

Abstract
Autophagy is a fundamental cellular degradation process which is essential for cell homeostasis, and dysfunctional autophagy has been associated with a variety of human diseases, such as cancer. Several autophagy chemical modulators have been applied in a number of preclinical or clinical trials against these autophagy related diseases, especially cancer. Small molecule vacuolin-1 potently and reversibly inhibits both endosomal-lysosomal trafficking and autophagosome-lysosome fusion, yet the molecular mechanisms underlying vacuolin-1 mediated autophagy inhibition remain unknown. Here, we first performed the virtual drug screening and identified 14 vacuolin-1 analogues as autophagy inhibitors. Based on these virtual screening results, we further designed and synthesized 17 vacuolin-1 analogues, and found that 13 of them are autophagy inhibitors and a couple of them are as potent as vacuolin-1. In summary, these studies expanded the pool of useful autophagy inhibitors and reveal the structural-activity relationship of vacuolin-1 analogues, which is useful for future development of vacuolin-1 analogues with high potency and for identification of the molecular targets of vacuolin-1.
Keywords: autophagy, vacuolin-1, LC3B-II, SAR, inhibitor
1. Introduction
Autophagy contains at least three types, including macroautophagy, microautophagy, and chaperone-mediated autophagy, and macroautophagy (hereafter referred as autophagy) is the most common and best studied form. Autophagy is initiated when misfolded proteins or damaged organelles are sequestered by double-membrane autophagosomes, and is completed when autophagosomes fuse with lysosomes to form autolysosomes, inside which the sequestered contents are degraded by lysosomal acidic hydrolases into amino acids or fatty acids and recycled back to cytosol [1,2]. Autophagy is essential to maintain the cellular homeostasis by preventing damaged and harmful substances from accumulating inside cells, and thus is involved in a wide variety of cellular processes, from cell proliferation, differentiation, apoptosis, development, ageing, to immunity [3,4,5]. The initiation, elongation, membrane closure, and maturation of autophagic process are tightly controlled by the complicated interplay among several core molecular machineries, including the Unc-51-like kinase (ULK) complex, the class III phosphatidylinositol 3-kinase (PtdIns3k) complex [6,7], and the two ubiquitin-like protein conjugation complexes, MAP1LC3B (microtubule associated protein 1 light chain 3B, herein referred simply as LC3B) and ATG12-ATG5-ATG16L complex (ATG, autophagy-related). The LC3B lipidation converts the cytosolic LC3B (LC3B-I) to the autophagic vesicle-associated form (LC3B-II). The lipidated LC3B-II showed a punctate staining pattern and had faster electrophoretic mobility compared with diffused LC3B-I, which have been commonly used to monitor the autophagic process.
Dysfunctional autophagy has been associated with a variety of human diseases, including neurodegenerative disorders, cardiovascular diseases, infection, and cancer [8,9,10]. On one hand, downregulated autophagy in normal tissue can lead to tumorigenesis due to the accumulation of misfolded proteins and damaged organelles inside cells. On the other hand, autophagy is upregulated in many kinds of cancers, which allow the cancer cells to meet the energy needs under the increased metabolic consumption and to survive in the hypoxic microenvironment [11,12]. Cancer cells also utilize autophagy to resist chemotherapy and radiation therapy, whereas inhibiting autophagy renders the cancer cells more sensitive to chemotherapy and radiation therapy [13,14]. Not surprisingly, a number of pre-clinical and clinical trials have been ongoing to evaluate the anti-cancer effects of chloroquine (CQ) or hydroxychloroquine (HCQ), the lysosomotropic agent that blocks the degradation of the products of autophagy by inhibiting lysosome function, in combination with chemotherapy or other agents [15]. Yet the therapeutic activity of CQ and HCQ seems to be partially due to the modulation of autophagy-unrelated mechanisms [16]. Therefore, compounds that regulate autophagy in a highly specific manner will certainly strengthen the clinical utility of this anti-cancer therapeutic paradigm.
Prompted by the fact that many available autophagy chemical modulators lack either potency or specificity [17], chemicals were developed for autophagy inhibition [18] and we set up assays to screen small molecules affecting autophagy. We found that the small chemical vacuolin-1, originally found to interfere with specific steps in membrane traffic, potently inhibited the fusion between autophagosomes and lysosomes, resulting in the accumulation of autophagosomes [19]. Vacuolin-1 treatment also blocked the fusion between endosomes and lysosomes, thus inhibiting general endosomal-lysosomal degradation. Moreover, we found that vacuolin-1 indirectly activates RAB5A to block autophagosome-lysosome fusion [20]. Recently, vacuolin-1 has also been shown to bind to PIKfyve in vitro, yet how vacuolin-1 regulates PIKfyve and whether it directly binds to PIKfyve in vivo remains unknown [21].
Since the molecular entities of vacuolin-1 remains to be identified, here we identified a number of vacuolin-1 structure analogues by combining virtual drug screening and chemical synthesis. By examining their effects on autophagy inhibition through western blot and confirming by fluorescence image-based assay, we studied the structure-activity relationship (SAR) of these analogues, and found that several vacuolin-1 analogues can potently inhibit autophagy.
