

Abstract
We describe here an atom efficient procedure to prepare selenol esters in good to excellent yields by reacting [(PhSe)2Zn] or [(PhSe)2Zn]TMEDA with acyl chlorides under “on water” conditions. The method is applicable to a series of aromatic and aliphatic acyl chlorides and tolerates the presence of other functionalities in the starting material.
Keywords: selenium, selenol esters, zinc, TMEDA, water
1. Introduction
Selenol esters are versatile tools in organic synthesis, once they can be easily converted to a sort of more complex molecules, acting as acyl-transfers, and in other important functional groups modifications [1]. Aromatic selenol esters, such as I, are stable liquid crystals with a large nematic mesophase range [2,3,4]. In contrast to their sulfur analogues, selenol esters only recently have attracted the attention to their pharmacological potential; the thiazolidine-4-carboselenoate II is a potent antioxidant [5], while the polyfunctionalized selenol esters III and IV presented high cytotoxic and antiproliferative activities against MCF-7 human cancer cells [6,7] (Figure 1).
Figure 1.

Examples of molecules bearing the selenol ester moiety.
The synthetic usefulness of selenol esters goes far beyond the transfer of acyl group [8], or its use as a protecting group for selenium compounds [9]. Selenol esters were explored in the total synthesis of several complex molecules [10,11,12,13,14], such as the marine alkaloid amphimedime [10], apparicine [11], (+)-geissoschizine [12], ciguatoxin CTX3C7d [13] and the peach moth (Carposia niponensis) pheromone [14]. More recently, selenol esters have gained even more importance, after the discovery that they can be used in native chemical ligation (NCL) reactions where the traditional thioester ligation chemistry is prohibitive [15,16].
As a consequence of the increasing demand for selenol esters, the number of methods to prepare this class of compounds has been increased along the years [1,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43] Among the strategies to prepare selenol esters there are the reactions of nucleophilic selenium reagents with an acyl group source, such as N-acyl benzotriazoles [17,18] activated carboxylic acids (using DCC [3] or PBu3 [4,5,19,20]), enol esters [21], anhydrides [22,23,24], esters [25], carbon monoxide [26,27], aldehydes [28,29] or acyl chlorides [2,7,30,31,32,33,34,35,36,37,38,39,40,41,42,43]. Other approaches to selenol esters involve the alkylation of selenocarboxylate anions with alkyl halides [6,44,45,46] or the acidic hydration of selenoalkynes [47,48]. The acyl substitution of acyl chlorides is by far the most explored method to access selenol esters, mainly because of its versatility, since the diversity of acyl chlorides that can be prepared is practically endless. The pivotal step in this reaction is the generation of the nucleophilic selenium species and a range of reagents have been used for this purpose, including Se°/ArMgBr [2], (NH2)2C=Se/Et3N [30], Se°/NaBH4 [6], InI/(PhSe)2 [31,39,40], Se°/LiAlH4 [32,37], (RSe)2/Zn/AlCl3 [33], Ph(NH2)C=Se [34], (Bu3Sn)2/(PhSe)2/hv [35,36], Se°/R2C=CZrCp2Cl [38], (RSe)2/Zn°/[bmim]PF6 [41], (PhSe)2/Hg°/dioxane [42] and (PhSe)2/SnCl2/CuBr2/[bmim]BF4 [43]. Despite these reaction systems afforded a range of selenol esters, they suffer from one or more of the following drawbacks: use of VOCs as solvent, strong bases, strong and moisture sensible reducing agents, expensive reagents and low atom-economy.
Significant progress towards the greenness of the synthesis of selenol esters has been recently described [49,50]. Braga and co-workers described the solvent-free, microwave accelerated reaction of diorganyl diselenides with acyl chlorides; good yields of selenol esters were obtained after irradiation at 80 °C for only 2 min [49]. In the same year, some of us described the use of the bench stable nucleophilic species, PhSeZnX (X=Br, Cl), in the synthesis of a range of selenol esters in good yields at room temperature and using water as medium [50]. It was observed that the reaction was accelerated when it was performed under “on water” conditions and, in addition, the water was reused for subsequent cycles of reactions.
Nine years ago, we introduced a simple method to prepare in situ nucleophilic sulfur and selenium reagents by reducing dichalcogenides with elemental zinc in an acidic biphasic system [51]. This protocol has been more recently used by us and others to effect a series of selenylation reactions involving nucleophilic substitutions, ring opening and hydrochalcogenations [51,52,53] and it was adopted by Flemer in the synthesis of peptides [54,55].
The actual reactive specie in this protocol was supposed to be a zinc bis-selenate [(PhSe)2Zn] probably in equilibrium with the corresponding selenol. Considering that [(PhSe)2Zn] is characterized by a higher atom economy respect to the PhSeZn-halides in the insertion of PhSe groups, the possibility to obtain it in a bench stable form is desirable. It could improve the versatility of its synthetic application (avoiding the strong acidic conditions of the biphasic system), controlling or preventing undesired side reactions like those observed when it was prepared and used in situ in the presence of THF [56].
2. Results
To better understand and prevent the side reaction observed in the presence of THF [56], DFT calculations were performed comparing [(PhCh)2Zn] (Ch=S, Se) with the previously reported PhChZnX (Ch=S, Se; X=Cl, Br) [57].
DFT-optimized geometries of the (PhCh)2Zn species (Ch=S, Se) show a nearly linear coordination at the Zn center. This geometry has been observed in the gas phase for ZnH2 and ZnCl2, [58] although several examples are reported of structurally characterized zinc(II) compounds featuring discrete linear E–Zn–E moieties with E=O [58,59,60], S [61,62], or Se [63]. On passing from Ch=S to Ch=Se, the natural charge polarization of the Ch–Zn bond decreases (1.233 and 1.093 e for Ch=S and Se, respectively), suggesting a higher reactivity of (PhS)2Zn in all conditions as compared to (PhSe)2Zn. The solvated species (PhCh)2Zn·2solv (solv=THF, H2O) were also successfully optimized. For both solvents, solvated species show a distortion of the Ch–Zn–Ch moiety towards the usually encountered tetrahedral coordination of the Zn center. A Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis allows evaluating the interaction energies between the Zn center and each solvent molecule by about 50, 58, 48, and 55 kcal·mol−1 for (PhS)2Zn·2THF, (PhS)2Zn·2H2O, (PhSe)2Zn·2THF, and (PhSe)2Zn·2H2O, respectively. Such interactions arise from the electron density donation from the oxygen LP of the solvent units to the antibonding empty Zn atomic orbitals (Figure 2) and, to a minor extent, to the Zn-Ch antibonding NBOs, and result in an overall electron transfer of 0.095, 0.105, 0.133, and 0.104 e for (PhS)2Zn·2THF, (PhS)2Zn·2H2O, (PhSe)2Zn·2THF, and (PhSe)2Zn·2H2O, respectively. Notably, the interactions between (PhCh)2Zn species and the solvent units are remarkably stronger than those calculated between PhChZnX (Ch=S, Se; X=Cl, Br and I) and the same solvents, which were calculated in about 18 kcal·mol−1 at the same level of theory.
Figure 2.

