1 expression in AML12 hepatocytes, in isolated primary hepatocytes, and in mice fed oral trehalose water ad libitum. Indeed, trehalose increased Ucp1 expression when compared with untreated controls (Figure 6(a)) in vitro and in vivo. We then determined the effect of trehalose on UCP1 protein levels in vitro and in vivo. To first establish the specificity of our antibody, we titrated the antibody and achieved a single 33-kDa UCP1 band in wild-type interscapular brown adipose tissue (iBAT) lysates from wild-type mice (Fig. S1A) at both low and high antibody dilutions. We further confirmed the specificity of the antibody in hepatocytes by immunoblot analysis of primary hepatocyte cultures with scrambled or Ucp1-directed siRNA prior to UCP1 immunoblotting. This demonstrated that Ucp1-specific siRNA diminished the 33 kDa immunoreactive band detected in hepatocytes (Fig. S1B). We then treated primary hepatocytes and wild-type animals with or without trehalose prior to UCP1 immunoblot analysis (Figure 6(b)). This again revealed enhanced hepatic UCP1 protein accumulation both in vitro and in vivo in response to trehalose treatment. By contrast, iBAT Ucp1 mRNA and protein levels did not change in response to trehalose feeding in vivo (Fig. S2A and B). We then examined whether Ucp1 upregulation required energetic substrate deficits induced by trehalose. Consistent with the hypothesis that Ucp1 is upregulated in response to cellular energy deficit, pyruvate treatment in hepatocytes partly reversed trehalose-induced Ucp1 mRNA accumulation (Figure 6(c)).
Figure 6.
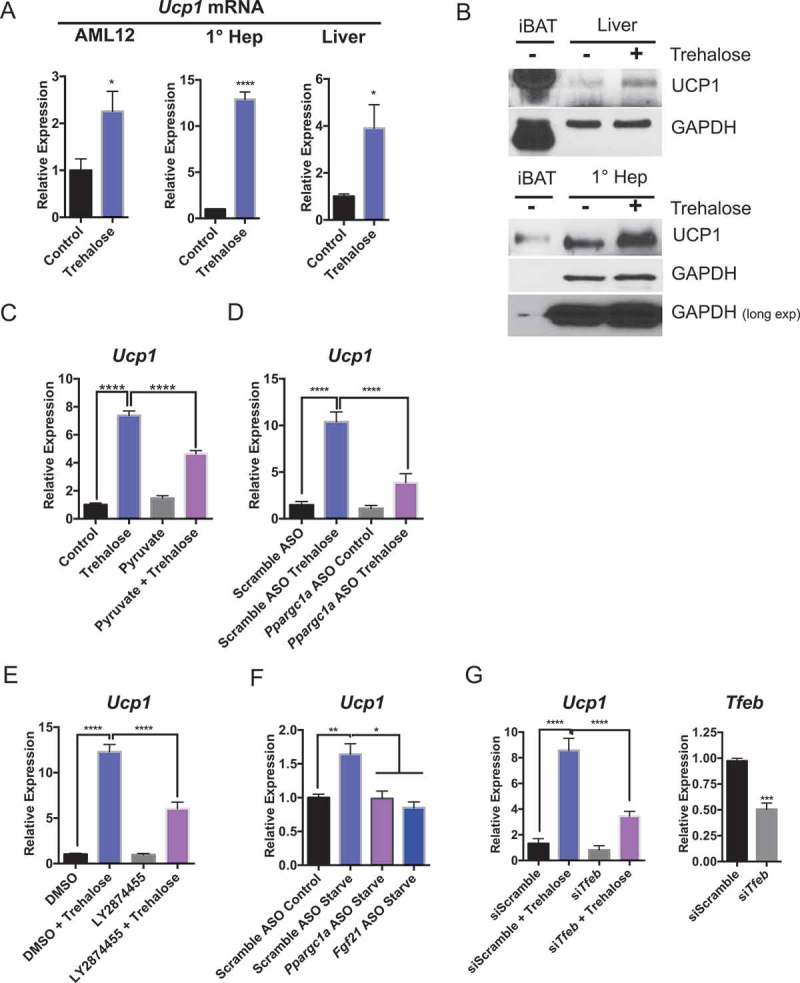
Trehalose upregulation of hepatocyte UCP1 is PPARGC1A- and TFEB-dependent. (A) Ucp1 mRNA accumulation in AML12 murine hepatocytes and primary murine hepatocytes treated with or without trehalose for 24 h (n = 3–9 cell cultures per group) and in liver from mice treated with or without oral trehalose (3% water, 5 days). (B) UCP1 immunoblot analysis in untreated iBAT or liver tissue from mice treated with or without trehalose (upper panels). UCP1 immunoblot analysis in untreated iBAT or primary hepatocytes treated with or without trehalose (24 h; lower panels). (C) Ucp1 mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of 5 mM pyruvate. (D) Ucp1 mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of scrambled or Ppargc1a-directed ASO. (E) Ucp1 mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of DMSO or LY2874455. (F) Ucp1 upregulation in primary hepatocytes incubated with low serum and low glucose (‘Starve’, 24 h) or regular growth media (‘Control’, 24 h) in the presence of scrambled or Ppargc1a- or Fgf21-directed ASO. (G) Ucp1 mRNA accumulation in primary hepatocytes treated with or without trehalose in the presence or absence of scrambled or Tfeb-directed siRNA (left). Tfeb mRNA abundance in Tfeb siRNA-treated hepatocytes (24 h; right). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 versus control by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests.
We quantified the role of FGF21 and PPARGC1A in trehalose-induced Ucp1 expression in vitro by treating primary hepatocytes with or without Ppargc1a-directed ASOs (Figure 6(d)) or with vehicle or LY2874455 (an inhibitor of FGFR1 [fibroblast growth factor receptor 1]-FGFR4; Figure 6(e)) prior to treatment with or without trehalose (24 h). Both Ppargc1a ASO and LY2874455 significantly blocked trehalose-induced Ucp1 expression when compared with control trehalose-treated cultures (Figure 6(d and e)). Accordingly, inhibition of PRKAA1 upstream of the PPARGC1A-FGF21 pathway in primary hepatocytes blocked trehalose-induced PPARGC1A, FGF21, and UCP1 protein accumulation (Fig. S3A). However, pan-PRKAA inhibition achieved by kinase-dead PRKAA1 overexpression blocked Ppargc1a and Fgf21 mRNA accumulation without detectable effects on Ucp1 gene induction (Fig. S3B). In contrast, Prkaa1-specfic siRNA significantly decreased total Prkaa1 expression without altering Ppargc1a, Fgf21, or Ucp1 expression (Fig. S3C). The data suggest that the PRKAA2 isoform selectivity exists downstream of trehalose to regulate Ppargc1a, Fgf21, and Ucp1 mRNA and protein induction.
