

Abstract
Successful pregnancy requires carefully-coordinated communications between the mother and fetus. Immune cells and cytokine signaling pathways participate as mediators of these communications to promote healthy pregnancy. At the same time, certain infections or inflammatory conditions in pregnant mothers cause severe disease and have detrimental impacts on the developing fetus. In this review, we examine evidence for the role of maternal and fetal immune responses affecting pregnancy and fetal development, both under homeostasis and following infection. We discuss immune responses that are necessary to promote healthy pregnancy and those that lead to congenital disorders and pregnancy complications, with a particular emphasis on the role of interferons and cytokines. Understanding the contributions of the immune system in pregnancy and fetal development provides important insights into the pathogenesis underlying maternal and fetal diseases and sheds insights on possible targets for therapy.
Keywords: interferon, cytokines, placenta, congenital diseases, autoimmunity, infections, innate immunity, maternal health, birth defect
Introduction
By the age of 5, approximately 8% of children are diagnosed with some form of birth defects (Moore et al., 2017b). Genetic abnormalities and environmental exposures such as smoking, alcohol, medications, and infections in utero are important causes of birth defects. However, the majority of birth defects (approximately 50–60%) have no known genetic or environmental causes (Moore et al., 2017b). The rate of failed pregnancies and pregnancies with complications is also remarkably high. The early stages of pregnancy have the high rate of failure of approximately 30% (Wilcox et al., 1988). About half of early pregnancy loss is due to abnormal chromosome numbers caused by meiotic failure, but many have no clear explanations (Moore et al., 2017b). Intrauterine growth restriction (IUGR) and preterm birth, the leading causes of perinatal mortality, are found in nearly 10% of pregnancies, each (Nardozza et al., 2017; Purisch and Gyamfi-Bannerman, 2017).
The immune system evolved to protect the host against invading pathogens. Protection of the developing fetus is critical for the success of a species. In mammals, immune protection of the fetus must be carried out without harming the mother harboring the fetus. Conversely, mothers carrying a semiallogeneic fetus need to tolerate and prevent immune-mediated damage of the fetus while protecting against pathogens. Thus, pregnancy is accompanied by dynamic alterations in the maternal and fetal immune responses dependent on the stage of pregnancy or development – a topic covered by previous reviews (Kollmann et al., 2017; Pazos et al., 2012). For species that spend a long gestational period with each pregnancy, the quality of the fetus must be ensured for its survival and reproductive success. Yet, the checkpoint mechanisms for ensuring quality control for the developing fetus is not well understood.
In this review, we examine the role of the immune system in both promoting successful pregnancy and in mediating congenital disorders and complications. Signaling pathways engaged by cytokines have a powerful ability to rapidly remodel tissue and alter cellular behavior. For example, upon binding to their receptor, the interferons (IFNs) induce hundreds of effectors, or interferon stimulated genes (ISGs), that act to restrict viral infections (Sadler and Williams, 2008). There are three types of IFNs: Type I IFNs (including IFN-α, β, ε, τ, and δ), type II IFN (IFN-γ), and type III IFNs (IFN- λ1, λ2 and λ3) (Sadler and Williams, 2008). These IFNs differ in both the cellular source and cellular targets based on receptor expressions (Schneider et al., 2014). Many of these IFNs are employed in normal pregnancy and development, as well as in defense against pathogens. Alterations or aberrant activation of immune responses during pregnancy can lead to pregnancy complications and congenital abnormalities. In this review, we put forth the hypothesis that aberrant expression of cytokines and IFNs is a common and critical mediator of congenital disorders and fetal loss that occurs in the settings of infections, chromosomal abnormalities, metabolic and autoimmune diseases.
Pregnancy complications during implantation and placental development
All stages of pregnancy, from implantation to parturition (Fig. 1), are susceptible to complications that result from fetal-intrinsic or extrinsic aberrations. Implantation and placental development are key vulnerability points during which pregnancy loss and complications arise. Implantation occurs at about day 8–9 in humans and day 4–5 after conception in mice. Just after hatching from the zona pellucida, the blastocyst, which is surrounded by a layer of trophoectoderm, adheres to the uterine epithelium (Aplin and Ruane, 2017) (Fig. 1A). This initial attachment is followed by trophoblast differentiation and invasion of the endometrium. There are high rates of implantation failure, both after natural conception and with in vitro fertilization treatments (Aplin and Ruane, 2017), which may be due to genetic abnormalities and failure to hatch, and maternal factors, including endometrial abnormalities, immunological imbalance at the endometrium, and abnormal blood clotting (Simon and Laufer, 2012).
Figure 1: Immune responses necessary for healthy pregnancy.
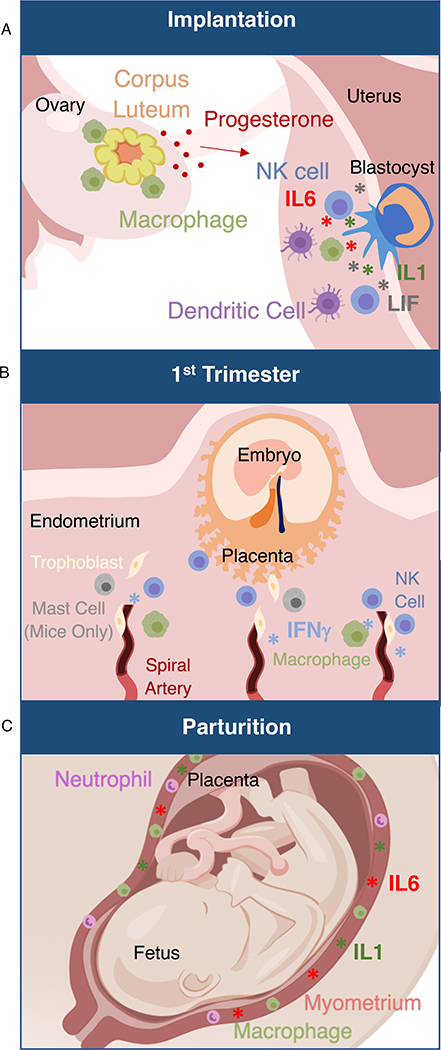
(A) The blastocyst attaches to and invades the maternal uterine endometrium. This process is accompanied by an evolutionary conserved inflammatory response including IL-6, IL-1, and LIF. Key cell types necessary for early pregnancy, as demonstrated by mouse models, include dendritic cells, which are essential for decidualization, and macrophages, which are essential for maintenance of the corpus luteum in the ovary. NK cells directly surround the trophoblasts after implantation.
(B) During development of the placenta in the first trimester, immune cells including NK cells and mast cells are necessary for uterine spiral artery remodeling, as demonstrated by mouse models. The cytokine IFN-γ is also necessary for this process in mouse models.
(C) Parturition is mediated by an inflammatory response. Macrophages and neutrophils infiltrate the uterine myometrium. IL-1β is secreted and induces muscle contraction. IL6 is necessary for on-time parturition in mouse models.
The first trimester is the critical period of placental development, and the origins of many common pregnancy complications can be traced back to abnormal placental development in the first trimester (Kroener et al., 2016). The two key processes of placental development are the villous and extravillous trophoblast formations. The chorionic villi are the site of gas and nutrient exchange between the maternal blood and fetal blood. This exchange occurs at the interface between fetal endothelial cells, which run through the core of the villi, and the maternal blood, in which the villi are bathed (Fig. 2A). The villi are covered with a layer of multinucleated fused syncytiotrophoblasts (SYN), which directly contact the maternal blood. Abnormal villous development can lead to early pregnancy loss, IUGR, and preeclampsia (defined clinically as elevated blood pressure and proteinuria or other signs of organ damage including impaired liver function, renal insufficiency, cerebral/ visual symptoms, or thrombocytopenia) (Huppertz et al., 2014).
Figure 2: Antimicrobial protective mechanisms at the placenta and decidua.
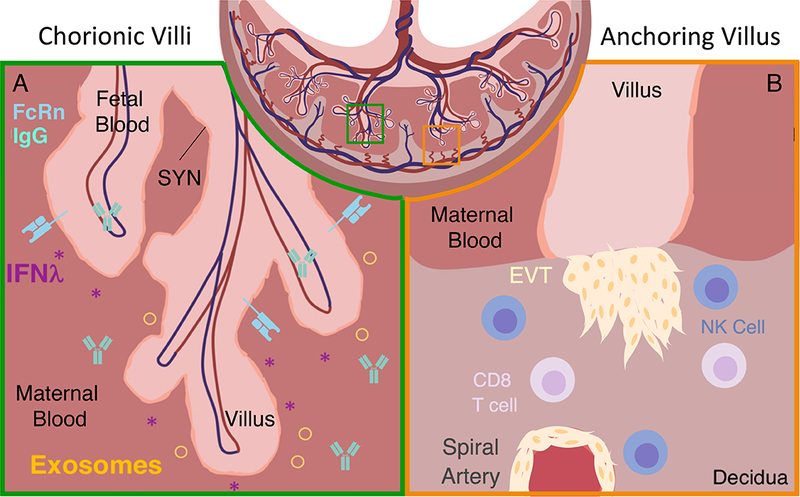
A) At the chorionic villi and intervillous space, syncytiotrophoblasts (SYN) constitutively express IFNλ and exosomes, which confer resistance to viruses and other known TORCH pathogens to SYNs and neighboring cells. Maternal IgG is transferred to the fetus using FcRn receptors starting at 13 weeks of gestation in humans.
B) At the anchoring villi and decidua, NK cells are capable of killing virus-infected cells and are located in close proximity to potential portals of viral entry. NK cells surround extravillous trophoblasts (EVTs) and spiral arteries. Pathogen specific CD8 T cells are enriched in the human decidua. They have the potential to be cytotoxic upon ex-vivo stimulation.
