

Abstract
The delivery of small interfering RNA (siRNA) remains a major hurdle for the clinical translation of RNA interference (RNAi) therapeutics. Due to its low valency and rigid nature, siRNA typically requires high excesses of cationic delivery materials to package it stably and deliver it to the cytoplasm of target cells, resulting in high toxicities and inefficient gene silencing in vivo. To address these challenges, we pair a polymeric form of siRNA, p-shRNA, with optimized biodegradable polycations to form stable complexes that induce far more potent gene silencing than with siRNA complexes. Furthermore, we unveil a set of design rules governing p-shRNA delivery, using degradable polycations containing hydrophobic and stabilizing polyethylene glycol domains that enable both stable condensation and efficient release inside cells. We demonstrate the therapeutic potential of this approach by silencing the oncogene STAT3 in a well-established B16F10 mouse melanoma model to significantly prolong survival. By blending nucleic acid engineering and polymer design, our system provides a potentially translatable platform for RNAi-based therapies.
Keywords: RNA interference, polycation, gene delivery, siRNA, poly(beta-amino ester)
Table of Contents Graphic
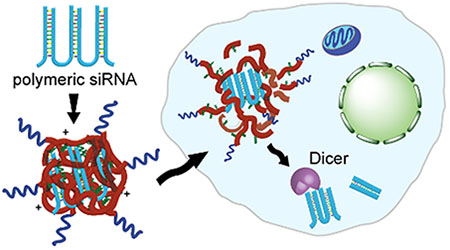
RNA interference (RNAi) provides a powerful therapeutic tool for specifically silencing genes implicated in disease. Despite its promise, clinical translation of RNAi has remained largely unsuccessful due to barriers in delivering small interfering RNA (siRNA) into target cells. Much effort has been devoted to developing delivery materials or methods that can protect siRNA from degradation, facilitate its uptake by diseased cells, promote escape from endosomes into the cytoplasm where RNAi occurs, or maximize RNA induced silencing complex (RISC) formation.1–4 However, the high rigidity and low valency of siRNA presents challenges in stable encapsulation, often necessitating large amounts of cationic delivery materials and poor in vivo silencing efficiency.5–7 To address these challenges, we previously adopted a polymeric form of siRNA, or periodic short hairpin RNA (p-shRNA), whose high valency and flexibility require far less polycationic carrier to condense into stable nanoparticles.8,9 Generated by rolling circle transcription (RCT) of a dumbbell DNA template, p-shRNA consists of siRNAs linked by short single stranded regions, and can be engineered to possess open ends for improved intracellular processing by Dicer into siRNAs, enhancing silencing efficacy when delivered in vitro with various commercial transfection reagents.9,10 With its superior silencing efficacy, along with much higher valency and flexibility compared to siRNA, op-shRNA represents a highly promising RNAi therapeutic that not only can be delivered more efficiently into target cells than siRNA, but also mediate more potent gene silencing in vivo. However, as commercial reagents are ill-suited for in vivo applications due to instability and toxicity, in this study we sought to design an optimal carrier for in vitro and in vivo delivery of op-shRNA, which can further guide future carrier design for other large RNA macromolecules that can be generated by RCT, thus extending the potential of these systems as a promising platform.
The known delivery challenges for siRNA can be extended to op-shRNA, but with some additional distinctive challenges related to the large size of these macromolecules, which range from roughly 200 up to 3000 bases. Delivery of op-shRNA with several polycations commonly used for gene delivery, including polyethyleneimine (PEI) of various molecular weights and architectures (linear and branched), poly-lysine, and poly-arginine, however, induces little to no gene silencing when delivered with these polymers, despite improved complexation compared to siRNA (Supplementary Figure S1). Since PEI is non-biodegradable and polypeptides contain stable amide bonds, we attributed this lack of silencing to possible inefficient release of op-shRNA from the complexes within cells due to stronger binding interactions with the multivalent RNA; the high molecular weight polyelectrolyte complex is extremely stable, and unlike packaging with lower molar mass 19–21 base pair siRNA, it is unable to release siRNA via charge-shift induced swelling and decomplexation at endosomal pH. To address this challenge while exploiting the improved gene silencing potency and complexation ability of op-shRNA, we hypothesized that rapidly degradable polycations would condense op-shRNA into stable nanoparticles and subsequently release it following intracellular uptake due to hydrolytic degradation (Figure 1).
Figure 1.

Schematic of encapsulation of open-ended periodic shRNA (op-shRNA) by a biodegradable polycation (e.g. poly(beta-amino ester)) containing hydrophobic domains to form a core complex, followed by addition of a PEG-conjugated polycation containing alkyl chains to stabilize the complex. Upon delivery into cells, degradation of the polycations in the complex efficiently releases op-shRNA, which is then processed by Dicer in the cytoplasm into siRNAs to mediate RNA interference.
To test our hypothesis, we chose to deliver op-shRNA with poly(beta-amino ester)s (PBAEs), a well-established class of biodegradable gene delivery vehicles.11,12 Synthesized by the Michael addition of bifunctional amines to diacrylates, PBAEs have been investigated in large combinatorial libraries, due to their facile synthesis and functional versatility.13–15 However, achieving significant gene silencing with PBAEs typically requires large excesses of polycation and high siRNA doses.16,17 Moreover, despite numerous high-throughput screenings to identify effective structures with high transfection efficiency and low cytotoxicity, there is still much to understand regarding structure-function relationships for PBAEs in delivering different nucleic acids, with slight structural variations exerting significant effects on efficiencies.18,19 As the high molecular weight and intracellular processing of op-shRNA requires distinct design rules, here we proposed to engineer a PBAE system for op-shRNA delivery that can stably complex op-shRNA while also enabling efficient cargo release inside cells, via factorial design, which can reveal key structure-function correlations for delivering such a large RNAi macromolecule. Furthermore, we extended our materials library to incorporate a polyethylene glycol (PEG)-conjugated PBAE, in which PEG serves as an end group that provides an outer stabilizing shell around the “core” op-shRNA/PBAE complex (Figure 1). Finally, we demonstrated the in vivo efficacy of our op-shRNA system by targeting signal transducer and activator of transcription 3 (STAT3), whose overexpression plays a crucial role in tumor progression and promoting an immunosuppressive tumor environment, in a well-established mouse melanoma model.20–22
Results and Discussion
Factorial Design of Biodegradable Polycations for op-shRNA Encapsulation
As the significantly greater size of op-shRNA compared to siRNA requires distinct design rules for encapsulation and delivery, we employed factorial design of rapidly biodegradable polycations to develop an effective nanocarrier for such a large RNAi macromolecule. Packaging op-shRNA with hydrolytically degradable PBAEs may enable efficient release of op-shRNA into the cytoplasm upon delivery into target cells. At the same time, the multivalency and flexibility of op-shRNA potentially requires far less excess polycation than typically required for PBAE-mediated siRNA delivery for efficient condensation into stable complexes, thereby reducing undesired toxicity and increasing in vivo silencing efficacy.