The data thus far indicated that trehalose induces a hepatocyte fasting state upon SLC2A inhibition. To determine the extent to which trehalose-induced signaling mechanisms are analogous to physiological fasting, we directly quantified hepatocyte Ucp1 mRNA abundance in primary hepatocyte cultures containing regular growth medium (4.5 g/L glucose, 10% FBS) or starvation medium (1.0 g/L glucose, 0.5% FBS) after transfecting either scrambled ASO or ASOs directed against Ppargc1a or Fgf21 (Figure 6(f)). Starvation medium in scrambled ASO-treated hepatocytes activated hepatocyte Ucp1 expression, and this was abrogated in cells with Ppargc1a- or Fgf21-directed ASOs. This suggested that fasting and trehalose treatment induce hepatocyte UCP1 via analogous signaling pathways.
Together, these data indicated that trehalose activates hepatic UCP1 protein and mRNA expression, and UCP1 expression depends upon energetic deficit as well as FGFR1-FGFR4 and PPARGC1A signaling. Because of the well-described interplay between TFEB and the PPARGC1A-FGF21-UCP1 pathways both during fasting, and in view of the PPARGC1A-FGF21-UCP1 pathway described here in trehalose-treated hepatocytes, we tested the effect of Tfeb knockdown on UCP1 upregulation. Tfeb-directed siRNA reduced Tfeb expression by approximately 50% (Figure 6(g)). Accordingly, Tfeb-directed siRNA partly attenuated trehalose-stimulated Ucp1 mRNA expression in comparison to scrambled siRNA-treated cultures treated with trehalose (Figure 6(g)).
Trehalose-induced fasting responses mediate changes in hepatocellular respiration
Trehalose blocked glycolytic flux and cellular respiration concomitant with activated fasting-mimetic signaling pathways. We aimed to determine if the elucidated signaling pathways directly modulated cellular metabolic alterations induced by trehalose. AML12 hepatocytes were treated 48 h with or without trehalose in the presence of LY2874455 to block FGFR1-FGFR4, or Fgf21-directed ASO, Ucp1-directed siRNA, or Tfeb-directed siRNA. Cells were then subjected to OCR and ECAR analysis. Trehalose significantly inhibited maximal respiration after FCCP treatment (Figure 7(a)), however, this was significantly reversed by LY2874455, and by genetic knockdown of Fgf21, Ucp1, and Tfeb. Remarkably, Tfeb knockdown had the most profound effect on maximal respiration in trehalose-treated hepatocyte cultures. Moreover, inhibition of Fgf21, Ucp1, and Tfeb also significantly reversed the trend toward lower basal OCR and ATP production in trehalose treated cultures (Figure 7(b and c)). In contrast, inhibition of FGFR1-FGFR4, Fgf21, Ucp1, and Tfeb did not restore ECAR inhibition in response to trehalose (Figure 7(d)).
Figure 7.
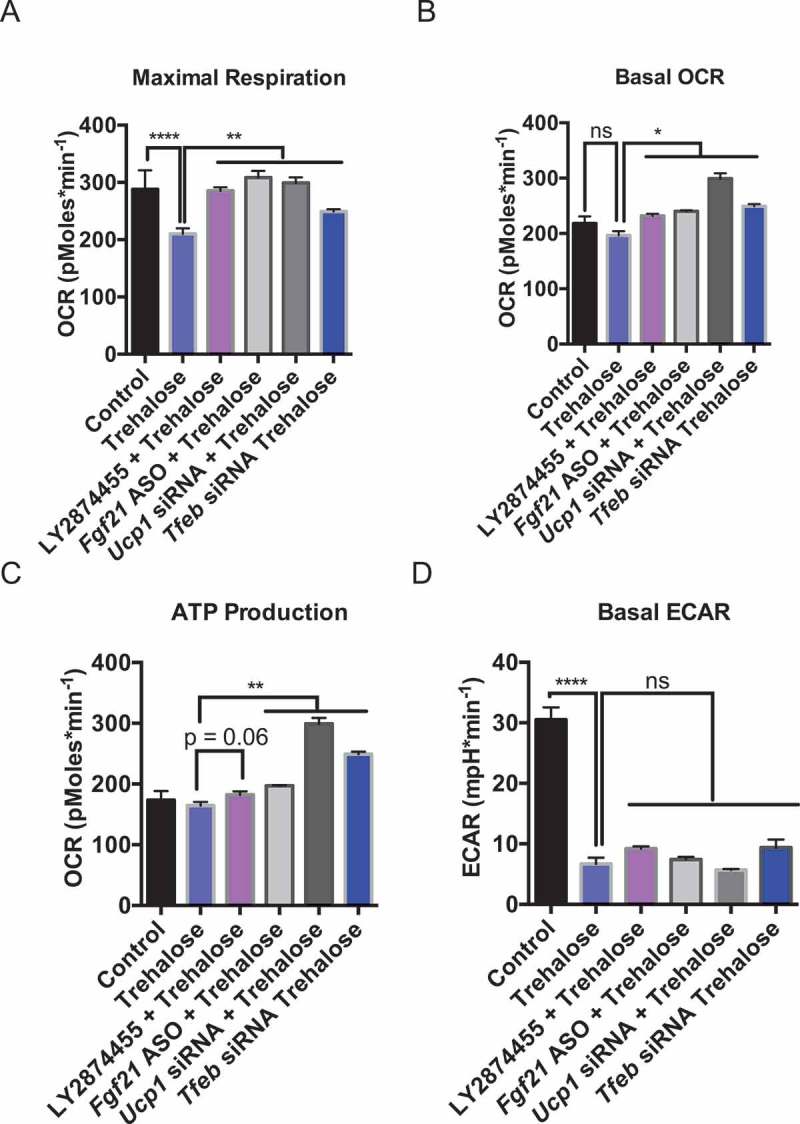
Trehalose-induced FGF21, TFEB, and UCP1 mediate changes in hepatocyte metabolism. (A-C). Quantification of maximal respiration (A), basal OCR (B), and ATP production (C) in cells treated with or without trehalose (48 h) in the presence or absence of DMSO or the FGFR1-FGFR4 inhibitor, LY2874455, or after knockdown of UCP1, FGF21, or TFEB (n = 6–12 cell cultures per group). (D) Quantification of basal ECAR in the presence or absence of TFEB, FGF21, or UCP1 knockdown, or after LY2874455 treatment in AML12 hepatocytes. * P < 0.05, ** P < 0.01, by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests. ns, not significant with p > 0.05 between bracketed groups.