The fetal extravillous trophoblasts invade the decidua and uterine myometrium, infiltrating the uterine vessels and glands to direct nutrients to the developing fetus (Fig 2B) (Moser and Huppertz, 2017). At the spiral arteries, they remodel the smooth muscles and elastic fibers and ultimately replace the endothelium of the blood vessels (Tessier et al., 2014). By 10–12 weeks, maternal blood is directed to the villous space, whereby nutrient and gas exchange between the mother and fetus occurs. Insufficient maternal artery remodeling can lead to preeclampsia, IUGR, and recurrent pregnancy loss (Tessier et al., 2014). Conversely, excessive placental invasion is associated with maternal anemia and postpartum hemorrhage (Tantbirojn et al., 2008; Tessier et al., 2014). We will discuss how immune cells and immune effectors play an important role in in regulating the extravillous trophoblast invasion, and inflammatory insults such as infections can disrupt placental development.
Mechanisms that protect the embryo against microbial threats
Despite some of the devastating effects of certain congenital infections, which we will discuss later, most pathogens are not able to reach the fetus (Arora et al., 2017; Robbins and Bakardjiev, 2012). The placenta provides a powerful physical barrier that protects the fetus against infection by pathogens. The SYNs are multinucleated cells that make up the barrier between maternal and fetal blood in the placenta (Arora et al., 2017). Upon differentiation, SYNs become highly resistant to viral infections: they do not express the entry receptors for viral pathogens including herpes simplex virus (HSV) and cytomegalovirus (CMV), and they have a dense cytoskeleton network of actin that help them to physically resist bacterial invasion such as Listeria monocytogenes (Robbins and Bakardjiev, 2012; Zeldovich et al., 2013). SYNs also constitutively produce exosomes and type III IFNs (IFN-λ) which confer antiviral resistance within themselves and in surrounding cells (Fig. 2A) (Bayer et al., 2016; Delorme-axford et al., 2013; Jagger et al., 2017). An alternative route by which pathogens may reach the fetus is from the uterus - infecting the extravillous trophoblasts and invading the anchoring villi (Tabata et al., 2016). Blocking this route of pathogen entry likely requires a different set of immunoprotective mechanisms. Effector-memory CD8+ T cells are present in the human endometrium, and some of these are pathogen-specific (van Egmond et al., 2016). While they are generally less cytotoxic than their peripheral counterparts, decidual natural killer (NK) cells are capable of killing human cytomegalovirus (HCMV)- infected cells, and decidual CD8+ T cells can degranulate and proliferate after in vitro activation (Fig. 2B) (Siewiera et al., 2013; van der Zwan et al., 2018). The mechanisms by which some pathogens are able to reach the fetus despite these barriers are still poorly understood.
In addition to providing a barrier to infection, the maternal immune response may provide further protection at the fetus. Maternal IgG antibodies are directly transferred to the fetus by FcRn receptors expressed by SYNs (Jennewein et al., 2017) (Fig. 2A). IgG transfer begins around 13 weeks of gestation, the beginning of the second trimester in humans (Palmeira et al., 2012). Whether transfer of innate inflammatory mediators from the mother to the fetus occurs is less well understood. Ex-vivo studies of term placentas indicate that transplacental transfer of most cytokines does not occur (Aaltonen et al., 2005). However, chronic maternal infection by human immunodeficiency virus 1 (HIV-1) and hepatitis B can lead to higher cytokine concentrations in the cord blood and altered fetal immune responses, indicating that the maternal immune responses can influence the fetus, perhaps due to placental cytokine production (Bunders et al., 2014; Hong et al., 2015). As we will discuss later, maternal IL-17 can directly affect fetal brain development (Choi et al., 2016).
The developing embryo also has cell-intrinsic mechanisms to protect itself from infection. One intriguing demonstration of this is that pluripotent stem cells, including embryonic stem cells, are resistant to viral infection due to constitutive expression of a subset of interferon stimulated genes (ISGs), including IFITM1 and IFITM3 (Wu et al., 2017). The developing embryo must not only protect itself from exogenous pathogens but also against endogenous genomic threats. Transposable elements, which make up 40% of the genome in mice and 44% of the genome in humans, must be actively suppressed to maintain genomic integrity (Rowe and Trono, 2011). The transposable elements can be classified as long terminal repeat (LTR) elements, including endogenous retroviruses (ERVs), and non-LTR elements, including long interspersed nuclear elements (LINEs). Gametes and early embryos are susceptible to transposable elements reactivation as the genome is globally demethylated during genome reprogramming (Rowe and Trono, 2011). PIWI-associated RNAs (piRNAs) are essential for suppressing transcription of transposable elements, particularly in the gonads and embryo (Siomi et al., 2011). piRNAs are 23–30 nucleotide small silencing RNAs that interact with the PIWI proteins, PIWI, AUB, and AGO3 to cleave their target mRNAs and impose transcriptional silencing by DNA methylation (Siomi et al., 2011). Loss of PIWI protein expression results in upregulation of transposable elements, subsequent double stranded DNA breaks, and a loss of fertility (Klattenhoff et al., 2007; Siomi et al., 2011). In Drosophila, PIWI protein expression is required for both oogenesis and spermatogenesis (Lin and Spradling, 1997). However, in mice, PIWI proteins are required for only spermatogenesis, and piRNA expression is low during oogenesis (Carmell et al., 2007). Moreover, expression of piRNAs and PIWI proteins in humans, macaques, and cows indicates that there may be a species-specific role for piRNAs in mammalian oogenesis (Roovers et al., 2015). ERVs are a collection of previously integrated exogenous retroviruses that accumulate over time in the genome of vertebrates. Recent studies identify a class of small RNAs, tRNA-derived fragments (tRFs), as another small RNA-mediated mechanism of suppressing ERV retrotransposition in the mouse pre-implantation embryo (Schorn et al., 2017). Two distinct classes of tRFs block translation and reverse transcription of retroviral RNAs (Schorn et al., 2017). Specific epigenetic silencing mechanisms are employed for ERV silencing during embryogenesis. Embryonic stem cells use ZFP809 to silence retroviral proviral transcription (Wolf and Goff, 2009). Other KRAB-ZFPs are also likely involved specifically during embryonic development in recruiting epigenetic machinery to silence ERV (Wiznerowicz et al., 2007). Thus, ISGs, epigenetic- and small RNA-mediated repression orchestrates intrinsic antiviral control in early stages of embryo development.
Immune pathways and cell types that promote normal pregnancy
Emerging evidence indicates a central role of the immune system in mediating successful pregnancy, from implantation to parturition. We will discuss a few examples of immune-mediated pregnancy processes. Implantation elicits an inflammatory reaction, where upregulation of inflammatory cytokines, including interleukin-6 (IL-6), IL-1, and leukemia inhibitory factor (LIF), are highly conserved across the therian mammals, including placental mammals and marsupials (Griffith et al., 2017) (Fig. 1A). LIF expression in the mother is essential for implantation in mice (Stewart et al., 1992). Various leukocytes are found in the decidua, including maternal NK cells, DCs, macrophages and lymphocytes (Liu et al., 2017). These cells directly interact with trophoblasts of the developing placenta, and deviations in cell numbers and functions are associated with pregnancy loss, preeclampsia, and preterm birth (Zenclussen and Hammerling, 2015). CD11 b+ macrophages are necessary for the maintenance of the corpus luteum in the ovary, which maintains early pregnancy by the secretion of progesterone (Fig. 1A): depletion of macrophages using CD11b-DTR mice results in failure of early implantation that can be rescued by progesterone (Care et al., 2013). Additionally, in vitro evidence shows that macrophages and trophoblasts interact closely and reciprocally regulate each other: macrophages promote trophoblast survival and differentiation, and trophoblasts regulate monocyte migration and cytokine production (Fest et al., 2007; Rozner et al., 2016). Dendritic cells (DCs) are also essential for early events of pregnancy: depletion of these cells using CD11c-DTR in mice leads to embryo resorption due to a failure of the decidua to develop, including impaired angiogenesis and decidual cell proliferation (Plaks et al., 2008). TGFβ and sFlt1 are two factors produced predominately by DCs and may be responsible for the impaired angiogenesis seen in the CD11 c-DTR mice. These functions are independent of the immune tolerance function of DCs, as the early implantation failure occurs in syngeneic pregnancy and when T cells are depleted. Thus, mouse models demonstrate that macrophages and DCs play distinct and essential roles in early pregnancy (Fig. 1A, B). While they are present in human pregnancy, whether they have similar roles in humans is unknown.
Remodeling of the maternal spiral arteries also requires immune cells. NK cells make up the predominant leukocyte population in the human decidua, and the NK cells are located in close proximity to the invading extravillous trophoblasts and surrounding the spiral arteries (Liu et al., 2017) (Fig. 1B). Mice that lack NK cells, IFN-γ, IFN-γ receptor (IFNGR), or STAT1 (transcription factor downstream of IFNγ receptor), do not have spiral artery remodeling, indicating an essential role for NK cells and IFNγ in this process (Ashkar et al., 2000). Mice lacking type I IFN receptor (IFNAR) also lack spiral artery remodeling, indicating a non-redundant roles of type I and type II IFNs (Murphy et al., 2009). Despite the abnormal spiral artery remodeling, pups of mice lacking IL-15, NK cells, IFNGR, or IFNAR have only mild growth restriction, but pregnancies are otherwise minimally affected (Barber and Pollard, 2003; Murphy et al., 2009). Uterine NK cells are essential for control of trophoblast invasion and produce a number of angiogenic factors (Hanna et al., 2006). A recent study also shows that a subset of NK cells may directly interact with the developing fetus by secreting growth-promoting factors including peiotrophin, osteoglycin, and osteopontin (Fu et al., 2017). Additionally, the Kitwsh/wsh mice, which are deficient in mast cells, have abnormal implantation sites and lack spiral artery remodeling (Woidacki et al., 2013). NK cells and mast cells are key regulators of uterine spiral artery remodeling, as demonstrated in mouse studies, a process that becomes aberrant in many pregnancy complications.