We first determined basic design rules for PBAE-mediated op-shRNA delivery by synthesizing a library of PBAE copolymer variants based on a base structure, poly-1 (Figure 2A). As studies have shown that introducing hydrophobic alkyl side chains into certain PBAE structures improves DNA and messenger RNA transfection efficiencies, we hypothesized that incorporation of alkyl chains into our PBAEs, along with tuning molecular weight, can exert a strong influence on op-shRNA silencing efficiency.23,24 Hydrophobicity was varied by incorporating differing mole percentages of dodecylamine (C12) in the total amine monomer amount, which includes 4,4’-trimethylenedipiperidine and dodecylamine, yielding random copolymers. As previous work has found that increasing the alkyl chain percentage past 30% did not further improve transfection efficiency with DNA or mRNA, we tested C12 feed percentages up to 30%.23 By varying the overall monomer stoichiometry in this step-growth polymerization, i.e., moles of 1,4-butanediol diacrylate to moles of total amine (1.02, 1.05, 1.1, and 1.2), we also investigated the effect of molecular weight on op-shRNA silencing efficiency. These base polymers were subsequently endcapped with diethylenetriamine to enhance binding interactions with the RNA and transfection efficiency. Quantification of nuclear magnetic resonance (NMR) spectra of the poly-1 structures confirmed that the actual percentages of C12 incorporated in the polymers matched the percent fed into the reaction (Supplementary Table 3).
Figure 2.

(A) Synthesis of poly-1 library, with varying molecular weights (tuned by monomer stoichiometry) and alkyl chain (C12) fractions. (B) Hydrodynamic size of op-shRNA/poly-1(30% C12) complex at a polymer:RNA weight ratio (w/w) of 5, as measured by dynamic light scattering. (C) GFP silencing in GFP-expressing HeLa cells by op-shRNA (20 nM, based on effective siRNA repeats) complexed with poly-1 library at polymer:RNA w/w 5, as a function of poly-1 molecular weight. Data are represented as the mean ± SEM (n = 3). (D) Release of RNA from op-shRNA complexes formed with 25 kDa branched PEI and 1.8 kDa linear PEI at N/P 10, and 0%C12 poly-1 and 30%C12 poly-1 at polymer:RNA w/w 10 after incubation in PBS, pH 7.4, at 37 °C, as measured by agarose gel electrophoresis.
Gel shift assays showed that our PBAEs complex op-shRNA very efficiently, fully condensing op-shRNA at much lower polymer to RNA weight ratios typically required for efficient siRNA delivery with PBAEs (Supplementary Figure S2).16,17,25,26 Furthermore, the poly-1 structures form compact nanoparticles with op-shRNA under 100 nm in diameter (Figure 2B and Supplementary Figure S3) at a polymer:RNA weight (w/w) ratio of 5. Screening this library for silencing of the reporter gene GFP in GFP-expressing HeLa cervical cancer cells showed that poly-1 structures with higher alkyl chain content exhibited greater silencing efficiency with op-shRNA, similar to previous observations with plasmid DNA.23 Comparison of poly-1 structures of similar molecular weights around 5 kDa revealed increasing silencing efficiency with increasing hydrophobicity. Furthermore, poly-1 structures of lower hydrophobicity (0% and 15% C12) demonstrated a negative correlation between molecular weight and silencing efficiency with op-shRNA. For 30% C12 poly-1 structures, which stayed within a lower, narrower molecular weight range, it was not possible to observe a clear trend; however, the highest molecular weight poly-1 containing 30% C12 achieved nearly 90% knockdown of GFP using very little polymer (polymer:RNA w/w 5). Poly-1 structures without any alkyl chains also demonstrated significantly lower cellular uptake than those containing alkyl chains, suggesting that one mechanism by which incorporation of alkyl chains into poly-1 improves silencing efficiency is by increasing cellular uptake, likely through enhanced interactions with the cell membrane (Supplementary Figure S3).
To determine whether our poly-1 structures outperformed commonly used PEI gene delivery vehicles by more efficient release of op-shRNA, we performed gel shift assays of op-shRNA complexes incubated at physiological pH and ionic conditions in phosphate buffered saline (PBS) at 37 °C over time. Both branched and linear PEI complexes with op-shRNA failed to release any RNA after 48 hours (Figure 2D). In contrast, poly-1 complexes released most of the encapsulated RNA by 48 hours, supporting our hypothesis that the biodegradability of poly-1 enables efficient silencing with op-shRNA, while the non-biodegradable PEI hinders release of op-shRNA into the cytoplasm (Figure 2D). Interestingly, the RNA was released in the form of short RNA strands, likely op-shRNA repeats refolded into short hairpin structures. Thus, while the high molecular weight op-shRNA is readily condensed by poly-1 into compact nanoparticles, once delivered into the target cell, the op-shRNA may be progressively released into the cytoplasm as refolded short hairpin RNAs that are then processed by Dicer into siRNAs. We further compared release by op-shRNA complexes formed with 0% C12 and 30% C12 poly-1 structures of similar molecular weights (~5 kDa). Complexes formed with poly-1 containing no alkyl side chains released a significant portion of the encapsulated RNA by 6 hours, with most of the RNA released by about 24 hours (Figure 2D). Incorporating alkyl side chains in poly-1 delayed release of RNA, with the majority of RNA remaining complexed after 24 hours (Figure 2D). The delayed RNA release by 30% C12 poly-1 over a later time window may therefore contribute to the superior silencing efficiency of 30% C12 compared to 0% C12 poly-1.