Drawings of the NBOs involved in the second order interaction between (PhSe)2Zn and the THF fragments in the compound (PhSe)2Zn·2THF at the DFT-optimized geometry (energy difference 0.7465 a.u.; interaction energy 25.77 kcal·mol−1). Left: filled NBO LP localized on the oxygen donor atom of one THF solvent fragment (NBO #127, 34.45% s and 65.54% p character). Right: virtual NBO LP* localized on the Zn center (NBO #124, 9.51% s and 90.35% p character). Cutoff value: 0.1 |e|. Hydrogen atoms have been omitted for clarity. (Grey = Carbon; Red = Oxygen; Yellow = Selenium; Violet = Zinc).
All (PhCh)2Zn species and the corresponding solvated forms show their HOMOs largely localized on the negatively charged Ch atoms (QS = −0.315, −0.377, and −0.367 |e| for (PhSe)2Zn, (PhCh)2Zn·2THF, and (PhCh)2Zn·2H2O; QSe = −0.236, −0.305, and −0.274 |e| for (PhSe)2Zn, (PhCh)2Zn·2THF, and (PhCh)2Zn·2H2O, respectively).
The LUMOs of (PhCh)2Zn species are antibonding MOs displaying a remarkable contribution from the zinc atomic orbitals. About solvated species, it is worth noting that both (PhS)2Zn·2THF and (PhSe)2Zn·2H2O feature empty molecular orbitals with large contributions from the virtual molecular orbitals of the solvent molecules, which are therefore sensibly stabilized. In particular, the LUMO+4 MO calculated for THF shows an antibonding character with respect to the C–O bonds Kohn-Sham (KS) eigenvalue ε = +0.1389 Hartree). This orbital contributes to the LUMO+14 and LUMO+15 of (PhS)2Zn·2THF and (PhSe)2Zn·2THF, respectively, which are stabilized in energy by 0.887 eV (ε = +0.1063 Hartree for both virtual MOs). The strength of the interaction between THF and (PhCh)2Zn accompanied by a low-lying MO with remarkable C–O antibonding character partly localized on the coordinated THF units may account for the ring opening reactions of coordinated solvents observed experimentally [56].
Therefore, we investigated the possibility to prepare an isolable zinc bis-selenate to be used in the synthesis of chalcogenol esters avoiding the presence of THF during the reaction with the acyl chlorides. Different conditions for the oxidative insertion of the zinc in the Se-Se bond starting from the commercially available diphenyl diselenide were explored (Table 1). The reduction of diselenides in organic solvents can be unequivocally evidenced by the discoloration of the originally yellow solution and it was observed that both the presence of catalytic amount of TFA (10 mol %) and the THF (or a 1:1 THF/water mixture) are mandatory for the reduction at reflux (Table 1, entries 4 and 5 respectively). Interestingly, the insertion did not occur in refluxing THF for 2 h (Table 1, entry 2) neither in water suspension at the same temperature (Table 1, entry 1), as well as in the presence of TFA (10 mol %) (Table 1, entry 3).
Table 1.
Synthesis of compound 1.
| Entry | Solvent | Additive | Time | T (°C) | Discoloration |
|---|---|---|---|---|---|
| 1 | H2O | none | 2 h | 70 | No |
| 2 | THF | none | 2 h | reflux | No |
| 3 | H2O | TFA 10 mol % | 1 h | 70 | No |
| 4 | THF | TFA 10 mol % | 20 min | reflux | Yes |
| 5 | H2O/THF | TFA 10 mol % | 20 min | 70 | Yes |
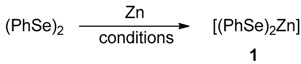
Using the conditions depicted in Table 1, entry 4, after the discoloration, the THF was removed under-reduced pressure, giving a whitish amorphous and, unfortunately, relatively unstable solid, which was used without further purification. The supposed formation of a polymeric form of the reagent 1 was confirmed by the presence of a broad signal at −41 ppm in the 77Se NMR. Similarly, starting from diphenyl disulfide, the zinc bis-thiolate 2 was formed in situ (Figure 3). In this case, the starting material is colorless and the reduction time was arbitrarily chosen according to that of the reduction of diphenyl diselenide to afford 1. A monomeric and more stable zinc selenate (3) can be prepared according to the literature using the bidentate TMEDA(N,N,N1,N1-tetramethyl-ethylendiamine) for the stabilization of the complex 3, with TMEDA in the place of THF [64].
Figure 3.

Reagents investigated in the present work.
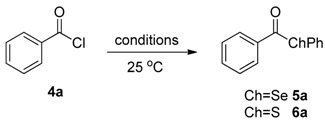
The reactivity of 1,3 and 2 with benzoyl chloride (4a) for the formation of the selenol ester 5a or thiol ester 6a, was evaluated in “on water” conditions (Table 2, entries 10–12) and these results were compared with some data recently reported using other zinc selenates, as well as different reaction conditions (Table 2, entries 1–3, 5–9). The bis-phenylselenate (1) showed an interesting reactivity affording, after 30 min, the formation of 5a in 83% yield (Table 2, entry 10). This result confirms that zinc selenates, as previously reported, are efficient selenenylating reagents for the on-water acyl substitution. In addition, the use of water as medium for the reaction with the acyl chloride prevents the undesired ring opening of THF [56], affording diphenyl diselenide and benzoic acid as side organic products of residual decomposition, and TMEDA when 3 was used. Reasonably the zinc derivatives are removed during the workup due to their water solubility. We also observed that the reactivity of 1 in THF depends on the acid used as catalyst in the oxidative zinc insertion into the Se-Se bond (Table 2, entries 3 and 4) and that the best results were obtained using TFA. When the reaction was performed using the TMEDA-stabilized zinc selenate 3, we observed only a decrease of the reactivity, obtaining a good conversion of 4a into 5a (71%) in 30 min (Table 2, entry 11). Interestingly, the sulfur-containing reagent 2 afforded only 50% of conversion (Table 2, entry 12) and this is consistent with DFT calculations, that evidenced a higher reactivity of the sulfur reagents. This aspect, probably, represents a problem for the stability of the zinc thiolate during the solvent evaporation or in the presence of water, leading to a faster decomposition of the reactant.
Table 2.
Zinc chalcogenates 1–3 in the preparation of chalcogenol esters.
| Entry | Reagent | Medium | Time (h) | Yield (%) a | ae (%) b | Reference |
|---|---|---|---|---|---|---|
| 1 | PhSeZnCl | THF | 24 | 25 | 66 | [50] |
| 2 | PhSeZnBr | THF | 24 | 30 | 60 | [50] |
| 3 | [PhSeZnSePh] 1 | THF | 3 | 32 c | 79.6 | [56] |
| 4 | [PhSeZnSePh] 1 | THF | 3 | 40 d | 79.6 | – |
| 5 | PhSZnBr | THF | 24 | 86 | 54.6 | [57] |
| 6 | [PhSeZnSePh/PhSeH] | HClacq/Et2O | 4 | 38 | – | [56] |
| 7 | PhSeZnCl | H2O | 3 | 60 | 66 | [50] |
| 8 | PhSeZnBr | H2O | 3 | 70 | 60 | [50] |
| 9 | PhSZnBr | H2O | 3 | 65 | 54.6 | [57] |
| 10 | [PhSeZnSePh] 1 | H2O | 0.5 | 83 | 79.6 | – |
| 11 | [PhSeZnSePh]TMEDA 3 | H2O | 0.5 | 66 | 77 | – |
| 12 | [PhSZnSPh] 2 | H2O | 0.5 | 50 | 76 | – |
a Conversion estimated by NMR; b Atom economy = m.w. of final product × 100/Σ (m.w. reactants); c 1 was prepared in the presence of 10 mol % of TfOH. Compound 5a was formed together with 34% PhC(O)O(CH2)4SePh and 28% PhC(O)O(CH2)4 O(CH2)4SePh; d 1 was prepared in the presence of 10 mol % of TFA. Compound 5a was formed together with 27% PhC(O)O(CH2)4SePh and 5% PhC(O)O(CH2)4 Cl.
