

Abstract
The genetic analysis of complex disorders has undoubtedly led to the identification of a wealth of associations between genes and specific traits. However, moving from genetics to biochemistry one gene at a time has, to date, rather proved inefficient and under-powered to comprehensively explain the molecular basis of phenotypes. Here we present a novel approach, weighted protein–protein interaction network analysis (W-PPI-NA), to highlight key functional players within relevant biological processes associated with a given trait. This is exemplified in the current study by applying W-PPI-NA to frontotemporal dementia (FTD): We first built the state of the art FTD protein network (FTD-PN) and then analyzed both its topological and functional features. The FTD-PN resulted from the sum of the individual interactomes built around FTD-spectrum genes, leading to a total of 4198 nodes. Twenty nine of 4198 nodes, called inter-interactome hubs (IIHs), represented those interactors able to bridge over 60% of the individual interactomes. Functional annotation analysis not only reiterated and reinforced previous findings from single genes and gene-coexpression analyses but also indicated a number of novel potential disease related mechanisms, including DNA damage response, gene expression regulation, and cell waste disposal and potential biomarkers or therapeutic targets including EP300. These processes and targets likely represent the functional core impacted in FTD, reflecting the underlying genetic architecture contributing to disease. The approach presented in this study can be applied to other complex traits for which risk-causative genes are known as it provides a promising tool for setting the foundations for collating genomics and wet laboratory data in a bidirectional manner. This is and will be critical to accelerate molecular target prioritization and drug discovery.
Keywords: weighted protein network, complex disorders, functional enrichment, frontotemporal dementia, protein−protein interactions, systems biology
Introduction
The genetic analysis of complex diseases has led to the identification of a wealth of associations between Mendelian genes or susceptibility loci (i.e., regions of DNA incorporating coding and noncoding variants) and specific traits or endophenotypes.1,2 While genetic association has greatly aided shedding light on disorders as diverse as heart disease and leprosy,3,4 such knowledge is still insufficient to fully explain disease pathogenesis. This is a critical issue considering that the final goal of biomedical research is that of understanding disease mechanisms and their associated molecular underpinnings to identify biomarkers or targets for disease diagnosis, prevention, or treatment.
In complex disorders many of the established mutations in Mendelian (familial) genes are rare and may present with incomplete penetrance.5,6 Conversely, the vast majority of cases are sporadic and are associated with the gradual and cumulative effect of susceptibility loci, that is, multiple variants with small effect size, the severity of which might be modulated, for example, by environmental factors.7−11 The implication of this is that in complex disorders a broad underlying genetic susceptibility architecture contributing to disease risk may be equally or even more relevant than just Mendelian inheritance.12,13 In addition the current absence of a straightforward translation of genetic knowledge into the functional landscape of biochemistry and cell biology14 represents a challenge that contributes to a substantive gap in our understanding of the molecular underpinnings of disease.
Moving from genetics to biochemistry one gene at a time has led to pivotal discoveries only in a limited number of cases, for example, that of Alzheimer’s disease (AD) with the development of the Amyloid cascade hypothesis, following mechanistic studies of mutations in the APP and the presenilin (PSEN1/2) genes.15 However, breakthroughs of this magnitude require decades of intense cell biology and animal-model-based studies, suggesting these approaches are still not entirely efficient as well as underpowered to comprehensively explain disease mechanisms in a timely manner.16
One way to overcome these limitations and increase analytical power and resolution is to integrate genetic information with other types of data.17 In the first instance, strategies have been and are being developed to better interpret genome-wide association studies to prioritize genes within significant loci, such as gene-based burden analyses,14,18,19 as well as to improve the statistical power of the genetic analysis by incorporating functional information, such as PINBA20 or ALIGATOR.21 Building from DNA to mRNA, the possibility arises of combining genetic with expression data moving from assessing the effect of single-SNPs on expression levels through expression quantitative trait loci analysis (eQTL)22 to more complex approaches such as weighted gene coexpression network analysis (WGCNA).23 The latter is a powerful bioinformatics method to cluster genes based on similar expression patterns and on the assumption that coexpressed genes are likely to be part of the same functional pathway(s).23 Although there is considerable promise in using different methods to expand on purely genetic data, validation at the protein level is necessary and required to understand the functional consequences of variation in genes. Such validation is normally carried out directly in the wet laboratory. In this scenario, bioinformatics coupled to protein–protein interaction (PPI) analyses represents an emerging and potentially powerful tool to further fine-tune genetic and expression analyses before wet laboratory work is performed,24,25 thus constituting a valuable support to guide cellular and biochemical investigations.26,27
In the current study, we used frontotemporal dementia (FTD) as a disease model to explore our proposed approach. FTD is the second most common form of young-onset dementia after AD, representing 10–20% of all dementias worldwide.28 FTD occurs in approximately 3–15/100 000 individuals aged mid/late-50s to early 60s,29 has an insidious onset, and is inherited in a Mendelian fashion in between 10 and 30% of cases depending on the population studied.30 Clinically, most cases present with behavioral variant syndrome (bvFTD), while a smaller proportion present with language impairment called primary progressive aphasia (PPA);31 there is also phenotypic (and genetic) overlap with motor neuron disease (FTD-MND) and atypical parkinsonian disorders.30 From the molecular pathology perspective, the majority of cases present with either tau (FTLD-tau) or ubiquitin/TDP-43 (FTLD-TDP) inclusions (≤40–50%), whereas a minority has FUS (≤10%; FTLD-FUS) or ubiquitin/p62 (≤1–2%; FTLD-UPS) inclusions.32 Mutations in three genes (MAPT, GRN, and C9orf72) explain ∼7–20% of all cases, while a handful of other genes (TDP-43, CHMP2B, VCP, FUS, SQSTM1, UBQLN2, IFT74, OPTN, DCTN1, and CHCHD10) are linked to <5% of all cases;30,31,33 the remaining cases (∼70%) appear idiopathic in nature. More recently, two genome-wide association studies (GWAS) revealed the modifying factor TMEM106B(34,35) and two further novel loci encompassing the HLA locus and the RAB38 and CTSC genes.36 Clearly, FTD is characterized by a broad range of clinical manifestations, pathological signatures, and genetic variability.37 Given the predominantly idiopathic nature of this disorder and the absence of a clear unidirectional relationship between the associated genes and the molecular pathology, the etiology of FTD appears to derive from a complex web of patho-mechanisms. Unfortunately, there is currently a critical void in the understanding of these mechanisms, one of the key reasons why both preventive and therapeutic strategies are completely absent in FTD.38
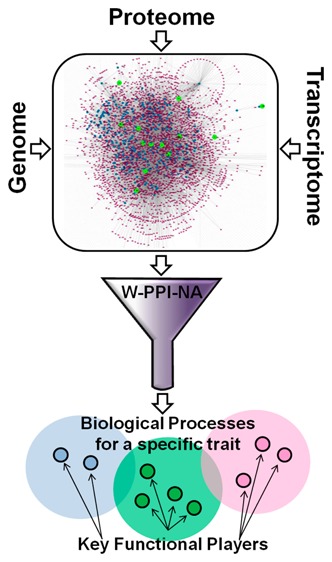
Here we present a novel pipeline to generate a weighted PPI network analysis (W-PPI-NA) to build and analyze the tissue-specific (brain) interactome (= totality of proteins interacting with a seed) of the known FTD associated genes/proteins (= seeds). The current work: (i) represents the development of a pipeline that uses genes known to be associated with a trait (i.e., FTD) and projects them in the protein domain; (ii) is based on current state of the art genetics and proteinomics; (iii) generates additional knowledge and fosters cross-disciplinary work (e.g., genomics, transcriptomics and proteinomics);17 and (iv) identifies proteins that represent the backbone of biological processes (BPs) likely impacted in FTD to be validated using biochemical and functional approaches.
Experimental Section
The FTD protein network (FTD-PN) was built in a multilayer fashion. FTD-spectrum genes were identified as seeds, and their protein–protein interactors (PPIs) were downloaded, filtered, and scored. The collection of all seeds’ interactors represents the first layer of the FTD-PN. The proteins in the first layer of the FTD-PN were then used each as a seed to download a second layer of PPIs and determine their reciprocal cross-interactions.
Meaning of terminology: the first layer of the FTD-PN contains all FTD seeds plus the first layer of interactors. The complete FTD-PN is composed of all FTD seeds, plus their first layer interactors, plus their second layer interactors. The complete interactome of each seed is composed of the seed under investigation, plus its first layer of interactors, plus the second layer of interactors.
The pipeline for building the network is described hereafter and is summarized in Figure 1.
Figure 1.
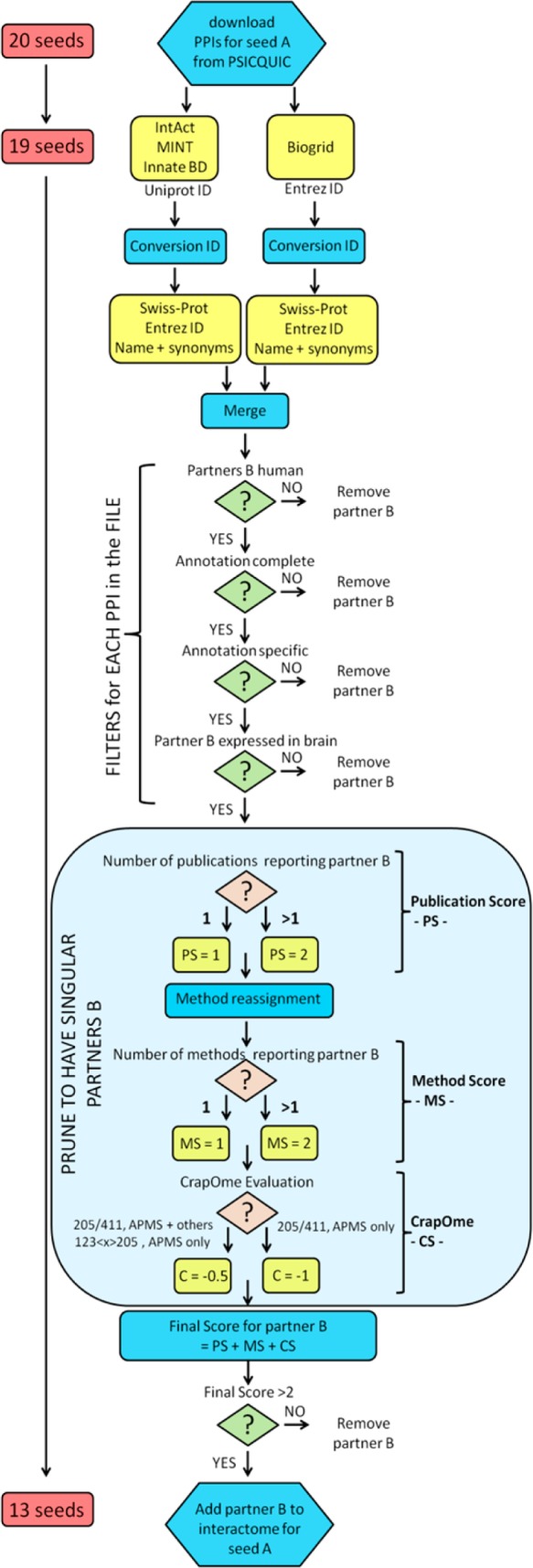
Workflow to generate and build the network.