Stabilizaton of op-shRNA complexes via PEGylated PBAEs
Despite the ability to use far less polymer for delivering op-shRNA, the poly-1/op-shRNA complexes still possess a highly positive surface charge that would not be suitable for systemic delivery, and they require further stabilization for in vivo applications. With the presence of alkyl chains in our optimized poly-1 structure, we hypothesized that adding a PEG-poly-1 copolymer containing alkyl chains to the complex would provide a stabilizing PEG shell that is stably associated with the core complex via hydrophobic interactions. (Figure 1). Incorporation of such an amphiphilic PEG-poly-1 could thereby increase colloidal stability while decreasing positive surface charge and cytotoxicity. To test this, we synthesized three PEG-poly-1 structures with high C12 feed percentages (40%, 70%, and 100% of total amine monomer, denoted PEG-40%C12, PEG-70%C12, and PEG-100%C12, respectively) to enable strong hydrophobic interactions with the poly-1 alkyl chains, by adding a monofunctional 5 kDa PEG-acrylate to the polymerization reaction to endcap the polyamine (Figure 3A). Quantification of NMR spectra showed that the actual percentage of C12 incorporated along the backbone decreased slightly from the feed percentage as the C12 feed percentage increased to 70%, but maintained the increasing trend of C12 fraction in the backbone (Supplementary Table 3).
Figure 3.

(A) Structure of PEG-poly-1 containing alkyl side chains. (B) Hydrodynamic sizes of op-shRNA complexes formed with 30%C12 poly-1 only (w/w 10) or poly-1 and PEG-poly-1 (poly-1:PEG-poly-1:RNA w/w 10:10:1) containing varying C12 fractions, before and after incubation in phosphate-buffered saline, pH 7.4, as measured by dynamic light scattering. (C) Zeta potential measurements of op-shRNA complexes formed with 30%C12 poly-1 only (w/w 10) or poly-1 and PEG-poly-1 (poly-1:PEG-poly-1:RNA w/w 10:10:1) containing varying C12 fractions, diluted in 10 mM HEPES. (D) Cryogenic transmission electron microscopy of op-shRNA complexes formed with 30%C12 poly-1 and PEG-100%C12 poly-1, at poly-1:PEG-poly-1:RNA w/w 10:10:1.
PEGylated complexes were formed by sequentially adding poly-1 (1.05–30%C12, the top-performing structure from our library) to op-shRNA, and then PEG-poly-1 to the core complex. At a 1:1 mass ratio of PEG-poly-1 to poly-1, addition of PEG-poly-1 dramatically reduced particle aggregation after incubation at physiological pH and ionic conditions in phosphate buffered saline (PBS) compared to unmodified core complexes (Figure 3B). These increases in colloidal stability were observed at various polymer to RNA weight ratios (Supplementary Figure S4). The positive surface charge of the complexes also decreased upon addition of PEG-poly-1, further confirming the presence of a PEG “shell” around the core complex (Figure 3C). Increasing the amount of PEG-poly-1 relative to core poly-1 did not further improve colloidal stability in PBS or decrease positive surface charge (Supplementary Figure S4); further experiments were therefore performed at a 1:1 PEG-poly-1:poly-1 mass ratio. The compact size of the complexes was retained with addition of PEG-poly-1, as evidenced by cryo-electron microscopy (Figure 3D).
We next assessed whether the addition of PEG-poly-1 would reduce any undesired cytotoxicity as well. At the same total polymer weight ratio to RNA, non-PEGylated poly-1 complexes showed significantly greater toxicity than complexes modified with PEG-poly-1 with 70% and 100% C12 (Supplementary Figure S5). Incorporation of PEG-40%C12, however, induced similar levels of cytotoxicity as non-PEGylated poly-1 complexes.
Having demonstrated improved colloidal stability with PEG-poly-1 addition, we next expanded our library, again using factorial design, to screen combinations of the core poly-1 and PEG-poly-1 structures. This second generation library consisted of four core polymers with the same alkyl chain percentage (30%, selected from first generation library) but varying alkyl chain length (C12 and C16) and molecular weight, and four PEGylated polymers (Figure 4A). For PEG-poly-1, we selected 70% and 100% alkyl chain feed content, which increased colloidal stability while decreasing cytotoxicity, and also varied alkyl chain length (C12 and C16). Transfections of these PEG-poly-1 complexes containing anti-luciferase op-shRNA into luciferase-expressing B16F10 melanoma cells revealed that adding PEG-poly-1 with 70% C12 or C16 significantly reduced silencing efficiency (Figure 4B). On the other hand, incorporation of PEG-poly-1 with 100% C12 or C16 yielded much higher silencing efficiencies, with C16 performing slightly better than C12 for most of the core polymers. For both C12 and C16-containing core polymers, higher molecular weight structures also outperformed lower molecular weight structures. The top-performing polymer combinations containing PEG-100%C12 or PEG-100%C16 showed superior silencing efficacies compared to the commercial transfection reagent TransIT-X2®, inducing over 80% specific knockdown at a dose of 20 nM op-shRNA while exhibiting little cytotoxicity (Figure 4B and Supplementary Figure S5). Notably, we were able to achieve high silencing levels at a much lower polymer:RNA weight ratio (core poly-1:PEG-poly-1:RNA w/w = 10:10:1) compared to previous studies on PBAE-mediated siRNA delivery, which have required weight ratios of 40 and above, including other alkyl chain-containing PBAEs.16,17,25,26
Figure 4.

(A) Factorial design of second-generation library of combinations of poly-1 and PEG-poly-1. (B) Luciferase knockdown in luciferase-expressing B16F10 melanoma cells by op-shRNA (20 nM) complexed with second-generation poly-1 and PEG-poly-1 library (poly-1:PEG-poly-1:RNA w/w = 10:10:1). Data are represented as the mean ± SEM (n = 3). **p<0.01, ***p<0.001 (comparison to TransIT-X2®). (C) Release of RNA from op-shRNA complexes formed with 30%C12 poly-1 and PEG-70%C12 or PEG-100%C12 poly-1 at poly-1:PEG-poly-1:RNA w/w 10:10:1, after incubation in PBS, pH 7.4, at 37 °C, as measured by agarose gel electrophoresis. (D) Confocal fluorescence microscopy of B16F10 cells at 24 hours after transfection with 50 nM op-shRNA complexes formed with 30%C12 poly-1 and PEG-70%C12 or PEG-100%C12 poly-1 at poly-1:PEG-poly-1:RNA w/w 10:10:1, with op-shRNA (red) labeled with Cy5.5, late endosomes (green) labeled by Rab7, and nuclei (blue) labeled by DAPI.