Interferons play central roles in pregnancy in distinct species. A prime example of this is the role of interferon-τ (IFNτ), a unique type I IFN, that is essential for pregnancy recognition in ungulates (Roberts, 2007). IFNτ is expressed by the early trophoectoderm and acts as a hormone of pregnancy recognition to rescue the corpus luteum. It is also thought to be important for implantation. While IFNτ expression is controlled transcriptionally by Ets2 and not by the IRF transcription factors downstream of viral sensors, it binds to IFNAR and has antiviral activity (Ealy et al., 2001). IFNα exposure of the endometrium in ewes is sufficient to prolong the estrous cycle with similar efficacy as IFNτ (Green et al., 2005). While no equivalent of IFNτ exists in humans, some IFNs are upregulated during implantation, indicating that there may be a common role for IFNs in some aspects of implantation (Roberts, 2007).
Post implantation, the maternal-fetal interface undergoes dynamic immunological changes (Prabhudas et al., 2015). Decidual stromal cells (DSCs) are a defining cell type in species with extended gestational periods and are critical for maintaining the unique immune environment of the decidua: they silence the chemokines CXCL9 and CXCL10 which recruit cytotoxic T cells, and they secrete IL-15 which is responsible for converting decidual NK cells and macrophages to decidual phenotypes, including reduced cytotoxicity and secretion of angiogenic factors (Ashkar et al., 2003; Chavan et al., 2016). Additional mechanisms exist in the uterus to prevent anti-fetal immune responses. DCs in the decidua are restricted from migrating to the lymph node (LN), likely due to their unresponsiveness to CCL21, the chemokine responsible for their migration to the LN (Collins et al., 2009). Meanwhile, regulatory T (Treg) cells expand during pregnancy in both humans and allogeneic mouse models, and these Treg cells have been shown to be fetus-specific in mouse models (Aluvihare et al., 2004; Feyaerts et al., 2017). Partial and transient ablation of Treg cells at midgestation is sufficient to lead to expansion of fetus-specific CD4 and CD8 T cells and loss of allogenic fetuses in mice (Rowe et al., 2011). Thus, the specialized local microenvironment of the placenta and developing fetus promotes tolerance to fetal antigens and downregulation of specific chemokines and cytokines.
Parturition is another key event in pregnancy that utilizes the immune system and is associated with a proinflammatory environment (Fig. 1C). Parturition during normal term pregnancy is associated with neutrophil and macrophage infiltration into the myometrium, an upregulation of inflammatory cytokines including IL-1β and IL-8 in the maternal-fetal tissues, and transcriptional changes in the decidua including upregulation of TNF signaling pathway, Nod-like receptors, and NFκB activation (Christiaens et al., 2008; Rinaldi et al., 2017). Cytokines including IL-1β may directly induce smooth muscle contraction in the uterus: in vitro studies show that it can induce calcium influx into myometrial smooth muscle, phosphodiesterase activity, and prostaglandin F2a production, all of which can contribute to muscle contraction (Oger et al., 2002; Tribe et al., 2003). Cervical remodeling and dilation are also accompanied by infiltrating leukocytes into the cervix (Yellon, 2017). Immunological changes in the decidua and myometrium may coordinate the timing of parturition, which requires integration of multiple signals from the mother and fetus (Menon et al., 2016). As seen during embryo implantation, uterine artery remodeling, and parturition, leukocytes and cytokines (Table 1) are essential mediators of coordinated tissue remodeling necessary for successful pregnancy.
Table 1:
Beneficial and pathological roles of cytokines in pregnancy
| Cytokine | Beneficial roles | Citation | Pathological roles | Citation |
|---|---|---|---|---|
| Type I IFNs | Pregnancy recognition in ungluates (IFN-τ) | (Roberts, 2007) | Block in placenta development after ZIKV infection | (Yockey et al., 2018) |
| Mediates uterine artery remodeling in mouse models | (Murphy et al., 2009) | Microcephaly and cranial calcifications after genetic overexpression, possibly viral infection | (Crow et al., 2015) | |
| Correlates with pregnancy complications in patients with SLE | (Andrade et al., 2015) | |||
| IFN-γ | Mediates uterine spiral artery remodeling | (Ashkar et al., 2000) | Prevents implantation and toxic to embryo | (Robertson et al., 2018) |
| Induces abnormal cerebellar development when overexpressed in mouse brains | (Wang et al., 2004) | |||
| Mediates placental damage after Malaria infection in mouse models | (Niikura et al., 2017) | |||
| Mediates fetal resorption after Toxplasma infection in mouse models | (Senegas et al., 2009) | |||
| IL-1β | Upregulated during implantation in an evolutionary- conserved manner | (Griffith et al., 2017) | Mediates abnormal lung development after peripartum inflammation in mouse models | (Hogmalm et al., 2014) |
| Mediator of myometrial contraction during labor | (Oger et al., 2002; Tribe et al., 2003) | Mediates neuronal damage due to inflammatory preterm birth in mouse models | (Leitner et al., 2014) | |
| Sufficient to induce | (Sadowsky et | |||
| preterm labor in non-human primate models | al., 2006) | |||
| IL-6 | Upregulated during implantation in an evolutionary- conserved manner | (Griffith et al., 2017) | Induces autism-like phenotype and abnormal brain development in mouse models | (Smith et al., 2007) |
| IL-15 | Required for uNK cell recruitment and uterine spiral artery remodeling in mouse models | (Ashkar et al., 2003) | ||
| IL-17 | Induces autism-like phenotype and abnormal brain development in mouse models | (Choi et al., 2016) | ||
| LIF | Upregulated during implantation in an evolutionary- conserved manner | (Griffith et al., 2017) | ||
| Required for successful implantation in mouse models | (Stewart et al., 1992) | |||
| TNFα | Toxic to early embryo development | (Robertson et al., 2018) | ||
| Sufficient to induce preterm labor in non-human primate models | (Sadowsky et al., 2006) | |||
| Sufficient to induce neural tube defects after systemic maternal injection | (Weldon Taubeneck et al., 1995) | |||
| Mediates fetal demise in mouse models of LPS and poly(I:C)-induced | (Carpentier et al., 2011; Thaxton et al., 2013) | |||
| fetal resorption. |
Host immune responses contribute to pregnancy complications associated with infections
The “TORCH” pathogens encompass a range of infectious agents that are known to cause congenital defects. They include protozoa (Toxoplasma gondii), bacteria (Listeria monocytogenes and Treponema pallidum), and viruses (rubella, CMV, HSV, varicella zoster virus, HIV, enteroviruses, parvovirus B19, and ZIKV) (Coyne and Lazear, 2016). In addition to congenital defects which we will discuss later, TORCH infections cause a number of different pregnancy complications including pregnancy loss, IUGR, and preterm birth (Cappelletti et al., 2017a; Goldenberg et al., 2010; Pereira et al., 2014) One of the newest TORCH pathogens, ZIKV, has raised public fear as it spread across the Americas in 2015–2017, resulting in over 3,000 estimated cases of microcephaly as well as miscarriage and fetal growth restriction (PAHO and WHO, 2017; Simões et al., 2016). Mouse models of ZIKV infection in pregnant dams have shown similar complications, with growth restriction, fetal demise, and placenta damage being most pronounced (Cugola et al., 2016; Miner et al., 2016; Szaba et al., 2018; Yockey et al., 2016). Recent studies in mouse and human tissue explant models indicate that other emerging neurotropic viruses related to ZIKV, including West Nile Virus and Powassan virus have the capacity to infect the placenta and fetal brain, leading to fetal demise (Platt et al., 2018). Thus, other viruses may have the potential to impact fetal development at an epidemic level.
Animal models of these infections indicate that pregnancy complications may be, at least in part, mediated by the immune response induced by the pathogen. A recent study has shown that type I IFN signaling through IFNAR within the fetus and fetal- derived placenta mediate the severe complications, including fetal demise and severe growth restriction following ZIKV infection of pregnant mice (Yockey et al., 2018). The detrimental effect of IFNAR signaling is mostly due to a block in placental development. Type II IFN, IFN-γ, may play a similar role: fetal resorption induced by Toxoplasma gondii infection is less severe in mice that lack IFNGR (Senegas et al., 2009). Infection can also lead to a break in the immune-tolerance towards the fetus. In mouse models of Listeria monocytogenes, infection is sufficient to recruit fetal-specific CD8+ T cells to the placenta and cause fetal demise (Chaturvedi et al., 2015). Thus, both innate and adaptive immune responses can contribute to fetal demise after congenital infection.
Malaria is another prevalent pathogen associated with poor pregnancy outcomes: women infected with P. falciparum or P. vivax are at an elevated risk of growth restriction and preterm birth (Moore et al., 2017a). Other studies show a correlation between poor pregnancy outcomes and systemic concentrations of cytokines and chemokines including TNF-α, IFN-γ, IL-10, and CXCL9 in women infected with malaria (Fried et al., 2017). Consistent with these findings, mouse models show that TLR4 signaling and IFN-γ signaling are mediators of abnormal development of the placenta labyrinth vasculature and fetal growth restriction caused by malaria infection (Barboza et al., 2017; Niikura et al., 2017). Thus, the inflammatory response to malaria appears to be a key mediator of malaria-related pregnancy complications.
Intrauterine infection and inflammation are important contributors to preterm birth, which is defined by birth before 37 weeks (Romero et al., 2014a). Around 25% of preterm births are associated with intrauterine infection, usually with vaginal microbes, indicating an ascending infection (Romero et al., 2014a). Extrauterine infections in the mother including pneumonia, pyelonephritis, and periodontal disease are also associated with preterm labor (Agrawal and Hirsch, 2012). Whether or not clinically-detectable infection is present, elevated concentrations of cytokines including IL-6, IL-1, IL-8, and TNF in amniotic fluid or in cervicovaginal lavage of patients is predictive of the onset of preterm labor (Agrawal and Hirsch, 2012; Holst et al., 2009). Recent studies have shown that a vaginal microbiome not dominated by Lactobacillus is associated with increased risk for preterm labor, but these findings are not consistent across all studies (Digiulio et al., 2015; Romero et al., 2014b). While the role of viral infections on preterm births are less clear, experimental evidence shows that coinfection with herpes viruses can lead to both ascending bacterial infection and an enhanced inflammatory responses to bacterial stimulation (Cross et al., 2017; Racicot et al., 2013). Collectively, emerging evidence indicates that the host immune response elicited by infection and possibly the vaginal microbiome of the pregnant mother may be an important contributor to the detrimental outcomes of congenital infections.