Download of the PPIs
PPIs were downloaded as MITAB 2.5 files (first layer on 10-Oct-2015, second layer on 17-Nov-2015) from the IntAct,39 Biogrid,40 InnateDB, InnateDB-all, InnateDB-IMEx,41 and MINT42 databases by means of the PSICQUIC platform (http://www.ebi.ac.uk/Tools/webservices/psicquic/view/main.xhtml) developed by the IMEX consortium.43 In a MITAB file, PPIs from peer-reviewed literature are reported in lists in which each row is an annotation of a binary interaction (protein A and protein B), reporting both the features of the interaction and the Pubmed identifier of the paper in which that interaction was reported. Different databases classify proteins using different identifiers (Swiss-Prot or Entrez gene ID), and thus raw data were processed to convert Entrez to Swiss-Prot identifiers. We created a protein-ID conversion algorithm by downloading files containing multiple protein-ID synonyms from the UniProt Web site (HUMAN_9606_idmapping_selected.tab downloaded from ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/idmapping/README, on 04-Sept-2015; SwissProt-UniProtKB-human-results.tab downloaded from http://www.uniprot.org/uniprot/?query=human&fil=organism%3A%22Homo+sapiens+%28Human%29+[9606]%22+AND+reviewed%3Ayes&sort=score, on 04-Oct-2015) (Supporting Information File S1). Entrez IDs matching to multiple Swiss-Prot identifiers (nonunivocal gene to protein conversions) were removed. Raw PPIs data were run against this protein-ID conversion algorithm, and nonmatched entries were removed (TrEMBL, nonprotein interactors [e.g., chemicals], obsolete Entrez, and Entrez matching to multiple Swiss-Prot identifiers). Raw PPIs annotations from different databases were finally merged into a single file for each seed.
Construction of the PPI Network
Each single file containing the PPIs of a seed underwent filtering: (i) All the annotations for which interactor A or interactor B were classified with a nonhuman taxid, or as chemicals, were removed. (ii) All the annotations with incomplete fields were removed (i.e., “non-assigned” interaction detection method or Pubmed-ID). (iii) All the annotations with misleading/generic specifications were removed (i.e., multiple Pubmed-ID in a single row, “biochemical”, “biophysical”, or “experimental” interaction detection method; examples of removed annotations are reported in Supporting Figure S1). (iv) All proteins whose entire transcript was not reported in brain (http://www.braineac.org/) were removed. Filtered files underwent reassignment of the “Interaction detection method” field to pool together similar methodologies by following the ontology at http://www.ebi.ac.uk/ontology-lookup/browse.do?ontName=MI&termId=MI:0001&termName=interaction%20detection%20method and as reported in Supporting Table S1. The interactions were then scored with an in-house developed pipeline taking into consideration the following parameters: (i) the number of different publications (publication score, PS): for the same interaction partners A–B reported multiple times, the PS score was 1 = only 1 Pubmed-ID, 2 = multiple Pubmed-IDs; (ii) the number of different methods (method score, MS): for the same interaction partners A–B reported multiple times, the MS score was 1 = only 1 method reported, 2 = multiple methods, and; (iii) the CrapOme (Contaminant Repository for Affinity Purification)44 score (CS) (for the first layer only). CrapOme is a database that lists proteins known to contaminate affinity purification coupled to mass spectrometry (APMS) experiments (http://www.crapome.org/, 411 data sets at the time of this analysis, 27-Oct-2015). In particular, because of high levels of false-positives that are present in the APMS (Pull Down, Tandem Affinity Purification, GST Pulldown, Affinity Chromatography, Affinity Technology, Chromatography technology, His pull down, and Interactome parallel affinity capture) data, the CS applies different weightings based on the likelihood of the protein to be a contaminant in APMS experiments. The CS was computed as follows: −1 = for a CrapOme ratio >205/411 (>50%) if only APMS has been used; −0.5 = for a CrapOme ratio >205/411 (>50%) if one other method was used alongside APMS; 0 = for a CrapOme ratio >205/411 (>50%) if more than two other methods have been used alongside APMS; −0.5 = for a CrapOme ratio between 123 < x > 205 (30% < x > 50%) if only APMS has been used. After computation of the final score (PS+MS+CS), all of the interactors with a final score of 2 or less were discarded.
Functional Enrichment Analysis and Replication
We performed enrichment analyses for Gene Ontology (GO) terms in g:Profiler (g:GOSt, http://biit.cs.ut.ee/gprofiler/)45 on 29-Sept-2016. g:Profiler settings were used as follows: enrichment for GO terms only (BPs, cellular components (CCs), and molecular functions (MFs)); Fisher’s one-tailed test as statistical method for enrichment, SCS-threshold as multiple testing correction; statistical domain size was only annotated genes; no hierarchical filtering was included. We repeated this analysis twice using all annotated genes (n = 18 531) or brain-specific expressed genes (n = 13 859) as statistical background.
The following proteins were excluded
from analysis because they were not identified by g:Profiler: ECM29,
LINC00312, and LPHN1. Enriched GO-BP terms were grouped into custom-made
semantic classes. Generic terms (classified in the semantic classes
of: Enzyme – Generic – Metabolism – Motility
– Muscle – Physiology – Protein Modification
– Response to Stimulus, and Virus) were discarded from further
analysis. For each semantic class, all of the contributing single
GO terms were merged to identify the list of proteins within the network
that contribute to the enrichment of that specific semantic class.
The protein list contributing to enrichment allowed us to calculate
the contribution of each interactome to the enrichment of that specific
semantic class. Considering the largest interactome in the network
(i.e., VCP, n = 2247 interactors) as a reference,
we fixed a custom threshold at 75% (= at least 3/4 of the VCP-interactors
within the VCP interactome) to indicate robust contribution of the
VCP interactome to enrichment of that specific semantic class. This
75% threshold was weighted, keeping into account the random possibility
of extracting proteins based on the size of each complete seed’s
interactome (relative ratio percentage). For example, for the interactome
of VCP (n = 2247 interactors), the threshold was
75%, while for that of TARDBP (n = 1002) it was 33.4%
(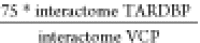 ). The same approach was adopted for any
other complete interactome, leading to the following relative thresholds:
C9orf72 and HLA-DRA 0.2% and 0.3% (thus excluded); CHMP2B 3.6%; DCTN1
5.5%; UBQLN2 5.2%; GRN 8.9%; ATXN2 10.4%; FUS 30.7%; OPTN 32.9%; TARDBP
33.4%; SQSTM1 46.2%; and MAPT 53.1%.
). The same approach was adopted for any
other complete interactome, leading to the following relative thresholds:
C9orf72 and HLA-DRA 0.2% and 0.3% (thus excluded); CHMP2B 3.6%; DCTN1
5.5%; UBQLN2 5.2%; GRN 8.9%; ATXN2 10.4%; FUS 30.7%; OPTN 32.9%; TARDBP
33.4%; SQSTM1 46.2%; and MAPT 53.1%.
To replicate the enrichment analysis results obtained from g:Profiler (positive control), we used two alternative online tools such as PANTHER (accessed on October 24, 2016; overrepresentation test with Bonferroni correction)46 and Webgestalt (accessed on October 24, 2016; hypergeometric test with Benjamini–Hochberg [BH] correction).47 Moreover, we generated the complete protein network for Albinism (A-PN) in the same fashion as for FTD-PN using TYR, SLC, 45A2, SLC24A5, TYRP1, OCA2, C10orf11, and GPR143 (https://ghr.nlm.nih.gov/condition/oculocutaneous-albinism#genes) as seeds and assessed the associated functional enrichment as a negative control. (Download and analysis were performed on May 9, 2016 through g:Profiler.)
Software
Data were handled, filtered, and scored through in-house R scripts (https://www.r-project.org/). For information and requests about the code(s), please contact the authors directly. The final network was visualized through the freely available Cytoscape 2.8.2 software48 and analyzed by the network analysis plug-in (http://www.cytoscape.org/). All graphs were composed through the Prism-GraphPad software (http://www.graphpad.com/scientific-software/prism/).
Results
Construction of the PPI FTD-PN: First Layer
We selected Mendelian (familial) and GWAS genes associated with the FTD-spectrum as seeds (Table 1) to build the FTD-PN. Of note, although pathogenic variants in TARDBP and FUS are more frequent in ALS (and ALS-FTD) cases, TDP-43 and FUS are pathological hallmarks of FTD subtypes and thus are likely to hold functional relevance in the pathogenesis of FTD; for this reason, they were included in the analyses. Overall, seven seeds were dropped in different stages of the pipeline (as explained in the following sections), and thus a total of 13 seeds (ATXN2, C9orf72, CHMP2B, DCTN1, FUS, GRN, HLA-DRA, MAPT, OPTN, SQSTM1, TARDBP, UBQLN2, and VCP) was included in the final FTD-PN.
Table 1. List of Seeds Used for Building the FTD-PN and Associated Features.
| gene name | frequency (Mendelian%) | pathology | inclusion in PPI network |
|---|---|---|---|
| C9orf72a | common (7–20%) | FTLD-TDP | YES |
| GRN | common (5–11%) | FTLD-TDP | YES |
| MAPT | common (2–11%) | FTLD-Tau | YES |
| ATXN2a | rare (<1%) | SNCA, polyGIn | YES |
| CHCHD10a | rare (<1%) | FTLD-TDP | NO |
| CHMP2B | rare (<1%) | FTLD-UPS | YES |
| DCTN1a | rare (<1%) | not understood; possibly FTLD-TDP | YES |
| FUSa | rare (<1%) | FTLD-FUS | YES |
| IFT74a | rare (<1%) | not known; possibly FTLD-TDP | NO |
| OPTNa | rare (<1%) | FTLD-TDP | YES |
| SQSTM1a | rare (<1%) | FTLD-TDP | YES |
| TARDBPa | rare (<1%) | FTLD-TDP | YES |
| UBQLN2a | rare (<1%) | FTLD-TDP | YES |
| VCP | rare (<1%) | FTLD-TDP | YES |
| BTNL2 | GWAS | not known | NO |
| CTSC | GWAS | not known | NO |
| HLA-DRA | GWAS | not known | YES |
| HLA-DRB5 | GWAS | not known | NO |
| RAB38 | GWAS | not known | NO |
| TMEM106B | GWAS | FTLD-TDP | NO |
Genes belonging to the FTD-ALS spectrum; ALS = amyotrophic lateral sclerosis.