To gain a mechanistic understanding of how incorporating PEG-100%C12 or PEG-100%C16 poly-1 retains high silencing activity by op-shRNA, we first measured cellular uptake of the op-shRNA complexes in B16F10 cells via flow cytometry, as addition of PEG often decreases nanoparticle uptake by cells despite improving colloidal stability.27 At 24 hours after transfection, complexes with added PEG-70%C12 or PEG-100%C12 showed similar cell uptake relative as the core complex consisting of only 30%C12 poly-1 at w/w 10 (Supplementary Figure S5). Thus, addition of PEG-poly-1 does not impair cellular uptake by op-shRNA/poly-1 complexes.
We next compared RNA release kinetics of complexes modified with PEG-70%C12 and PEG-100%C12. Addition of PEG-70%C12 to the core complex resulted in earlier release of more RNA by 24 hours of incubation in PBS at 37 °C (Figure 4C). On the other hand, the PEG-100%C12/poly-1 complexes maintained a similarly slow release profile as the core complex, with most of the RNA released by 48 hours. These observations suggest that while PEG-70%C12 complexes maintained colloidal stability in PBS over several hours, addition of PEG-100%C12 keeps op-shRNA complexed over a longer period of time, maintaining a tighter or more stable condensation of RNA than PEG-70%C12. Thus, possible premature release of RNA from PEG-70%C12/poly-1 complexes before the complexes reach the cytoplasm may contribute to their low silencing efficiency.
We further examined endosomal escape of the PEGylated complexes via confocal microscopy. At 24 hours post-transfection of B16F10 cells, both PEG-70%C12 and PEG-100%C12 complexes showed low degrees of colocalization with the late endosomal marker Rab7, suggesting that the complexes were able to escape the endosomal pathway into the cytoplasm (Figure 4D). These results therefore suggest similar endosomal escape efficiencies between PEG-70%C12 and PEG-100%C12 complexes. However, op-shRNA prematurely released from PEG-70%C12 complexes before endosomal escape may undergo partial degradation within the endosomes, with fewer intact op-shRNA units reaching the cytoplasm to successfully mediate silencing. On the other hand, the majority of op-shRNA in PEG-100%C12 complexes may be released after reaching the cytoplasm over an extended period of time, enabling stronger silencing as the RNA is gradually released.
Therapeutic targeting of STAT3 by op-shRNA/PEG-PBAE system
To demonstrate therapeutic efficacy, we targeted STAT3, whose persistent activation in many cancers contributes to tumor progression and immune evasion, with our best-performing PEG-poly-1/op-shRNA system, consisting of 1.05–30%C12 as the core polymer and PEG-100%C16.20–22 Transfection of B16F10 cells with 20 nM op-shRNA (based on effective siRNA repeats) yielded over 70% knockdown of STAT3 at the mRNA level, compared to about 50% knockdown with an equivalent dose of siRNA packaged with the same carrier (Figure 5A). This contrast in silencing efficiency was even more pronounced at the protein level, for both tyrosine-phosphorylated STAT3, the activated protein form, and total STAT3 (Figure 5B). Having verified STAT3 knockdown in vitro, we evaluated therapeutic efficacy in a syngeneic B16F10 melanoma model. After primary subcutaneous tumors were established in C57BL/6 mice, four treatments of our top performing PEG-poly-1 system encapsulating anti-STAT3 or scrambled op-shRNA or siRNA (5 μg RNA per tumor) were administered every three days, with concurrent tumor size measurements as a disease progression indicator (Figure 5C). Treatment with anti-STAT3 op-shRNA significantly decreased tumor growth rates and prolonged survival compared to all other control groups (Figure 5D, E). In contrast, mice treated with anti-STAT3 siRNA showed no improvements in tumor progression rate or survival. We further verified STAT3 knockdown via immunohistochemical staining of isolated tumors from a separate cohort of mice injected twice with the same treatment groups (Figure 5F). Consistent with the tumor burden progression and survival trends, STAT3 expression in the tumors of mice treated with anti-STAT3 op-shRNA was significantly lower than in all other control groups. We further observed reduced cell proliferation only in tumors treated with anti-STAT3 op-shRNA, as evidenced by lower Ki-67 expression (Figure 5F). However, active caspase 3 expression levels were similar among all treatment groups, indicating insignificant levels of cell apoptosis. Our results therefore suggest that our op-shRNA system induces a therapeutic effect by mitigating the tumorigenic activity of STAT3 via specific silencing and by inhibiting tumor cell proliferation.28
Figure 5.

(A, B) Knockdown of STAT3 by anti-STAT3 op-shRNA and siRNA complexed with top-performing poly-1/PEG-poly-1 combination at the messenger RNA (A) and protein (B) levels. Messenger RNA levels were measured by quantitative PCR with normalization to β-actin messenger RNA, and tyrosine-phosphorylated and total STAT3 protein levels were measured by Western blotting with λ-tubulin as an internal loading control. Data are represented as the mean ± SEM (n = 3). **p<0.01. (C) Treatment regime for C57BL/6 mice bearing subcutaneous B16F10 melanoma tumors. Mice received intratumoral injections of 5 μg anti-STAT3 or scrambled op-shRNA or siRNA complexed with 30%C12 poly-1/PEG-100%C16 poly-1, every three days for four times (poly-1:PEG-poly-1:RNA w/w = 10:10:1). (D) Tumor sizes, measured every three days before injection on the same day until some mice with large tumor burdens in control groups were euthanized. (E) Kaplan-Meier survival curve for the different treatment groups. Treatment with anti-STAT3 op-shRNA significantly prolonged survival compared to all other groups. Vehicle control, scrambled op-shRNA, and anti-STAT3 op-shRNA (n=8); anti-STAT3 siRNA and scrambled siRNA (n=7). *, p<0.05 (comparison of anti-STAT3 op-shRNA with scrambled op-shRNA and vehicle control groups). (F) STAT3 and Ki-67 expression in tumor samples, detected by immunohistochemical staining 24 hours after three treatments of 5 μg anti-STAT3 or scrambled op-shRNA or siRNA complexed with 30%C12 poly-1/PEG-100%C16 poly-1.
As long double-stranded RNAs can act as immunostimulatory agents, we also explored the potential ability of our op-shRNA/poly-1 system to activate innate immunity.29 Comparison of the various PEG-poly-1 structures combined with 30%C12 poly-1 showed that PEG-100%C12 and PEG-100%C16 systems induced the greatest activation of NF-κB and AP-1 in RAW-Blue™ mouse macrophage reporter cells, consistent with their superior silencing efficiencies (Supplementary Figure S8). Interestingly, op-shRNA complexed with these two PEG-poly-1 carriers triggered significantly greater stimulation than high molecular weight poly(I:C), commonly used as a vaccine adjuvant, as well as duplex siRNA.30 Since innate immune activation can trigger cancer cell death and antitumor adaptive immune responses, the ability of op-shRNA to act as an innate immune agonist when delivered with our poly-1 carriers can potentially act synergistically with silencing of a therapeutic target.31 Indeed, combining innate immune agonists with siRNA-mediated silencing has been demonstrated to induce synergistic therapeutic effects in several preclinical models.32.33 Future work will tune both op-shRNA and polycation structures to exploit innate immune activation by op-shRNA systems in concert with RNAi for potent antitumor responses.