From the results reported in Table 2, it appears clear that zinc-bis-chalcogenates are considerably more atom-efficient respect other similar reagents (compare entries 10,11 vs. 7,8; and 12 vs. 9) and this is an important parameter to be evaluated in terms of “greenness” of a synthetic process.
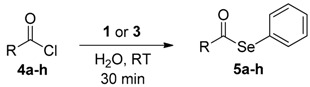
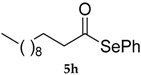
The best conditions optimized for 1 and 3 were applied to a series of acyl chlorides 4a–h, affording the corresponding selenol esters 5a–h; the conversion rate as well as the isolate yields are reported in Table 3. All the final products were fully characterized by GC-MS, 1H- and 13C-NMR after purification by flash chromatography (For 1H- and 13C-NMR of the purified compounds see Supplementary Materials).
Table 3.
Synthesis of selenol esters 5.
| Entry | Substrate 4 | Product 5 | Conv, % of 4 in 5 (Yield, %) of 5 Using 1 |
Conv, % of 4 in 5 Using 3 |
|---|---|---|---|---|
| 1 |  |
 |
84 (80) | 66 |
| 2 | 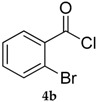 |
 |
81 (53) | 80 |
| 3 | 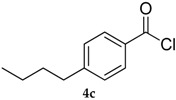 |
 |
71 (57) | 40 |
| 4 | 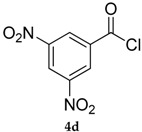 |
 |
92 (90) | 80 |
| 5 | 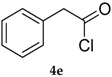 |
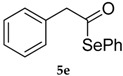 |
83 (64) | 62 |
| 6 | 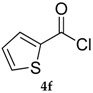 |
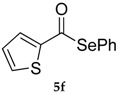 |
80 (65) | 35 |
| 7 | 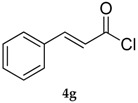 |
 |
8 | 76 |
| 8 | 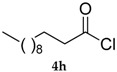 |
 |
75 (69) | 80 |
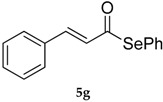
Results collected in Table 3 indicate that the reaction works well with various acyl chlorides (4a–h), including aromatic and aliphatic ones, with the only exception of cinnamic derivative 4g, that afforded only in traces of the corresponding selenol ester 5g (Table 3, entry 7). In contrast to the previously described biphasic system [56], this approach tolerates functional groups sensitive to reduction, allowing the preparation of selenol ester 4d (R=3,5(NO2)2C6H3) in excellent yields (Table 3, entry 4). Noteworthily, it can be observed that acyl chlorides bearing electron-withdrawing groups gave better yields, probably because of the more pronounced electrophilic character of the carboxylic carbon.
In order to clarify the nature of the species that are involved in the reaction mechanism leading to 5a, the possible 1:1 initial intermediate model species formed by the interaction between (PhSe)2Zn 1 and PhCOCl 4a was optimized. The only optimized geometry of the adducts shows PhCOCl interacting with the zinc atom of (PhSe)2Zn through its carbonyl group, thus distorting the Ch–Zn–Ch group towards a roughly trigonal geometry. A natural population analysis shows that the interaction results in a charge-transfer (CT) from PhCOCl to (PhSe)2Zn, whose LUMO is partly localized on the positively charged metal ion (QZn = +0.857). The CT-interaction results in an increase in the positive charge on the acyl carbon and the polarization of the C=O bond (|ΔQCO| = 1.083, 1.177 for PhCOCl; and PhSeZnSePh·PhCOCl, respectively). Therefore, the activated carbonyl group in PhCOCl might be more suitable to undergo a nucleophilic attack by the chalcogen donor of a second molecule of (PhCh)2Zn.
The TMEDA-stabilized reagent 3 was investigated using the same set of acyl chlorides 4a–h and, generally, it confirmed a slightly/moderate reduced reactivity with a superimposable trend respect to the nature of the substrate. The only exception was observed for acyl chloride 4g, that using TMEDA afforded selenol ester 5g in 80% yield (Table 3, entry 7).
Considering that the complex 3 is reported in literature as particularly stable and it is sufficiently soluble in several organic solvents, we envisioned the possibility to perform a ”one-pot” synthesis and chromatographic purification of the selenol ester 5a following the setup reported in Figure 4. A pump fluxes petroleum ether through a Flash Pack Jones Chromatography apparatus silica gel column packed with 5 g of silica flash (40–63 µm) and, at the head of the column, was poured the reagent 3 (0.25 mmol), mixed with 500 mg of the same silica. The column was conditioned with the solvent and the substrate (0.5 mmol), dissolved in DCM, was injected. After the column, the eluent was fractioned and the fractions were isolated and characterized by NMR. The selenol ester 5a was obtained as pure compound (separated from both, diphenyl diselenide and benzoic acid) in a yield comparable to that observed in the bench condition (60%). This protocol allowed to bypass the workup of the reaction saving a considerable amount of organic solvents (15 mL of EtOAc), brine solution (20 mL) and Na2SO4, reducing the production of wastes.
Figure 4.

One-pot synthesis and chromatographic purification of the selenol ester 5a.