Innate immune activation is sufficient to cause fetal loss and preterm birth in animal models
Mouse models show that innate immune signals, independent of infectious pathogens, are sufficient to cause miscarriage and preterm birth. A number of studies show that injection of PAMPs or recombinant cytokines in pregnant mice leads to fetal demise and preterm birth and has been a tool to study some of the mechanisms underlying pregnancy complications. Injection of poly(I:C), a double stranded RNA viral mimic, in early pregnancy (E6.5) in mice reveals that uterine NK (uNK) cells induce fetal demise in an NKG2D-dependent manner (Thaxton et al., 2013). Poly(I:C) induces proliferation and activation of uNK cells and expression of the activating ligand, NKG2D, on uterine macrophages, resulting in abnormal trophoblast migration, increased apoptosis of placental cells, and fetal resorption. Of interest, we found that this model of poly(I:C)- induced fetal death is also dependent on IFNAR expression by the mother (Yockey et al., 2018). It is unclear where IFN signaling is acting on this NK-mediated pathway. In contrast, intraperitoneal LPS injection during early pregnancy (E7.0) shows a role for nitric oxide, presumably produced by infiltrating macrophages, in fetal resorption (Ogando et al., 2003). LPS, a component of the cell wall of gram negative bacteria, is recognized by the maternal tissues (via TLR4) to induce TNFα, which acts on the placenta and/or fetus to induce placenta necrosis in mice (Carpentier et al., 2011). The immunomodulatory cytokine IL-10, on the other hand, plays a protective role in preventing fetal resorption after intraperitoneal injection of LPS or CpG (TLR9 agonist) (Murphy et al., 2005; Thaxton et al., 2009). NK cells or F4/80+ macrophages mediate the fetal resorption after LPS or CpG treatment, respectively, in IL-10-deficient mice, indicating distinct cellular mechanisms based on the type inflammatory of stimulus. Prior to implantation, cytokines may also directly impact the embryo: TNF-α, IFN-γ, and TRAIL, have been shown to be toxic to the early embryo, inducing apoptosis and impaired growth (Robertson et al., 2018).
Microbial ligands or cytokines, used experimentally to mimic infections or conditions found in preterm labor, are sufficient to induce preterm labor when injected later in pregnancy (Agrawal and Hirsch, 2012). Direct intramniotic infusion of IL-1β and TNF-α, but not IL-6 or IL-8, are also sufficient to induce preterm labor in non-human primate models (Sadowsky et al., 2006). In pregnant mice inoculated intravaginally with LPS, complement activation leads to macrophage-mediated remodeling of the cervix and preterm birth (Gonzalez et al., 2011). Mice lacking the complement receptor C5aR are resistant to LPS-induced preterm labor but still deliver at term, indicating that C5aR is not required for term labor (Gonzalez et al., 2013). In this regard, TLR4 and IL-6 appear to be mediators of on-time labor: in addition to being protective of preterm labor, genetic deletion or inhibitors of TLR4 or IL-6 lead to delayed parturition and increased perinatal mortality (Robertson et al., 2010; Wahid et al., 2015). The context in which type I IFN signaling occurs is also critical: blocking signaling through IFNAR or suppressing type I IFNs increases sensitivity to intraperitoneal-challenge LPS-mediated preterm birth (Racicot et al., 2016). Conversely, type I IFNs mediate preterm birth in the case of intraperitoneal poly(I:C) injection and maternal viral infection (Cappelletti et al., 2017b). These animal studies collectively highlight a role for distinct inflammatory cells and pathways that underlie fetal loss and preterm labor.
Chronic maternal diseases leading to pregnancy complications
Maternal autoimmune disease is another known risk factor for pregnancy complications. Since many autoimmune diseases affect women of child-bearing age, the effect of these diseases on pregnancy is of high relevance. Two of the commonly recognized autoimmune diseases associated with pregnancy complications are systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APS). SLE is characterized by systemic inflammation, most commonly affecting the skin and kidney. Women with SLE have an increased risk of preeclampsia, preterm delivery, fetal growth restriction and fetal loss (Bundhun et al., 2017). Children born to mothers with SLE may also have an increased risk of neurodevelopment disorders and congenital heart defects (Vinet et al., 2014, 2015). Anti-phospholipid antibody syndrome (APS) is characterized by presence of one or more of the anti-phospholipid antibodies and episodes of thrombosis or recurrent miscarriage (Schreiber et al., 2018). Many patients with SLE also have antiphospholipid antibodies. In addition to recurrent miscarriages, women with APS have an increased risk of preeclampsia, growth restriction, and still birth (Saccone et al., 2017; Schreiber et al., 2018).
A number of different inflammatory mechanisms have been implicated in the poor pregnancy outcomes in individuals with SLE and APS, beyond hypercoagulability alone. Antibodies against cardiolipin, beta2-glycoprotein, and lupus anticoagulant are most common antibodies present in APS, and the concentrations of antibodies correlate with the risk of miscarriage (Santos et al., 2017). A number of in vitro studies have shown these antibodies can directly affect trophoblast function by decreasing proliferation, inhibiting syncytialization, and decreasing hormone production (Tong et al., 2015). Antibodies against beta2-glycoprotein, which is expressed by trophoblasts, directly induce inflammatory cytokine expression and cell death of trophoblasts in vitro (Mulla et al., 2009). Elevated concentrations of complement factors in the serum and deposition of complement in the decidua, are also predictive of poor pregnancy outcomes in patients and mediate disease in mouse models of APS (Cohen et al., 2011; Girardi et al., 2003; Kim et al., 2018). These mouse models, which involve induction of fetal demise and growth restriction after passive transfer of human antiphospholipid antibodies, also show a role for neutrophils and TNFα in mediating fetal demise (Berman et al., 2005; Girardi et al., 2003). In addition to antibody- and complement- mediated mechanisms, interferons may also be mediators of poor outcomes in SLE and APS patients. Elevated type I IFN activity, a signature of SLE, is one of the factors that correlates with development of preeclampsia in SLE patients (Andrade et al., 2015). The authors suggest that this clinical correlation may be due to impaired placental vasculature remodeling in the setting of elevated IFNα concentrations. The high risk pregnancy for individuals with autoimmune disease extends beyond SLE and APS: rheumatoid arthritis is associated with low birth weight, mothers with systemic sclerosis have higher rates of preterm delivery and IUGR, and those with Sjogren’s disease have higher rates of miscarriage, preterm delivery, and low birth weight (Gupta and Gupta, 2017; Ostensen and Clowse, 2013).
In addition to classic states of maternal inflammation including infection and autoimmunity, the role of inflammation in metabolic diseases including obesity and diabetes is increasingly being recognized. Obesity leads to a state of chronic low-grade inflammation, which has been shown to contribute to many of the health consequences including insulin resistance and type II diabetes (Reilly and Saltiel, 2017). Obese individuals have an increased risk of many pregnancy complications including preeclampsia, stillbirth, miscarriage and pre-term birth (Kalliala et al., 2017). Children of obese mothers are at an increased risk for a number of complications, ranging from macrosomia (above average infant size), neurodevelopmental/psychiatric disorders (including autism spectrum disorder, schizophrenia, attention deficit hyperactivity disorder, and poor cognitive performance), congenital malformations including congenital heart defects, and asthma (Edlow, 2017; Godfrey et al., 2017; Persson et al., 2017). Global transcriptional analyses of placenta of obese women identify inflammation and immune responses among the top dysregulated pathways (Altmäe et al., 2017). Analyses of the placentas of obese women show increased IL-1β and IL-8 expression (Aye et al., 2014; Roberts et al., 2011). Obese women also have reduced numbers of uNK cells, altered gene expression of uNK cell, and delayed uterine artery remodeling during early pregnancy (Perdu et al., 2016). Fetuses of mothers with obesity have increased markers of inflammation in the cord blood including increased C-reactive protein (CRP), IL-6 and TNFα concentrations (Dosch et al., 2016; Wilson et al., 2015). While, there is a consistent association between maternal body mass index and expression of the inflammatory marker, CRP, there is still no consensus between studies on the changes in systemic cytokine expression during pregnancies in obese women (Pendeloski et al., 2017). Animal studies using mouse and nonhuman primate models support this correlation between obesity, increased inflammatory markers in the mother or placenta, and poor pregnancy outcomes including stillbirth and fetal resorption (Frias et al., 2011; Mahany et al., 2018). More studies are needed to investigate the causal and mechanistic link between increased inflammation in obesity and associated pregnancy complications. Collectively, pregnancy complications and congenital diseases associated with chronic autoimmune and obesity correlate with elevated expression of type I IFNs, IL-6 and IL-1β (Fig. 3, Table 1).
Figure 3: Pathological effects of cytokines in pregnancy.