Conclusions
In summary, we have engineered a stable and efficient delivery system that maximizes the in vivo efficacy of op-shRNA by using rationally designed biodegradable polymers. As the large size of op-shRNA presents a different set of delivery requirements compared to siRNA or other nucleic acids, commonly used polycationic gene delivery vehicles such as PEI and other polymers that have successfully delivered nucleic acids in vivo may be ineffective with op-shRNA, especially in achieving efficient cargo release inside cells. Thus, our PBAE-based vehicle proves highly promising due to its ability to both stably condense op-shRNA and efficiently release it into the cytoplasm. By employing factorial design, we identified several important factors in PBAE structure that affect op-shRNA silencing efficiency, including molecular weight and hydrophobicity. Optimization of these design factors and addition of a stabilizing PEGylated PBAE that associates strongly with the core complex via hydrophobic interactions enable potent silencing using low amounts of polymer, by forming a stable nanoparticle that can still efficiently dissociate within cells to release op-shRNA. These structure-function relationships can guide future optimization of PBAE systems for op-shRNA delivery, such as incorporating different hydrophobic side groups and varying amine densities along the backbone. Moreover, such factorial designs can potentially prove useful for engineering PBAEs to deliver other large nucleic acids such as DNA and messenger RNA. Combined with the versatility of rolling circle transcription for generating high molecular weight RNA macromolecules, our systematically optimized PBAE platform can be used to deliver op-shRNA targeting multiple genes of interest, and potentially periodic microRNAs or anti-microRNA therapeutics for inhibiting oncogenic microRNAs.34 We can further enhance protection from nuclease degradation by incorporating chemically modified nucleotides in op-shRNA. Our synergistic pairing of op-shRNA with rationally designed PBAEs can potentially provide a transformative platform for RNAi therapeutic delivery.
Methods and Materials
Chemicals.
Monomers were obtained from Alfa Aesar (Ward Hill, MA, USA), and the diethylenetriamine endcap and solvents were obtained from Sigma Aldrich (St. Louis, MO, USA). PEG-acrylate was obtained from Laysan Bio (Arab, AL, US).
Synthesis of op-shRNA.
Custom PAGE-purified, 5’-phosphorylated DNA oligos were purchased from Integrated DNA Technologies (Coralville, IA, US) and resuspended in RNase-free water. DNA oligo was diluted to 3 μM in NEBNext® Quick Ligation Reaction Buffer (New England Biolabs, Ipswich, MA, US) and heated at 95 °C for 2 minutes before allowing to slowly cool to room temperature. DNA oligos were then ligated with 1.5 μL T4 DNA ligase (400,000 units/mL, New England Biolabs, Ipswich, MA, US) to give a total ligation reaction volume of 120 μL. After incubating for two hours at room temperature, the ligation reaction was mixed with 12.5 μL ribonucleotide solution mix ((New England Biolabs, Ipswich, MA, US), 1.5 μL water, 6 μL MgCl2 (200 mM), 20 μL 10× T7 RNA polymerase reaction buffer, and 40 μL T7 RNA polymerase (50,000 units/mL, Lucigen, Middleton, WI, US). Transcription was allowed to proceed for 48 hours at 37 °C before addition of 20 μL of 0.5 M ethylenediaminetetraacetic acid (EDTA). The p-shRNA was purified with Advantec disposable ultrafiltration units (200 kDa molecular weight cutoff; Cole-Parmer, Vernon Hills, IL, US). Open-ended p-shRNA was formed by incubating p-shRNA with RNase T1 (ThermoFisher Scientific, Cambridge, MA, US) at a ratio of 1 unit RNase T1:2 μg RNA in 20 mM HEPES (pH 7.5) and 1 mM EDTA at 37 °C for two hours, followed by purification with 40 kDa Zeba spin desalting columns (ThermoFisher Scientific).
Poly(beta-amino ester) synthesis.
PBAEs were synthesized following previous procedures.35 Briefly, 1,4-butanediol diacrylate, 4,4’-trimethylenedipiperidine, and 1-dodecylamine or hexadecylamine were separately dissolved in tetrahydrofuran (first generation poly-1 library) or toluene (second generation poly-1 library) and then added together under a nitrogen atmosphere. After stirring at 90 °C for 48 hours (50 °C for THF), the reaction mixture was precipitated in cold hexanes to collect the base polymer, which was subsequently dried under vacuum. Endcapped PBAEs were synthesized by addition of 4 mmol diethylenetriamine (Sigma Aldrich) to 1 g base polymer dissolved in tetrahydrofuran. After stirring for 24 hours at room temperature, endcapped polymer was precipitated in cold hexanes and washed thoroughly before drying under vacuum.
Synthesis of PEG-poly-1. PEG-poly-1 was synthesized in toluene using the same procedure, with the addition of methoxy-PEG-acrylate (5 kDa) to the reaction mixture (1 eq. diacrylate, 1.1 eq. total amine, 0.01 eq. PEG-acrylate).
Polymer characterization.
Gel permeation chromatography was performed on a Waters system with a Malvern Viscotek™ TDA 305 triple detection system and Styragel columns (Waters), with tetrahydrofuran as the mobile phase. 1H NMR spectra were obtained with a Bruker Advance 400 MHz NMR spectrometer at 25 °C. Molecular weights were determined based on a linear polystyrene standard.
Cell lines.
GFP-expressing HeLa cells (Cell Biolabs, San Diego, CA, US), B16F10 cells (American Type Culture Collection, Rockville, MD, US), and RAW-Blue™ mouse macrophage reporter cells (InvivoGen, San Diego, CA) were cultured in DMEM (Invitrogen, Grand Island, NY, US) with 10% fetal bovine serum and 1% penicillin/streptomycin. B16F10 cells stably expressing GFP and luciferase (B16F10-GFP-luc) were made by stable transfection by the lentivirus pSICO-GFP-luciferase-puro. Cells were verified to be mycoplasma-free by the High Throughput Screening facility at the Koch Institute for Integrative Research (Cambridge, MA, US).