2. Results and Discussion
2.1. Identification of Novel Vacuolin-1 Analogues by Virtual Drug Screening
Since the molecular targets of vacuolin-1 remains unknown, we performed a 2D similarity search from ZINC database which contains over 13 million compounds and J & K Chemical database which contains more than 700,000 molecules by the Pipeline Pilot 7.5 software, and identified 14 vacuolin-1 analogues with the 1,3,5-triazine scaffold (Table 1). We subsequently analyzed the effects of these compounds on autophagy in tandem fluorescence-tagged LC3B (tfLC3B) expressing HeLa cells by both western blot and confocal image analyses. As shown in Figure 1 and Figure S1, except VS11, the rest of the analogues all induced the accumulation of both lapidated LC3B-II and SQSTM1, an autophagy substrate, in a dose dependent manner, albeit exhibiting different potency. In addition, co-treatment of cells with the vacuolin-1 analogues (10 µM) and bafilomycin (BAF) (100 nM), an inhibitor of the vacuolar proton pump that blocks the fusion of autophagosomes with lysosomes, failed to further induce the accumulation of LC3B-II and SQSTM1 as compared to either drug alone (Figure 2A). Likewise, treatment of cells with these vacuolin-1 analogues strongly induced the yellow LC3B-II puncta (representing the autophagosomes), not red-only LC3B-II puncta (representing autolysosomes) (Figure 2B and Figure S2). Moreover, the induced LC3B-II puncta did not colocalize with lysosome-associated membrane protein 1 (LAMP1) (Figure 3). Taken together, these data indicate that the identified vacuolin-1 analogues via drug virtual screening, like vacuolin-1, can potently inhibit the fusion between autophagosome and lysosomes, resulting in the accumulation of autophagosomes thus blocking autophagic flux.
Table 1.
Structure-activity relationship (SAR) of selected compounds from virtual screening a.
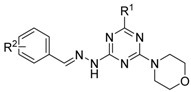
| Compound | R1 | R2 | 0.1 µM | 1 µM | 10 µM | 100 µM |
|---|---|---|---|---|---|---|
| Vacuolin-1 | N,N-diphenylamino | 3-I | + | ++ | +++ | ++++ |
| VS1 | N,N-diphenylamino | 4-F | + | + | + | |
| VS2 | 4-Cl-phenylamino | 4-Cl | − | + | + | |
| VS3 | 4-Cl-phenylamino | 3-I | + | ++ | ++ | ++ |
| VS4 | 4-F-phenylamino | 3-I | + | ++ | ++ | ++ |
| VS5 | 4-Cl-phenylamino | 4-dimethylamino | + | ++ | ++ | ++ |
| VS6 | 4-Cl-phenylamino | 3-NO2 | + | +++ | +++ | +++ |
| VS7 | 4-Cl-phenylamino | 4-NO2 | − | − | + | + |
| VS8 | 4-OH-phenylamino | 2,4-dimethyl | + | ++ | ++ | ++ |
| VS9 | morpholino | H | − | + | + | |
| VS10 | morpholino | 4-Cl | − | − | + | |
| VS11 | morpholino | 4-Br | − | − | − | |
| VS12 | morpholino | 4-I | − | − | + | |
| VS13 | morpholino | 3,4-di-Cl | − | − | + | |
| VS14 | morpholino | 3-Br-6-OMe | − | + | + |
a . λ0: control group. “−”: λ0 ≤ 1.1; “+”: 1.1 < λ/λ0 ≤ 3; “++”: 3 < λ/λ0 ≤ 7; “+++”: 7 < λ/λ0 ≤ 10; “++++”: 10 < λ/λ0.
Figure 1.

Vacuolin-1 analogues identified via virtual screening induced the accumulation of both LC3B-II and SQSTM1 in HeLa cells in a dose dependent manner after a 6 h treatment.
Figure 2.

Vacuolin-1 analogues identified via virtual screening inhibited autophagic flux in HeLa cells. (A) Treatment of HeLa cells with vacuolin-1 analogues (10 µM) and BAF (100 nM) failed to further increase the accumulation of both LC3B-II and SQSTM1 as compared to either drug alone. (B) Vacuolin-1 (10 µM) or VS6 (10 µM) induced the accumulation of yellow LC3B-II puncta in RFP-GFP-LC3B expressing HeLa cells. Scale bar = 10 µm.
Figure 3.

Vacuolin-1 analogues (10 µM) markedly induced GFP-LC3B-II puncta in HeLa cells, which were not colocalized with RFP-LAMP1. Scale bar = 10 µm. *, paired t-test, p < 0.05 compared to control group.
Comparing with vacuolin-1, VS1–VS8 whose R1 groups were aromatic, maintained comparable autophagy inhibitory activity, whereas VS9–VS14 which bearing two morpholino groups showed obviously decreased potency. The difference of VS6 and VS7 suggested that electron-withdrawing R2 substituted at the meta-position is more important for the autophagy inhibitory effects than para-position.
2.2. Identification of Novel Vacuolin-1 Analogues by Chemical Synthesis
To further explore the SAR of vacuolin-1 on autophagy, we performed a systematic modification. The modifications could be divided into three parts based on the structure of vacuolin-1: the N,N-diphenylamino group modified analogues A1–A6, the morpholino group modified analogues B1–B5, and the aromatic moiety attached to hydrazine (henceforward hydrazine area) modified analogues C1–C6. The synthesis of vacuolin-1 analogues (Scheme 1) was carried out from trichlorotriazine to afford dichlorotriazine derivatives (1a–1g). Desired monoaddition products were obtained by controlling temperature and the excess amount of cyanuric chloride. Then 1a–1f were converted to monochlorotriazines (2a–2f and 2h–2k) after reacting with aliphatic amines with yields ranging from 67% to 86%. Compound 2g was obtained from the reaction of 1g with the corresponding aromatic amine under reflux due to low reactivity of the amine. Introduction of hydrazine on suitably derivatized monochlorotriazines provided 3a–3k. Notably, the use of excess hydrazine hydrate converted the alkynyl group in 2e to an alkyl group in 3e. Condensation of 3a–3k with 3-iodobenzaldehyde in the presence of acetic acid respectively provided vacuolin-1, A1–A6, B1, B2, B4, and B5. Deprotection of the Boc group in B2 by TFA successfully produced B3. The reactions of intermediate 3a with aldehydes gave C1–C4. Diphenyl substituent analogue C5 was prepared from C4 via Suzuki cross coupling with phenylboronic acid. Coupling of 3a with hydrocinnamoyl chloride afforded target C6.