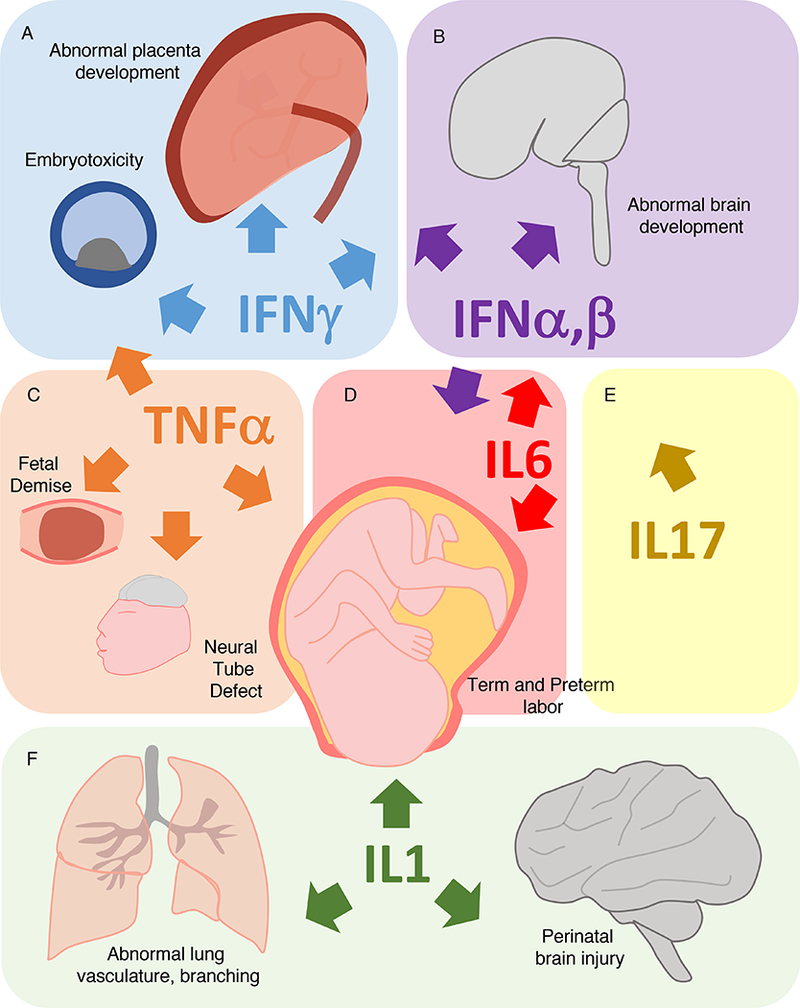
Aberrant expression of IFN-γ, IFN-β, TNF-α, IL6, IL17, and IL-1β can lead to developmental failure in multiple organ systems.
(A) Exposure of embryos to IFN-γ in culture is toxic, and systemically elevated IFN-γ at the time of implantation inhibits implantation. Mouse models implicate that IFN-γ responsiveness after malaria and toxoplasma infection mediates some of the placental defects and that IFN-γ overexpression leads to abnormal brain development in mouse models.
(B) Type I IFNs (including IFNα and IFNβ) mediate abnormal placental development after ZIKV infection, as demonstrated by mouse models. Humans with overexpression of type I IFNs due to interferonopathies have abnormal brain development, similar to “TORCH” infections, implicating IFNs as mediators of abnormal brain development.
(C) Exposure of embryos to TNF-α can induce a block in development. TNF-α is a mediator of fetal demise in mouse models of immune stimulation (CpG, LPS, and Poly(I:C)). TNF-α injection in mice can cause neural tube defects. Intraamniotic infusion of TNF-α is sufficient to induce preterm birth in non-human primate models.
(D) IL-6 (upstream of IL-17) induces abnormal brain development and behavior in mouse models of maternal immune activation. IL-6 mediates on time and preterm parturition in mouse models.
(E) IL17 (downstream of IL6) induces abnormal brain development and behavior in mouse models of maternal immune activation.
(F) IL-1 may induce preterm birth, as amniotic IL-1β administration is sufficient to induce preterm labor in non-human primates. Mouse models reveal that IL-1 may mediate defects associated with peripartum intrauterine inflammation including abnormal lung development associated with bronchopulmonary dysplasia and brain injury.
Inflammatory pathways that disrupt fetal brain development
Fetal development can also be impacted by inflammation. Nervous system development appears to be exquisitely sensitive to inflammatory insults throughout pregnancy. In the case of the “TORCH” infections, some of the common neurological defects seen include microcephaly, cranial calcifications, ventriculomegaly, ocular defects, and sensorineural hearing loss (Adams Waldorf and McAdams, 2013; Coyne and Lazear, 2016). Many of these presentations, including microcephaly, are similar among pathogens including CMV, rubella, rubeola (measles) and varicella zoster, Toxoplasma, HSV, and Zika virus (Devakumar et al., 2017). Similar clinical presentation across a range of viral and protozoan pathogens suggests a common underlying mechanism. This possibility is supported by the findings that fetuses with genetic defects leading to type I IFN overproduction can present as if they have a congenital viral infection, a disease referred to as pseudo-TORCH syndrome or Aicardi-Goutieres syndrome (AGS) (Crow and Manel, 2015). Most of the type I interferonopathies are due to defects in genes that are necessary for nucleic acid degradation, recognition and modification, including TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, IFIH1 and ADAR; or negative regulation of the IFN pathway in the case of USP18 (Crow and Manel, 2015; Meuwissen et al., 2016). Infants with AGS suffer from severe encephalopathy with seizures, cortical blindness, spasticity, and psychomotor retardation (Crow et al., 2015). AGS patients have elevated IFN concentrations in their blood and cerebral spinal fluid (CSF) and increased expression of ISGs in their blood, and the concentration of interferon in the CSF predicts disease severity, suggesting a role for elevated IFN in disease pathogenesis (Crow et al., 2015).
Recent studies show that in people with Down’s syndrome, or trisomy 21, many ISGs are overexpressed, likely due to an additional copy of the IFN receptor genes on chromosome 21 (Sullivan et al., 2016). Partial IFNAR and IFNGR deficiency leads to improved growth and neuron viability in a mouse model of trisomy 21, raising the possibility that some of the complications associated with trisomy 21, including developmental delay and growth restriction, may be mediated by IFN (Maroun et al., 2000). Taken together, these findings point towards hyperactive interferon signaling being a potential driver of brain pathology and abnormal brain development after congenital infections and genetic diseases.
IFNγ is primarily produced by T and NK cells and induces some overlapping effectors with type I IFNs. Mouse models demonstrate a direct role for IFNγ in altering brain development. Genetic overexpression of IFNγ in astrocytes leads to abnormal cerebellar development and ataxia in mice (Wang et al., 2004). IFNγ induces expression of Shh signaling demonstrating a direct interaction with a key neural developmental pathway (Wang et al., 2003). Other studies implicate IFNγ in vitro and in vivo in impaired neural progenitor differentiation (Ahn et al., 2015). In these studies, IFNγ acts by downregulating neurogenin 2, an important regulator of neuronal differentiation and proneural factor (Ahn et al., 2015). Thus, IFNγ overexpression during development can disrupt developmental pathways and differentiation in neurons.
Systemic maternal immune responses have the potential to be teratogenic: LPS or CpG administration during early pregnancy (starting E8.5 or E6.5 in mice, respectively) lead to neural tube defects, including exencephaly, in mice (Thaxton et al., 2009; Zhao et al., 2008). Excess reactive oxygen species contribute to the LPS-mediated defects, and folic acid administration can protect these mice from neural tube defects (Zhao et al., 2008, 2013). Interestingly, folic acid supplementation reduces the production of IL-1β, TNFα, and IL-6 made in the response to LPS administration. LPS-induced neural tube defects and resorption were also partially rescued by depletion of NK cells and in mice lacking IL-15 signaling, which lack uterine NK cells (Lee et al., 2013). TNFα administration alone is sufficient to induce neural tube defects in mice, potentially through its role in altering zinc metabolism (Weldon Taubeneck et al., 1995). Animal models demonstrate that a systemic maternal immune response is sufficient to induce neural tube defects, but the exact mechanisms and the respective contributions of cytokines, oxidative damage, and nutrient imbalances to these phenotypes are still unclear.
In addition to overt structural malformations, maternal immune activation (MIA) during pregnancy is associated with an increased risk of psychiatric diseases including schizophrenia and autism spectrum disorder (Patterson, 2009). One retrospective study estimates that maternal exposure to influenza virus can account for as many as 20% of cases of schizophrenia (Brown et al., 2004). Abnormal behavior analogous to schizophrenia and autism, including inability to inhibit the startle response, has been recapitulated in animals by injection of influenza virus, poly(I:C), LPS, or recombinant IL-6 (Patterson, 2009; Smith et al., 2007). The poly(I:C)-induced autism-like phenotype leads to specific defects in the dysgranular zone of the somatosensory cortex and is mediated by IL-17 (Choi et al., 2016; Yim et al., 2017) (Fig. 3). IL-17 produced by maternal T helper-17 (Th17) cells presumably crosses the placenta to trigger IL-17R in the fetal neurons, resulting in cortical and behavioral abnormalities. Maternal gut microbiota, such as segmented filamentous bacteria (SFB), promote the development of Th17 cells and predispose the fetus to neurodevelopmental disorders following poly(I:C) challenge in mice (Kim et al., 2017). Recent studies of healthy human subjects show a correlation between maternal IL-6 concentrations during pregnancy with neonatal brain connectivity and executive memory at 2 years (Rudolph et al., 2018). This indicates that baseline inflammation of the mother may impact brain connectivity even without overt disease. In addition to the direct action of cytokines on the fetal brain, MIA can modulate fetal neurodevelopment through inducing changes in placental tryptophan metabolism and increased serotonin production (Goeden et al., 2016). Thus, during MIA, inflammatory mediators released by the mother may have either a direct or an indirect effect on the fetal brain development.
Perinatal inflammation is also recognized as an important risk factor for brain injury and development disorders including cerebral palsy, white matter injury, and autism (Boyle et al., 2017). Elevated cytokines IL-6 and IL-1β in the amniotic fluid as well as placental inflammation are predictors of brain injury in premature infants (Boyle et al., 2017) (Fig. 3). Animal models have helped to confirm this connection and identify some of the inflammatory pathways that contribute to abnormal brain development. Induction of preterm birth by intrauterine LPS exposure leads to abnormal neuronal morphology and decreased dendrites compared to preterm birth induced by progesterone inhibition with RU486 (Burd et al., 2010). Inhibition of IL-1 signaling using an IL-1 receptor antagonist eliminates the abnormal morphology and dendrite numbers (Leitner et al., 2014). IL-1 may act to promote an adaptive immune response: depletion of maternal CD8+ T cells reverses the cortical density and behavioral abnormalities associated with inflammation- induced preterm delivery (Lei et al., 2017). A recent study shows that γδ T cells, which are among the first T cells to develop, may contribute to brain injury: γδ T cell infiltration is observed in postmortem analysis of human fetuses with white matter brain injury as well as in sheep and mouse models of perinatal brain injury due to hypoxia (Albertsson et al., 2018). Depletion of γδ T cells in mouse models is sufficient to reduce brain injury (Albertsson et al., 2018). Thus, the brain is susceptible to developmental disruption by increased expression of type I immune cytokines including type I and type II IFNs, IL-17 and IL-1β (Fig. 3).