The first-layer interactome for each seed was built based on biochemical/physical interactions, as reported in peer-reviewed literature (see pipeline in Figure 1).
To collect PPI data, we used a set of publicly available, manually curated databases that annotate published protein interactions (see the Experimental Section), while we excluded databases implementing text mining algorithms or de novo prediction of PPIs. Similarly, we ignored inferred (but not experimentally validated) spoke expanded cocomplexes. Curation procedures in databases within the IMEX consortium are based on ad hoc and harmonized protocols to guarantee accuracy and comprehensive literature coverage.43 We downloaded PPIs from the unique platform PSICQUIC (the seed CHCHD10 was dropped in this stage because no human protein interactors were found), and PPIs for each seed, gathered from different databases, were merged into a final library. Of note, despite the effort directed toward harmonizing annotations within the IMEX databases, they are not yet fully superimposable; in particular, the most relevant problem is the type of identifiers through which interaction partners are annotated. To overcome this issue we developed a protein-ID conversion algorithm to interconvert Swiss-Prot and Entrez identifiers. The application of this conversion algorithm led to the exclusion of annotations reporting TrEMBL identifiers (nonmanually annotated UniProt codes), obsolete Entrez, and nonunivocal Entrez. This first pruning step was deliberately stringent to remove old or inaccurate annotations. Ubiquitin(s) (UBB-P0CG47, UBC-P0CG48, UBD-O15205) are normally conjugated to proteins to flag them for degradation (thus likely to introduce biased interactions); therefore, they were excluded from the FTD-PN. We performed additional quality control (QC) by means of in-house developed filters to remove incomplete or misleading annotations (see the Experimental Section). Subsequently, we scrutinized the taxid of the interaction partners to remove nonhuman PPI data; finally, we applied an additional expression filter (EF) to remove protein partners that were not documented to be expressed in human brain. Subsequently, we scored the interactome of each seed to remove those interactors lacking reproducibility (thus not sufficiently reliable for further functional analysis) by means of a publication score (PS; number of publications supporting the interaction) and a method score (MS; number of different experimental detection methods). We considered at risk all interactions detected via mass spectrometry or pull-down techniques, and thus for these we generated a penalizing score based on the CrapOme project (CS; see the Experimental Section). The final score was computed as a sum of PS, MS, and CS. All of the interactors with a final score ≤2 were removed for likely low reliability, leading to the exclusion of six seeds (BTNL2, CTSC, HLA-DRB5, IFT74, RAB38, and TMEM106B). The merging of the remaining 13 interactomes generated the first layer of the FTD-PN (Figure 2A) constituted of 283 single nodes and 307 undirected edges. Calculating the number of proteins shared across different interactomes (Figure 2B) revealed overlap between multiple interactomes. Only the interactomes of C9orf72 and HLA-DRA appeared as disconnected components of the network, probably due to the small number of annotated interactors (<4) for either protein.
Figure 2.
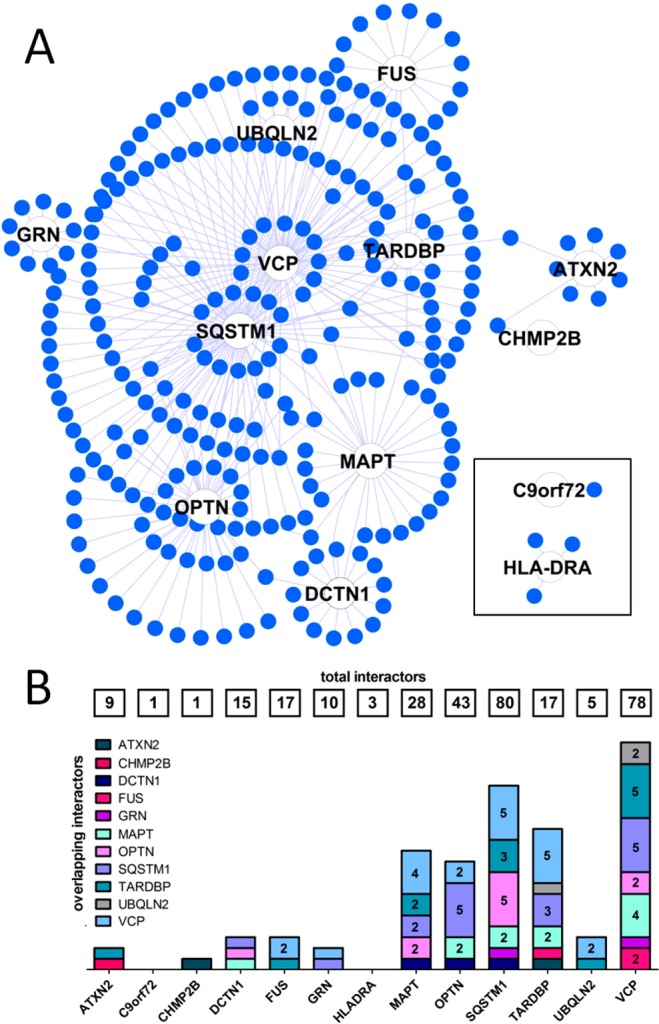
Topological features of the first layer of the FTD-PN. (A) Organic layout of the first layer of the FTD-PN. Seeds are represented by a white node. The closer their position within the organic layout to the center, the higher the number of nodes/interactors they share with other seeds (i.e., number of cross-interactions between single interactomes). (B) Level of overlap across interactomes. Numbers in the boxes at the top indicate the total dimension (n of interactors) of each single interactome. Numbers in the bars identify the number of shared nodes/interactors between two interactomes (where the absence of number indicates one shared node only). The interactome of each seed is color-coded.
Construction of the PPI FTD-PN: Second Layer
To minimize any potential bias associated with the first layer network due to strong influence of the seeds (i.e., seed centrality), we built the second layer around each node of the first layer following the same procedure described in the previous section. After merging all single interactomes the complete FTD-PN held 4198 single nodes and 10754 edges (Figure 3). There were no disconnected components in the second layer, showing increased cohesion of the FTD-PN. One important feature of the second layer is that it is not directly influenced by the seeds because the nodes/interactors within this layer are extracted on the basis of all proteins of the first layer but not on the seeds directly as they no longer act as hubs (with this role being substituted by the first-layer genes/proteins). Moreover, we identified many cross-interactions between single interactomes due to a high number of nodes/interactors shared between interactomes. The mean connections for each single node (n of edges/n of nodes) increased from 1.08 in the first layer to 2.56 with the addition of the second layer; similarly, the average number of neighbors increased from 2.141 to 4.621.
Figure 3.

Topological features of the second layer of the FTD-PN. Organic layout of the first layer and second layer of the FTD-PN. The seeds are represented by green nodes, the first layer interactors are highlighted in blue, and the second layer interactors are evidenced in purple.
The complete FTD-PN (first + second layers interactors) represents the state-of-the-art protein interactors gravitating around the FTD-spectrum genes/proteins. We calculated overlap between the interactomes considering the number of interactors shared between each interactome of the single seeds (Figure 4A). We also extracted inter-interactome hubs (IIHs) defined as interactors shared across a minimum of eight different interactomes (8/13 = 50 + 10%), thus bridging at least 60% of the entire network (= inter-interactome degree) (Figure 4B,C, Table 2, and Supporting Figures S2–27).
Figure 4.
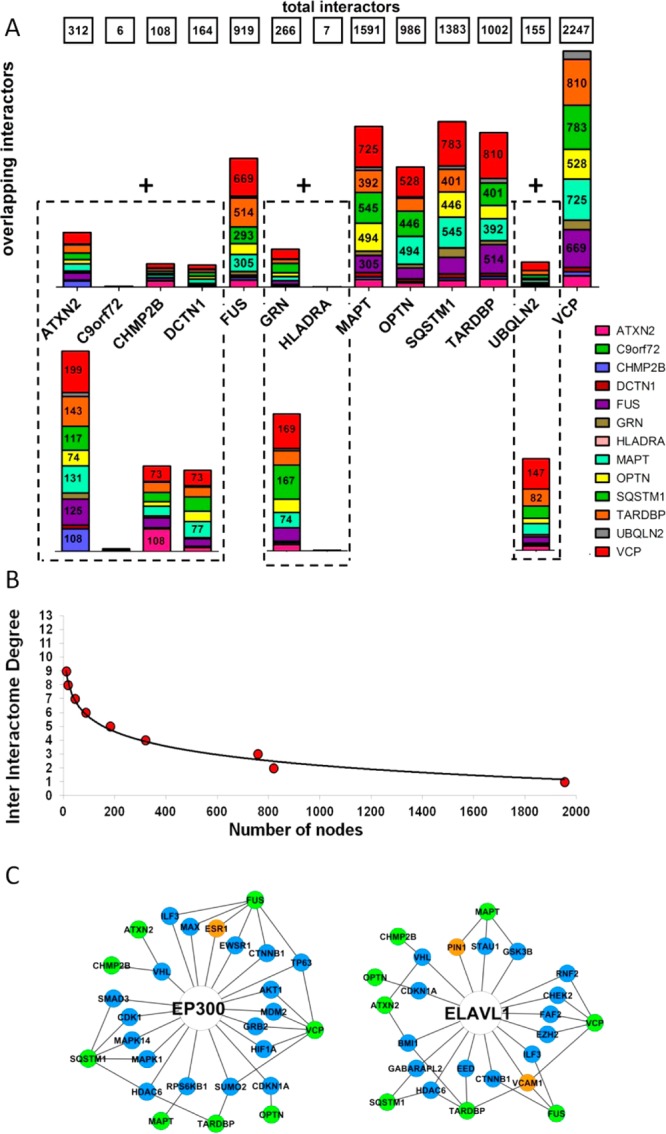
Level of overlap across interactomes in the first and second layers. (A) Level of overlap across interactomes with two different resolutions to appreciate the architecture of both the large (top) and small (bottom) interactomes. Numbers in the boxes at the top indicate the total dimension (n of interactors) of each single interactome. Numbers in the bars identify the number of shared nodes between two interactomes (where the absence of number indicates <230 [top] and 70 [bottom] shared nodes). (B) Inter-interactomes degree distribution: number of nodes (x axis) as a function of the number of interactomes they belong to (y axis): 29 inter-interactome hubs are shared among 8 to 9 interactomes out of 13 total interactomes (>60%). (C) Example of the IIHs with their surrounding interactors and degree of connectivity across the interactomes of multiple seeds. Color code: green, seeds; blue, first-layer interactors; orange, other IIHs.