Hepatocyte-selective FGF21 knockdown partly abrogates trehalose-induced thermogenesis in vivo
We tested the role of hepatocyte-selective FGF21 in mediating trehalose-induced thermogenesis by treating WT mice with scrambled or Fgf21-directed ASO 3 weeks before feeding 3% trehalose water ad libitum. Fgf21-directed ASO blocked mRNA expression and protein in liver, but not in epididymal white adipose tissue (eWAT) (Figure 8(a)), iBAT, or skeletal muscle (Figure S4). Oral trehalose significantly increased Fgf21 mRNA and circulating peptide in scrambled ASO-treated mice (Figure 8(b and c)), but both mRNA and circulating peptide levels were blocked in vivo by Fgf21 ASO treatment. In parallel, Fgf21 ASO partially but significantly blocked 5-day oral trehalose-induced dark and light cycle thermogenesis (Figure 8(d and e)). This suggested that hepatocyte FGF21 mediates at least part of the trehalose-induced thermogenic response.
Figure 8.
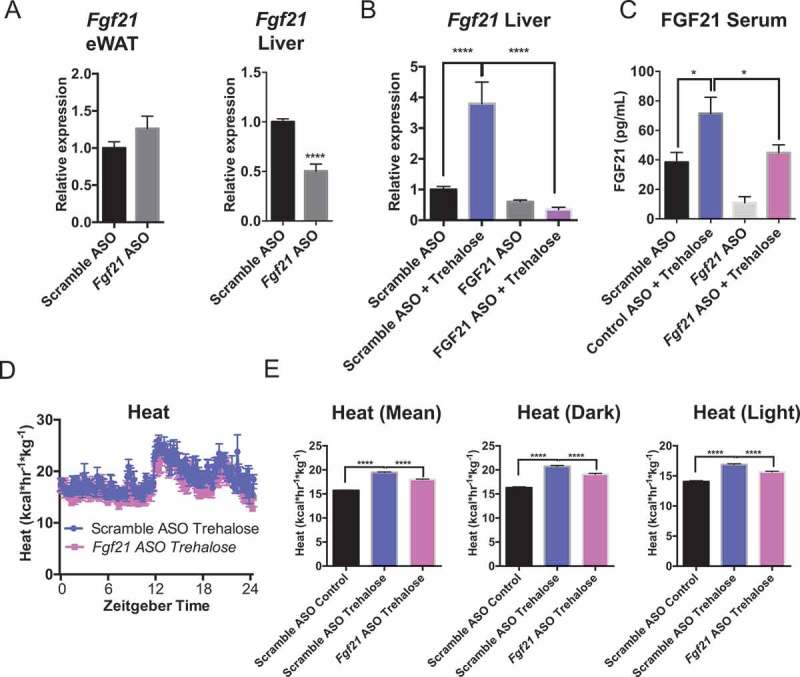
Trehalose-induced thermogenesis is partly FGF21-dependent. (A) qPCR analysis of Fgf21 mRNA in epididymal white adipose tissue (eWAT) and liver after scrambled or Fgf21-directed antisense oligonucleotide (ASO) treatment 2 wk prior to trehalose treatment and sacrifice. (B) Hepatic Fgf21 mRNA quantification in livers derived from mice treated with or without trehalose and with or without Fgf21 ASO. (C) Circulating FGF21 peptide quantification by colorimetric ELISA in serum from mice treated with or without trehalose and with or without Fgf21 ASO. (D) Heat generation over time for trehalose-treated mice pre-treated with scrambled or Fgf21-specific antisense oligonucleotide (ASO). (E) Mean areas under the total heat-time curve, or during dark- (1801–0600 h) or light cycles (0601 – 1800 h). * and ****, P < 0.05 and 0.0001 vs. scramble ASO + trehalose by T-test with Bonferroni-Dunn post hoc correction for n = 4 mice per treatment group.
Hepatocyte-selective TFEB knockdown abrogates trehalose-induced thermogenesis in vivo
Given observations that 1) FGF21 knockdown partly blocked trehalose-induced thermogenesis, 2) TFEB is a primary regulator of hepatic and whole-body metabolism during starvation, regulating pathways proximal and parallel to Fgf21 induction, and 3) TFEB knockdown had the most profound effect in reversing trehalose effects on hepatocellular respiration, we tested the in vivo effects of hepatocyte TFEB knockdown on trehalose-induced thermogenesis. Wild-type mice were treated with oral trehalose (3% water, 5 days ad libitum) 10 days after tail-vein injection of adeno-associated virus serotype 8 (AAV8) encoding scrambled or Tfeb-directed shRNA. Knockdown of approximately 60% in AAV8-shTfeb-treated mice was confirmed by qPCR analysis in liver tissue (Figure 9(a)). Similarly, Tfeb expression by qPCR analysis was not altered in eWAT or in iBAT (Figure 9(b)) or skeletal muscle (not shown) of mice treated with Tfeb shRNA-encoding AAV8 compared with scrambled shRNA-treated controls. Similarly, TFEB protein was knocked down in liver tissue, but not in eWAT, iBAT, or skeletal muscle in AAV8-shTfeb-treated mice versus controls (Fig. S5A). Trehalose feeding induced both Ppargc1a and Ucp1 expression in eWAT of mice treated with scrambled shRNA-encoding AAV8. In contrast, hepatocyte Tfeb knockdown significantly blocked eWAT Ucp1 and Ppargc1a induction (Figure 9(c)), although hepatic Tfeb knockdown did not significantly alter trehalose-induced hepatic Ucp1 (Fig. S5B). Nevertheless, Tfeb knockdown in vivo blocked trehalose-induced enhancement of whole-body thermogenesis (Figure 9(d)), oxygen consumption (Figure 9(e)), CO2 production (Figure 9(f)) and circulating FGF21 peptide (Figure 9(g)).