Scheme 1.

Reagents and conditions: (i) Amine, acetone, K2CO3, 0–5 °C →r.t, 65–94%; (ii) Amine, acetone, 0 °C →r.t, 67–86%. (for 1g: K2CO3; reflux; 42%); (iii) Dioxane, 80% hydrazine hydrate (m/m), r.t; (iv) Aldehyde, ethanol, CH3COOH, r.t, 55%–93%; (v) THF, acyl chloride, 0 °C →r.t, 73%; (vi) Phenylboronic acid, Pd(PPh3)4, Na2CO3, ethanol/ toluene/H2O (1:5:2, v/v), Ar, reflux, 12 h. 77%; (vii) DCM, TFA, r.t, 95%.
Subsequently, we again analyzed the effects of these 17 vacuolin-1 analogues on autophagy in tfLC3B-expressing HeLa cells by both western blot and confocal image analyses. As shown in Figure 4, S3, and Table 2, analogues B3, B4, B5, and C5 failed to induce the accumulation of both LC3B-II and SQSTM1 at all doses, analogues B1, C2, and C6 only weakly induced the accumulation of LC3B-II and SQSTM1, analogues A1, A2, A3, A6, C1, and C3 showed good potency, and analogues A4 and A5 were as potent as vacoulin-1. Similarly, the positive analogues, e.g., A5 and others, markedly induced the yellow LC3B-II puncta in HeLa cells (Figure 5A and Figure S4), and co-treatment of cells with these analogues and bafilomycin (BAF) failed to further induce the accumulation of LC3B-II and SQSTM1 as compared to either drug alone (Figure 5B). Therefore, these data indicate that the synthesized vacuolin-1 analogues are autophagy inhibitors with good potency comparable to vacuolin-1.
Figure 4.

Synthesized vacuolin-1 analogues induced the accumulation of both LC3B-II and SQSTM1 in HeLa cells in a dose dependent manner after a 6 h treatment.
Table 2.
SAR of synthesized compounds a.
| Compound | 0.1 µM | 1 µM | 10 µM | 100 µM | Compound | 0.1 µM | 1 µM | 10 µM | 100 µM |
|---|---|---|---|---|---|---|---|---|---|
| vacuolin-1 | + | ++ | +++ | ++++ | B3 | − | − | * | |
| A1 | + | ++ | ++ | ++ | B4 | − | − | − | − |
| A2 | + | ++ | ++ | ++ | B5 | − | − | − | − |
| A3 | + | ++ | ++ | ++ | C1 | + | + | ++ | ++ |
| A4 | + | + | +++ | +++ | C2 | − | − | − | + |
| A5 | + | +++ | ++++ | ++++ | C3 | − | − | ++ | ++ |
| A6 | + | + | ++ | ++ | C4 | − | − | − | + |
| B1 | − | + | + | + | C5 | − | − | − | − |
| B2 | − | − | + | + | C6 | − | − | − | ++ |
a
. λ0: control group. “−”: λ/λ0 ≤ 1.1; “+”: 1.1 < λ/λ0 ≤ 3; “++”: 3 < λ/λ0 ≤ 7; “+++”: 7 < λ/λ0 ≤ 10; “++++”: 10 < λ/λ0. * Toxic, 10 µM caused cell death.
Figure 5.

Synthesized vacuolin-1 analogues inhibited autophagic flux in HeLa cells. (A) A5 (1 µM), not C5 (1 µM), induced the accumulation of yellow LC3B-II puncta in RFP-GFP-LC3B expressing HeLa cells. Scale bar = 10 µm. (B) Treatment of HeLa cells with vacuolin-1 analogues (1 µM) and BAF (100 nM) failed to further increase the accumulation of both LC3B-II and SQSTM1 as compared to either drug alone.
Consistent with virtual screening results (Figure 1 and Table 1), removing one phenyl from diphenylamino group (A2–A6) maintained the ability to inhibit autophagy (Figure 4 and Table 2). The results not only suggested that aromatic R1 is important to keep autophagy inhibitory activity, but also implied that modification is tolerable on the phenyl group, particularly, analogues A5 exhibited slightly stronger inhibitory potency than vacuolin-1 (Figure 4 and Table 2). Notably, small changes in morpholine caused a great loss of potency (B1–B5), indicating the crucial role of the morpholine ring in autophagy inhibition. As for the substituent in hydrazine area (C1–C6), moderate changes were acceptable, while bulky substituents killed the drug effects. The structure-activity relationship of vacuolin-1 analogues as autophagy inhibitors is summarized in Figure 6.
Figure 6.

Structure-activity relationship for vacuolin-1 analogues as autophagy inhibitors.
3. Experimental Section
3.1. 2D Similarity Virtual Screening
The research was performed on ZINC database and two collections of the J & K Chemical database (K66-X4436 EXPRESS-Pick Collection and L62-X6857 2013feb_sc1). Calculated by Pipeline Pilot 7.5 software (Accelrys, San Diego, CA, USA), the similarity of molecules and vacuolin-1 will be scored by Tanimoto coefficient described by FCFP_4 fingerprint. Molecules with Tanimoto great than 0.6 were screened out.