Inflammatory pathways that disrupt development of other organs
Heart development is also susceptible to disruption by different states of inflammation. While defects in heart development are less common amongst congenital infections, patients with congenital rubella syndrome have high rates of heart defects including patent ductus arteriosus (PDA), an abnormal connection between the aorta and pulmonary arteries, and pulmonary artery stenosis, a narrowing of the pulmonary artery (Oster et al., 2010). Of the TORCH pathogens, why only rubella infection results in cardiac defects is unknown. Chorioamnionitis, or infection of the fetal membranes, is also associated with an increased risk for PDA (Park et al., 2015).
A classic example of immune-mediated congenital heart defects is the case of congenital heart block (CHB) that occurs in the fetus of mothers with autoimmune diseases. CHB occurs when the maternal autoimmune antibodies, anti-Ro and anti-La, cross the placental barrier and attack the conduction system of the fetal heart (Brito- zeron et al., 2015). These autoantibodies commonly occur in mothers with SLE, and 1–2% of women with anti-Ro antibodies will have babies with CHB. The most common clinical finding of CHB is irreversible atrioventricular node block (Brito-zerón et al., 2015). The atrioventricular node is the pacemaker of the heart and controls passage of current to the ventricles. The exact mechanism by which the conducting system of the developing fetal heart is specifically damaged by anti-Ro antibody is poorly understood as most of the target antigens are intracellular (Ambrosi et al., 2014).
Lung development spans the entire gestational period, with the formation of alveoli occurring just weeks before birth (Kalikkot et al., 2017). Bronchopulmonary dysplasia (BPD) is common in premature infants and results when there is an early block in lung development. While there are genetic susceptibilities, bacterial infection of the amnion and chorion is an important contributor to BPD (Kalikkot et al., 2017). Intrauterine LPS administration leads to abnormal lung development, with decreased airway branching and perinatal death, in a macrophage-dependent manner (Blackwell et al., 2011). Constitutive activation of NFκB in macrophages is sufficient to reproduce these effects. Inhibition of IL-1β and inflammasome activity is sufficient to inhibit the effects of LPS- induced lung development defects (Hogmalm et al., 2014). Interestingly, the cell type in which NFκB is activated influences the subsequent defects: when NFκB is constitutively activated in mesenchymal cells, vascular development in the lung is abnormal (Mccoy et al., 2017). One of the possible mediators of abnormal lung development after LPS stimulation is inhibition of Shh signaling, which is critical for lung development (Collins et al., 2012). Thus, animal models of BPD have provided insights into the mechanisms of disease: including NFκB activation and IL-1 expression (Fig. 3).
In addition to brain, heart, and lung, other organ systems can be affected by inflammation as well. For example, congenital varicella infection is associated with limb and urogenital anomalies. Congenital Treponema pallidum, or syphilis, infection is associated with abnormal teeth and bone development (Adams Waldorf and McAdams, 2013). Overall, human disease and animal models demonstrate a key role of cytokines, whether induced by infection, inflammation or genetic abnormalities, in disrupting development of specific organs.
Unanswered questions and future directions
While many questions remain, we highlight a few that may have potential therapeutic benefits. First, a long-standing question has been why some maternal infections, but not others, lead to congenital diseases (Arora et al., 2017). Must the pathogen infect the placenta or the fetus in order to cause developmental defects? How much cytokines, locally or systemically, are required to induce placental insufficiency, IUGR, or abnormal brain development? What are the cellular sources and targets of cytokines in the placenta and the fetus? What are the molecular mechanisms of cytokine-induced dysfunction in target cells?
Second, how might we use our knowledge of toxicity associated with inflammatory cytokines and IFNs to develop therapies to prevent pregnancy complications and birth defects? Is it advantageous to interfere with these inflammatory responses? In the case of congenital infection, blocking type I or type II IFNs may have a detrimental impact on the mother, as the pathogen will replicate and spread to cause severe infection (Racicot et al., 2017). However, if we can understand the ISGs involved in mediating placental insufficiency and abnormal brain development vs. those involved in antimicrobial defense, we may be able to selectively target the pathogenic ISGs while leaving the protective ISGs intact. Similarly, it will be important to delineate which aspects of cytotoxic responses to pathogens by NK cells and CD8+ T cells at the maternal-fetal interface control pathogens or contribute to pathology. Since many pregnancy complications and birth defects have no known genetic or environmental causes, identification of common “developmentally-toxic ISGs” that underlie infectious and non- infectious causes of birth defects could serve as targets for future therapy.
Concluding remarks
Healthy pregnancy requires tightly coordinated immune responses. Cytokines and IFNs are critical mediators of healthy pregnancy, for their ability to drastically alter cellular function, migration, cell-cell communication, proliferation, and gene expression. However, when dysregulated or inappropriately expressed, these have the potential to act as teratogens and disrupt fetal and placental developmental pathways, leading to birth defects and pregnancy complications. Investigating the ways in which different immune signaling pathways impact pregnancy and fetal development could provide important insights into congenital disorders and possible therapeutics to prevent pregnancy complications.
Acknowledgements:
The authors received funding from the NIH AI054359, R01EB000487, R01AI127429 and R21AI131284 (to AI). AI is an Investigator of the Howard Hughes Medical Institute. LJY was supported in part by T32GM007205 and F30 HD094717. Biorender was used to make figures 1 and 2. Due to space constraint, we regret our inability to cite all relevant studies.
Footnotes
Cytokines and interferons are necessary to promote healthy pregnancy. Yet in settings of infection, metabolic and autoimmune diseases, they can cause detrimental pregnancy outcomes. Here, Yockey and Iwasaki review evidence for the role of maternal and fetal immune responses in pregnancy and fetal development and highlight areas of clinical relevance.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaltonen R, Heikkinen T, and Hakala K (2005). Transfer of Proinflammatory Cytokines. Obs. Gynecol 106, 802–807. [DOI] [PubMed] [Google Scholar]
- Adams Waldorf KM, and McAdams RM (2013). Influence of infection during pregnancy on fetal development. Reproduction 146, R151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal V, and Hirsch E (2012). Intrauterine infection and preterm labor. Semin Fetal Neonatal Med 17, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Lee J, and Kim S (2015). Interferon-gamma inhibits the neuronal differentiation of neural progenitor cells by inhibiting the expression of Neurogenin2 via the JAK / STAT1 pathway. Biochem. Biophys. Res. Commun 466, 52–59. [DOI] [PubMed] [Google Scholar]
- Albertsson A, Zhang X, Vontell R, Bi D, Bronson RT, Supramaniam V, Baburamani AA, Hua S, Nazmi A, Cardell S, et al. (2018). gd T Cells Contribute to Injury in the Developing Brain. Am. J. Pathol 188, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmäe S, Segura MT, Esteban FJ, Bartel S, Brandi P, Irmler M, Beckers J, Demmelmair H, López-Sabater C, Koletzko B, et al. (2017). Maternal pre-pregnancy obesity is associated with altered placental transcriptome. PLoS One 12, e0169223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluvihare VR, Kallikourdis M, and Betz AG (2004). Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol 5, 266–271. [DOI] [PubMed] [Google Scholar]
- Ambrosi A, Sonesson S, and Wahren-herlenius M (2014). Molecular mechanisms of congenital heart block. Exp. Cell Res. 325, 2–9. [DOI] [PubMed] [Google Scholar]
- Andrade D, Kim M, Blanco LP, Karumanchi SA, Koo GC, Redecha P, Kirou K, Alvarez AM, Mulla MJ, Crow MK, et al. (2015). Interferon-a and Angiogenic Dysregulation in Pregnant Lupus Patients Who Develop Preeclampsia. Arthritis and Rheumatatology 67, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplin JD, and Ruane PT (2017). Embryo – epithelium interactions during implantation at a glance. J. Cell Sci 130, 15–22. [DOI] [PubMed] [Google Scholar]
- Arora N, Sadovsky Y, Dermody TS, and Coyne CB (2017). Microbial Vertical Transmission during Human Pregnancy. Cell Host Microbe 21, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkar AA, Black GP, Wei Q, He H, Liang L, Head JR, and Croy BA (2003). Assessment of Requirements for IL-15 and IFN Regulatory Factors in Uterine NK Cell Differentiation and Function During Pregnancy. J. Immunol 171, 2937–2944. [DOI] [PubMed] [Google Scholar]
- Ashkar BAA, Santo JP Di, and Croy BA (2000). Interferon-gamma Contributes to Initiation of Uterine Vascular Modification, Decidual Integrity-gamma, and Uterine Natural Killer Cell Maturation during Normal Murine Pregnancy. J Exp Med 192, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye ILMH, Lager S, Ramirez VI, Gaccioli F, Dudley DJ, Jansson T, and Powell TL (2014). Increasing Maternal Body Mass Index Is Associated with Systemic Inflammation in the Mother and the Activation of Distinct Placental Inflammatory Pathways. Biol. Reprod 90, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber EM, and Pollard JW (2003). The Uterine NK Cell Population Requires IL-15 but These Cells Are Not Required for Pregnancy nor the Resolution of a Listeria monocytogenes Infection. J. Immunol 171, 37–46. [DOI] [PubMed] [Google Scholar]