Figure 9.
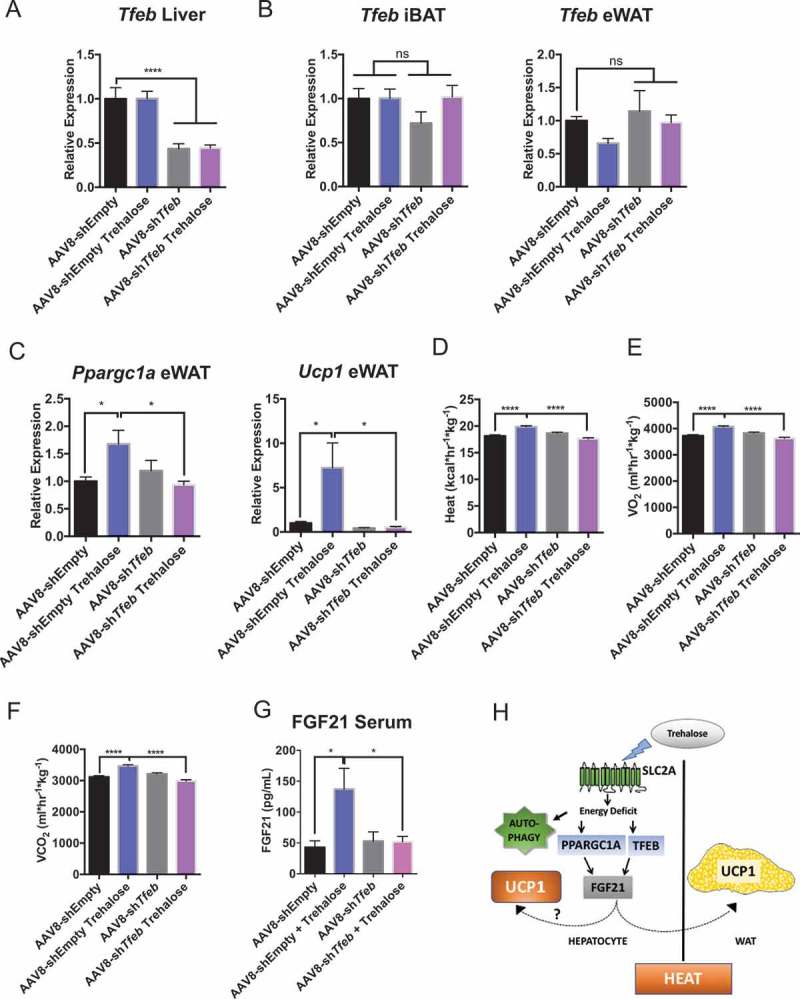
Hepatocyte-selective TFEB knockdown attenuates trehalose-induced thermogenesis in vivo. (A) Hepatic Tfeb mRNA abundance in Tfeb shRNA-treated mice (2 wk) treated with or without 5-day 3% trehalose water (n = 4–8 mice per group). (B) Interscapular brown adipose tissue (iBAT) and epididymal white adipose tissue (eWAT) expression of Tfeb in mice treated with AAV8 encoding Tfeb shRNA or empty vector with or without trehalose treatment. (C) eWAT Ppargc1a and Ucp1 mRNA abundance in response to AAV8-mediated Tfeb shRNA delivery and 5-day trehalose treatment. (D-F). Heat production (D), oxygen consumption (E), and CO2 production (F) in mice infected with AAV8 encoding empty vector or Tfeb-directed shRNA following treatment with sterile water or trehalose (5 days). (G) Circulating FGF21 peptide quantification by colorimetric ELISA in serum from mice treated with or without trehalose and with or without Tfeb-directed shRNA. (H) Working model of trehalose-induced thermogenesis. Trehalose induces hepatic fasting responses via TFEB and PPARGC1A to enhance FGF21 and downstream UCP1 in the hepatocyte and in eWAT. Autophagy appears to act independent of this thermogenic pathway. * P < 0.05, ** P < 0.01, **** P < 0.0001 by Student’s T-test with Bonferroni-Dunn post-hoc correction for multiple tests.
Discussion
Trehalose is a promising cardiometabolic and anti-neurodegenerative therapeutic with mechanisms that have yet to be revealed [19,26]. Seminal work indicated this was an MTOR-independent, autophagy-inducing agent in the contexts of neurodegeneration, atherosclerosis, and obesity [24,33]. Our prior data demonstrated that trehalose initiates canonical hepatic glucose pseudofasting responses: AMPK activation and MTOR signaling inhibition [20,34]. Subsequent studies demonstrated that trehalose administration recapitulates macrophage TFEB overexpression to suppress atherogenesis [27]. Our work here now provides direct evidence regarding critical involvement of hepatic TFEB and FGF21 in trehalose-induced thermogenesis (Model in Figure 9(h)). Together, an overarching theme emerges: trehalose blocks hepatocyte metabolism to activate canonical adaptive fasting signals. This includes both AMPK activation and MTOR suppression in liver [20,25], and moves downstream to the MTOR target, TFEB [11,53], as a key mediator of distal hepatic fasting transcriptional responses.
Similar to the effects of trehalose treatment and hepatic SLC2A blockade, physiological fasting robustly induces hepatic FGF21 release. In rodents, FGF21 induces thermogenesis, although teleological explanations for this well-described phenomenon are elusive [54–57]. However, in the specific context of hepatocyte SLC2A blockade we cannot say that the trehalose response is precisely equivalent to the physiological fasting response. For example, many of the key mediators we defined within the trehalose response also parallel cold exposure responses (e.g. eWAT Ucp1, eWAT Ppargc1a, hepatic Fgf21) and ketogenic diet (e.g. dietary glucose restriction [58,59]). Given that the hepatic glucose-specific fasting response is not yet fully understood, understanding the precise pathways of trehalose signaling will aid us in understanding unique and highly analogous pathways to be targeted in metabolic disease.