3.2. Chemistry
All of chemicals and solvents used were obtained from J & K or Sigma. The solvents were dried by standard procedures.1H NMR spectra and 13C NMR spectra were recorded at 400 MHz using Bruker Avance III spectrometer (Bruker BioSpin AG, Fällanden, Switzerland) in CDCl3 or DMSO-d6 solution with tetramethylsilane as the internal standard and chemical shift values were given in ppm. The NMR data was processed by software MestReNova (Ver.6.1.0, Mestrelab research S.L., Santiago de Compostela, Spain). The high resolution mass (HRMS) was measured on an FT-MS-Bruker APEX IV mass spectrometer.
3.2.1. General Procedure for the Synthesis of 1a–1g
An amine (1 eq.) in acetone solution was added dropwise to a stirred solution of cyanuric chloride (1.5 eq.) and K2CO3 (1 eq.) in acetone at 0–5 °C. The resulting mixture was stirred at room temperature overnight. Ice water was added to the solution. The precipitate was collected by filtration and dried. The crude product was further purified by column chromatography.
3.2.2. General Procedure for the Synthesis of 2a–2k
General procedure for 2a–2f and 2h–2k: 1a–1g or 2h–2k (1 eq.), K2CO3 (1 eq.) were suspended in DCM, cooled to 0–5 °C with an ice bath while stirred. Amine solution (1.2 eq., 0.3 mol/L solution of DCM) was added dropwise. The resulting mixture was stirred at room temperature for an additional 30 min, monitored by TLC until the starting material disappeared. The solvent was evaporated and the residue was purified by column chromatography.
Procedure for 2g: 1g (1 eq.) was dissolved in acetone, K2CO3 (1 eq.) and amine (1.2 eq.) were added. The resulting mixture was stirred and refluxed overnight. The solvent was evaporated and partitioned between ethyl acetate and water. The organic phase was dried over with Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography.
3.2.3. General Procedure for the Synthesis of 3a–3k
2a–2k was dissolved in dioxane. Excess hydrazine hydrate (80%, m/m) was added to the solution while stirring. The reaction was monitored by TLC. After the staring material disappeared, water was added and then extracted using ethyl acetate (60 mL × 3). The combined organic phase was then washed with water, dried over with Na2SO4, filtered and concentrated in vacuo. The resulting solid (3a–3k) was used in the next condensing step without further purification.
3.2.4. General Procedure for the Synthesis of Vacuolin-1, A1–A6, B1–B5 and C1–C6
The reaction mixture under the conditions mentioned in Scheme 1 was stirred, and then evaporated to dryness under reduced pressure. The residue was purified by column chromatography.
2-N,N-Diphenylamino-6-(2-(3-iodobenzylidene)hydrazinyl)-4-morpholino-1,3,5-triazine (vacuolin-1). White solid. Mp 187.3–188.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.59 (br s, 1H), 7.97 (s, 1H), 7.64 (d, J = 7.6 Hz, 2H), 7.54 (d, J = 6.4 Hz, 1H), 7.33 (dt, J = 15.2, 7.6 Hz, 8H), 7.21 (t, J = 7.0 Hz, 2H), 7.08 (t, J = 7.8 Hz, 1H), 3.69 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 166.0, 165.2, 164.3, 143.8, 140.9, 138.2, 136.6, 135.5, 130.2, 128.7, 128.0, 126.2, 125.7, 94.5, 66.8, 43.6. HRMS (MS-ES+) m/z Calcd. for C26H25IN7O [M + H]+: 578.1165; found 578.1165.
6-(2-(3-Iodobenzylidene)hydrazinyl)-2-(N-(3-methoxyphenyl)-N-phenylamino)-4-morpholino-1,3,5-triazine (A1). White solid. Yield: 72%. Mp 187.3–188.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.01 (s, 1H), 7.89 (s, 1H), 7.68 (d, J = 7.3 Hz, 1H), 7.54 (s, 1H), 7.38 (t, J = 7.1 Hz, 2H), 7.24 (m, 5H), 6.91 (s, 1H), 6.86–6.77 (m, 2H), 3.73 (s, 3H), 3.56 (s, 8H). 13C NMR (101 MHz, DMSO-d6) δ 166.2, 165.1, 164.7, 159.9, 145.4, 144.3, 138.0, 137.8, 131.2, 129.7, 129.1, 128.5, 126.1, 124.8, 120.7, 114.5, 111.6, 105.1, 95.6, 66.4, 55.7, 43.6. HRMS (MS-ES+) m/z Calcd. for C27H27IN7O2 [M + H]+: 608.1271; found 608.1271.
6-(2-(3-Iodobenzylidene)hydrazinyl)-4-morpholino-2-(N-(naphthalen-1-yl)amino)-1,3,5-triazine (A2). Light pink solid. Yield: 93%. Mp 225.5–226.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 9.64 (s, 1H), 8.58 (br s, 1H), 8.17 (s, 1H), 8.14 (s, 1H), 7.93–7.79 (m, 4H), 7.74 (d, J = 7.4 Hz, 1H), 7.70 (d, J = 5.4 Hz, 1H), 7.49 (s, 1H), 7.36 (t, J = 7.2 Hz, 1H), 7.25 (t, J = 7.4 Hz, 1H), 3.82 (s, 4H), 3.70 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.8, 164.8, 164.4, 140.3, 137.4, 137.4, 134.8, 134.2, 133.8, 130.8, 128.9, 128.0, 126.0, 125.8, 125.5, 125.5, 124.8, 123.5, 122.7, 95.2, 66.0, 43.2. HRMS (MS-ES+) m/z Calcd. for C24H23IN7O [M + H]+: 552.1009; found 552.1004.