Table 2.
| ID/gene name | functional association with dementia | name | IID | score | layer | overlap with WGCNA |
|---|---|---|---|---|---|---|
| Q92905 COPS5 CSN5, JAB1a | increased levels in Alzheimer’s brains, positive correlation with Abeta processing (2013)54 | COP9 signalosome complex subunit 5 | 9 | 4 | I | COPS proteins are subunits of the COP9 signalosome complex. In the WGCNA COPS3 was significantly coexpressed with VCP in frontal cortex. |
| P03372 ESR1 ESR, NR3A1 | estrogen stimulation exerts a protective role against Alzheimer’s dementia63 | estrogen receptor | 9 | 4 | I | NA |
| P08238 HSP90AB1 HSP90B, HSPC2, HSPCB | not reported | heat shock protein HSP 90-beta | 9 | 4 | I | NA |
| Q9UNE7 STUB1 CHIP, PP1131 | not reported | E3 ubiquitin-protein ligase CHIP | 9 | 4 | I | NA |
| P00533 EGFR ERBB, ERBB1, HER1 | not reported | epidermal growth factor receptor | 9 | 4 | II | NA |
| P02751 FN1 FN | identified as disease marker in the plasma of Alzheimer’s61 | fibronectin | 9 | 4 | II | NA |
| P07900 HSP90AA1 HSP90A, HSPC1, HSPCA | not reported | heat shock protein HSP 90-alpha | 9 | 4 | II | NA |
| P11142 HSPA8 HSC70, HSP73, HSPA10 | downregulation in hippocampus, entorhinal, and auditory cortices of Alzheimer’s60 | heat shock cognate 71 kDa protein | 9 | 4 | II | In the WGCNA, HSPA13 was significantly coexpressed with UBQLN2 in frontal and temporal cortex. HSPA 13 and 8 are part of the family of the heat shock proteins (HSP 70 kDa), activated in response to stress. |
| Q8WUM4 PDCD6IP AIP1, ALIX, KIAA1375 | not reported | programmed cell death 6-interacting protein | 9 | 4 | II | NA |
| P04637 TP53 P53 | genetic variation in TP53 was associated with Alzheimer’s disease48 | cellular tumor antigen p53 | 9 | 4 | II | NA |
| P55072 VCP | mutated in familial FTD50 | transitional endoplasmic reticulum ATPase | 9 | 4 | seed | NA |
| P05067 APP A4, AD1a | pathological hallmark of Alzheimer’s disease47 | amyloid beta A4 protein | 8 | 4 | I | In the WGCNA work, we found APP to be coexpressed within the C9orf72 module in frontal and temporal cortices. |
| Q16658 FSCN1 FAN1, HSN, SNL | decrease protein levels in the Alzheimer’s white matter proteome57 | fascin | 8 | 4 | II | NA |
| P63244 GNB2L1 HLC7, PIG21 | knock-down induces learning and memory impairment in mice55 | receptor of activated protein C kinase 1 | 8 | 4 | I | NA |
| Q13547 HDAC1 RPD3L1 | histone acetylation was proposed as a strategy to address synaptic loss in Alzheimer’s and FTD53 | histone deacetylase 1 | 8 | 4 | II | NA |
| P34932 HSPA4 APG2 | not reported | heat shock 70 kDa protein 4 | 8 | 4 | I | NA |
| P42858 HTT HD, IT15 | pathological hallmark of Huntington’s corea46 | huntingtin | 8 | 4 | I | NA |
| Q13526 PIN1 | rare variants may be related to Alzheimer’s disease; decrease in expression may be associated with FTD49 | peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 | 8 | 4 | I | NA |
| P19320 VCAM1 L1CAM | plasma levels increased in late onset Alzheimer’s disease ot vascular dementia, but not in cerebrovascular disease withouth dementia62 | vascular cell adhesion protein 1 | 8 | 4 | I | NA |
| P63104 YWHAZ | not reported | 14–3–3 protein zeta/delta | 8 | 4 | I | NA |
| P24941 CDK2 CDKN2 | elevated protein levels in Alzheimer’s periferal lymphocytes58 | cyclin-dependent kinase 2 | 8 | 4 | II | NA |
| Q15717 ELAVL1 HURa | implicated in amiloid precursor protein (APP) processing56 | ELAV-like protein 1 | 8 | 4 | II | In the WGCNA, we found ELAVL1 to be coexpressed with C9orf72. |
| Q09472 EP300 P300a | controls the espression level of Alzheimer’s genes and was therefore proposed as therapeutic target (curcumin)52 | histone acetyltransferase p300 | 8 | 4 | II | In the WGCNA, EP300 was significantly coexpressed with MAPT in frontal and temporal cortices. |
| P33993 MCM7 CDC47, MCM2 | not reported | DNA replication licensing factor MCM7 | 8 | 4 | II | NA |
| P29590 PML MYL, PP8675, RNF71 TRIM19 | not reported | protein PML | 8 | 4 | II | NA |
| P23396 RPS3 OK/SW-cl.26 | not reported | 40S ribosomal protein S3 | 8 | 4 | II | NA |
| P17987 TCP1 CCT1, CCTA | brain protein level increased in Alzheimer’s mouse model (J20)59 | T-complex protein 1 subunit alpha | 8 | 4 | II | NA |
| Q13049 TRIM32HT2A | mediator of HIV-1 associated neurological disorder via alteration of neural precursor cells51 | E3 ubiquitin-protein ligase TRIM32 | 8 | 4 | II | NA |
| Q71U36 TUBA1A TUBA3 | not reported | tubulin alpha-1A chain | 8 | 4 | II | NA |
Genes also found in previous WGCNA study.53
Functional Enrichment Analysis
We assessed GO terms for the complete (first + second layers) FTD-PN using gProfiler and grouped all of the significantly enriched GO-BP terms (p < 0.05) into semantic classes. Nonspecific semantic classes were dropped due to their negligible biological meaning (see Experimental Section), leading to the removal of 25.6% of all enriched terms. The analysis of the structure and content of the remainder and more specific semantic classes (74.4%) revealed notable BPs, depicted in Figure 5A (and Supporting Figures S28–S40), collectively implicating: (i) adhesion, (ii) cell cycle, (iii) cell death, (iv) chromatin metabolism, (v) development, (vi) DNA metabolism, (vii) RNA metabolism, (viii) immune system, (ix) signaling, (x) stress, (xi) transport, (xii) protein localization, and (xiii) waste disposal. The complete list of enriched GO terms can be found in Supporting Information File S2.
Figure 5.

Functional enrichment analysis. Functional enrichment for the entire network (A) and for the inter-interactome hubs (B) to extrapolate the functions that are driven by proteins shared at least by 60% of the interactomes.
The semantic classes contributing to (ii), (iii), (iv), (vi), and (x) (see above and Figure 5A) revealed convergent information, particularly that cell cycle-related activities included phase transition and DNA damage checkpoints, while stress-related cell death indicated apoptotic processes subsequent to DNA damage, ER, or oxidative stress. Concomitantly, chromatin and DNA metabolism indicated histone modification and DNA damage checkpoints (and associated repair or induction of cell death). Interestingly, these combinations suggest an univocal process such as that of DNA damage/integrity check followed by either DNA damage repair or cell death (apoptosis) as relevant BPs underlined by elements of the FTD-PN.
The semantic classes contributing to (v) and (xi) indicated that neuronal and glial development were particularly enriched for terms indicating axonal and neuronal projection morphogenesis, while vesicles transport pointed to vesicle trafficking along the cytoskeleton as well as to endocytosis/exocytosis/secretion. Taken together, these semantic classes suggest the critical importance of elements of the cytoskeleton (as implied by the known genes MAPT and DCTN1) for the overall neuronal development and homeostasis as well as for the support of vesicle transport, likely indicating an effect on exo/endocytic as well as secretion (neurotransmission) pathways.49,50
The semantic classes contributing to (vii) RNA metabolism strongly pointed toward gene expression modulation via RNA polymerase-I and -II (pol-I, pol-II) and RNA stabilization/localization that could represent a mechanism that activates stress-related responses.51
Finally, the semantic classes contributing to (xiii) waste disposal specifically pointed to degradative pathways referring to the ubiquitin-proteasome system (UPS) and unfolded protein response (UPR). Such semantic classes have previously been highlighted in the context of neurodegenerative disorders, including FTD,52,53 and the current work confirms and further expands on this sensitive and critical BP.
Provided the extent of BPs implicated by the entire FTD-PN (first + second layers), we ran the functional enrichment analysis exclusively using the IIHs as an input to highlight the semantic classes driven by proteins shared across the majority (>60%) of the interactomes. Remarkably, the 29 IIHs globally replicated the functional annotation analysis of the entire FTD-PN supporting the following semantic classes: (i) adhesion, (ii) cell cycle, (iii) cell death, (v) development, (vi) DNA metabolism, (vii) RNA metabolism, (ix) signaling, (x) stress, (xii) protein localization, and (xiii) waste disposal (Figure 5B and Supporting Information File S3). Although we observed a reduction in the total amount of BP-GO terms (due to the massive reduction in the number of input terms from 4198 to 29), several semantic classes including DNA/RNA metabolism and stress were nearly completely retained.
Of note, when we reran the functional annotation analyses by reducing the reference panel to genes only expressed in brain (n = 13 859), we could appreciate almost negligible differences (see Supporting Tables S2 and S3).
Positive and Negative Control
Replication in Webgestalt and Panther (Positive Control)
The results of the functional enrichment analysis were replicated to a large extent when applying different portals (which use different statistical algorithms); in particular, 98.3 and 73.1% of GO terms found in PANTHER (the Web site is updated to 2014) and WebGestalt (the Web site is updated to 2012), respectively, were completely superimposable with those of gProfiler (the Web site is updated to 2016) (Supporting Table S4).