3. Materials and Methods
Reactions were conducted in a round bottom flask and were stirred with Teflon-coated magnetic stirring bars at 800 rpm. Solvents and reagents were used as received unless otherwise noted. Analytical thin-layer chromatography (TLC) was performed on silica gel 60 F254 precoated aluminum foil sheets and visualized by UV irradiation or by KMnO4 staining. Kieselgel 60 (70–230 mesh) silica gel was used for column chromatography. NMR experiments were conducted at 25 °C with a DPX 200 spectrometer (Bruker, Faellanden, Switzerland).) operating at 200 MHz for 1H, 50.31 MHz for 13C experiments or with a Bruker DRX spectrometer (Bruker, Faellanden, Switzerland) operating at 400 MHz for 1H, 100.62 MHz for 13C and 76.31 MHz for 77Se. Chemical shifts (δ) are reported in parts per million (ppm), relative to TMS (δ = 0.0 ppm) and the residual solvent peak of CDCl3 (δ = 7.26 and 77.00 ppm in 1H- and 13C-NMR, respectively) and PhSeSePh δ = 464 ppm in 77Se. Data are reported as chemical shift (multiplicity, coupling constants where applicable, number of hydrogen atoms, and assignment where possible). Abbreviations are: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), dd (doublet of doublet), dt (doublet of triplet), tt (triplet of triplet), m (multiplet), br. s (broad signal). Coupling constants (J) are quoted in Hertz (Hz) to the nearest 0.1 Hz. GC-MS analyses were carried out with an HP-6890 gas chromatography (dimethyl silicone column, 12.5 m) equipped with an HP-5973 mass-selective detector (Hewlett-Packard, Waldbronn, Germany). Acyl chlorides used for obtaining the selenol esters 5a, 5e, 5f and 5h are commercially available; acyl chlorides used for obtaining the selenol esters 5b, 5c, 5d and 5g were synthesized according to the literature [65]. Density Functional Theory (DFT) calculations were performed with the commercial suite of software Gaussian09 [66]. All calculations were carried out with the mPW1PW hybrid functional [67] and the full-electron Ahlrichs double-ζ basis sets with polarization functions (pVDZ) for all atomic species [67]. NBO populations [68,69,70] and Wiberg bond indices [71] were calculated at the optimized geometries, which were verified by harmonic frequency calculations. The results of the calculations were examined with GaussView 5 [72] and Molden 5.3 [73] programs.
General Procedure for the Synthesis of Selenol Esters 5a–h
To a water suspension of the zinc complexes 1 or 3 (0.5 mmol, 6 mL of H2O), acyl chloride 4 (1 mmol) was added at room temperature and under stirring. After 30 min, the reaction mixture was extracted with ethyl acetate (3 × 10 mL), washed with brine (2 × 20 mL), dried with Na2SO4 and concentrated under vacuum to obtain a residue that was purified by flash chromatography.
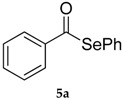
Se-Phenyl benzoselenoate (5a) [40] was purified eluting with 20% DCM in petroleum ether. Yellow solid. m.p.: 39°–40 °C (Lit.: [38] 37°–38 °C). 1H-NMR (CDCl3, ppm) δ: 7.96–7.92 (m, 2H, H-Ar), 7.63–7.42 (m, 8H, H-Ar) ppm. 13C-NMR (CDCl3, ppm) δ: 193.5, 138.5, 136.4, 133.9, 129.4, 129.1, 128.9, 127.4, 125.8 ppm. CG-MS: m/z (%) = 262 (1) [M+], 157 (5), 105 (100), 77 (50), 51 (14).
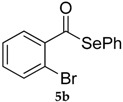
Se-Phenyl 2-bromobenzoselenoate (5b) [74] was purified eluting with 5% EtOAc in petroleum ether. Yellow oil. 1H-NMR (CDCl3, ppm) δ: 7.72–7.6 (m, 4H, H-Ar), 7.45–7.34 (m, 5H, H-Ar) ppm. 13C-NMR (CDCl3, ppm) δ: 194.4, 140.6, 135.8, 134.3, 132.6, 129.5, 129.2, 128.8, 127.3, 126.6, 118.0 ppm.77Se-NMR (CDCl3, ppm) δ: 662.1 ppm. CG-MS m/z (%) = 340 (1) [M+], 232 (3), 183 (100), 157 (54), 76 (16), 50 (9).
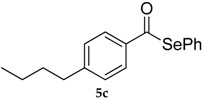
Se-Phenyl 4-butylbenzoselenoate (5c) [50] was purified eluting with 20% DCM in petroleum ether. Yellow oil. 1H-NMR (CDCl3, ppm) δ: 7.85 (d, J = 8.1 Hz, 2H, H-Ar), 7.61–7.57 (m, 2H, H-Ar), 7.44–7.41 (m, 3H, H-Ar), 7.31–7.26 (m, 2H, H-Ar), 2.67 (t, J = 7,8 Hz, 2H, CH2), 1.61 (quin, J = 8.15 Hz, 2H, CH2), 1.36 (sex, J = 7.6 Hz, 2H, CH2), 0.95 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C-NMR (CDCl3, ppm) δ: 192.7, 149.8, 136.4, 136.2, 129.3, 129.0, 127.5, 126.0, 35.8, 33.1, 22.3, 13.9 ppm. 77Se-NMR (CDCl3, ppm) δ: 661.0 ppm. CG-MS m/z (%) = 318 [M+], 161 (100), 91 (30).
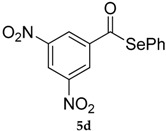
Se-Phenyl-3,5-dinitrobenzoselenoate (5d) [50] was purified eluting with 5% EtOAc in petroleum ether. Yellow solid. m.p.: 148–150 °C (Lit.: [48] 148°–150°). 1H-NMR (CDCl3, ppm) δ: 9.28 (t, J = 2.05 Hz, 1H, H-Ar), 9.04 (d, J = 2.06 Hz, 2H, H-Ar), 7.7–7.4 (m, 5H, H-Ar) ppm. 13C-NMR (CDCl3, ppm) δ: 190.4, 148.9, 141.5, 136.1, 130.07, 129.9, 126,7, 124.1, 122.6 ppm. 77Se-NMR (CDCl3, ppm) δ: 662.3 ppm.
Se-Phenyl 2-phenylethaneselenoate (5e) [75] was purified eluting with 20% DCM in petroleum ether. Yellow solid. m.p.: 41–43 °C (Lit.: [59] 41°–43 °C). 1H-NMR (CDCl3, ppm) δ: 7.45–7.35 (m, 2H, H-Ar), 7.34–7.26 (m, 8H, H-Ar), 3.88 (s, 2H, CH2) ppm. 13C-NMR (CDCl3, ppm) δ: 198.9, 135.8, 132.6, 130.1, 129.3, 128.9, 128.8, 127.8, 126.6, 53.6 ppm. CG-MS m/z (%) = 276 [M+], 157 (22), 119 (26), 91 (100), 65 (26).
Se-Phenyl thiophene-2-carboselenoate (5f) [22] was purified eluting with 5% EtOAc in petroleum ether. Yellow oil. 1H-NMR (CDCl3, ppm) δ: 7.88 (dd, J = 1.2, 3.9 Hz, 1H, H-Ar), 7.71 (dd, J = 1.2, 4.96, Hz, 1H, H-Ar), 7.63–7.58 (m, 2H, H-Ar), 7.44–7.41 (m, 3H, H-Ar), 7.17 (dd, J = 3.9, 4.96 Hz, 1H, H-Ar) ppm. 13C-NMR (CDCl3, ppm) δ: 183.6, 143.1, 136.3, 133.7, 132.0, 129.4, 129.2, 128.0, 125.5 ppm. CG-MS m/z (%) = 268 (1) [M+], 157 (16), 111 (100), 83 (20).
(E)-Se-Phenyl 3-phenylprop-2-eneselenoate (5g) [65] was purified eluting with 5% EtOAc in petroleum ether. Yellow solid. m.p.: 79–80 °C (Lit.: [57] 81°–82 °C).1H-NMR (CDCl3, ppm) δ: 7.58–7.54 (m, 5H, H-Ar), 7.43–7.40 (m, 6H, H-Ar), 6.78 (d, J = 15.0 Hz, 1H, CH) ppm. 13C-NMR (CDCl3, ppm) δ: 190.8, 141.1, 135.9, 133.9, 130.9, 129.4, 129.1, 129.0, 128.6, 128.1, 126.3 ppm. 77Se-NMR (CDCl3, ppm) δ: 663.6 ppm. CG-MS m/z (%) = 288 (1) [M+], 157 (14), 131 (100), 103 (55), 77 (36).