2-(N-(3-Ethoxycarbonyl)phenylamino-6-(2-(3-iodobenzylidene)hydrazinyl)-4-morpholino-1,3,5-triazine (A3). White solid. Yield: 75%. Mp 194.9–195.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 8.47 (s, 1H), 8.02 (s, 1H), 7.76–7.57 (m, 4H), 7.51 (d, J = 6.9 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.05 (t, J = 7.7 Hz, 1H), 4.36 (q, J = 7.0 Hz, 2H), 3.91 (s, 4H), 3.78 (s, 4H), 1.38 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.5, 165.2, 164.5, 164.3, 141.4, 139.4, 138.5, 136.1, 135.6, 131.0, 130.2, 128.8, 126.6, 124.2, 124.0, 121.2, 94.6, 66.9, 61.1, 44.1, 14.5. HRMS (MS-ES+) m/z Calcd. for C23H25IN7O3 [M + H]+: 574.1064; found 574.1085.
2-N-Hexyl-N-phenylamino-6-(2-(3-iodobenzylidene)hydrazinyl)-4-morpholino-1,3,5-triazine (A4). White solid. Yield: 72%. Mp 140.3–141.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.98 (s, 1H), 7.64 (m, 2H), 7.55 (d, J = 6.7 Hz, 1H), 7.39 (t, J = 7.4 Hz, 2H), 7.27 (s, 2H), 7.07 (t, J = 7.7 Hz, 1H), 3.95 (t, J = 8.0 Hz, 2H), 3.90–3.55 (m, 8H), 1.63 (t, J = 6.4 Hz, 2H), 1.35–1.24 (m, 6H), 0.86 (t, J = 5.5 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 165.5, 165.2, 164.2, 143.4, 140.4, 140.4, 138.1, 136.8, 135.5, 130.1, 128.7, 127.9, 126.1, 94.4, 66.9, 49.9, 43.7, 31.6, 28.0, 26.6, 22.6, 14.0. HRMS (MS-ES+) m/z Calcd. for C26H33IN7O [M + H]+: 586.1791; found 586.1796.
2-(N-(4-Ethoxy-4-oxobutyl)-N-phenylamino)-6-(2-(3-iodobenzylidene)hydrazinyl)-4-morpholino-1,3,5-triazine (A5). White solid. Yield: 92%. Mp 146.5–147.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 7.99 (s, 1H), 7.65 (d, J = 7.8 Hz, 2H), 7.57 (d, J = 4.0 Hz, 1H), 7.40 (t, J = 7.7 Hz, 2H), 7.28 (dd, J = 9.5, 5.6 Hz, 3H), 7.09 (t, J = 7.8 Hz, 1H), 4.11 (m, 2H), 4.05 (t, J = 7.1 Hz, 2H), 3.78 (br s, 8H), 2.38 (t, J = 7.4 Hz, 2H), 2.04–1.94 (m, 2H), 1.23 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.2, 165.6, 165.2, 164.2, 143.1, 140.5, 138.1, 136.7, 135.5, 130.2, 128.9, 127.9, 126.3, 126.1, 94.4, 66.9, 60.4, 49.1, 43.7, 31.7, 23.5, 14.3. HRMS (MS-ES+) m/z Calcd. for C26H31IN7O3 [M + H]+: 616.1533; found 616.1526.
2-(N-(4-Cyanobutyl)-N-phenylamino)-6-(2-(3-iodobenzylidene)hydrazinyl)-4-morpholino-1,3,5-triazine (A6). White solid. Yield: 84%. Mp 112.5–113.8 °C 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.95 (s, 1H), 7.66–7.48 (m, 3H), 7.35 (t, J = 6.2 Hz, 2H), 7.27–7.17 (m, 3H), 7.05 (t, J = 8.0 Hz, 1H), 4.04 (s, 2H), 3.85–3.53 (m, 8H), 2.44 (s, 2H), 1.77 (d, J = 4.7 Hz, 2H), 1.70 (d, J = 4.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 165.5, 164.9, 164.0, 142.6, 140.5, 137.8, 136.5, 135.2, 130.1, 128.7, 127.7, 126.2, 125.9, 119.5, 94.3, 66.5, 48.0, 43.5, 26.8, 22.3, 16.6. HRMS (MS-ES+) m/z Calcd. for C25H28IN8O [M + H]+: 583.1431; found 583.1447.
(S)-2-N,N-Diphenylamino-6-(2-(3-iodobenzylidene)hydrazinyl)-4-(3-methylmorpholino)-1,3,5-triazine (B1). White solid. Yield 70%. Mp 219.6–210.5 °C. 1H NMR (400 MHz, CDCl3) δ 9.09 (br s, 1H), 7.96 (s, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 6.2 Hz, 2H), 7.34 (m, J = 7.9 Hz, 8H), 7.20 (t, J = 6.5 Hz, 2H), 7.07 (t, J = 7.8 Hz, 1H), 4.94–2.99 (m, 7H), 1.23 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 165.9, 164.9, 164.2, 143.8, 141.0, 138.1, 136.7, 135.5, 130.2, 128.7, 127.9, 126.1, 125.7, 94.6, 71.0, 67.0, 46.3, 38.6, 14.5. HRMS (MS-ES+) m/z Calcd. for C27H27IN7O [M + H]+: 592.1322; found 592.1323.
4-(4-(t-Butyloxycarbonyl)piperazin-1-yl)-2-N,N-diphenylamino-6-(2-(3-iodobenzylidene)hydrazinyl)-1,3,5-triazine (B2). White solid. Yield 83%. Mp 238.9–240.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.36 (br s, 1H), 8.00 (s, 1H), 7.71 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.56 (s, 1H), 7.41–7.31 (m, 8H), 7.24 (t, J = 7.1 Hz, 2H), 7.11 (t, J = 7.8 Hz, 1H), 3.87 (s, 2H), 3.51 (s, 4H), 3.39 (s, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 166.1, 165.2, 164.3, 154.8, 143.8, 140.8, 138.2, 135.5, 130.2, 128.7, 128.0, 126.2, 125.7, 94.5, 80.0, 43.0, 28.4. HRMS (MS-ES+) m/z Calcd. for C31H34IN8O2 [M + H]+: 677.1849; found 677.1865.