In conjunction with TFEB, we delineate a trehalose-induced PPARGC1A-FGF21-UCP1 regulatory pathway that mimics glucose and serum deprivation as a model of physiological fasting (Figure 5(h)). Consistent with this notion, oxygen consumption rate analysis confirmed that trehalose time-dependently blocked glycolytic flux (Figure 3(e)) and cellular basal and maximal cellular respiration (Figure 3(a-d)). Blocking any part of the TFEB-PPARGC1A- FGF21-UCP1 pathway via antisense or pharmacological strategies restored cellular respiratory capacity and enhanced hepatocyte ATP production. In light of our data here that 1) hepatic TFEB knockdown did not completely reduce hepatic Ucp1 expression, 2) eWAT Ucp1 was also upregulated in response to trehalose feeding in vivo (Figure. 9(c)), and 3) eWAT ‘browning’ is a well-established source of heat generation downstream of hepatic TFEB and FGF21 29, we acknowledge that the extent to which hepatic UCP1 participates in heat generation per se, (as proposed in [50–52,60]) versus signal transduction, anti-oxidant, or other functions specifically remains to be fully dissected.
Concomitant with trehalose-mediated activation of TFEB in hepatic and extrahepatic tissues is activation of the PPARGC1A-FGF21 signaling pathway in vitro and in vivo. The data are consistent with our prior reports that trehalose drives PRKAA1 phosphorylation and MTOR blockade [20,25], each of which is reported to regulate PPARGC1A-FGF21 during fasting [49,61–63]. We demonstrated an analogous role for PRKAA1 in regulating the PPARGC1A-FGF21 cascade in response to trehalose here (Figure S3). The broader implications of these data are perhaps more intriguing. In view of data that trehalose is a hepatic SLC2A inhibitor that is largely restricted in bioavailability to the enterohepatic circulation [20,27], we propose that pharmacological hepatic pseudofasting is sufficient to reproduce parts of the hepatic and extrahepatic therapeutic effects of generalized caloric restriction. The alternative hypothesis, that low circulating trehalose concentrations that escape the enterohepatic circulation might themselves have important extrahepatic action, remains under investigation by ours and other groups.
Mechanistically, the preponderance of prior work either associates or directly demonstrates that the pro-metabolic effects of trehalose depend on autophagic induction using autophagy-deficient mouse models (e.g. Atg16l1HM mouse hepatocytes or atg7-null mice [19,20,22–24,27,28,33]). Here, we provide data that autophagy-deficient mice, Atg16l1HM, Becn1±, and epg5 null mice, are indistinguishable from WT controls in their response to trehalose induction of thermogenesis. Therefore, trehalose likely induces part of its therapeutic effects independent of autophagic flux. The implication of these data for subsequent studies utilizing trehalose as a pharmacological tool should reasonably account for the possibility that any effects might be unrelated to autophagic flux. More specifically, the desirable effects of trehalose might be attributable to the glucose fasting response per se.
The therapeutic implications of this work are that inducing hepatic glucose fasting mechanisms may be sufficient to induce the manifold benefits of whole-organism macronutrient fasting, such as thermogenesis and FGF21 release. Yet a targeted approach that mimics glucose fasting only in the liver may circumvent some of the undesirable aspects of other glucose deprivation methods, such as ketogenic diet intermittent fasting, and caloric restriction, both of which are practically unsustainable in a clinical setting, and each of which is associated with micronutrient deficiencies and other complications [64–68]. Future efforts aiming to determine whether signals from the fasting liver have extrahepatic effects that extend beyond the pro-thermogenic effects demonstrated herein are warranted. Regardless, trehalose stands at present as a promising pre-clinical therapeutic against neurodegenerative and cardiometabolic disease. Elucidating the fasting-mimetic, pro-autophagic, and other properties of this disaccharide will not only advance our understanding of the pathogenesis of the conditions treated, but also may identify further pathways to be networked or leveraged therapeutically.
Materials and methods
Mice
Wild-type C57BL/6J-strain mice (Jackson Laboratory, 000664) and Becn1+/- mice (Jackson Laboratory, 018429) aged 6–8 weeks were obtained directly from the Jackson Laboratory. Upon arrival, mice were equilibrated for a minimum of 7 days in the specific pathogen-free vivarium prior to initiating metabolic measurements. All in vivo experimental procedures were performed in strict accordance with IACUC guidelines at Washington University School of Medicine.
Trehalose (Sigma Aldrich, T9531) was ≥ 99% purity by HPLC. 3% trehalose water (w:v) fed ad libitum was used in all in vivo experiments.
Adeno-associated viruses overexpressing either empty vector (AAV8-shEmpty) (Vector Biolabs, 7077) or Tfeb-directed shRNA (AAV8-shTfeb) (Vector Biolabs, shAAV-281,149) were obtained as ready-to-use viral stock. 1011 viral particles were injected via tail vein 10–14 days prior to trehalose initiation. Tfeb expression was quantified in pilot experiments by qRT-PCR analysis of hepatic, skeletal muscle, brown and epididymal white adipose tissues 10 days post-injection to ascertain selective hepatic Tfeb knockdown.
Antisense oligonucleotides (ASOs) were obtained directly from IONIS Pharmaceuticals. Fgf21 ASO (IONIS Pharmaceuticals, ION-256,617) and Ppargc1a (IONIS Pharmaceuticals, ION-185,538) as ready-to-inject oligomers, and were used precisely as described [68,69].
Indirect calorimetry
Indirect calorimetry was carried out using a TSE Phenomaster (TSE Systems; Chesterfield, MO) precisely as described previously [21,70]. Briefly, mice were placed in indirect calorimeters for at least 28 h. The first 4 h in the cage were unmeasured in order to allow for acclimation time. Thereafter, the oxygen and CO2 consumption and production were quantified for a minimum of one light cycle (0601–1800 h) and for one dark cycle (1801–0600 h). VO2, VCO2, heat, movement and respiratory exchange ratio (RER) were automatically calculated using TSE Systems Phenomaster software.
Quantitative real-time RT-PCR (qpcr)
qPCR was performed with primer sequences and method precisely as described [70]. Primer sequences are as shown in Table 1. All primers were custom-synthesized oligonucleotides obtained from Integrated DNA Technologies.