Se-Phenyl dodecaneselenoate (5h) [76] was purified eluting with 20% DCM in petroleum ether. Yellow oil. 1H-NMR (CDCl3, ppm) δ: 7.55–7.53 (m, 2H, H-Ar), 7.42–7.39 (m, 3H, H-Ar), 2.73 (t, J = 7.5 Hz, 2H, CH2C(O)), 1.73 (quin, J = 7.4 Hz; 2H, CH2), 1.4–1.3 (m, 16H, CH2), 0.94–0.90 (t, J = 6.5 Hz, 3H, CH3) ppm. 13C-NMR (CDCl3, ppm) δ: 200.3, 135.7, 129.2, 128.7, 126.5, 47.5, 31.8, 29.5, 29.3, 29.26, 29.17, 28.8, 25.3, 22.6, 14.0 ppm. CG-MS m/z (%) = 340 (2) [M+], 183 (100), 157 (34), 109 (20), 85 (27), 71 (30), 57 (43).
4. Conclusions
In conclusion, we report here that the side reactivity of zinc bis-selenates with the solvent (THF) during the reaction with acyl chlorides can be rationalized by DFT calculations demonstrating that the interactions between (PhSe)2Zn species and the solvent units are remarkably stronger than those calculated between PhSeZnCl and PhSeZnBr and THF. Even if water seems to be equally able to coordinate the Zn, it was not possible to perform the oxidative zinc insertion directly in “on water conditions”. Nevertheless, after the removal of THF, the solid product obtained by the reduction of PhSeSePh with Zn in the presence of a catalytic amount of TFA (formally [(PhSe)2Zn]) can be efficiently used for the synthesis of selenol esters starting from the corresponding acyl chlorides and using water as reaction medium. Furthermore, the complex [(PhSe)2Zn]-TMEDA, prepared according to literature, showed a similar reactivity in water having the additional advantage to be bench stable. To the best of our knowledge, this is the first example that report the use of [(PhSe)2Zn]-TMEDA as nucleophilic reagent and the possibility to use it in a one pot reaction-purification process is an intriguing aspect that is currently under deep investigation by some of us. It is important to underline that the reagents reported in the present work (1, 2, and 3) are more atom-efficient not only relative to the previously reported zinc-halo-selenates, but also among most of the alternative known methods for the synthesis of selenol esters. During the preparation of this manuscript a further synthesis of these compounds appeared starting from anhydrides, evidencing the current interest in this class of derivatives [24]. Nevertheless, in our opinion, both in terms of atom economy and general applicability, it can be claimed that the use of acyl chloride results largely preferable if compared to the anhydrides.
Acknowledgments
Financial support from the Dipartimento di Scienze Farmaceutiche, Università degli Studi di Perugia “Fondo per il sostegno della Ricerca di Base 2014—Composti Organici ed Ibridi Inorgano-Organici del Selenio nella Sintesi Eco-Compatibile di Molecole Bioattive”. L.S. was fellow with a Regione Umbria, project “POR Umbria 2007–2013,”. Eder Joao Lenardao was Bolsista CAPES—Processo no. BEX 6391/14-1 and Mrs. Vargas was in CAPES Sandwich program with a Doctorate scholarship (PDSE process 99999.001342/2014-02). This research was undertaken as part of the scientific activity of the international multidisciplinary “SeS Redox and Catalysis” network. M.A. and V.L. thank the Università degli Studi di Cagliari for financial support.
Supplementary Materials
Copies of the 1H and 13C-NMR spectra are available online. Optimized geometries in orthogonal Cartesian format, Mulliken and Natural charges, and thermochemical data for all investigated compounds are available from M.A. upon request.
Author Contributions
C.S. and G.P. conceived and designed the experiments; J.P.V. and B.M. performed the experiments; C.S., L.S. and E.J.L. analyzed the data; V.L. and M.A. designed and executed DFT calculations; C.S., L.S., E.J.L., and V.L. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest and the founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Footnotes
Sample Availability: Original samples (due to their instability) are not available but can be provided upon request.
References
- 1.Fujiwara S., Kambe N. Thio-, Seleno-, and Telluro-Carboxylic Acid Esters. In: Kato S., editor. Chalcogenocarboxylic Acid Derivatives. Springer; Berlin/Heidelberg, Germany: 2005. pp. 87–140. [Google Scholar]
- 2.Pakzad B., Praefcke K., Simon H. 5H-[1]Benzoselenino[2,3-b]pyridine—A Novel Heterocyclic Ring System. Angew. Chem. Int. Ed. Eng. 1977;16:319–320. doi: 10.1002/anie.197703191. [DOI] [Google Scholar]
- 3.Rampon D.S., Rodembusch F.S., Gonçalves P.F.B., Lourega R.V., Merlo A.A., Schneider P.H. An evaluation of the chalcogen atom effect on the mesomorphic and electronic properties in a new homologous series of chalcogeno esters. J. Braz. Chem. Soc. 2010;21:2100–2107. doi: 10.1590/S0103-50532010001100011. [DOI] [Google Scholar]
- 4.Rampon D.S., Rodembusch F.S., Schneider J.M.F.M., Bechtold I.H., Gonçalves P.F.B., Merlo A.A., Schneider P.H. Novel selenoesters fluorescent liquid crystalline exhibiting a rich phase polymorphism. J. Mater. Chem. 2010;20:715–722. doi: 10.1039/B917366H. [DOI] [Google Scholar]
- 5.Victoria F.N., Martinez D.M., Castro M., Casaril A.M., Alves D., Lenardão E.J., Salles H.D., Schneider P.H., Savegnago L. Antioxidant properties of (R)-Se-aryl thiazolidine-4-carboselenoate. Chem. Biol. Interact. 2013;205:100–107. doi: 10.1016/j.cbi.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 6.Sanmartín C., Plano D., Domínguez E., Font M., Calvo A., Prior C., Encío I., Palop J.A. Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents. Molecules. 2009;14:3313–3338. doi: 10.3390/molecules14093313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domínguez-Álvarez E., Plano D., Font M., Calvo A., Prior C., Jacob C., Palop J.A., Sanmartín C. Synthesis and antiproliferative activity of novel selenoester derivatives. Eur. J. Med. Chem. 2014;73:153–166. doi: 10.1016/j.ejmech.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 8.Makriyannis A., Guenther W.H.H., Mautner H.G. Selenol esters as specific reagents of the acylation of thiol groups. J. Am. Chem. Soc. 1973;95:8403–8406. doi: 10.1021/ja00806a033. [DOI] [Google Scholar]