Specificity Control (Negative Control)
To evaluate the sensitivity and specificity of the BPs associated with the FTD-PN, we sought to apply the same pipeline to a group of genes associated with a different trait (albinism). On the basis of literature reports, we selected seven seeds (TYR, SLC45A2, SLC24A5, TYRP1, OCA2, C10orf11, and GPR143; see the Experimental Section) and built the albinism-PN that included 79 proteins (first + second layers). Functional enrichment analysis for the albinism-PN versus the 29 FTD-IIHs led to substantially different GO terms (Supporting Table S5), as no reference to adhesion, cell cycle, DNA damage control, cell death, regulation of gene expression, stress, and waste disposal was seen. The only overlapping terms were referring to signaling, protein localization, and intracellular transport (including, however, only very general terms). Conversely, 27.3% of the GO enriched terms were exclusive to the albinism-PN, including pigment metabolism and biosynthesis. This indicates that the pipeline per se is likely a sensitive and specific tool to perform trait-specific functional assessments on the basis of known genes associated with that trait.
Portions of the Network Contributing to the Functional Enrichment
We scrutinized and collapsed all GO-BP terms supporting the semantic classes derived from the IIHs and compared them with the corresponding semantic classes from the FTD-PN (first + second layers). We then extracted the complete list of proteins directly responsible for those BPs (Supporting Table S6). Subsequently, we then assigned the proteins to their original seed-specific interactome and determined the percentage by which each interactome contributed to the enrichment calculating the relative ratio (see Experimental Section) between the size of each seed-specific interactome and the entire FTD-PN. The size of the C9orf72 and HLA-DRA interactomes was too small to possibly contribute to any enriched term, and, similarly, CHMP2B only had one interactor in the first layer; therefore, these seeds were excluded to prevent unspecific functional annotation results. For all of the other seeds (ATXN2, DCTN1, FUS, GRN, MAPT, OPTN, SQSTM1, TARDBP, UBQLN2, and VCP) results are fully shown in Supporting Figures S41–S45, while the most interesting semantic classes (see Discussion) are shown in Figure 6.
Figure 6.

Graphs showing the contribution of the interactome (first + second layers) of each seed toward the enrichment of specific semantic classes. The semantic classes are part of functional processes indicating DNA damage response, gene expression regulation, apoptosis, and waste disposal/ER stress. The gray bars indicate the weighted threshold for each single interactome. Points above that threshold are considered to highly contribute toward enrichment. Interactomes contributing to enrichment are indicated by the arrows.
It is noteworthy that, on one hand, the contribution of some seed’s interactomes to specific semantic classes confirmed expected BPs based on previous knowledge (e.g., growth factor signaling [interactome of GRN], gene expression regulation, and translation [interactomes of FUS and TARDBP] or waste/ER stress [interactomes of VCP and UBQLN2]), showing that our pipeline is specific and sensitive. On the other hand, it indicated association of novel BPs with FTD as we observed: (i) cell death (apoptosis) through the interactomes of ATXN2, DCTN1, GRN, UBQLN2, OPTN, SQSTM1, and TARDBP; (ii) DNA damage response through the interactomes of ATXN2, FUS, UBQLN2, MAPT, OPTN, and SQSTM1; and (iii) DNA damage repair through the interactomes of ATXN2, TARDBP, and UBQLN2.
Biological Meaning(s)
To gather the biological message of all of the analyses shown above, we first grouped similar or convergent semantic classes into “functional blocks”, as follows (Figure 7A): (i) “DNA damage response” (made of DNA damage response, DNA damage checkpoint G1, cell cycle checkpoint, DNA damage checkpoint, and intrinsic apoptosis after DNA damage); (ii) “apoptosis” (made of apoptotic signaling and cell death intrinsic apoptosis); (iii) “phase transition” (made of mitotic phase transition, G2-M phase transition, and G1-S phase transition); (iv) “DNA repair” (made of DNA damage repair); (v) “development/proliferation epithelium” (made of epithelial cell proliferation, cell-matrix adhesion, and growth factor signaling); (vi) “waste disposal/ER stress” (made of ER transport, ER stress, ER ubiquitin-proteasome, UPR, response to oxidative stress, and response to ionizing radiation); (vii) “gene expression regulation” (made of gene expression, translation, transcription, and transcription-RNA pol II); and (viii) “protein localization” (made of protein localization to nucleus and protein localization to membrane).
Figure 7.

Contribution of each interactome (first + second layers) toward the enrichment of functional blocks. (A) Each gray box indicates high contribution toward enrichment. The interactome contributes to the enrichment of at least half of the semantic classes within the functional block. (B) Overlaps between functional blocks in W-PPI-NA and WGCNA.53 The number of identical nodes (proteins or genes) in the W-PPI-NA and WGCNA contributing to the enrichment of each semantic class was calculated (number in red). The percentage of common nodes was computed against W-PPI-NA (percentage on the left) and against WGCNA (percentage on the right). (BLACK) and (PURPLE) refer to the modules of the WGCNA analysis.53
Second, we calculated the relevance of each single interactome toward the enrichment of each single functional block considering as relevant an enrichment of at least half of the semantic classes within the functional block. Results are summarized in Figure 7A. On the basis of this analysis, these functional blocks (see Figure 7A) indicated susceptibility processes shared by at least 60% of the FTD-PN, highlighting processes of critical relevance and clearly worthy of attention as well as further investigation and characterization at the molecular level. To put these data better into context, we evaluated the groups of genes contributing to the enrichment of similar functional blocks in our previous WGCNA study53 searching for overlaps with the list of proteins found in the current W-PPI-NA (Figure 7B). We identified the highest level of overlap for three critical functional blocks: DNA damage response, gene expression regulation, and cell waste disposal. The identical nodes defining the overlapping genes/proteins between WGCNA and W-PPI-NA (Supporting Table S7) represent the elements where the transcriptome (WGCNA) and the proteome (W-PPI-NA) fully match in a cross-supportive fashion, lending support to the inference that those very elements are key functional factors in FTD to be carried forward in the cell biology setting for hypothesis-driven functional validation.
Of note, four IIHs (cross-supportive between WGCNA and W-PPI-NA data) were found among those three critical functional blocks: (1) EP300 (DNA damage control and gene expression regulation), (2) APP and ELAVL1 (gene expression regulation), and (3) VCP (waste disposal).
Discussion
In this study, we report on a coherent pipeline, W-PPI-NA, that we developed to filter and combine knowledge generated within the related yet distinct fields of genomics and proteinomics. Our approach is based on the analysis of the BPs implied by known PPIs of genes associated with a trait or disease (FTD in the current work). This is an important step to better understand the function(s) of disease-associated genes and highlight potential risk processes as well as pertaining key functional proteins.
As reported above, we constructed the FTD-PN incorporating 4198 interactors for 13 seeds (ATXN2, C9orf72, CHMP2B, DCTN1, FUS, GRN, HLA-DRA, MAPT, OPTN, SQSTM1, TARDBP, UBQLN2, and VCP). We first analyzed the FTD-PN to identify IIHs, the nodes that bridge ≥60% of the complete network identifying 29 IIHs (Table 2) that likely hold functional significance in the disease context. Interestingly, we were able to verify that several of such IIHs (COPS5, ESR1, FN1, HSPA8, TP53, VCP, APP, FSCN1, GNB2L1, HDAC1, HTT, PIN1, VCAM1, CDK2, ELAVL1, EP300, TCP1, and TRIM32; 18/29 [60%]) have already been reported to be broadly associated with dementia (across the spectrum of AD, vascular, and HIV-1-induced dementia). It is remarkable to note that evidence of association was based on either genetic studies for TP53, VCP, APP, HTT, and PIN154−57 or proven molecular mechanisms for COPS5, VCP, APP, GNB2L1, HDAC1, HTT, EP300, and TRIM3254,55,57−63 or verified protein expression changes for COPS5, HSPA8, FSCN1, PIN1, CDK2, ELAVL1, and TCP1.56,62,64−68 In addition, some IIHs such as FN1, VCAM1, and CDK2 were reported for a role as biomarkers for disease onset and progression,66,69,70 while ESR1, HDAC1, and EP300 were reported for a role as potential therapeutic targets.60,61,71 Provided all this, we expect that the remainder of the IIHs (HSP90AB1, STUB1, EGFR, HSP90AA1, PDCD6EP, HSPA4, YWHAZ, MCM7, PML, RPS3, and TUBA1A, for which no link to dementia is established yet) might hold relevance to FTD or dementias and thus should be prioritized in both genetic reassessments as well as cell biology work to explore and validate this possibility. Of further note, we not only here observed that five IIHs (COPS5, HSPA8, APP, ELAVL1, and EP300) replicated results from our previous WGCNA study (Table 2),53 but also intriguingly noted that ELAVL1 and EP300 appeared to be important elements among the relevant functional blocks (i.e., DNA damage control and gene expression regulation; see further below) associated with the FTD-PN. This supports their relevance as core key regulators of FTD-associated processes and makes them likely functional biomarkers as well as potential targets for therapeutic intervention. In this respect, all the more, EP300 is among the targets of curcumin, which is currently undergoing clinical evaluation for mild cognitive impairment (MCI) (clinical trial no. NCT01383161).60,72
As a further intrinsic validation of the inference power of our approach, we found that newly reported genes (that we did not include as seeds in our W-PPI-NA because the current study design was developed prior their disclosure) such as TBK1 (TANK binding kinase 1)73 and CCNF (cyclin F)74 are indeed part of the FTD-PN and fall within the interactomes of seeds including OPTN (first layer), SQSTM1 (second layer), and VCP (second layer) for TBK1 and VCP (second layer) for CCNF.
This all together reinforces the view that our pipeline is able to extract, integrate, and order scattered or isolated multidisciplinary types of data contributing to the generation of novel and more comprehensive awareness about the implication of genes/proteins in the pathobiology of a trait.
From a functional perspective, we assessed BPs for the entire list of 4198 nodes and subsequently the 29 IIHs. The first analysis revealed up to 13 global BPs (Figure 5A); the secondary analysis supported these results (Figure 5B), strongly implying the following susceptibility processes: (i) DNA damage response (related to DNA damage checks, cell death [induced apoptosis] and DNA repair); (ii) regulation of gene expression; and (iii) waste disposal/ER stress (particularly, referring to the ubiquitin/proteasome and unfolded protein response). Even more strikingly, we observed that these three functional blocks (DNA damage response, gene expression regulation, and waste disposal/ER stress) highlighted by our current W-PPI-NA replicated results previously obtained by gene coexpression data through WGCNA (Figure 7B).53 We therefore gather that those three functional blocks are, collectively, the most common BPs associated with the FTD-PN and are therefore likely to represent the core functional architecture impacted in FTD (reflecting the underlying genetic architecture contributing to disease).