Table 1.
Primers used for qRT-PCR analysis. Sequences are listed 5ʹ-3ʹ.
| Target | Species | Forward (5ʹ to 3ʹ) | Reverse (5ʹ to 3ʹ) |
|---|---|---|---|
| Actb | Mouse | GAT TAC TGC TCT GGC TCC TAG | GAC TCA TCG TAC TCC TGC TTG |
| Fgf21 | Mouse | TAC ACA GAT GAC GAC CAA GA | GGC TTC AGA CTG GTA CAC AT |
| Ppargc1a | Mouse | ACA CCG CAA TTC TCC CTT GT | CGG CGC TCT TCA ATT GCT TT |
| Tfeb | Mouse | GAG CTG GGA ATG CTG ATC C | GGG ACT TCT GCA GGT CCT T |
| Ucp1 | Mouse | AGG CTT CCA GTA CCA TTA GGT | CTG AGT GAG GCA AAG CTG ATT T |
Cell culture
AML12 murine hepatocytes were obtained directly from the American Type Culture Collection (ATCC, CRL-2254). Cultures were maintained in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (DMEM/F12; ThermoFisher Scientific, 11,320–033) supplemented with 10% fetal bovine serum (Gibco, 26,140–079) and insulin-transferrin-selenium solution (Sigma Aldrich, I1884) through passage 20 according to ATCC specifications. Primary murine hepatocytes were cultured as we have described [20,25,70]. Antisense oligonucleotides (ASOs) were obtained from IONIS Pharmaceuticals, directed against Fgf21 ASO (IONIS Pharmaceuticals, ION-256,617) and Ppargc1a (IONIS Pharmaceuticals, ION-185,538). siRNA directed against Tfeb (Santa Cruz Biotechnology, sc-38,510), Ucp1 (Santa Cruz Biotechnology, SC42681), and Prkaa1 (Santa Cruz Biotechnology, sc-29,674) were obtained from Santa Cruz Biotechnology. LY2874455 inhibitor was purchased from Cayman Chemical (Cayman Chemical, 20,843). All nucleotides were transfected using Lipofectamine 3000 (ThermoFisher Scientific, L3000001) precisely according to manufacturer instructions.
Immunoblotting
Immunoblotting was performed as described [71]. PPARGC1A antibody was a gift provided by Alan L Schwartz (Washington University School of Medicine) [72], originally generated in the laboratory of Daniel Kelly [73]. Antibodies from Cell Signaling Technology were: anti-phospho-PRKAA1 (T172; 2535); anti-PRKAA1 (5832S); anti-ACTB (4970); anti-GAPDH (5174); and anti-VCL (13901S). Anti-FGF21 (ab64857) and anti-UCP1 antisera (ab155117) were from Abcam.
In vitro metabolism (seahorse XF96) assays
Seahorse assays were performed using a Seahorse XF96 analyzer Mito Stress Test Kit (Agilent Technologies, 103,015–100) precisely according to manufacturer specifications. This kit provides the oligomycin, FCCP, rotenone and antimycin used in each assay. Hepatocyte cultures were seeded at 20,000 cells per well prior to assay.
Enzyme-linked immunosorbent assays (ELISA)
Total FGF21 ELISAs (Millipore, EZRMFGF21-26K) in serum obtained from experimental mice were performed precisely according to manufacturer specifications.
Statistical analysis
All data were analyzed using GraphPad Prism 6.0. P < 0.05 was defined as statistically significant after post hoc correction for multiple comparisons. Specific statistical tests applied are noted in the Figure Legends.
Funding Statement
This work was supported by the Children’s Discovery Institute [MI-FR-2014-426, MD-FR-2017-593]; Washington University NIGMS Institutional Training Grant in Cell and Molecular Bioscience [T32GM007067];National Science Foundation (NSF) [DGE-1143954];HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) [R01DK100644];U.S. Department of Defense (DOD) [W81XWH-17-1-0133];Washington University Child Health Research Center [K12HD076224];Robert Wood Johnson Foundation (RWJF) [AMFDP73315];Washington University Digestive Disease Research Center [P30DK52574];Washington University Diabetes Research Center [P30DK020579];HHS | NIH | National Center for Advancing Translational Sciences (NCATS) [UL1TR002345].
Abbreviations
- AAV8
adeno-associated virus serotype 8
- ASO
antisense oligonucleotide
- ATG16L1
autophagy related 16-like 1 (S. cerevisiae)
- BECN1
beclin 1, autophagy related
- ECAR
extracellular acidification rate
- EPG5
ectopic P-granules autophagy protein 5 homolog (C. elegans)
- eWAT
epididymal white adipose tissue
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- FGF21
fibroblast growth factor 21
- FGFR
fibroblast growth factor receptor
- iBAT
interscapular brown adipose tissue
- LEP
leptin
- MTOR
mechanistic target of rapamycin kinase
- OCR
oxygen consumption rate
- PPARGC1A
peroxisome proliferative activated receptor, gamma, coactivator 1 alpha
- PRKAA1/AMPK
protein kinase, AMP-activated, alpha 1 catalytic subunit
- SLC2A
solute carrier family 2 (facilitated glucose transporter)
- TFEB
transcription factor EB
- UCP1
uncoupling protein 1 (mitochondrial, protein carrier)
Acknowledgments
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Medical Research Program under Award No. W81XWH-17-1-0133. Opinions, interpretations, conclusions and reccomendations are those of the author and are not necessarily endorsed by the Department of Defense. This work was also supported by grants from the NIH/National Center for Advancing Translational Sciences (NCATS) grant #UL1TR002345, the Children’s Discovery Institute (MI-FR-2014-426) AGA-Gilead Sciences Research Scholar Award in Liver Disease, the Washington University Digestive Disease Research Core Center (BJD) (P30DK52574), the Washington University Diabetes Research Center (P30DK020579), Nutrition and Obesity Research Center (P30DK056341), and the Robert Wood Johnson Foundation. BJD is a Scholar of the Washington University Child Health Research Center (K12HD076224), and of the Children’s Discovery Institute (MI-FR-2014-426). IUM was supported by R01DK100644. ALM was supported by the Washington University Spencer T. Olin Fellowship, the Washington University NIGMS Institutional Training Grant in Cell and Molecular Biosciences (T32GM007067), and National Science Foundation Graduate Student Fellowship (DGE-1143954). We thank Herbert Virgin at Washington University School of Medicine, for the Atg16l1HM mouse line, and Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences) for the epg5 null mice used in our studies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Longo VD, Mattson MP.. Fasting: molecular mechanisms and clinical applications. Cell Metab [Internet] 2014;19:181–192. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Seyer P, Vallois D, Poitry-Yamate C, et al. Hepatic glucose sensing is required to preserve β cell glucose competence. J Clin Invest [Internet] 2013;123:1662–1676. cited 2015 April 14 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3613916&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sengupta S, Peterson TR, Laplante M, et al. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature [Internet] 2010;468:1100–1104. Available from. [DOI] [PubMed] [Google Scholar]