Complex formation and characterization.
Polymers were dissolved in sodium acetate buffer (50 mM, pH 5), then diluted and added to op-shRNA at the desired polymer:RNA ratio, immediately followed by thorough pipette mixing. The number average hydrodynamic sizes and zeta potentials of complexes were measured by dynamic light scattering using a Malvern Zetasizer Nano ZS90 particle analyzer (λ = 633 nm). Cryogenic transmission electron microscopy was performed using a JEOL 2100 FEG instrument. For release assays, complexes were diluted five-fold in PBS, pH 7.4, and incubated at 37 °C, with aliquots taken at the indicated timepoints and subsequently snap frozen until assaying. RNA release was assayed by gel electrophoresis using a 1% E-Gel EX agarose gel (Thermo Fisher Scientific).
In vitro transfections.
5,000 B16F10-GFP-Luc or HeLa-GFP cells were seeded in DMEM with 10% FBS in a white 96-well plate and allowed to adhere overnight. Complexes were formed as described for characterization and equilibrated at room temperature for at least ten minutes, before adding to the cells at the desired final concentration (based on effective siRNA repeats in op-shRNA). The medium was replaced with fresh culture medium 24 hours post-transfection. GFP expression of HeLa-GFP cells was measured by flow cytometry 48 hours post-transfection using a BD FACSCalibur equipped with a high throughput sampler. Luciferase expression of B16F10-GFP-Luc cells was measured 48 hours post-transfection with the Firefly Luciferase Assay Kit 2.0 (Biotium, Fremont, CA, US), following the manufacturer’s instructions, and normalized to the number of viable cells, as measured by the CellTiter-Fluor Cell Viability Assay (Promega, Madison, WI, US). For cell uptake studies, Cy5- or Cy5.5-labeled op-shRNA was synthesized by adding 1 μL Cy5-uridine-5’-triphosphate (Enzo Life Sciences, Inc., Farmingdale, NY) or 5-aminoallylcytidine-5’-triphosphate (TriLink Biotechnologie, San Diego, CA), respectively, was added to each 200 μL RCT reaction. Cy5.5 N-hydroxysuccinimide ester (Lumiprobe, Hunt Valley, MD) was subsequently added to the amine-labeled p-shRNA to make Cy5.5-labeled p-shRNA, followed by washing via centrifugal filtration to remove excess dye, before treating with RNase T1. Cy5- or Cy5.5-labeled op-shRNA complexed with poly-1 were transfected into HeLa-GFP or B16F10 cells, respectively, in a 96-well plate. Cy5 or Cy5.5 fluorescence was measured 24 hours post-transfection by flow cytometry using a BD FACSCalibur or BD FACSCelesta.
Cell proliferation assays.
Cells were transfected as described above, and at 48 hours post-transfection, were measured for mitochondrial dehydrogenase activity in an MTT assay. Cells were incubated with MTT (0.5 mg/mL) for 1 hour at 37 °C, followed by solubilization of the purple MTT product in dimethyl sulfoxide. Absorbance was measured at 570 nm (reference 630 nm) with a Tecan plate reader.
Confocal microscopy.
20,000 B16F10 cells were seeded in 8-well chamber slides and transfected as described above with 50 nM Cy5.5-labeled op-shRNA complexed with poly-1 and PEG-poly-1. At 24 hours post-transfection, cells were fixed with 4% paraformaldehyde and permeablized with 0.4% Triton-X, followed by overnight staining with anti-Rab7 (Cell Signaling, Catalog #9367, 1:200 dilution) at 4 °C. Secondary antibody staining was performed with Alexa Fluor 488-conjugated anti-rabbit IgG, F(ab’)2 fragment (Cell Signaling, Catalog #4412, 1:2000 dilution) for 1 hour at room temperature, followed by DAPI staining before imaging.
In vitro innate immune activation.
10,000 RAW-Blue™ cells were seeded in each well of a 96-well plate and transfected as described above with 1 μg/mL op-shRNA, high molecular weight poly(I:C) (InvivoGen), or siRNA complexed with poly-1 and PEG-poly-1 (poly-1:PEG-poly-1:RNA w/w = 10:10:1). At 24 hours post-transfection, expression of secreted embryonic alkaline phosphatase was measured by the QUANTI-Blue™ assay according to the manufacturer’s instructions.
Quantitative PCR.
Transfections of anti-STAT3 and scrambled op-shRNA were performed as described above in B16F10 cells. Total RNA was extracted 48 hours post-transfection, by an RNeasy Plus Mini kit (Qiagen, Hilden, Germany) and then converted to cDNA using an Ecodry cDNA synthesis kit (Clontech, Mountain View, CA, US). The cDNA was amplified with a LightCycler® 480 SYBR Green I Master reagent and quantified by a Roche LightCycler 480 Real-Time PCR System. Primer sequences used for detection were: mouse actin forward: tggcgcttttgactcaggat; mouse actin reverse: gggatgtttgctccaaccaa; mouse STAT3 forward: ggatcgctgaggtacaaccc; mouse STAT3 reverse: gtcaggggtctcgactgtct A common 2−ΔΔCT method was applied to data with automatic removal of background fluorescence by the qPCR-associated software.
Western blot.
48 hours after transfection, cells were lysed in a lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% Sodium Deoxycholate, 1 mM EDTA, 0.1% SDS, protease inhibitors). Proteins were separated by 4–15% SDS-PAGE and subsequently transferred to a nitrocellulose membrane (Fisher Scientific). The membranes were incubated overnight at 4 °C with the primary antibodies anti-tubulin (clone G-8, 1:1000, Santa Cruz Biotechnology, Dallas, TX, US), anti-phosphorylated STAT3 (Tyr705, clone D3A7, 1:2000, Cell Signaling) and anti-STAT3 (clone 124H6, 1:2000, Cell Signaling) in 5% bovine serum albumin in TBST buffer (25 mM Tris pH 7.4, 150 mM NaCl, 2.5 mM KCl, 0.05% Tween-20) at 4°C overnight, followed by a one-hour incubation with secondary horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG (Santa Cruz) and extensive washes with TBS containing 0.05% Tween-20. Phosphorylated STAT3, total STAT3, and tubulin proteins were detected by enhanced chemiluminescence (Pierce).
Animal experiments.