DNA damage response is probably the most biologically intriguing of the functional blocks that we identify here. DNA damage response is highly related to terms such as DNA damage checkpoint, DNA repair, and damage-associated cell death to name a few. DNA damage response controls the detection of DNA damage (e.g., single- and double-strand breaks as consequence of oxidative stress) and activates the protection signaling cascade prior to determining whether a cell undergoes damage repair or apoptosis;75 the signaling cascade is clearly mediated by the activation or modulation of a particular pattern of ad hoc expression of genes, an observation that might explain why we find regulation of gene expression to be one of the other major impacted functional blocks. This is relevant because it highlights a link between DNA damage response and neuronal homeostasis and longevity, reiterating a concept recently promoted.53 Finally, we found novel insight into additional supportive disease-associated processes such as (i) ubiquitin-mediated labeling of damage within DNA damage response signaling cascades52,76,77 and (ii) the DNA damage response (again) likely associated with neuronal death through abortive re-entering in cell cycle.78,79 This suggests the possibility that the three major functional blocks, which at first appear to be separate and acting independently to influence disease pathobiology, might instead be acting in a synergistic fashion.
It is important to note the limitations that apply to W-PPI-NA. PPI data are gathered from wet-lab approaches that are neither high-throughput nor hypothesis-free and are therefore biased by a restricted selection of proteins chosen for the investigation based on research-driven priorities. This may lead to an increased rate of type-I errors, with some protein interactions reported incorrectly. To minimize the impact of this issue, we built the first layer of the network as a basis for the second layer to (i) increase the dimension of the network (and therefore increase the power to infer functional enrichment), (ii) dilute the above-mentioned bias, and (iii) minimize the impact of seed-centrality, an issue that is intrinsic in networks of this kind (i.e., built on the basis of trait-specific known associated genes). At the same time, however, introducing the second layer may also lead to a potential specificity issue for the trait under study. To address this concern, we successfully implemented: (i) a positive control (replication of enrichment results by means of multiple enrichment portals) and (ii) a negative control (applying the same approach to a different trait, with different seeds, to evaluate the extent of potential spurious functional enrichment annotations). In addition, we are aware that the bias applying to the PPIs also applies to the functional enrichment analysis because GO is founded on the current literature, and some proteins might not be comprehensively annotated, and thus the enrichment analysis is based on partial annotation data. Finally, our findings are in silico; therefore, they will need to be assessed and validated via functional analysis of the putative susceptibility processes and pathways.
Conclusions
The current study indicates that the W-PPI-NA pipeline can be adopted to study, in silico, the protein network (and associated biological functions) of a trait (i.e., FTD) starting from disease-associated genes/loci (as designated seeds). W-PPI-NA contributes to address major challenges in biomedicine, vide the need to identify susceptibility processes and prioritize molecular targets provided by modern genetics for functional validation.
All this taken together opens the way for at least two further opportunities: The first is pathways analysis as W-PPI-NA highlights not only susceptible processes but also the associated key players (i.e., specific proteins). These can then be further explored using in silico approaches to identify risk-pathways through the use of ad hoc portals (including KEGG, Reactome, to name a few) and validating such pathways in vitro through hypothesis-driven cell biology investigations. The second is the integration of risk-pathways with pharmacogenomics data: In this scenario, any drug or compound that already modulates a particular target (or process) might represent a potential therapeutic measure for the trait under study.
In summary, the current work is promising for multiple reasons, not only for setting the basis to start understanding human disease passing from the genomic suite to the laboratory but also for supporting drug discovery and prospective implementation for patient benefit.
Acknowledgments
We acknowledge generous research support from the Michael J. Fox Foundation, Parkinson’s UK, and the Rosetrees Trust. This work was supported in part by the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson’s Disease Consortium (UKPDC) whose members are from the UCL Institute of Neurology, the University of Sheffield, and the MRC Protein Phosphorylation Unit at the University of Dundee, by a MRC Programme grant (to J.H. and P.A.L.) MR/N026004/1, and by a MRC New Investigator Research Grant (MR/L010933/1) to P.A.L. P.A.L. is a Parkinson’s UK research fellow (grant F1002). R.C.L. is funded by Parkinson’s UK (grant G-1307) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research. R.F. is supported by funding from Alzheimer’s Society (grant number 284).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00934.
The authors declare no competing financial interest.
Supplementary Material
References
- Welter D.; MacArthur J.; Morales J.; Burdett T.; Hall P.; Junkins H.; Klemm A.; Flicek P.; Manolio T.; Hindorff L.; Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42 (D1), D1001–6. 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood T. A.; Lazzeroni L. C.; Calkins M. E.; Freedman R.; Green M. F.; Gur R. E.; Gur R. C.; Light G. A.; Nuechterlein K. H.; Olincy A.; Radant A. D.; Seidman L. J.; Siever L. J.; Silverman J. M.; Stone W. S.; Sugar C. A.; Swerdlow N. R.; Tsuang D. W.; Tsuang M. T.; Turetsky B. I.; Braff D. L. Genetic assessment of additional endophenotypes from the Consortium on the Genetics of Schizophrenia Family Study. Schizophr Res. 2016, 170 (1), 30–40. 10.1016/j.schres.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. Genetics of coronary artery disease. Circ. Res. 2014, 114 (12), 1890–903. 10.1161/CIRCRESAHA.114.302692. [DOI] [PubMed] [Google Scholar]
- Sauer M. E.; Salomao H.; Ramos G. B.; D’Espindula H. R. S.; Rodrigues R. S.; Macedo W. C.; Sindeaux R. H.; Mira M. T. Genetics of leprosy: expected and unexpected developments and perspectives. Clin Dermatol 2015, 33 (1), 99–107. 10.1016/j.clindermatol.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Burgess D. J. Human genetics: Loss-of-function variants--not always what they seem. Nat. Rev. Genet. 2016, 17 (5), 251. 10.1038/nrg.2016.33. [DOI] [PubMed] [Google Scholar]
- Chen R.; Shi L.; Hakenberg J.; Naughton B.; Sklar P.; Zhang J.; Zhou H.; Tian L.; Prakash O.; Lemire M.; Sleiman P.; Cheng W. Y.; Chen W.; Shah H.; Shen Y.; Fromer M.; Omberg L.; Deardorff M. A.; Zackai E.; Bobe J. R.; Levin E.; Hudson T. J.; Groop L.; Wang J.; Hakonarson H.; Wojcicki A.; Diaz G. A.; Edelmann L.; Schadt E. E.; Friend S. H. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 2016, 34 (5), 531–8. 10.1038/nbt.3514. [DOI] [PubMed] [Google Scholar]
- Manolio T. A.; Collins F. S.; Cox N. J.; Goldstein D. B.; Hindorff L. A.; Hunter D. J.; McCarthy M. I.; Ramos E. M.; Cardon L. R.; Chakravarti A.; Cho J. H.; Guttmacher A. E.; Kong A.; Kruglyak L.; Mardis E.; Rotimi C. N.; Slatkin M.; Valle D.; Whittemore A. S.; Boehnke M.; Clark A. G.; Eichler E. E.; Gibson G.; Haines J. L.; Mackay T. F.; McCarroll S. A.; Visscher P. M. Finding the missing heritability of complex diseases. Nature 2009, 461 (7265), 747–53. 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O.; Hechter E.; Sunyaev S. R.; Lander E. S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (4), 1193–8. 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O.; Schaffner S. F.; Samocha K.; Do R.; Hechter E.; Kathiresan S.; Daly M. J.; Neale B. M.; Sunyaev S. R.; Lander E. S. Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (4), E455–64. 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. D. Finding the epistasis needles in the genome-wide haystack. Methods Mol. Biol. 2015, 1253, 19–33. 10.1007/978-1-4939-2155-3_2. [DOI] [PubMed] [Google Scholar]
- Eichler E. E.; Flint J.; Gibson G.; Kong A.; Leal S. M.; Moore J. H.; Nadeau J. H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010, 11 (6), 446–50. 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotogora J. Penetrance and expressivity in the molecular age. Genet. Med. 2003, 5 (5), 347–52. 10.1097/01.GIM.0000086478.87623.69. [DOI] [PubMed] [Google Scholar]