- [4].Tan VP, Miyamoto S.. Nutrient-sensing mTORC1: integration of metabolic and autophagic signals. J Mol Cell Cardiol [Internet] 2016:1–11. Available from http://www.sciencedirect.com/science/article/pii/S0022282816300025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xiao B, Sanders MJ, Underwood E, et al. Structure of mammalian AMPK and its regulation by ADP. Nature [Internet] 2011;472:230–233. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lage R, Diéguez C, Vidal-Puig A, et al. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–549. [DOI] [PubMed] [Google Scholar]
- [7].Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell [Internet] 2008;30:214–226. Available from http://linkinghub.elsevier.com/retrieve/pii/S109727650800169X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Potthoff MJ, Finck BN. Head over hepatocytes for FGF21. Diabetes. 2014;63:4013–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol [Internet] 2013;15:647–658. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23604321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Holmes D. Metabolism: fasting induces FGF21 in humans. Nat Rev Endocrinol [Internet] 2016;12:3 Available from. [DOI] [PubMed] [Google Scholar]
- [13].Xu J, Lloyd DJ, Hale C, et al. FGF21 reverses hepatic steatosis, increases energy expenditure and improves insulin sensitivity in diet-induced obese mice. Diabetes [Internet] 2009;58:250–259. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18840786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim KH, Lee MS. FGF21 as a mediator of adaptive responses to stress and metabolic benefits of anti-diabetic drugs. J Endocrinol. 2015;226:R1–16. [DOI] [PubMed] [Google Scholar]
- [15].Potthoff MJ, Inagaki T, Satapati S, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A [Internet] 2009;106:10853–10858. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19541642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gaich G, Chien JY, Fu H, et al. The effects of LY2405319, an FGF21 Analog, in obese human subjects with type 2 diabetes. Cell Metab [Internet] 2013;18:333–340. Available from. [DOI] [PubMed] [Google Scholar]
- [17].Li Y, Wong K, Giles A, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Villarroya F, Cereijo RR, Villarroya J, et al. Brown adipose tissue as a secretory organ. Nat Rev Endocrinol [Internet] 2017;13:26–35. Available from. [DOI] [PubMed] [Google Scholar]
- [19].Mardones P, Rubinsztein DC, Hetz C. Mystery solved: trehalose kickstarts autophagy by blocking glucose transport. Sci Signal [Internet] 2016;9:fs2LP–fs2. Available from http://stke.sciencemag.org/content/9/416/fs2.abstract [DOI] [PubMed] [Google Scholar]
- [20].DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal [Internet] 2016;9:ra21–ra21. Available from: http://stke.sciencemag.org/content/9/416/ra21.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].DeBosch BJ, Chen Z, Finck BN, et al. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Mol Endocrinol [Internet] 2013;27:1887–1896. DOI: 10.1210/me.2013-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Castillo K, Nassif M, Valenzuela V, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9:1308–1320. [DOI] [PubMed] [Google Scholar]
- [23].Aguib Y, Heiseke A, Gilch S, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5:361–369. [DOI] [PubMed] [Google Scholar]
- [24].Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J Biol Chem [Internet] 2007;282:5641–5652. cited 2014 October 6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17182613 [DOI] [PubMed] [Google Scholar]
- [25].Mayer AL, Higgins CB, Heitmeier MR, et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep [Internet] 2016;6:38586 Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron [Internet] 2017;93:1015–1034. Available from. [DOI] [PubMed] [Google Scholar]
- [27].Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun [Internet] 2017;8:15750 DOI: 10.1038/ncomms15750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Arai C, Arai N, Mizote A, et al. Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance. Nutr Res. 2010;30:840–848. [DOI] [PubMed] [Google Scholar]
- [29].Cornu M, Oppliger W, Albert V, et al. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc Natl Acad Sci U S A [Internet] 2014;111:11592–11599. Available from http://stke.sciencemag.org/content/vj/pnas/111/32/11592.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jo YH, Buettner C. Why leptin keeps you warm. Mol Metab [Internet] 2014;3:779–780. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Richards AB, Krakowka S, Dexter LB, et al. Trehalose: A review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem Toxicol. 2002;40:871–898. [DOI] [PubMed] [Google Scholar]
- [32].Krüger U, Wang Y, Kumar S, et al. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging [Internet] 2016;33:2291–2305. Available from. [DOI] [PubMed] [Google Scholar]
- [33].Zhang X, Chen S, Song L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Salem M, Ammitzboell M, Nys K, et al. ATG16L1: A multifunctional susceptibility factor in crohn disease. Autophagy. 2015;11:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ [Internet] 2011;18:571–580. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature [Internet] 2008;456:259–263. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang C, Symington JW, Mysorekar IU. ATG16L1 and pathogenesis of urinary tract infections. Autophagy. 2012;8:1693–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Piano Mortari E, Folgiero V, Marcellini V, et al. The Vici syndrome protein EPG5 regulates intracellular nucleic acid trafficking linking autophagy to innate and adaptive immunity. Autophagy. 2017;1–16. DOI: 10.1080/15548627.2017.1389356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhao H, Zhao YG, Wang X, et al. Mice deficient in epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. 2013;200:731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhao YG, Zhao H, Sun H, et al. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy. 2013;9:1258–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang Z, Miao G, Xue X, et al. The vici syndrome protein EPG5 Is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell [Internet] 2016;63:781–795. Available from. [DOI] [PubMed] [Google Scholar]
- [42].Divakaruni AS, Paradyse A, Ferrick DA, et al. Analysis and interpretation of microplate-based oxygen consumption and pH data. 1st ed. Amsterdam: Elsevier Inc; 2014. DOI: 10.1016/B978-0-12-801415-8.00016-3 [DOI] [PubMed] [Google Scholar]