- Barboza R, Lima FA, Reis AS, Murillo OJ, Peixoto PM, Bandeira CL, Fotoran WL, Roberto L, Wunderlich G, Bevilacqua E, et al. (2017). TLR4- Mediated Placental Pathology and Pregnancy Outcome in Experimental Malaria. Sci. Rep 7, 8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ETDA, Cherry S, Sadovsky Y, and Coyne CB (2016). Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 19, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman J, Girardi G, and Salmon JE (2005). TNF-α Is a Critical Effector and a Target for Therapy in Antiphospholipid Antibody-Induced Pregnancy Loss. J. Immunol 174, 485–490. [DOI] [PubMed] [Google Scholar]
- Blackwell TS, Hipps AN, Han W, Barham WJ, Michael C, Yull FE, and Prince LS (2011). NF- κ B Signaling in Fetal Lung Macrophages Disrupts Airway Morphogenesis. J Immunol 187, 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AK, Rinaldi SF, Norman JE, and Stock SJ (2017). Preterm birth: Inflammation, fetal injury and treatment strategies. J. Reprod. Immunol 119, 62–66. [DOI] [PubMed] [Google Scholar]
- Brito-zerón P, Izmirly PM, Ramos-casals M, Jill P, and Khamashta MA (2015). The clinical spectrum of autoimmune congenital heart block. Nat. Rev. Rheumatol 11, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, and Susser ES (2004). Serologic Evidence of Prenatal Influenza in the Etiology of Schizophrenia. Arch Gen Psychiatry 61, 774–780. [DOI] [PubMed] [Google Scholar]
- Bunders MJ, Hamme JL Van, Jansen MH, Boer K, Kootstra NA, and Kuijpers TW (2014). Fetal exposure to HIV-1 alters chemokine receptor expression by CD4+T cells and increases susceptibility to HIV-1. Sci. Rep 4, 6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundhun PK, Soogund MZS, and Huang F (2017). Impact of systemic lupus erythematosus on maternal and fetal outcomes following pregnancy: A meta-analysis of studies published between years 2001–2016. J. Autoimmun 79, 17–27. [DOI] [PubMed] [Google Scholar]
- Burd I, Bentz AI, Chai J, Gonzalez J, Monnerie H, Roux PD Le, Cohen AS, Yudkoff M, and Elovitz MA (2010). Inflammation-induced Preterm Birth Alters Neuronal Morphology in the Mouse Fetal Brain. J. Neurosci. Res 1881, 1872–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelletti M, Bella S Della, Ferrazzi E, Mavilio D, and Divanovic S (2017a). Inflammation and preterm birth. J. Leukoc. Biol 99, 67–78. [DOI] [PubMed] [Google Scholar]
- Cappelletti M, Presicce P, Lawson MJ, Chaturvedi V, Stankiewicz TE, Vanoni S, Harley ITW, McAlees JW, Giles DA, Moreno-Fernandez ME, et al. (2017b). Type I interferons regulate susceptibility to 7inflammation-induced preterm birth. JCI Insight 2, e91288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Care AS, Diener KR, Jasper MJ, Brown HM, Ingman WV, and Robertson SA (2013). Macrophages regulate corpus luteum development during embryo implantation in mice. J. Clin. Invest 123, 3472–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Girard A, Van De Kant HJG, Bourc’his D, Bestor TH, Rooij DG De, and Hannon GJ (2007). MIWI2 Is Essential for Spermatogenesis and Repression of Transposons in the Mouse Male Germline. Dev. Cell 12, 503–514. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Dingman AL, and Palmer TD (2011). Placental TNF- alpha Signaling in Illness-Induced Complications of Pregnancy. AJPA 178, 2802–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi V, Ertelt JM, Jiang TT, Kinder JM, Xin L, Owens KJ, Jones HN, and Way SS (2015). CXCR3 blockade protects against Listeria monocytogenes infection - induced fetal wastage. J. Clin. Invest 125, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan AR, Bhullar BS, and Wagner GP (2016). What was the ancestral function of decidual stromal cells? A model for the evolution of eutherian pregnancy. Placenta 40, 40–51. [DOI] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, and Huh JR (2016). The maternal interleukin-17a pathway in mice promotes autismlike phenotypes in offspring. Science (80-. ). 351, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaens I, Zaragoza DB, Guilbert L, Robertson SA, Mitchell BF, and Olson DM (2008). Inflammatory processes in preterm and term parturition. J. Reprod. Immunol 79, 50–57. [DOI] [PubMed] [Google Scholar]
- Cohen D, Buurma A, Goemaere NN, Girardi G, Le Cessie S, Scherjon S, Bloemenkamp KWM, De Heer E, Bruijn JA, and Bajema IM (2011). Classical complement activation as a footprint for murine and human antiphospholipid antibody- induced fetal loss. J. Pathol 225, 502–511. [DOI] [PubMed] [Google Scholar]
- Collins JJP, Kuypers E, Nitsos I, Pillow JJ, Polglase GR, Kemp MW, Newnham JP, Cleutjens JP, Frints SGM, Kallapur SG, et al. (2012). LPS- induced chorioamnionitis and antenatal corticosteroids modulate Shh signaling in the ovine fetal lung. Am J Physiol Lung Cell Mol Physiol 70, 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MK, Tay C, and Erlebacher A (2009). Dendritic cell entrapment within the pregnant uterus inhibits immune surveillance of the maternal / fetal interface in mice. J. Clin. Invest 119, 2062–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, and Lazear HM (2016). Zika virus — reigniting the TORCH. Nat. Rev. Microbiol 14, 707–715. [DOI] [PubMed] [Google Scholar]
- Cross SN, Potter JA, Aldo P, Young J, Pitruzzello M, Tong M, Guller S, Carla V, Mor G, Abrahams VM, et al. (2017). Viral Infection Sensitizes Human Fetal Membranes to Bacterial Lipopolysaccharide by MERTK Inhibition and Inflammasome Activation. J. Immunol 199, 2885–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, and Manel N (2015). Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Schmidt JL, Szynkiewicz M, Forte GMA, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. (2015). Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASE H2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet 167A, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JLM, Guimarães KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al. (2016). The Brazilian Zika virus strain causes birth defects in experimental models. Nature 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-axford E, Donker RB, Mouillet J, Chu T, and Bayer A (2013). Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci U S A 110, 12048–12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devakumar D, Bamford A, Ferreira MU, Broad J, Rosch RE, Groce N, Breuer J, Cardoso MA, Copp AJ, Alexandre P, et al. (2017). Infectious causes of microcephaly: epidemiology, pathogenesis, diagnosis, and management. Lancet Infect. Dis 3099, 1–13. [DOI] [PubMed] [Google Scholar]
- Digiulio DB, Callahan BJ, Mcmurdie PJ, Costello EK, and Lyell DJ (2015). Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci U S A 112, 11060–11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosch NC, Guslits EF, Weber MB, Murray SE, Ha B, Coe CL, Auger AP, and Kling PJ (2016). Maternal obesity affects inflammatory and iron indices in umbilical cord blood. J. Pediatr 172, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ealy AD, Larson SF, Liu L, Alexenko AP, Winkelman GL, Kubisch HM, Bixby JA, and Roberts RM (2001). Polymorphic Forms of Expressed Bovine Interferon-tau Genes: Relative Transcript Abundance during Early Placental Development, Promoter Sequences of Genes and Biological Activity of Protein Products. Endocrinology 142, 2906–2915. [DOI] [PubMed] [Google Scholar]
- Edlow AG (2017). Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat Diagn 37, 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Egmond A, van der Keur C, Swings GMJS, Scherjon SA, and Claas FHJ (2016). The possible role of virus-specific CD8+ memory T cells in decidual tissue. J. Reprod. Immunol 113, 1–8. [DOI] [PubMed] [Google Scholar]
- Fest S, Aldo PB, Abrahams VM, Visintin I, Alvero A, Chen R, Chavez SL, Romero R, and Mor G (2007). Trophoblast – Macrophage Interactions: a Regulatory Network for the Protection of Pregnancy. Am. J. Reprod. Immunol 57, 55–66. [DOI] [PubMed] [Google Scholar]
- Feyaerts D, Benner M, Cranenbroek Van B, and Heijden, Van Der OWH (2017). Human uterine lymphocytes acquire a more experienced and tolerogenic phenotype during pregnancy. Sci. Rep 7, 2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, and Grove KL (2011). Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152, 2456–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried M, Kurtis JD, Swihart B, Pond-tor S, Barry A, Sidibe Y, Gaoussou S, Traore M, Keita S, Mahamar A, et al. (2017). Systemic Inflammatory Response to Malaria During Pregnancy Is Associated With Pregnancy Loss and Preterm Delivery. Clin. Infect. Dis 65, 1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Zhou Y, Ni X, Tong X, Xu X, Dong Z, and Sun R (2017). Natural Killer Cells Promote Fetal Development through the Secretion of Growth-Promoting Factors Article Natural Killer Cells Promote Fetal Development through the Secretion of Growth- Promoting Factors. Immunity 47, 1100–1113.e6. [DOI] [PubMed] [Google Scholar]
- Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, et al. (2003). Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Invest 112, 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VWV, Eriksson JG, and Broekman BFP (2017). Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol 15, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeden N, Velasquez J, Arnold KA, Chan Y, Lund BT, Anderson GM, and Bonnin A (2016). Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J. Neurosci 36, 6041–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg RL, Mcclure EM, Saleem S, and Reddy UM (2010). Infection-related stillbirths. Lancet 375, 1482–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J, Romero R, and Girardi G (2013). Comparison of the mechanisms responsible for cervical remodeling in preterm and term labor. J Reprod Immunol 97, 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JM, Franzke C, Yang F, and Romero R (2011). Complement Activation Triggers Metalloproteinases Release Inducing Cervical Remodeling and Preterm Birth in Mice. AJPA 179, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MP, Spate LD, Bixby JA, Ealy AD, and Roberts RM (2005). A Comparison of the Anti-Luteolytic Activities of Recombinant Ovine Interferon- Alpha and -Tau in Sheep. Biol. Reprod 73, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Chavan AR, Protopapas S, Maziarz J, and Romero R (2017). Embryo implantation evolved from an ancestral inflammatory attachment reaction. Proc Natl Acad Sci U S A 114, E6566–E6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, and Gupta N (2017). Sjögren Syndrome and Pregnancy: A Literature Review. Perm. J 21, 16–047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Goldman-wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-yaron S, Prus D, Cohen-daniel L, Arnon TI, Manaster I, et al. (2006). Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med 12, 1065–1074. [DOI] [PubMed] [Google Scholar]