2-N,N-Diphenylamino-6-(2-(3-iodobenzylidene)hydrazinyl)-4-(piperazin-1-yl)-1,3,5-triazine (B3). White solid. Yield 90%. Mp 210.9–211.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.95 (s, 2H), 8.02 (s, 1H), 7.92 (s, 1H), 7.71 (d, J = 7.9 Hz, 1H), 7.43–7.28 (m, 8H), 7.25 (t, J = 6.6 Hz, 2H), 7.19 (t, J = 7.9 Hz, 1H), 4.11–3.47 (m, 4H), 3.12 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.2, 165.0, 164.7, 158.7, 144.2, 141.3, 138.0, 137.8, 134.8, 131.3, 129.2, 128.5, 126.2, 95.6, 42.8. HRMS (MS-ES+) m/z Calcd. for C26H26IN8O [M + H]+: 577.1325; found 577.1320.
2-N,N-Diphenylamino-4-((6-hydroxyhexyl)amino)-6-(2-(3-iodobenzylidene)hydrazinyl)-1,3,5-triazine (B4). White solid. Yield 60%. Mp 87.3–88.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (m, 1H), 7.98 (m, 2H), 7.68 (d, J = 6.7 Hz, 1H), 7.49 (d, J = 6.8 Hz, 1H), 7.35 (d, J = 7.2 Hz, 4H), 7.30 (d, J = 6.8 Hz, 4H), 7.22–7.14 (m, 3H), 4.35 (s, 1H), 3.40–3.36 (m, 2H), 2.94 (d, J = 5.7 Hz, 1H), 1.58–0.77 (m, 10H). 13C NMR (101 MHz, DMSO-d6) δ 166.4, 166.2, 164.5, 144.6, 140.1, 138.0, 137.7, 134.1, 131.2, 129.0, 128.6, 126.8, 125.8, 95.7, 61.2, 33.1, 29.7, 26.8, 25.7, 19.4. HRMS (MS-ES+) m/z Calcd. for C28H31IN7O [M + H]+: 608.1635; found 608.1649.
2-N,N-Diphenylamino-4-((2-hydroxyethyl)amino)-6-(2-(3-iodobenzylidene)hydrazinyl)-1,3,5-triazine (B5). White solid. Yield 55%. Mp 229.5–231.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 8.00 (m, 2H), 7.69 (d, J = 7.6 Hz, 1H), 7.50 (d, J = 6.3 Hz, 1H), 7.37 (t, J = 7.6 Hz, 4H), 7.29 (d, J = 7.7 Hz, 4H), 7.25–7.15 (m, 3H), 4.49 (t, J = 4.8 Hz, 1H), 3.52 (s, 1H), 3.33 (m, 2H), 3.05 (dd, J = 11.5, 5.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 166.3, 164.5, 144.7, 144.5, 140.8, 138.0, 134.2, 131.3, 131.2, 129.3, 129.1, 128.8, 128.6, 126.8, 126.0, 125.9, 95.7, 60.3, 43.3. HRMS (MS-ES+) m/z Calcd. for C24H23IN7O [M + H]+: 552.1009; found 552.1011.
2-N,N-Diphenylamino-6-(2-((1H-indol-3-yl)methylene)hydrazinyl)-4-morpholino-1,3,5-triazine (C1). White solid. Yield 93%. Mp 273.6–274.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.24 (s, 1H), 7.71 (d, J = 6.7 Hz, 1H), 7.61 (s, 1H), 7.45–7.29 (m, 8H), 7.21–7.10 (m, 2H), 6.90 (t, J = 6.9 Hz, 1H), 6.72 (s, 2H), 6.47 (d, J = 7.9 Hz, 1H), 6.34 (t, J = 7.3 Hz, 1H), 3.69–3.39 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 165.0, 164.5, 144.7, 140.0, 139.9, 137.4, 129.0, 128.6, 125.8, 124.7, 123.0, 122.8, 120.6, 113.0, 111.9, 66.4, 43.6. HRMS (MS-ES+) m/z Calcd. for C28H27N8O [M + H]+: 491.2308; found 491.2306.
2-N,N-Diphenylamino-4-morpholino-6-(2-(pyridin-4-ylmethylene)hydrazinyl)-1,3,5-triazine (C2). White solid. Yield 70%. Mp 262.3–263.7 °C. 1H NMR (400 MHz, CDCl3) δ 9.03 (s, 1H), 8.60 (d, J = 5.4 Hz, 2H), 7.74 (s, 1H), 7.48 (d, J = 4.6 Hz, 2H), 7.42–7.30 (m, 8H), 7.23 (t, J = 7.1 Hz, 2H), 4.03–3.47 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 166.0, 165.1, 164.3, 150.1, 143.8, 142.0, 139.6, 128.7, 128.0, 125.8, 120.8, 66.8, 43.6. HRMS (MS-ES+) m/z Calcd. for C25H25N8O [M + H]+: 453.2151; found 453.2143.
2-N,N-Diphenylamino-4-morpholino-6-(2-(thiophen-2-ylmethylene)hydrazinyl)-1,3,5-triazine (C3). White solid. Yield 73%. Mp 242.8–243.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.29 (s, 1H), 7.53 (d, J = 4.8 Hz, 1H), 7.36 (t, J = 7.5 Hz, 4H), 7.29 (d, J = 7.5 Hz, 4H), 7.23–7.16 (m, 3H), 7.10–7.04 (m, 1H), 3.97–3.44 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 166.3, 165.01, 164.5, 144.5, 140.7, 138.1, 129.1, 129.0, 128.5, 128.1, 127.9, 125.9, 66.4, 43.6. HRMS (MS-ES+) m/z Calcd. for C24H24N7OS [M + H]+: 458.1763; found 458.1765.