Animal studies were carried out in accordance with established institutional guidelines and protocols approved by the MIT Divison of Comparative Medicine. Seven-to-eight-week-old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, US) were inoculated subcutaneously with 0.5 million B16F10 cells. Starting at 7 days post-inoculation, when the tumors had grown to an average size of 3 mm, the mice received intratumoral injections of 5 μg op-shRNA or siRNA complexed with poly-1 and PEG-poly-1 (10:10 polymer:RNA w/w). Complexes were formed as in in vitro transfections, at higher concentrations for a final injection volume of 40 μL after diluting to 5% glucose. Injections were performed every three days for a total of four times for the survival study. A vehicle control group receiving only acetate buffer with 5% glucose was included. Tumor size measurements for each treatment group were taken before each treatment on the same day, until several mice in the control groups needed to be euthanized due to tumor burden. Tumor volume was calculated by V = (L × W × W)/2, where V is tumor volume, W is tumor width, and L is tumor length. Survival was monitored until the last mouse was euthanized, and treatment efficacies were assessed by plotting a Kaplan-Meier survival curve. The number of mice in each treatment group were n=8 for anti-STAT3 op-shRNA, scrambled op-shRNA, and vehicle, and n=7 for anti-STAT3 siRNA and scrambled siRNA. Mice were euthanized when tumor size reached over 1 cm in diameter.
Immunohistochemistry staining.
B16F10 tumors were isolated from C57BL/6 mice 48 hours after the last of two treatments of op-shRNA or siRNA (10 μg) complexed with poly-1/PEG-poly-1. Tumors were formalin-fixed for 24 hours and then paraffin-embedded. Heat-induced epitope retrieval was carried out for all formalin-fixed paraffin embedded slides at 97 °C for 20 minutes in citrate buffer, pH 6. Staining was performed by an automated machine (ThermoScientific IHC Autostainer 360) via the following steps: endogenous peroxidase blocking for 10 minutes, protein blocking for 30 minutes, primary antibody incubation for 60 minutes, labeled polymer for 15 minutes, and DAB for 10 minutes. Species-specific polymer was used as a secondary antibody (Biocare Medical, Pacheco, CA, US). The Swanson Biotechnology Center at the Koch Institute (MIT) performed the entire IHC procedure. Primary antibodies used for staining were STAT3 (Cell Signaling, Catalog #9139, 1:600 dilution), Ki-67 (BD Biosciences, Catalog #550609, 1:50 dilution), and cleaved caspase-3 (Cell Signaling, Catalog #9664, 1:100 dilution), Images were taken by a Leica Aperio Slide Scanner (Leica Biosystems, Buffalo Grove, IL, US) and processed using ImageScope (Leica Biosystems).
Statistical analysis.
All statistical analyses were performed with GraphPad Prism 5.0. Statistical significance was determined by Student’s t test or a one-way ANOVA followed by a Tukey post test.
Supplementary Material
Acknowledgements
This work was supported by the Congressionally Directed Medical Research Program (CDMRP) Ovarian Cancer Research Program Teal Innovator Award from the Department of Defense (DoD) (13–1-0151, PTH), the National Science Foundation Graduate Research Fellowship (CW), the Koch Institute Ludwig Center for Molecular Oncology Graduate Fellowship (CW), and the Koch Institute Quinquennial Postdoctoral Fellowship (JL). We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the flow cytometry, histology, nanotechnology materials, and animal imaging and preclinical testing facilities. We also thank Dr. Erik Dreaden and Dr. Mohi Quadir for valuable discussions, and Dr. John Martin for assistance with polymer characterization.
ASSOCIATED CONTENT
Supporting Information.
Additional figures, including physiochemical characterization of complexes, cytotoxicity, cellular uptake, in vitro immunostimulatory activity, materials characterization, and DNA template and siRNA sequences. The Supporting Information is available free of charge on the Internet at https://pubs.acs.org.
References
- 1.Wittrup A; Lieberman J Knocking Down Disease: a Progress Report on siRNA Therapeutics. Nat. Rev. Genet 2015, 16, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorenzer C; Dirin M; Winkler A-M; Baumann V; Winkler J Going beyond the Liver: Progress and Challenges of Targeted Delivery of siRNA Therapeutics. J. Controlled Release 2015, 203, 1–15. [DOI] [PubMed] [Google Scholar]
- 3.Kim HJ; Kim A; Miyata K; Kataoka K Recent Progress in Development of siRNA Delivery Vehicles for Cancer Therapy. Adv. Drug Delivery Rev 2016, 104, 61–77. [DOI] [PubMed] [Google Scholar]
- 4.Li J; Wu C; Wang W; He Y; Elkayam E; Joshua-Tor L; Hammond PT Structurally Modulated Codelivery of siRNA and Argonaute 2 for Enhanced RNA Interference. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 2696–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozielski KL; Tzeng SY; Green JJ Bioengineered Nanoparticles for siRNA Delivery. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2013, 5, 449–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gary DJ; Puri N; Won Y-Y Polymer-Based siRNA Delivery: Perspectives on the Fundamental and Phenomenological Distinctions from Polymer-Based DNA Delivery. J. Controlled Release 2007, 121, 64–73. [DOI] [PubMed] [Google Scholar]
- 7.Scholz C; Wagner E Therapeutic Plasmid DNA versus siRNA Delivery: Common and Different Tasks for Synthetic Carriers. J. Controlled Release 2012, 161, 554–565. [DOI] [PubMed] [Google Scholar]
- 8.Shopsowitz KE; Wu C; Liu G; Dreaden EC; Hammond PT Periodic-shRNA Molecules are Capable of Gene Silencing, Cytotoxicity and Innate Immune Activation in Cancer Cells. Nucleic Acids Res 2016, 44, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C; Shopsowitz KE; Hammond PT Engineering Periodic shRNA for Enhanced Silencing Efficacy. Mol. Ther 2016, 24, 1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seyhan AA; Vlassov AV; Johnston BH RNAi from Multimeric shRNAs Generated by Rolling Circle Transcription. Oligonucleotides 2006, 363, 353–363. [DOI] [PubMed] [Google Scholar]
