

ABSTRACT
Mitophagy is a main type of selective autophagy, via which damaged mitochondria are selectively degraded via the autophagic pathway. The protein kinase PINK1 and E3 ubiquitin ligase PRKN are the most well studied regulators of mitophagy, via a feedforward mechanism involving ubiquitin phosphorylation (p-Ser65-Ub) and accumulation at the damaged mitochondria. However, it is unknown whether there is a protein phosphatase against PINK1-mediated phosphorylation of ubiquitin. We recently reported that PTEN-L, a newly identified PTEN isoform, is a novel negative regulator of mitophagy through dephosphorylation of p-Ser65-Ub. Our data demonstrate that a significant portion of PTEN-L localizes at the outer mitochondrial membrane and is able to prevent PRKN’s mitochondrial translocation, reduce the phosphorylation of PRKN, impair its E3 ligase activity as well as maintain PRKN in a closed/inactive status. Moreover, we found that PTEN-L dephosphorylates p-Ser65-Ub to disrupt the feedforward mechanism of mitophagy. Our findings suggest that PTEN-L acts as a brake in the regulation of mitophagy.
Abbreviations: ATR: alternatively translated region; CCCP: carbonylcyanide 3-chlorophenylhydrazone; DUBs: deubiquitinating enzymes; MFN2: mitofusion2; MS/MS: tandem mass spectrometry; mtDNA: mitochondrial DNA; MTS: mitochondrial targeting sequences; O/A: oligomycin and antimycin A; PINK1: PTEN induced putative kinase 1; PRKN/parkin: parkin RBR E3 ubiquitin protein ligase; PTEN: phosphatase and tensin homolog; PTEN-L: phosphatase and tensin homolog-long; Ub: ubiquitin; USP: ubiquitin-specific proteases; YFP: yellow fluorescence protein.
KEYWORDS: Mitophagy, PRKN, phosphatase, PTEN-L, ubiquitin
Mitophagy is probably the most well-studied type of selective autophagy, via which damaged mitochondria are degraded and cleared through lysosomal degradation. Defective mitophagy has been linked to the pathogenesis of neurodegeneration, like Parkinson disease. In the past decade the molecular mechanisms underlying mitophagy have been extensively studied. One key pathway involves a protein kinase, PINK1 (PTEN induced putative kinase 1), and an E3 ligase, PRKN/parkin (parkin RBR E3 ubiquitin protein ligase). When mitochondria are damaged, PINK1 is stabilized and activated on mitochondria and then phosphorylates both ubiquitin (p-Ser65-Ub) and PRKN (p-Ser65-PRKN). Such phosphorylation is thought to play a key role in triggering mitophagy. Nowadays, negative regulatory mechanisms against the role of PRKN’s E3 ligase activity in mitophagy have been reported, with the discovery of a few deubiquitinating enzymes (DUBs), such as USP8, USP15 and USP30. However, it is still unknown whether there is any protein phosphatase against PINK1-mediated phosphorylation of ubiquitin and/or PRKN. In this study, we found that PTEN-L acts as a protein phosphatase to dephosphorylate p-Ser65-Ub, leading to a series of events, including blockage of PRKN’s mitochondrial translocation, reduction of p-Ser65-PRKN level and suppression of PRKN’s E3 ligase activity, and eventually interruption of the feedforward mechanisms and suppression of mitophagy.
PTEN (phosphatase and tensin homolog) is an important tumor suppressor to inhibit the phosphoinositide 3-kinase (PI3K)-AKT pathway via its lipid phosphatase activity. Recently, a new isoform of PTEN, termed PTEN-Long or PTENα (referred to as PTEN-L hereafter), has been identified. PTEN-L shares the same mRNA with PTEN, but it translates from a non-AUG start codon (CUG), adding an ATR region (with 173 amino acids) at the N terminus. Intriguingly, PTEN-L is reported to be a mitochondrial protein that functions to regulate mitochondrial energy metabolism. However, the potential regulatory effects of PTEN-L on mitophagy have not been reported.
To understand the possible regulatory role of PTEN-L in mitophagy, we first carefully examined the distribution of PTEN-L in mitochondria [1]. We observed that a significant proportion of PTEN-L is present in the mitochondrial fraction in both cultured cells and in various mouse tissues, whereas PTEN is mostly in the cytosol. Through a topology assay and immuno-gold electron microscopy experiment, we further confirmed that PTEN-L localizes on the outer mitochondrial membrane. At present, the exact molecular mechanisms underlying PTEN-L’s mitochondrial localization are not known, because there are no mitochondrial targeting sequences (MTS) in the ATR region of PTEN-L.