6-(2-(3,5-Dibromobenzylidene)hydrazinyl)-2-N,N-diphenylamino-4-morpholino-1,3,5-triazine (C4). White solid. Yield 72%. Mp 225.4–226.7 °C. 1H NMR (400 MHz, CDCl3) δ 8.64 (br s, 1H), 7.65 (s, 2H), 7.59 (s, 1H), 7.39–7.28 (m, 8H), 7.22 (t, J = 6.9 Hz, 2H), 3.98–3.42 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 166.0, 165.1, 164.3, 143.7, 139.4, 138.2, 134.4, 128.7, 128.3, 127.9, 125.8, 123.1, 66.8, 43.6. HRMS (MS-ES+) m/z Calcd. for C26H24Br2N7O [M + H]+: 608.0409; found 608.0410.
2-N,N-Diphenylamino-4-morpholino-6-(2-((1,1':3',1''-terphenyl)-5'-ylmethylene)hydrazinyl)-1,3,5-triazine (C5). White solid. Yield 77%. Mp 225.4–226.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1H), 8.24 (s, 1H), 7.86 (s, 1H), 7.82–7.74 (m, 5H), 7.54 (t, J = 7.0 Hz, 4H), 7.44 (t, J = 7.1 Hz, 2H), 7.39–7.28 (m, 8H), 7.17 (t, J = 5.7 Hz, 2H), 3.83–3.36 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 165.0, 164.8, 144.5, 142.2, 141.8, 140.2, 137.0, 129.4, 129.0, 128.5, 128.3, 127.4, 126.3, 125.9, 124.1, 66.3, 43.6. HRMS (MS-ES+) m/z Calcd. for C38H34N7O [M + H]+: 604.2825; found 604.2826.
2-N,N-Diphenylamino-4-morpholino-6-(2-(3-phenylpropanoyl)hydrazinyl)-1,3,5-triazine (C6). White solid. Yield 73%. Mp 174.6–175.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.16 (br s, 1H), 7.34–7.27 (m, 7H), 7.23–7.14 (m, 8H), 6.61 (br s, 1H), 3.78–3.47 (m, 8H), 2.96–2.79 (m, 2H), 2.51–2.12 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 178.2, 167.5, 166.0, 164.9, 143.8, 140.7, 128.6, 128.3, 128.0, 127.6, 126.3, 125.6, 124.9, 66.7, 43.5, 35.8, 31.2. HRMS (MS-ES+) m/z Calcd. for C28H30N7O2 [M + H]+: 496.2461; found 496.2462.
3.3. Autophagy Inhibitory Activity Measurement
Cell culture—HeLa cells were maintained in DMEM (Invitrogen, 12800-017) plus 10% fetal bovine serum and penicillin/streptomycin at 5% CO2 and 37 °C.
Antibodies and reagents—LC3B (Novus, Littleton, CO, USA, NB100-2220; 1:1000 for the Western blotting analysis (WB)), SQSTM1 (Novus, NBP1-48320; 1:1000 WB), EGFR (Santa Cruz Biotechnology, Santa Cruz, CA, USA, SC-03; 1:250 WB), LAMP1 (Cell Signaling Technology, 9091; 1:500 immunofluorescence analysis), GAPDH antibodies (Sigma, Saint Louis, MO, USA, G8795; 1:5000 WB). BAF (Sigma-Aldrich, B1793), and Vacuolin-1 (Santa Cruz Biotechnology, SC-216045).
GFP-LC3B and RFP-GFP-LC3B Lentivirus Production and Infection—GFP-LC3B and RFP-GFP-LC3B (tfLC3B) cDNA was cloned into pLenti-CMV-Puro-DEST (Addgene). The lentivirus production was performed in 293T cells. For infection, cells were plated at a density of 3 × 105 cells/well in 6-well plates. The experiment was performed as described previously [20].
Western blot analysis—HeLa cells were treated with or without indicated vacuolin-1 analogues for 6h before lyzed by EBC lysis buffer (50 mM HEPES at pH 7.5, 0.15 M NaCl, 1 mM EDTA, 1% (m/m) NP-40, 150 μM PMSF, 10 mM NaF, 10 ng/mL leupeptin, 1 mM DTT, and 1 mM sodium vanadate). Protein concentrations of the cell lysates were determined by Bradford protein assay. 30 or 40 µg of cell lysates per lane were subjected to electrophoresis on 8%, 10%, or 12% SDS polyacrylamide gels, transferred to PVDF membrane, blocked with 5% (m/m) milk in TBST, and incubated with the primary antibody (anti-LC3B, Novus Biologicals; anti-SQSTM1, Novus Biologicals; anti-GAPDH, Sigma-Aldrich). After washing with TBST three times, the blots were probed with a secondary antibody (1:5000 dilution) for detection by chemiluminescence.
Confocal imanging analysis—RFP-GFP-LC3B or GFP-LC3B/RFP-Lamp1 expressing HeLa cells were treated with the indicated drugs for 6 h. Images were then captured under a Zeiss LSM710 confocal microscope with a Plan-Apochromot × 63/1.40 oil DIC objective. Images were processed and analyzed with Zeiss ZEN software (Zeiss, Göttingen, Germany).