- 9.Reinerth W.A., Tour J.M. Protecting Groups for Organoselenium Compounds. J. Org. Chem. 1998;63:2397–2400. doi: 10.1021/jo972144a. [DOI] [Google Scholar]
- 10.Subramanyam C., Noguchi M., Weinreb S.M. An approach to amphimedine and related marine alkaloids utilizing an intramolecular Kondrat’eva pyridine synthesis. J. Org. Chem. 1989;54:5580–5585. doi: 10.1021/jo00284a035. [DOI] [Google Scholar]
- 11.Bennasar M.-L., Zulaica E., Solé D., Roca T., García-Díaz D., Alonso S. Total Synthesis of the Bridged Indole Alkaloid Apparicine. J. Org. Chem. 2009;74:8359–8368. doi: 10.1021/jo901986v. [DOI] [PubMed] [Google Scholar]
- 12.Martin S.F., Chen K.X., Eary C.T. An Enantioselective Total Synthesis of (+)-Geissoschizine. Org. Lett. 1999;1:79–82. doi: 10.1021/ol990554a. [DOI] [PubMed] [Google Scholar]
- 13.Inoue M., Yamashita S., Ishihara Y., Hirama M. Two Convergent Routes to the Left-Wing Fragment of Ciguatoxin CTX3C Using O, S-Acetals As Key Intermediates. Org. Lett. 2006;8:5805–5808. doi: 10.1021/ol062350h. [DOI] [PubMed] [Google Scholar]
- 14.Sviridov A.F., Ermolenko M.S., Yashunskii D.V., Kochetkov N.K. Selenoesters in organic synthesis. 2. New synthesis of saturated and α,β-unsaturated ketones and the synthesis of the Peach moth pheromone (Carposia niponensis) Bull. Acad. Sci. USSR Div. Chem. Sci. 1985;34:1514–1518. doi: 10.1007/BF00950160. [DOI] [Google Scholar]
- 15.Durek T., Alewood P.F. Preformed Selenoesters Enable Rapid Native Chemical Ligation at Intractable Sites. Angew. Chem. Int. Ed. Eng. 2011;50:12042–12045. doi: 10.1002/anie.201105512. [DOI] [PubMed] [Google Scholar]
- 16.Ghassemian A., Vila-Farrés X., Alewood P.F., Durek T. Solid phase synthesis of peptide-selenoesters. Bioorg. Med. Chem. 2013;21:3473–3478. doi: 10.1016/j.bmc.2013.03.076. [DOI] [PubMed] [Google Scholar]
- 17.Gais H.-J. Synthesis of Thiol and Selenol Esters from Carboxylic Acids and Thiols or Selenols, Respectively. Angew. Chem. Int. Ed. Eng. 1977;16:244–246. doi: 10.1002/anie.197702441. [DOI] [Google Scholar]
- 18.Lin S.-M., Zhang J.-L., Chen J.-X., Gao W.-X., Ding J.-C., Su W.-K., Wu H.-Y. An approach to the synthesis of thioesters and selenoesters promoted by Rongalite®. J. Braz. Chem. Soc. 2010;21:1616–1620. doi: 10.1590/S0103-50532010000900003. [DOI] [Google Scholar]
- 19.Grieco P.A., Jaw J.Y., Claremon D.A., Nicolaou K.C. N-Phenylselenophthalimide. A useful reagent for the facile transformation of (1) carboxylic acids into either selenol esters or amides and (2) alcohols into alkyl phenyl selenides. J. Org. Chem. 1981;46:1215–1217. doi: 10.1021/jo00319a037. [DOI] [Google Scholar]
- 20.Grieco P.A., Yokoyama Y., Williams E. Aryl selenocyanates and aryl thiocyanates: Reagents for the preparation of activated esters. J. Org. Chem. 1978;43:1283–1285. doi: 10.1021/jo00400a070. [DOI] [Google Scholar]
- 21.Gais H.-J., Lied T. A New Method for Selective Activation of Amino, Hydroxy, and Mercapto Carboxylic Acids at the Carboxyl Group: Preparation of Thiol and Selenol Esters. Angew. Chem. Int. Ed. Eng. 1978;17:267–268. doi: 10.1002/anie.197802671. [DOI] [Google Scholar]
- 22.Dan W., Deng H., Chen J., Liu M., Ding J., Wu H. A new odorless one-pot synthesis of thioesters and selenoesters promoted by Rongalite®. Tetrahedron. 2010;66:7384–7388. doi: 10.1016/j.tet.2010.07.023. [DOI] [Google Scholar]
- 23.Perin G., Silveira M.B., Barcellos A.M., Araujo D.R., Jacob R.G., Barcellos T., Lenardão E.J. Rongalite®/PEG-400 as reducing system in the synthesis of new glycerol-derived selenol esters using anhydrides and bis-(2,2-dimethyl-1,3-dioxolanylmethyl)diselenide as substrates. Arkivoc. 2016;2017:138. [Google Scholar]
- 24.Temperini A., Piazzolla F., Minuti L., Curini M., Siciliano C. General, Mild, and Metal-Free Synthesis of Phenyl Selenoesters from Anhydrides and Their Use in Peptide Synthesis. J. Org. Chem. 2017;82:4588–4603. doi: 10.1021/acs.joc.7b00173. [DOI] [PubMed] [Google Scholar]
- 25.Sviridov A.F., Ermolenko M.S., Yashunskii D.V., Kochetkov N.K. Selenoesters in organic synthesis. 1. Conversion of mixed carboxylic acid esters to selenoesters. Bull. Acad. Sci. USSR Div. Chem. Sci. 1985;34:1509–1513. doi: 10.1007/BF00950159. [DOI] [Google Scholar]
- 26.Maeda H., Fujiwara S., Shin-Ike T., Kambe N., Sonoda N. Carbonylation of Acidic Hydrocarbons with Selenium and Carbon Monoxide. A Novel Method for Synthesis of Selenol Esters. J. Am. Chem. Soc. 1996;118:8160–8161. doi: 10.1021/ja9616995. [DOI] [Google Scholar]
- 27.Nishiyama Y., Tokunaga K., Kawamatsu H., Sonoda N. Synthesis of selenol esters: Palladium-catalyzed coupling of phenyl tributylstannyl selenide with aryl iodides and carbon monoxide. Tetrahedron Lett. 2002;43:1507–1509. doi: 10.1016/S0040-4039(02)00048-5. [DOI] [Google Scholar]
- 28.Inoue T., Takeda T., Kambe N., Ogawa A., Ryu I., Sonoda N. Synthesis of Thiol, Selenol, and Tellurol Esters from Aldehydes by the Reaction with iBu2AlYR (Y=S, Se, Te) J. Org. Chem. 1994;59:5824–5827. doi: 10.1021/jo00098a053. [DOI] [Google Scholar]
- 29.He C., Qian X., Sun P. Syntheses of thiol and selenol esters by oxidative coupling reaction of aldehydes with RYYR (Y=S, Se) under metal-free conditions. Org. Biomol. Chem. 2014;12:6072. doi: 10.1039/C4OB01159G. [DOI] [PubMed] [Google Scholar]