- [43].Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim KH, Jeong YT, Oh H, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med [Internet] 2013;19:83–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23202295 [DOI] [PubMed] [Google Scholar]
- [45].Markan KR, Naber MC, Ameka MK, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63:4057–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Settembre C, Ballabio A. Lysosome: regulator of lipid degradation pathways. Trends Cell Biol [Internet] 2014;24:743–750. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Siddiqui A, Bhaumik D, Chinta SJ, et al. Mitochondrial quality control via the PGC1α-TFEB signaling pathway is compromised by parkin Q311X mutation but independently restored by Rapamycin. J Neurosci [Internet] 2015;35:12833–12844. Available from http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.0109-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Puigserver P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. Int J Obes [Internet] 2005;29:S5–9. DOI: 10.1038/sj.ijo.0802905 [DOI] [PubMed] [Google Scholar]
- [49].Ishigaki Y, Katagiri H, Yamada T, et al. Dissipating excess energy stored in the liver is a potential treatment strategy for diabetes associated with obesity. Diabetes. 2005;54:322–332. [DOI] [PubMed] [Google Scholar]
- [50].Stoner HB. The role of the liver in non-shivering thermogenesis in the rat. J Physiol [Internet] 1973;232:285–296. Available from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1350455/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Adachi A, Fwnahashi M, Ohga J. Hepatic Thermoogenesis relation to food intake in the conscious rat. Brain Res Bull. 1991;27:529–533. [DOI] [PubMed] [Google Scholar]
- [52].Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB open. EMBO J [Internet] 2012;31:1095–1108. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hondares E, Rosell M, Gonzalez FJ, et al. Hepatic FGF21 expression is induced at birth via PPAR?? in response to milk intake and contributes to thermogenic activation of Neonatal Brown Fat. Cell Metab [Internet] 2010;11:206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fisher M, Kleiner S, Douris N, et al. adaptive thermogenesis FGF21 regulates PGC-1a and browning of white adipose tissues in adaptive thermogenesis. Genes Dev [Internet] 2012;26:271–281. Available from: http://www.luminpdf.com/files/5951223/Fisher_FM_2012.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fu T, Seok S, Choi S, et al. MicroRNA 34a inhibits beige and brown fat formation in obesity in part by suppressing adipocyte fibroblast growth factor 21 signaling and SIRT1 function. Mol Cell Biol [Internet] 2014;34:4130–4142. Available from DOI: 10.1128/MCB.00596-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Potthoff MJ. FGF21 and metabolic disease in 2016: A new frontier in FGF21 biology. Nat Rev Endocrinol [Internet] 2016;13:74–76. DOI: 10.1038/nrendo.2016.206 [DOI] [PubMed] [Google Scholar]
- [57].Cyphert HA, Ge X, Kohan AB, et al. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem. 2012;287:25123–25138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Domouzoglou EM, Maratos-Flier E. Fibroblast growth factor 21 is a metabolic regulator that plays a role in the adaptation to ketosis. Am J Clin Nutr. 2011;93:901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nedergaard J, Golozoubova V, Matthias A, et al. UCP1 : the only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim Biophys Acta. 2001;1504:82–106. [DOI] [PubMed] [Google Scholar]
- [60].Estall JL, Ruas JL, Soo C, et al. PGC-1a negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erba axis. Proc Natl Acad Sci U S A [Internet]. 2009:106:22510-22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chau MDL, Gao J, Yang Q, et al. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci U S A [Internet] 2010;107:12553–12558. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2906565&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Berglund ED, Kang L, Lee-Young RS, et al. Glucagon and lipid interactions in the regulation of hepatic AMPK signaling and expression of PPARalpha and FGF21 transcripts in vivo. Am J Physiol Endocrinol Metab. 2010;299:E607–14. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [63].Kang HC, Chung DE, Kim DW, et al. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia. 2004;45:1116–1123. [DOI] [PubMed] [Google Scholar]
- [64].Damms-Machado A, Weser G, Bischoff SC. Micronutrient deficiency in obese subjects undergoing low calorie diet. Nutr J [Internet] 2012;11:34 DOI: 10.1186/1475-2891-11-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Martin B, Mattson MP, Maudsley S. Caloric restriction and intermittent fasting: two potential diets for successful brain aging. Ageing Res Rev. 2006;5:332–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Stunkard AJ, Rush J. Dieting and depression reexamined. A critical review of reports of untoward responses during weight reduction for obesity. Ann Intern Med. 1974;81:526–533. [DOI] [PubMed] [Google Scholar]
- [67].Wl B, Gj A. Similarities of carbohydrate deficiency and fasting: I. weight loss, electrolyte excretion, and fatigue. Arch Intern Med [Internet] 1963;112:333–337. Available from. [DOI] [PubMed] [Google Scholar]
- [68].Soufi N, Hall AM, Chen Z, et al. Inhibiting monoacylglycerol acyltransferase 1 ameliorates hepatic metabolic abnormalities but not inflammation and injury in mice. J Biol Chem. 2014;289:30177–30188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Mayer AL, Zhang Y, Feng EH, et al. Enhanced hepatic PPAR a activity links GLUT8 deficiency to augmented peripheral fasting responses in male mice. Endocrinology 2018;159:2110–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].DeBosch BJ, Chen Z, Saben JL, et al. Glucose transporter 8 (GLUT8) mediates fructose-induced de Novo lipogenesis and macrosteatosis. J Biol Chem [Internet] 2014;289:10989–10998. cited 2014 August 12 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4036240&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].DeBosch BJ, Kluth O, Fujiwara H, et al. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun. 2014;5:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Trausch-Azar J, Leone TC, Kelly DP, et al. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1?? via the N-terminal pathway. J Biol Chem. 2010;285:40192–40200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Leone TC, Lehman JJ, Finck BN, et al. PGC-1a deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:0672–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.