- 11.Lynn DM; Langer R Degradable Poly (β-amino esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA. J. Am. Chem. Soc 2000, 122, 10761–10768. [Google Scholar]
- 12.Akinc A; Anderson DG; Lynn DM; Langer R Synthesis of Poly(beta-amino ester)s Optimized for Highly Effective Gene Delivery. Bioconjugate Chem 2003, 14, 979–988. [DOI] [PubMed] [Google Scholar]
- 13.Anderson DG; Lynn DM; Langer R Semi-Automated Synthesis and Screening of a Large Library of Degradable Cationic Polymers for Gene Delivery. Angew. Chem., Int. Ed 2003, 42, 3153–3158. [DOI] [PubMed] [Google Scholar]
- 14.Lynn DM; Anderson DG; Putnam D; Langer R Accelerated Discovery of Synthetic Transfection Vectors: Parallel Synthesis and Screening of a Degradable Polymer Library. J. Am. Chem. Soc 2001, 123, 8155–8156. [DOI] [PubMed] [Google Scholar]
- 15.Eltoukhy AA; Siegwart DJ; Alabi CA; Rajan JS; Langer R; Anderson DG Effect of Molecular Weight of Amine End-Modified Poly(β-amino ester)s on Gene Delivery Efficiency and Toxicity. Biomaterials 2012, 33, 3594–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozielski KL; Tzeng SY; Hurtado de Mendoza BA; Green JJ Bioreducible Cationic Polymer-Based Nanoparticles for Efficient and Environmentally Triggered Cytoplasmic siRNA Delivery to Primary Human Brain Cancer Cells. ACS Nano 2014, 8, 3232–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jere D; Xu C-X; Arote R; Yun C-H; Cho M-H; Cho C-S Poly(beta-amino ester) as a Carrier for si/shRNA Delivery in Lung Cancer Cells. Biomaterials 2008, 29, 2535–2547. [DOI] [PubMed] [Google Scholar]
- 18.Sunshine J; Green JJ; Mahon KP; Yang F; Eltoukhy AA; Nguyen DN; Langer R; Anderson DG Small-Molecule End-Groups of Linear Polymer Determine Cell-Type Gene-Delivery Efficacy. Adv. Mater 2009, 21, 4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bishop CJ; Ketola T-M; Tzeng SY; Sunshine JC; Urtti A; Lemmetyinen H; Vuorimaa-Laukkanen E; Yliperttula M; Green JJ The Effect and Role of Carbon Atoms in PBAEs for DNA Binding and Gene Delivery. J. Am. Chem. Soc 2013, 135, 6951–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu H; Lee H; Herrmann A; Buettner R; Jove R Revisiting STAT3 Signalling in Cancer: New and Unexpected Biological Functions. Nat. Rev. Cancer 2014, 14, 736–746. [DOI] [PubMed] [Google Scholar]
- 21.Miklossy G; Hilliard TS; Turkson J Therapeutic Modulators of STAT Signalling for Human Diseases. Nat. Rev. Drug Discovery 2013, 12, 611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H; Kortylewski M; Pardoll D Crosstalk between Cancer and Immune Cells : Role of STAT3 in the Tumour Microenvironment. Nat. Rev. Immunol 2007, 7, 41–51. [DOI] [PubMed] [Google Scholar]
- 23.Eltoukhy AA; Chen D; Alabi CA; Langer R; Anderson DG Degradable Terpolymers with Alkyl Side Chains Demonstrate Enhanced Gene Delivery Potency and Nanoparticle Stability. Adv. Mater 2013, 25, 1487–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaczmarek JC; Patel AK; Kauffman KJ; Fenton OS; Webber MJ; Heartlein MW; DeRosa F; Anderson DG Polymer-Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem., Int. Ed 2016, 55, 13808–13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzeng SY; Hung BP; Grayson WL; Green JJ Cystamine-Terminated Poly(beta-amino ester)s for siRNA Delivery to Human Mesenchymal Stem Cells and Enhancement of Osteogenic Differentiation. Biomaterials 2012, 33, 8142–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong CA; Eltoukhy AA; Lee H; Langer R; Anderson DG; Nam YS Dendrimeric siRNA for Efficient Gene Silencing. Angew. Chem., Int. Ed 2015, 54, 6740–6744. [DOI] [PubMed] [Google Scholar]
- 27.Suk JS; Xu Q; Kim N; Hanes J; Ensign LM PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Delivery Rev 2016, 99, 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alshamsan A; Hamdy S; Samuel J; El-Kadi AOS; Lavasanifar A; Uludağ H The Induction of Tumor Apoptosis in B16 Melanoma Following STAT3 siRNA Delivery with a Lipid-Substituted Polyethylenimine. Biomaterials 2010, 31, 1420–1428. [DOI] [PubMed] [Google Scholar]
- 29.Yu M; Levine SJ Toll-like Receptor 3, RIG-I-like Receptors and the NLRP3 Inflammasome: Key Modulators of Innate Immune Responses to Double-stranded RNA Viruses. Cytokine Growth Factor Rev 2011, 22, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bianchi F; Pretto S; Tagliabue E; Balsari A; Sfondrini L Exploiting Poly(I:C) to Induce Cancer Cell Apoptosis. Cancer Biol. Ther 2017, 18, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corrales L; Matson V; Flood B; Spranger S; Gajewski TF Innate Immune Signaling and Regulation in Cancer Immunotherapy. Cell Res 2017, 27, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kortylewski M; Swiderski P; Herrmann A; Wang L; Kowolik C; Kujawski M; Lee H; Scuto A; Liu Y; Yang C; Deng J; Soifer HS; Raubitschek A; Forman S; Rossi JJ; Pardoll DM; Jove R; Yu H In Vivo Delivery of siRNA to Immune Cells by Conjugation to a TLR9 Agonist Enhances Antitumor Immune Responses. Nat. Biotechnol 2009, 27, 925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poeck H; Besch R; Maihoefer C; Renn M; Tormo D; Morskaya SS; Kirschnek S; Gaffal E; Landsberg J; Hellmuth J; Schmidt A; Anz D; Bscheider M; Schwerd T; Berking C; Bourquin C; Kalinke U; Kremmer E; Kato H; Akira S; Meyers R; Hacker G; Neuenhahn M; Busch D; Ruland J; Rothenfusser S; Prinz M; Hornung V; Endres S; Tüting T; Hartmann G 5’-Triphosphate-siRNA: Turning Gene Silencing and Rig-I Activation against Melanoma. Nat. Med 2008, 14, 1256–1263. [DOI] [PubMed] [Google Scholar]
- 34.Garzon R; Marcucci G; Croce CM Targeting microRNAs in Cancer: Rationale, Strategies and Challenges. Nat. Rev. Drug Discovery 2010, 9, 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith RC; Leung A; Kim B-S; Hammond PT Hydrophobic Effects in the Critical Destabilization and Release Dynamics of Degradable Multilayer Films. Chem. Mater 2009, 21, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.