- 30.Kumar A.A., Illyés T.Z., Kövér K.E., Szilágyi L. Convenient syntheses of 1,2-trans selenoglycosides using isoselenuronium salts as glycosylselenenyl transfer reagents. Carbohydr. Res. 2012;360:8–18. doi: 10.1016/j.carres.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 31.Ranu B.C., Mandal T. Indium(I) Iodide-Promoted Cleavage of Diaryl Diselenides and Disulfides and Subsequent Condensation with Alkyl or Acyl Halides. One-Pot Efficient Synthesis of Diorganyl Selenides, Sulfides, Selenoesters, and Thioesters. J. Org. Chem. 2004;69:5793–5795. doi: 10.1021/jo0493727. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S., Tripathi S.K., Singh H.B., Wolmershäuser G. Synthesis, reactivity, electrochemical and crystallographic studies of diferrocenoyl diselenide and ferrocenoyl selenides. J. Organomet. Chem. 2004;689:3046–3055. doi: 10.1016/j.jorganchem.2004.06.054. [DOI] [Google Scholar]
- 33.Movassagh B., Mirshojaei F. Synthesis of Selenol Esters from Acid Chlorides and Organic Diselenides in the Presence of the Zn/AlCl 3 System. Monatsh. Chem. 2003;134:831–835. doi: 10.1007/s00706-002-0545-0. [DOI] [Google Scholar]
- 34.Takido T., Toriyama M., Yamashita K., Suwa T., Seno M. A Novel Synthesis of Selenides and Selenol Esters Using Liquid-Liquid Phase-Transfer Catalysis. Phosphorus Sulfur Silicon Relat. Elem. 2003;178:319–326. doi: 10.1080/10426500307940. [DOI] [Google Scholar]
- 35.Beletskaya I.P., Sigeev A.S., Peregudov A.S., Petrovskii P.V. Synthesis of selenoesters. Mendeleev Commun. 2000;10:127–128. doi: 10.1070/MC2000v010n04ABEH001317. [DOI] [Google Scholar]
- 36.Beletskaya I.P., Sigeev A.S., Peregudov A.S., Petrovskii P.V. Tributylstannyl Aryl Selenides as Efficient Arylselenating Agents in the Synthesis of Seleno Esters. Russ. J. Org. Chem. 2001;37:1703–1709. doi: 10.1023/A:1013913816069. [DOI] [Google Scholar]
- 37.Koketsu M., Asada H., Ishihara H. Synthesis of Selenol Esters Using Acyl Halides and a Novel Selenating Reagent, LiAlHSeH. Phosphorus. Sulfur. Silicon Relat. Elem. 2004;179:591–595. doi: 10.1080/10426500490422254. [DOI] [Google Scholar]
- 38.Huang X., Wang J.-H. A Convenient and Stereoselective Synthesis of (E)-Vinylseleno Zirconocenes and (E)-Vinylic Selenol Esters. Synlett. 1999;1999:569–570. doi: 10.1055/s-1999-2698. [DOI] [Google Scholar]
- 39.Munbunjong W., Lee E.H., Ngernmaneerat P., Kim S.J., Singh G., Chavasiri W., Jang D.O. Indium-mediated cleavage of diphenyl diselenide and diphenyl disulfide: Efficient one-pot synthesis of unsymmetrical diorganyl selenides, sulfides, and selenoesters. Tetrahedron. 2009;65:2467–2471. doi: 10.1016/j.tet.2009.01.072. [DOI] [Google Scholar]
- 40.Marin G., Braga A.L., Rosa A.S., Galetto F.Z., Burrow R.A., Gallardo H., Paixão M.W. Efficient synthesis of selenol esters from acid chlorides mediated by indium metal. Tetrahedron. 2009;65:4614–4618. doi: 10.1016/j.tet.2009.03.069. [DOI] [Google Scholar]
- 41.Narayanaperumal S., Alberto E.E., Gul K., Kawasoko C.Y., Dornelles L., Rodrigues O.E.D., Braga A.L. Zn in ionic liquid: An efficient reaction media for the synthesis of diorganyl chalcogenides and chalcogenoesters. Tetrahedron. 2011;67:4723–4730. doi: 10.1016/j.tet.2011.04.018. [DOI] [Google Scholar]
- 42.Silveira C.C., Braga A.L., Larghi E.L. Synthesis of Thiol, Selenol, and Tellurol Esters by the Reaction of Organochalcogeno Mercurials with Acid Chlorides. Organometallics. 1999;18:5183–5186. doi: 10.1021/om990589e. [DOI] [Google Scholar]
- 43.Gul K., Narayanaperumal S., Dornelles L., Rodrigues O.E.D., Braga A.L. Bimetallic system for the synthesis of diorganyl selenides and sulfides, chiral β-seleno amines, and seleno- and thioesters. Tetrahedron Lett. 2011;52:3592–3596. doi: 10.1016/j.tetlet.2011.05.003. [DOI] [Google Scholar]
- 44.Abdo M., Knapp S. Biomimetic Seleninates and Selenonates. J. Am. Chem. Soc. 2008;130:9234–9235. doi: 10.1021/ja8034448. [DOI] [PubMed] [Google Scholar]
- 45.Fujiwara S., Asai A., Shin-ike T., Kambe N., Sonoda N. A New Synthesis of Selenol Esters via Carbophilic Addition of Organocopper Reagents to Carbonyl Selenide. J. Org. Chem. 1998;63:1724–1726. doi: 10.1021/jo9717189. [DOI] [Google Scholar]
- 46.Nanami M., Ando H., Kawai Y., Koketsu M., Ishihara H. Stereoselective synthesis of various α-selenoglycosides using in situ production of α-selenolate anion. Tetrahedron Lett. 2007;48:1113–1116. doi: 10.1016/j.tetlet.2006.12.056. [DOI] [Google Scholar]
- 47.Sheng S., Liu X. One-Pot Synthesis of Selenoesters from Alkynyl Aryl Selenides. Org. Prep. Proced. Int. 2002;34:499–502. doi: 10.1080/00304940209355767. [DOI] [Google Scholar]
- 48.Braga A.L., Martins T.L., Silveira C.C., Rodrigues O.E. Synthesis of chalcogenol esters from chalcogenoacetylenes. Tetrahedron. 2001;57:3297–3300. doi: 10.1016/S0040-4020(01)00200-9. [DOI] [Google Scholar]
- 49.Godoi M., Ricardo E.W., Botteselle G.V., Galetto F.Z., Azeredo J.B., Braga A.L. Synthesis of selenol esters from diorganyl diselenides and acyl chlorides under solvent-free conditions and microwave irradiation. Green Chem. 2012;14:456. doi: 10.1039/c1gc16243h. [DOI] [Google Scholar]
- 50.Santi C., Battistelli B., Testaferri L., Tiecco M. On water preparation of phenylselenoesters. Green Chem. 2012;14:1277. doi: 10.1039/c2gc16541d. [DOI] [Google Scholar]
- 51.Santi C., Santoro S., Testaferri L., Tiecco M. A Simple Zinc-Mediated Preparation of Selenols. Synlett. 2008;2008:1471–1474. doi: 10.1055/s-2008-1078408. [DOI] [Google Scholar]
- 52.Tidei C., Sancineto L., Bagnoli L., Battistelli B., Marini F., Santi C. A recyclable biphasic system for stereoselective and easily handled hydrochalcogenations. Eur. J. Org. Chem. 2014;2014:5968–5975. doi: 10.1002/ejoc.201402668. [DOI] [Google Scholar]