- Cooper D. N.; Krawczak M.; Polychronakos C.; Tyler-Smith C.; Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013, 132 (10), 1077–130. 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury M. J.; Gwinn M.; Yoon P. W.; Dowling N.; Moore C. A.; Bradley L. The continuum of translation research in genomic medicine: how can we accelerate the appropriate integration of human genome discoveries into health care and disease prevention?. Genet. Med. 2007, 9 (10), 665–74. 10.1097/GIM.0b013e31815699d0. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J.; Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8 (6), 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde T. E. Overcoming translational barriers impeding development of Alzheimer’s disease modifying therapies. J. Neurochem. 2016, 139 (Suppl 2), 224–236. 10.1111/jnc.13583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C. K.; D A.; Vandrovcova J.; Hardy J.; Wood N. W.; Lewis P. A.; Ferrari R. Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2016, bbw114. 10.1093/bib/bbw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Abecasis G. R.; Boehnke M.; Lin X. Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet. 2014, 95 (1), 5–23. 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Z.; McRae A. F.; Nyholt D. R.; Medland S. E.; Wray N. R.; Brown K. M.; Hayward N. K.; Montgomery G. W.; Visscher P. M.; Martin N. G.; Macgregor S. A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 2010, 87 (1), 139–45. 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Matsushita T.; Madireddy L.; Mousavi P.; Baranzini S. E. PINBPA: cytoscape app for network analysis of GWAS data. Bioinformatics 2015, 31 (2), 262–4. 10.1093/bioinformatics/btu644. [DOI] [PubMed] [Google Scholar]
- Holmans P.; Green E. K.; Pahwa J. S.; Ferreira M. A.; Purcell S. M.; Sklar P.; Owen M. J.; O’Donovan M. C.; Craddock N. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 2009, 85 (1), 13–24. 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A.; Trabzuni D.; Guelfi S.; Varghese V.; Smith C.; Walker R.; De T.; Hardy J.; Ryten M.; Trabzuni D.; et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 2014, 17 (10), 1418–28. 10.1038/nn.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P.; Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiassian S. D.; Menche J.; Chasman D. I.; Giulianini F.; Wang R.; Ricchiuto P.; Aikawa M.; Iwata H.; Muller C.; Zeller T.; Sharma A.; Wild P.; Lackner K.; Singh S.; Ridker P. M.; Blankenberg S.; Barabasi A. L.; Loscalzo J. Endophenotype Network Models: Common Core of Complex Diseases. Sci. Rep. 2016, 6, 27414. 10.1038/srep27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh G. C.; Porras P.; Aranda B.; Hermjakob H.; Orchard S. E. Analyzing protein-protein interaction networks. J. Proteome Res. 2012, 11 (4), 2014–31. 10.1021/pr201211w. [DOI] [PubMed] [Google Scholar]
- Wu G.; Feng X.; Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010, 11 (5), R53. 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak N. N.; Gandal M. J.; Geschwind D. H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 2015, 16 (8), 441–58. 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D.; Snowden J. S.; Gustafson L.; Passant U.; Stuss D.; Black S.; Freedman M.; Kertesz A.; Robert P. H.; Albert M.; Boone K.; Miller B. L.; Cummings J.; Benson D. F. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998, 51 (6), 1546–54. 10.1212/WNL.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Rabinovici G. D.; Miller B. L. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010, 24 (5), 375–98. 10.2165/11533100-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer J. D.; Warren J. D. Phenotypic signatures of genetic frontotemporal dementia. Curr. Opin. Neurol. 2011, 24 (6), 542–9. 10.1097/WCO.0b013e32834cd442. [DOI] [PubMed] [Google Scholar]
- Ferrari R.; Thumma A.; Momeni P.. Molecular Genetics of Frontotemporal Dementia. In eLS; John Wiley & Sons, Ltd: Chichester, U.K., 2013. [Google Scholar]
- Halliday G.; Bigio E. H.; Cairns N. J.; Neumann M.; Mackenzie I. R.; Mann D. M. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol. 2012, 124 (3), 373–82. 10.1007/s00401-012-1030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannwarth S.; Ait-El-Mkadem S.; Chaussenot A.; Genin E. C.; Lacas-Gervais S.; Fragaki K.; Berg-Alonso L.; Kageyama Y.; Serre V.; Moore D. G.; Verschueren A.; Rouzier C.; Le Ber I.; Auge G.; Cochaud C.; Lespinasse F.; N’Guyen K.; de Septenville A.; Brice A.; Yu-Wai-Man P.; Sesaki H.; Pouget J.; Paquis-Flucklinger V. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137 (8), 2329–45. 10.1093/brain/awu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming Y.; Cruchaga C. TMEM106B: a strong FTLD disease modifier. Acta Neuropathol. 2014, 127 (3), 419–22. 10.1007/s00401-014-1249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin V. M.; Sleiman P. M.; Martinez-Lage M.; Chen-Plotkin A.; Wang L. S.; Graff-Radford N. R.; Dickson D. W.; Rademakers R.; Boeve B. F.; Grossman M.; Arnold S. E.; Mann D. M.; Pickering-Brown S. M.; Seelaar H.; Heutink P.; van Swieten J. C.; Murrell J. R.; Ghetti B.; Spina S.; Grafman J.; Hodges J.; Spillantini M. G.; Gilman S.; Lieberman A. P.; Kaye J. A.; Woltjer R. L.; Bigio E. H.; Mesulam M.; Al-Sarraj S.; Troakes C.; Rosenberg R. N.; White C. L. 3rd; Ferrer I.; Llado A.; Neumann M.; Kretzschmar H. A.; Hulette C. M.; Welsh-Bohmer K. A.; Miller B. L.; Alzualde A.; Lopez de Munain A.; McKee A. C.; Gearing M.; Levey A. I.; Lah J. J.; Hardy J.; Rohrer J. D.; Lashley T.; Mackenzie I. R.; Feldman H. H.; Hamilton R. L.; Dekosky S. T.; van der Zee J.; Kumar-Singh S.; Van Broeckhoven C.; Mayeux R.; Vonsattel J. P.; Troncoso J. C.; Kril J. J.; Kwok J. B.; Halliday G. M.; Bird T. D.; Ince P. G.; Shaw P. J.; Cairns N. J.; Morris J. C.; McLean C. A.; DeCarli C.; Ellis W. G.; Freeman S. H.; Frosch M. P.; Growdon J. H.; Perl D. P.; Sano M.; Bennett D. A.; Schneider J. A.; Beach T. G.; Reiman E. M.; Woodruff B. K.; Cummings J.; Vinters H. V.; Miller C. A.; Chui H. C.; Alafuzoff I.; Hartikainen P.; Seilhean D.; Galasko D.; Masliah E.; Cotman C. W.; Tunon M. T.; Martinez M. C.; Munoz D. G.; Carroll S. L.; Marson D.; Riederer P. F.; Bogdanovic N.; Schellenberg G. D.; Hakonarson H.; Trojanowski J. Q.; Lee V. M. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 2010, 42 (3), 234–9. 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R.; Hernandez D. G.; Nalls M. A.; Rohrer J. D.; Ramasamy A.; Kwok J. B.; Dobson-Stone C.; Brooks W. S.; Schofield P. R.; Halliday G. M.; Hodges J. R.; Piguet O.; Bartley L.; Thompson E.; Haan E.; Hernandez I.; Ruiz A.; Boada M.; Borroni B.; Padovani A.; Cruchaga C.; Cairns N. J.; Benussi L.; Binetti G.; Ghidoni R.; Forloni G.; Galimberti D.; Fenoglio C.; Serpente M.; Scarpini E.; Clarimon J.; Lleo A.; Blesa R.; Waldo M. L.; Nilsson K.; Nilsson C.; Mackenzie I. R.; Hsiung G. Y.; Mann D. M.; Grafman J.; Morris C. M.; Attems J.; Griffiths T. D.; McKeith I. G.; Thomas A. J.; Pietrini P.; Huey E. D.; Wassermann E. M.; Baborie A.; Jaros E.; Tierney M. C.; Pastor P.; Razquin C.; Ortega-Cubero S.; Alonso E.; Perneczky R.; Diehl-Schmid J.; Alexopoulos P.; Kurz A.; Rainero I.; Rubino E.; Pinessi L.; Rogaeva E.; St George-Hyslop P.; Rossi G.; Tagliavini F.; Giaccone G.; Rowe J. B.; Schlachetzki J. C.; Uphill J.; Collinge J.; Mead S.; Danek A.; Van Deerlin V. M.; Grossman M.; Trojanowski J. Q.; van der Zee J.; Deschamps W.; Van Langenhove T.; Cruts M.; Van Broeckhoven C.; Cappa S. F.; Le Ber I.; Hannequin D.; Golfier V.; Vercelletto M.; Brice A.; Nacmias B.; Sorbi S.; Bagnoli S.; Piaceri I.; Nielsen J. E.; Hjermind L. E.; Riemenschneider M.; Mayhaus M.; Ibach B.; Gasparoni G.; Pichler S.; Gu W.; Rossor M. N.; Fox N. C.; Warren J. D.; Spillantini M. G.; Morris H. R.; Rizzu P.; Heutink P.; Snowden J. S.; Rollinson S.; Richardson A.; Gerhard A.; Bruni A. C.; Maletta R.; Frangipane F.; Cupidi C.; Bernardi L.; Anfossi M.; Gallo M.; Conidi M. E.; Smirne N.; Rademakers R.; Baker M.; Dickson D. W.; Graff-Radford N. R.; Petersen R. C.; Knopman D.; Josephs K. A.; Boeve B. F.; Parisi J. E.; Seeley W. W.; Miller B. L.; Karydas A. M.; Rosen H.; van Swieten J. C.; Dopper E. G.; Seelaar H.; Pijnenburg Y. A.; Scheltens P.; Logroscino G.; Capozzo R.; Novelli V.; Puca A. A.; Franceschi M.; Postiglione A.; Milan G.; Sorrentino P.; Kristiansen M.; Chiang H. H.; Graff C.; Pasquier F.; Rollin A.; Deramecourt V.; Lebert F.; Kapogiannis D.; Ferrucci L.; Pickering-Brown S.; Singleton A. B.; Hardy J.; Momeni P. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014, 13 (7), 686–99. 10.1016/S1474-4422(14)70065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer J. D.; Rosen H. J. Neuroimaging in frontotemporal dementia. Int. Rev. Psychiatry 2013, 25 (2), 221–9. 10.3109/09540261.2013.778822. [DOI] [PubMed] [Google Scholar]
- Riedl L.; Mackenzie I. R.; Forstl H.; Kurz A.; Diehl-Schmid J. Frontotemporal lobar degeneration: current perspectives. Neuropsychiatr Dis Treat 2014, 10, 297–310. 10.2147/NDT.S38706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S.; Ammari M.; Aranda B.; Breuza L.; Briganti L.; Broackes-Carter F.; Campbell N. H.; Chavali G.; Chen C.; del-Toro N.; Duesbury M.; Dumousseau M.; Galeota E.; Hinz U.; Iannuccelli M.; Jagannathan S.; Jimenez R.; Khadake J.; Lagreid A.; Licata L.; Lovering R. C.; Meldal B.; Melidoni A. N.; Milagros M.; Peluso D.; Perfetto L.; Porras P.; Raghunath A.; Ricard-Blum S.; Roechert B.; Stutz A.; Tognolli M.; van Roey K.; Cesareni G.; Hermjakob H. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42 (Database issue), D358–63. 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz B. J.; Stark C.; Tyers M. The GRID: the General Repository for Interaction Datasets. Genome Biol. 2003, 4 (3), R23. 10.1186/gb-2003-4-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer K.; Foroushani A. K.; Laird M. R.; Chen C.; Sribnaia A.; Lo R.; Winsor G. L.; Hancock R. E.; Brinkman F. S.; Lynn D. J. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013, 41 (D1), D1228–33. 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata L.; Briganti L.; Peluso D.; Perfetto L.; Iannuccelli M.; Galeota E.; Sacco F.; Palma A.; Nardozza A. P.; Santonico E.; Castagnoli L.; Cesareni G. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40 (D1), D857–61. 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S. Molecular interaction databases. Proteomics 2012, 12 (10), 1656–62. 10.1002/pmic.201100484. [DOI] [PubMed] [Google Scholar]