To explore the role of PTEN-L in mitophagy, we first transiently overexpressed PTEN-L or PTEN in HeLa-YFP-PRKN cells, and found that only PTEN-L, but not PTEN, can block mitophagy induced by the mitochondrial damaging agent CCCP, evidenced by reduced degradation of various mitochondrial proteins. Consistently, stable overexpression of PTEN-L prevents the degradation of mitochondrial proteins and mtDNA caused by various mitochondrial damaging agents, including CCCP, oligomycin and antimycin A (O/A), and valinomycin, whereas depletion of PTEN-L promotes mitophagy. Next, we systematically evaluated the effects of PTEN-L on PRKN and found that: (i) PTEN-L, but not PTEN, is able to block PRKN translocation from the cytosol to mitochondria; (ii) overexpression of PTEN-L prevents, while depletion of PTEN-L promotes, PRKN’s E3 ligase activity measured by PRKN’s auto-ubiquitination and poly-ubiquitination of outer mitochondrial membrane proteins (TOMM20 and MFN2); (iii) PTEN-L interacts with PRKN and maintains PRKN in a closed/inactive conformation status by stabilizing the interaction between PRKN’s UBL and RING1 domain; and (iv) overexpression of PTEN-L markedly reduces the phosphorylation of PRKN measured by Phos-tag gels and by MS/MS analysis. In this study, we are unable to establish whether PTEN-L is a direct phosphatase for p-Ser65-PRKN, mainly due to some technical difficulties including the lack of workable of p-Ser65-PRKN antibodies.
The most important finding of our study is that PTEN-L is the direct protein phosphatase for dephosphorylating p-Ser65-Ub in vivo and in vitro. We first showed that overexpression of PTEN-L markedly reduces p-Ser65-Ub levels based on western blot and immunostaining after CCCP or O/A treatment, whereas depletion of PTEN-L results in the opposite effects. Next, we used an in vitro dephosphorylation assay to demonstrate that PTEN-L can dephosphorylate p-Ser65-mono-Ub, p-Ser65-Tetra-Ub, and p-Ser65-poly-Ub. Third, and more importantly, PTEN-L is able to dephosphorylate the p-Ub chains binding to PRKN measured by MS/MS analysis. It is also relevant to note that overexpression of PTEN-L reduces the basal level of p-Ser65-Ub, whereas depletion of PTEN-L causes the opposite effect, which suggests that PTEN-L can also target the pre-existing p-Ser65-Ub on mitochondria.
The presence of a specific protein phosphatase against PINK1-mediated ubiquitin and PRKN phosphorylation has long been speculated. Identification of PTEN-L as a novel protein phosphatase for dephosphorylation of p-Ser65-Ub thus addressed this critical question. The model of how PTEN-L regulates mitophagy is illustrated in Figure 1. We think that PTEN-L can block the feedforward mechanism of mitophagy via dephosphorylation of p-Ser65-Ub, based on the critical role of p-Ser65-Ub in recruiting and activating PRKN. Interestingly, dephosphorylation of p-Ser65-Ub by PTEN-L may also promote the function of DUBs, another important group of negative regulators of mitophagy, based on the fact that those DUBs selectively target unphosphorylated ubiquitin. Thus, our study not only provides an important missing piece in the molecular mechanisms of mitophagy, but also suggests that PTEN-L may serve as a molecular target for development of novel interventional approaches in the regulation of mitophagy.
Figure 1.
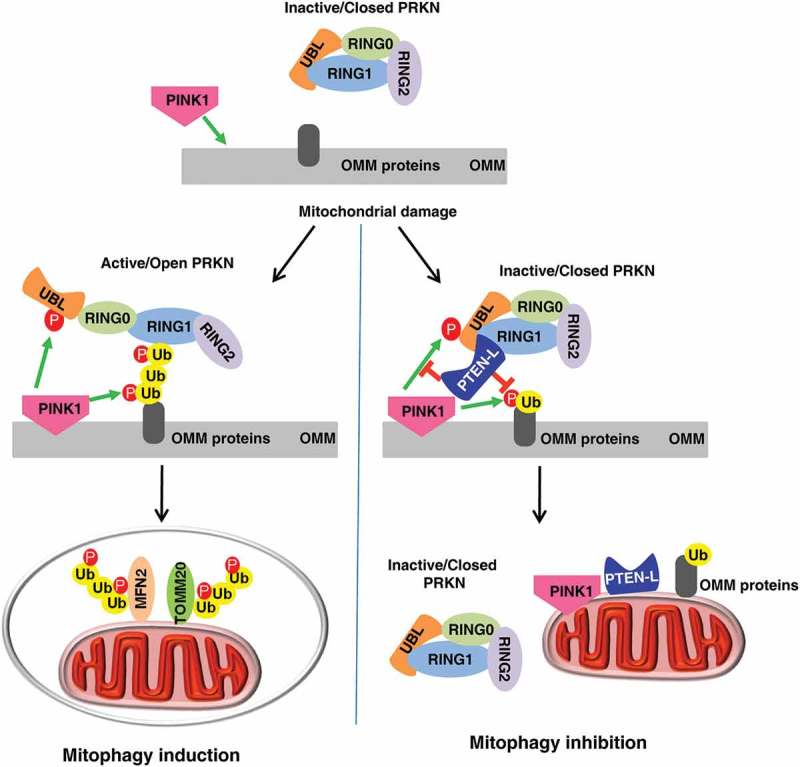
Illustration of how PTEN-L negatively regulates mitophagy.
Funding Statement
This work was supported by the MOH | National Medical Research Council (NMRC) [grant numbers CIRG/1430/2015 and CIRG/1373/2013].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Wang L, Cho YL, Tang Y, et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018;28:787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]