- 53.Salman S., Schwab R., Alberto E., Vargas J., Dornelles L., Rodrigues O., Braga A. Efficient Ring Opening of Protected and Unprotected Aziridines Promoted by Stable Zinc Selenolate in Ionic Liquid. Synlett. 2011;2011:69–72. doi: 10.1002/chin.201119173. [DOI] [Google Scholar]
- 54.Flemer S. Fmoc-Sec(Xan)-OH: Synthesis and utility of Fmoc selenocysteine SPPS derivatives with acid-labile sidechain protection. J. Pept. Sci. 2015;21:53–59. doi: 10.1002/psc.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flemer S. A comprehensive one-pot synthesis of protected cysteine and selenocysteine SPPS derivatives. Protein Pept. Lett. 2014;21:1257–1264. [PubMed] [Google Scholar]
- 56.Bellino G., Scisciani M., Vargas J.P., Sancineto L., Bagnoli L., Marini F., Lüdtke D.S., Lenardao E.J., Santi C. Reaction of Acyl Chlorides with in situ Formed Zinc Selenolates: Synthesis of Selenoesters versus Ring-Opening Reaction of Tetrahydrofuran. J. Chem. 2016;2016:2849140. doi: 10.1155/2016/2849140. [DOI] [Google Scholar]
- 57.Sancineto L., Tidei C., Bagnoli L., Marini F., Lippolis V., Arca M., Lenardão E.J., Santi C. Synthesis of Thiol Esters Using PhSZnBr as Sulfenylating Agent: A DFT-Guided Optimization of Reaction Conditions. Eur. J. Org. Chem. 2016;2016:2999–3005. doi: 10.1002/ejoc.201600366. [DOI] [Google Scholar]
- 58.Anantharaman G., Chandrasekhar V., Nehete U.N., Roesky H.W., Vidovic D., Magull J. New Polyhedral Zinc Siloxanes: Synthesis and X-ray Crystal Structures of Zn8Me7(dioxane)2(O3SiR)3 and [Zn7Me2(THF)5(O3SiR)4][R = (2,6-i-Pr2C6H3)N(SiMe3)] Organometallics. 2004;23:2251–2256. doi: 10.1021/om049863h. [DOI] [Google Scholar]
- 59.Chen W.-T., Zeng X.-R., Liu D.-S., Ying S.-M., Liu J.-H. An Unprecedented 2D 4f-3d-5d Multimetal-Isonicotinic Acid Complex: Synthesis, Structural Characterization and Magnetic Properties. Chin. J. Chem. 2008;26:1678–1682. doi: 10.1002/cjoc.200890303. [DOI] [Google Scholar]
- 60.Chen W.-T., Zeng X.-R., Liu D.-S., Ying S.-M., Liu J.-H. A novel 2-D 5d-4f-3d trimetal-isonicotinic acid complex: Synthesis and characterisation. J. Chem. Res. 2009:705–709. doi: 10.3184/030823409X12571600412723. [DOI] [Google Scholar]
- 61.Ellison J.J., Power P.P. Synthesis and Characterization of New Thiolato Derivatives of Lithium, Magnesium, and Zinc: Examples of Two-Coordinate Lithium and Zinc Species Ligated by Sulfur. Inorg. Chem. 1994;33:4231–4234. doi: 10.1021/ic00097a008. [DOI] [Google Scholar]
- 62.Nguyen T., Panda A., Olmstead M.M., Richards A.F., Stender M., Brynda M., Power P.P. Synthesis and Characterization of Quasi-Two-Coordinate Transition Metal Dithiolates M(SAr*)2(M=Cr, Mn, Fe, Co, Ni, Zn; Ar*=C6H3-2,6(C6H2-2,4,6-Pri3)2. J. Am. Chem. Soc. 2005;127:8545–8552. doi: 10.1021/ja042958q. [DOI] [PubMed] [Google Scholar]
- 63.Ellison J.J., Ruhlandt-Senge K., Hope H.H., Power P.P. Synthesis and Characterization of the New Selenolate Ligand -SeC6H3-2,6-Mes2 (Mes=C6H2-2,4,6-Me3) and Its Two-Coordinate Zinc and Manganese Derivatives: Factors Affecting Bending in Two-Coordinate Metal Complexes with Aryl-Substituted Ligands. Inorg. Chem. 1995;34:49–54. doi: 10.1021/ic00105a012. [DOI] [Google Scholar]
- 64.Jun Y., Koo J.-E., Cheon J. One-step synthesis of size tuned zinc selenide quantum dots via a temperature controlled molecular precursor approach. Chem. Commun. 2000:1243–1244. doi: 10.1039/b002983l. [DOI] [Google Scholar]
- 65.Kudelko A., Wróblowska M. An efficient synthesis of conjugated 5-aryl-1,3,4-oxadiazoles from 3-heteroarylacrylohydrazides and acid chlorides. Tetrahedron Lett. 2014;55:3252–3254. doi: 10.1016/j.tetlet.2014.04.029. [DOI] [Google Scholar]
- 66.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian 09, Revision D.01 2009. Gaussian, Inc.; Wallingford, CT, USA: [Google Scholar]
- 67.Adamo C., Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998;108:664–675. doi: 10.1063/1.475428. [DOI] [Google Scholar]
- 68.Reed A.E., Weinhold F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983;78:4066–4073. doi: 10.1063/1.445134. [DOI] [Google Scholar]
- 69.Reed A.E., Weinstock R.B., Weinhold F. Natural population analysis. J. Chem. Phys. 1985;83:735–746. doi: 10.1063/1.449486. [DOI] [Google Scholar]
- 70.Reed A.E., Curtiss L.A., Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988;88:899–926. doi: 10.1021/cr00088a005. [DOI] [Google Scholar]
- 71.Wiberg K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron. 1968;24:1083–1096. doi: 10.1016/0040-4020(68)88057-3. [DOI] [Google Scholar]
- 72.Dennington R., Keith T., Millam J. GaussView. Gaussian, Inc.; Wallingford, CT, USA: 2009. Version 5. [Google Scholar]
- 73.Schaftenaar G., Noordik J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000;14:123–134. doi: 10.1023/A:1008193805436. [DOI] [PubMed] [Google Scholar]
- 74.Wang L., Zhang Y. Water Accelerated Sm/TMSCl Reductive Cleavage of the Se-Se Bond: Synthesis of Selenoesters and Selenoformates. Synth. Commun. 1999;29:3107–3115. doi: 10.1080/00397919908085934. [DOI] [Google Scholar]
- 75.Ren K., Wang M., Liu P., Wang L. Iron-Catalyzed Synthesis of Selenoesters from Diselenides and Acyl Chlorides or Acid Anhydrides in the Presence of Magnesium Dust. Synthesis. 2010;2010:1078–1082. doi: 10.1002/chin.201031192. [DOI] [Google Scholar]
- 76.Zhang S., Zhang Y. Reductive Cleavage of Se–Si Bond in Arylselenotrimethylsilane: A Novel Method for the Synthesis of Selenoesters. Synth. Commun. 1998;28:3999–4002. doi: 10.1080/00397919808004959. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.