4. Conclusions
Here we identified a number of novel vacuolin-1 analogues by combining virtual drug screening and chemical synthesis, and found that several of these analogues, e.g., VS6, A4, and A5, are as potent as vacuolin-1 in inhibiting autophagy (Table 1 and Table 2). Therefore, our study expanded the pool of useful autophagy inhibitors, which could be applied to dissect the mechanism of autophagy or endosomal trafficking and be used as potential therapeutic agents in autophagy related diseases, e.g., cancer. In addition, the SAR profile of vacuolin-1 analogues as autophagy inhibitors provided by the current study (Figure 6) could aid us to synthesize photoaffinity analogues or analogues with an alkyne tag for identifying its intracellular molecular targets.
Acknowledgments
This work was supported by Hong Kong Research Grant Council (RGC) grants (785213 and 17126614), CAS-Croucher Funding Scheme, Guangdong-Hong Kong joint innovation Research Scheme (#2016A050503010), and Shenzhen government research grant (JCYJ20160229165235739) to JY, National Natural Science Foundation of China (NSFC) (#31501116) to YL, and NSFC (#91213302 and #81673279) to LZ.
Supplementary Materials
Supplementary materials are available online.
Author Contributions
L.Z., J.B., C.C. and Y.L. designed the research; C.C. wrote the manuscript and prepared the data, Figures and Tables; Y.L. prepared the Figures; C.C. synthesized the compounds in the article; Y.L., H.M.S., J.G. and L.Z. did the measurement of inhibitory activities; S.Z. did the virtual screening; J.Y. and L.Z. commented and revised on the manuscript. All authors reviewed and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds A1–A6, B1–B5 and C1–C6 are available from the authors.
References
- 1.Yang Z., Klionsky D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reggiori F., Ungermann C. Autophagosome maturation and fusion. J. Mol. Biol. 2017;429:486–496. doi: 10.1016/j.jmb.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Zeng M., Zhou J.N. Roles of autophagy and mtor signaling in neuronal differentiation of mouse neuroblastoma cells. Cell. Signal. 2008;20:659–665. doi: 10.1016/j.cellsig.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Markaki M., Tavernarakis N. The role of autophagy in genetic pathways influencing ageing. Biogerontology. 2011;12:377–386. doi: 10.1007/s10522-011-9324-9. [DOI] [PubMed] [Google Scholar]
- 5.Heath R.J., Xavier R.J. Autophagy, immunity and human disease. Curr. Opin. Gastroenterol. 2009;25:512–520. doi: 10.1097/MOG.0b013e32833104f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J.M., Jung C.H., Seo M., Otto N.M., Grunwald D., Kim K.H., Moriarity B., Kim Y.M., Starker C., Nho R.S., et al. The ulk1 complex mediates mtorc1 signaling to the autophagy initiation machinery via binding and phosphorylating atg14. Autophagy. 2016;12:547–564. doi: 10.1080/15548627.2016.1140293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petibone D.M., Majeed W., Casciano D.A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017;37:23–37. doi: 10.1002/jat.3393. [DOI] [PubMed] [Google Scholar]
- 8.Maiese K. Targeting molecules to medicine with mtor, autophagy and neurodegenerative disorders. Br. J. Clin. Pharmacol. 2016;82:1245–1266. doi: 10.1111/bcp.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Meyer G.R., Martinet W. Autophagy in the cardiovascular system. Biochim. Biophys. Acta. 2009;1793:1485–1495. doi: 10.1016/j.bbamcr.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Kondo Y., Kanzawa T., Sawaya R., Kondo S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 11.Degenhardt K., Mathew R., Beaudoin B., Bray K., Anderson D., Chen G., Mukherjee C., Shi Y., Gelinas C., Fan Y., et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eskelinen E.L. The dual role of autophagy in cancer. Curr. Opin. Pharmacol. 2011;11:294–300. doi: 10.1016/j.coph.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Koustas E., Karamouzis M.V., Mihailidou C., Schizas D., Papavassiliou A.G. Co-targeting of egfr and autophagy signaling is an emerging treatment strategy in metastatic colorectal cancer. Cancer Lett. 2017;396:94–102. doi: 10.1016/j.canlet.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 14.Lu Z., Xu N., He B., Pan C., Lan Y., Zhou H., Liu X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017;36:43. doi: 10.1186/s13046-017-0512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Towers C.G., Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016;14:15–23. doi: 10.1016/j.ebiom.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura T., Takabatake Y., Takahashi A., Isaka Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y.T., Tan H.L., Shui G., Bauvy C., Huang Q., Wenk M.R., Ong C.N., Codogno P., Shen H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class i and iii phosphoinositide 3-kinase. J. Biol. Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson E., Leung E., Matuszek A.M., KrogsgaardLarsen N., Furkert D.P., Brimble M.A., Richardsona A., Reynisson J. Virtual screening for novel atg5–atg16 complex inhibitors for autophagy modulation. Med. Chem. Commun. 2015;6:239–246. doi: 10.1039/C4MD00420E. [DOI] [Google Scholar]
- 19.Feng Y., Yu S., Lasell T.K., Jadhav A.P., Macia E., Chardin P., Melancon P., Roth M., Mitchison T., Kirchhausen T. Exo1: A new chemical inhibitor of the exocytic pathway. Proc. Natl. Acad. Sci. USA. 2003;100:6469–6474. doi: 10.1073/pnas.0631766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Y., Dong S., Hao B., Li C., Zhu K., Guo W., Wang Q., Cheung K.H., Wong C.W., Wu W.T., et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating rab5a. Autophagy. 2014;10:1895–1905. doi: 10.4161/auto.32200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sano O., Kazetani K., Funata M., Fukuda Y., Matsui J., Iwata H. Vacuolin-1 inhibits autophagy by impairing lysosomal maturation via pikfyve inhibition. FEBS Lett. 2016;590:1576–1585. doi: 10.1002/1873-3468.12195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.