- Mellacheruvu D.; Wright Z.; Couzens A. L.; Lambert J. P.; St-Denis N. A.; Li T.; Miteva Y. V.; Hauri S.; Sardiu M. E.; Low T. Y.; Halim V. A.; Bagshaw R. D.; Hubner N. C.; Al-Hakim A.; Bouchard A.; Faubert D.; Fermin D.; Dunham W. H.; Goudreault M.; Lin Z. Y.; Badillo B. G.; Pawson T.; Durocher D.; Coulombe B.; Aebersold R.; Superti-Furga G.; Colinge J.; Heck A. J.; Choi H.; Gstaiger M.; Mohammed S.; Cristea I. M.; Bennett K. L.; Washburn M. P.; Raught B.; Ewing R. M.; Gingras A. C.; Nesvizhskii A. I. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10 (8), 730–6. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J.; Arak T.; Vilo J. g:Profiler--a web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res. 2011, 39 (Web Server issue), W307–15. 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P. D.; Campbell M. J.; Kejariwal A.; Mi H.; Karlak B.; Daverman R.; Diemer K.; Muruganujan A.; Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003, 13 (9), 2129–41. 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Duncan D.; Shi Z.; Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013, 41 (W1), W77–83. 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13 (11), 2498–504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binotti B.; Jahn R.; Chua J. J., Functions of Rab Proteins at Presynaptic Sites. Cells 2016, 5, ( (1), ).7. 10.3390/cells5010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. A.; Banerjee R.; Gunawardena S., Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics. Mar. Drugs 2016, 14, ( (5), ).102. 10.3390/md14050102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nadal E.; Ammerer G.; Posas F. Controlling gene expression in response to stress. Nat. Rev. Genet. 2011, 12 (12), 833–45. 10.1038/nrg3055. [DOI] [PubMed] [Google Scholar]
- Cohen-Kaplan V.; Livneh I.; Avni N.; Cohen-Rosenzweig C.; Ciechanover A. The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int. J. Biochem. Cell Biol. 2016, 79, 403–418. 10.1016/j.biocel.2016.07.019. [DOI] [PubMed] [Google Scholar]
- Ferrari R.; Forabosco P.; Vandrovcova J.; Botia J. A.; Guelfi S.; Warren J. D.; Momeni P.; Weale M. E.; Ryten M.; Hardy J. Frontotemporal dementia: insights into the biological underpinnings of disease through gene co-expression network analysis. Mol. Neurodegener. 2016, 11, 21. 10.1186/s13024-016-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan C.; Jethi S.; Kalsi N.; Purohit R. Biophysical Aspect of Huntingtin Protein During polyQ: An In Silico Insight. Cell Biochem. Biophys. 2016, 74 (2), 129–39. 10.1007/s12013-016-0728-7. [DOI] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992, 256 (5054), 184–5. 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Maruszak A.; Safranow K.; Gustaw K.; Kijanowska-Haladyna B.; Jakubowska K.; Olszewska M.; Styczynska M.; Berdynski M.; Tysarowski A.; Chlubek D.; Siedlecki J.; Barcikowska M.; Zekanowski C. PIN1 gene variants in Alzheimer’s disease. BMC Med. Genet. 2009, 10, 115. 10.1186/1471-2350-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder R.; Watts G. D.; Mehta S. G.; Evert B. O.; Broich P.; Fliessbach K.; Pauls K.; Hans V. H.; Kimonis V.; Thal D. R. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann. Neurol. 2005, 57 (3), 457–61. 10.1002/ana.20407. [DOI] [PubMed] [Google Scholar]
- Fatima M.; Kumari R.; Schwamborn J. C.; Mahadevan A.; Shankar S. K.; Raja R.; Seth P. Tripartite containing motif 32 modulates proliferation of human neural precursor cells in HIV-1 neurodegeneration. Cell Death Differ. 2016, 23 (5), 776–86. 10.1038/cdd.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan C.; Jethi S.; Kalsi N.; Purohit R. Biophysical Aspect of Huntingtin Protein During polyQ: An In Silico Insight. Cell Biochem. Biophys. 2016, 74, 129. 10.1007/s12013-016-0728-7. [DOI] [PubMed] [Google Scholar]
- Lu X.; Deng Y.; Yu D.; Cao H.; Wang L.; Liu L.; Yu C.; Zhang Y.; Guo X.; Yu G. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer’s disease. PLoS One 2014, 9 (7), e103067. 10.1371/journal.pone.0103067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerappan C. S.; Sleiman S.; Coppola G. Epigenetics of Alzheimer’s disease and frontotemporal dementia. Neurotherapeutics 2013, 10 (4), 709–21. 10.1007/s13311-013-0219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Dey D.; Carrera I.; Minond D.; Bianchi E.; Xu S.; Lakshmana M. K. COPS5 (Jab1) protein increases beta site processing of amyloid precursor protein and amyloid beta peptide generation by stabilizing RanBP9 protein levels. J. Biol. Chem. 2013, 288 (37), 26668–77. 10.1074/jbc.M113.476689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Chen X.; Song Y.; Zhang Y.; Zhou L.; Wan L. Deficit of RACK1 contributes to the spatial memory impairment via upregulating BECLIN1 to induce autophagy. Life Sci. 2016, 151, 115–21. 10.1016/j.lfs.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Amadio M.; Pascale A.; Wang J.; Ho L.; Quattrone A.; Gandy S.; Haroutunian V.; Racchi M.; Pasinetti G. M. nELAV proteins alteration in Alzheimer’s disease brain: a novel putative target for amyloid-beta reverberating on AbetaPP processing. J. Alzheimers Dis. 2009, 16 (2), 409–19. 10.3233/JAD-2009-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castao E. M.; Maarouf C. L.; Wu T.; Leal M. C.; Whiteside C. M.; Lue L. F.; Kokjohn T. A.; Sabbagh M. N.; Beach T. G.; Roher A. E. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochem. Int. 2013, 62 (2), 145–56. 10.1016/j.neuint.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Kwon Y. A.; Ahn I. S.; Kim S.; Kim S.; Jo S. A.; Kim D. K. Overexpression of Cell Cycle Proteins of Peripheral Lymphocytes in Patients with Alzheimer’s Disease. Psychiatry Invest. 2016, 13 (1), 127–34. 10.4306/pi.2016.13.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson R. A.; Lange M. B.; Sultana R.; Galvan V.; Fombonne J.; Gorostiza O.; Zhang J.; Warrier G.; Cai J.; Pierce W. M.; Bredesen D. E.; Butterfield D. A. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-beta peptide of amyloid precursor protein. Neuroscience 2011, 177, 207–22. 10.1016/j.neuroscience.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva P. N.; Furuya T. K.; Braga I. L.; Rasmussen L. T.; Labio R. W.; Bertolucci P. H.; Chen E. S.; Turecki G.; Mechawar N.; Payao S. L.; Mill J.; Smith M. C. Analysis of HSPA8 and HSPA9 mRNA expression and promoter methylation in the brain and blood of Alzheimer’s disease patients. J. Alzheimers Dis. 2014, 38 (1), 165–70. 10.3233/JAD-130428. [DOI] [PubMed] [Google Scholar]
- Sattlecker M.; Kiddle S. J.; Newhouse S.; Proitsi P.; Nelson S.; Williams S.; Johnston C.; Killick R.; Simmons A.; Westman E.; Hodges A.; Soininen H.; Kloszewska I.; Mecocci P.; Tsolaki M.; Vellas B.; Lovestone S.; Dobson R. J. Alzheimer’s disease biomarker discovery using SOMAscan multiplexed protein technology. Alzheimer's Dementia 2014, 10 (6), 724–34. 10.1016/j.jalz.2013.09.016. [DOI] [PubMed] [Google Scholar]
- Zuliani G.; Cavalieri M.; Galvani M.; Passaro A.; Munari M. R.; Bosi C.; Zurlo A.; Fellin R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J. Neurol. Sci. 2008, 272 (1–2), 164–70. 10.1016/j.jns.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Lan Y. L.; Zhao J.; Li S. Update on the neuroprotective effect of estrogen receptor alpha against Alzheimer’s disease. J. Alzheimers Dis. 2015, 43 (4), 1137–48. 10.3233/JAD-141875. [DOI] [PubMed] [Google Scholar]
- Lim G. P.; Chu T.; Yang F.; Beech W.; Frautschy S. A.; Cole G. M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21 (21), 8370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C.; Ravenscroft T. A.; Sanchez-Contreras M.; Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J. Neurochem. 2016, 138 (Suppl 1), 32–53. 10.1111/jnc.13622. [DOI] [PubMed] [Google Scholar]
- Williams K. L.; Topp S.; Yang S.; Smith B.; Fifita J. A.; Warraich S. T.; Zhang K. Y.; Farrawell N.; Vance C.; Hu X.; Chesi A.; Leblond C. S.; Lee A.; Rayner S. L.; Sundaramoorthy V.; Dobson-Stone C.; Molloy M. P.; van Blitterswijk M.; Dickson D. W.; Petersen R. C.; Graff-Radford N. R.; Boeve B. F.; Murray M. E.; Pottier C.; Don E.; Winnick C.; McCann E. P.; Hogan A.; Daoud H.; Levert A.; Dion P. A.; Mitsui J.; Ishiura H.; Takahashi Y.; Goto J.; Kost J.; Gellera C.; Gkazi A. S.; Miller J.; Stockton J.; Brooks W. S.; Boundy K.; Polak M.; Munoz-Blanco J. L.; Esteban-Perez J.; Rabano A.; Hardiman O.; Morrison K. E.; Ticozzi N.; Silani V.; de Belleroche J.; Glass J. D.; Kwok J. B.; Guillemin G. J.; Chung R. S.; Tsuji S.; Brown R. H. Jr.; Garcia-Redondo A.; Rademakers R.; Landers J. E.; Gitler A. D.; Rouleau G. A.; Cole N. J.; Yerbury J. J.; Atkin J. D.; Shaw C. E.; Nicholson G. A.; Blair I. P. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. 10.1038/ncomms11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai A. The contribution of the DNA damage response to neuronal viability. Antioxid. Redox Signaling 2007, 9 (2), 211–8. 10.1089/ars.2007.9.211. [DOI] [PubMed] [Google Scholar]
- Citterio E. Fine-tuning the ubiquitin code at DNA double-strand breaks: deubiquitinating enzymes at work. Front. Genet. 2015, 6, 282. 10.3389/fgene.2015.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M.; Boddy M. N. Cooperativity of the SUMO and Ubiquitin Pathways in Genome Stability. Biomolecules 2016, 6 (1), 14. 10.3390/biom6010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade J. M.; Ovejero-Benito M. C. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 2015, 14 (5), 712–20. 10.1080/15384101.2015.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T. Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol. Neurobiol. 2012, 46 (1), 125–35. 10.1007/s12035-012-8262-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.