- Hogmalm A, Bry M, Strandvik B, and Bry K (2014). IL-1beta expression in the distal lung epithelium disrupts lung morphogenesis and epithelial cell differentiation in fetal mice. Am J Physiol Lung Cell Mol Physiol 306, L23–L34. [DOI] [PubMed] [Google Scholar]
- Holst R-M, Hagberg H, Wennerholm U-B, Skogstrand K, Thorsen P, and Jacobsson B (2009). Prediction of Spontaneous Preterm Delivery in Women With Preterm Labor: Analysis of Multiple Proteins in Amniotic and Cervical Fluids. Obstet. Gynecol 114, 268–277. [DOI] [PubMed] [Google Scholar]
- Hong M, Sandalova E, Low D, Gehring AJ, Fieni S, Amadei B, Urbani S, Chong Y, Guccione E, and Bertoletti A (2015). Trained immunity in newborn infants of HBV-infected mothers. Nat. Commun 6, 6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppertz B, Ghosh D, and Sengupta J (2014). An integrative view on the physiology of human early placental villi. Prog. Biophys. Mol. Biol 114, 33–48. [DOI] [PubMed] [Google Scholar]
- Jagger BW, Miner JJ, Cao B, Arora N, Smith AM, Kovacs A, Mysorekar IU, Coyne CB, and Diamond MS (2017). Gestational Stage and IFN- l Signaling Regulate ZIKV Infection In Utero. Cell Host Microbe 22, 366–376.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein MF, Abu-raya B, Jiang Y, and Alter G (2017). Transfer of maternal immunity and programming of the newborn immune system. Semin Immunopathol 39, 605–613. [DOI] [PubMed] [Google Scholar]
- Kalikkot R, Cuevas M, and Shivanna B (2017). Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir. Med 132, 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliala I, Markozannes G, Gunter MJ, Paraskevaidis E, Gabra H, Mitra A, Terzidou V, Bennett P, Martin-hirsch P, Tsilidis KK, et al. (2017). Obesity and gynaecological and obstetric conditions: umbrella review of the literature. BMJ 359, j4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Guerra MM, Kaplowitz E, Laskin CA, Petri M, Branch DW, Lockshin MD, Sammaritano LR, Merrill JT, Porter TF, et al. (2018). Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and / or antiphospholipid antibodies. Ann Rheum Dis 77, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Littman DR, Choi GB, and Huh JR (2017). Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Bratu DP, Mcginnis-schultz N, Koppetsch BS, Cook HA, and Theurkauf WE (2007). Drosophila rasiRNA Pathway Mutations Disrupt Embryonic Axis Specification through Activation of an ATR / Chk2 DNA Damage Response. Dev. Cell 12, 45–55. [DOI] [PubMed] [Google Scholar]
- Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, and Levy O (2017). Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 46, 350–363. [DOI] [PubMed] [Google Scholar]
- Kroener L, Wang ET, and Pisarska MD (2016). Predisposing Factors to Abnormal First Trimester Placentation and the Impact on Fetal Outcomes. Semin Reprod Med 34, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AJ, Kandiah N, Karimi K, and Clark DA (2013). Interleukin-15 is required for maximal lipopolysaccharide-induced abortion. J. Leukoc. Biol 93, 905–912. [DOI] [PubMed] [Google Scholar]
- Lei J, Xie L, Zhao H, Gard C, Clemens JL, Mclane MW, Feller MC, Ozen M, Novak C, Alshehri W, et al. (2017). Maternal CD8 + T- cell depletion alleviates intrauterine induced perinatal brain injury. Am J Reprod Immunol 79, e12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner K, Shammary M Al, Mclane M, Johnston MV, Elovitz MA, and Burd I (2014). IL-1 Receptor Blockade Prevents Fetal Cortical Brain Injury but Not Preterm Birth in a Mouse Model of Inflammation-Induced Preterm Birth and Perinatal Brain Injury. Am. J. Reprod. Immunol 71, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, and Spradling AC (1997). A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 124, 2463–2476. [DOI] [PubMed] [Google Scholar]
- Liu S, Diao L, Huang C, Li Y, Zeng Y, and Kwak-kim JYH (2017). The role of decidual immune cells on human pregnancy. J. Reprod. Immunol 124, 44–53. [DOI] [PubMed] [Google Scholar]
- Mahany EB, Han X, Borges BC, Da Silveira Cruz-Machado S, Allen SJ, Garcia-Galiano D, Hoenerhoff MJ, Bellefontaine NH, and Elias CF (2018). Obesity and High-Fat Diet Induce Distinct Changes in Placental Gene Expression and Pregnancy Outcome. Endocrinology 159, 1718–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun LE, Heffernan TN, and Hallam DM (2000). Partial IFN- a / b and IFN- g Receptor Knockout Trisomy 16 Mouse Fetuses Show Improved Growth and Cultured Neuron Viability. J. Interf. Cytokine Res. 203, 197–203. [DOI] [PubMed] [Google Scholar]
- Mccoy AM, Herington JL, Stouch AN, Mukherjee AB, Lakhdari O, Blackwell TS, and Prince LS (2017). IKK b Activation in the Fetal Lung Mesenchyme Alters Lung Vascular Development but Not Airway Morphogenesis. Am. J. Pathol 187, 2635–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R, Bonney EA, Condon J, Mesiano S, and Taylor RN (2016). Novel concepts on pregnancy clocks and alarms: redundancy and synergy in human parturition. Hum. Reprod. Update 22, 535–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen MEC, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, Unen L Van, Heijsman D, Goldmann T, et al. (2016). Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 213, 1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, et al. (2016). Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 165, 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Simpson JA, Wiladphaingern J, Min AM, Pimanpanarak M, Paw MK, Raksuansak J, Pukrittayakamee S, Fowkes FJI, White NJ, et al. (2017a). Influence of the number and timing of malaria episodes during pregnancy on prematurity and small-for-gestational-age in an area of low transmission. BMC Med. 15, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KL, Persaud T, and Torchia MG (2017b). Human Birth Defects. In The Developing Human, pp. 457–486. [Google Scholar]
- Moser G, and Huppertz B (2017). Implantation and extravillous trophoblast invasion : From rare archival specimens to modern biobanking. Placenta 56, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulla MJ, Brosens JJ, Chamley LW, Giles I, Pericleous C, Rahman A, Joyce SK, Panda B, Paidas MJ, and Abrahams VM (2009). Antiphospholipid Antibodies Induce a Pro-Inflammatory Response in First Trimester Trophoblast Via the TLR4 / MyD88 Pathway. Am. J. Reprod. Immunol 62, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SP, Fast LD, Hanna NN, Sharma S, Murphy SP, Fast LD, Hanna NN, and Sharma S (2005). Uterine NK Cells Mediate Inflammation-Induced Fetal Demise in IL-10-Null Mice. J. Immunol 175, 4084–4090. [DOI] [PubMed] [Google Scholar]
- Murphy SP, Tayade C, Ashkar AA, Hatta K, Zhang J, and Croy BA (2009). Interferon Gamma in Successful Pregnancies. Biol. Reprod 859, 848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardozza LMM, Caetano ACR, Zamarian ACP, Mazzola JB, Silva CP, Marcal VMG, Lobo TF, Peixoto AB, and Júnior EA (2017). Fetal growth restriction : current knowledge. Arch. Gynecol. Obstet 295, 1061–1077. [DOI] [PubMed] [Google Scholar]
- Niikura M, Inoue SI, Mineo S, and Asahi H (2017). IFNGR1 signaling is associated with adverse pregnancy outcomes during infection with malaria parasites. PLoS One 12, e0185392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando DG, Paz D, Cella M, and Franchi AM (2003). The fundamental role of increased production of nitric oxide in lipopolysaccharide-induced embryonic resorption in mice. Reproduction 125, 95–110. [DOI] [PubMed] [Google Scholar]
- Oger S, Mehats C, Dallot E, Ferre F, and Leroy M-J (2002). Interleukin-1beta Induces Phosphodiesterase 4B2 Expression in Human Myometrial Cells through a Prostaglandin E2- and Cyclic Adenosine 3’,5’-Monophosphate-Dependent Pathway. J. Endocrinol. Metab 87, 5524–5531. [DOI] [PubMed] [Google Scholar]
- Ostensen M, and Clowse M (2013). Pathogenesis of pregnancy complications in systemic lupus erythematosus. Curr Opin Rheumatol 25, 591–596. [DOI] [PubMed] [Google Scholar]
- Oster ME, Riehle-colarusso T, and Correa A (2010). An Update on Cardiovascular Malformations in Congenital Rubella Syndrome. Birth Defects Res. 88, 1–8. [DOI] [PubMed] [Google Scholar]
- PAHO, and WHO (2017). Zika suspected and confirmed cases reported by countries Sand territories in the Americas Cumulative cases, 2015–2017. Updated as of 21 December 2017.
- Palmeira P, Quinello C, Ana L, Zago A, and Carneiro-sampaio M (2012). IgG Placental Transfer in Healthy and Pathological Pregnancies. Clin. Dev. Immunol 2012, 985646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Choi Y, Kim KS, and Kim S (2015). Chorioamnionitis and Patent Ductus Arteriosus: A Systematic Review and Meta- Analysis. PLoS One 10, e0138114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH (2009). Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav. Brain Res 204, 313–321. [DOI] [PubMed] [Google Scholar]
- Pazos M, Sperling RS, Moran TM, and Kraus T. a. (2012). The influence of pregnancy on systemic immunity. Immunol. Res 54, 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendeloski KPT, Ono E, Torloni MR, Mattar R, and Daher S (2017). Maternal obesity and inflammatory mediators: A controversial association. Am. J. Reprod. Immunol 77, 1–8. [DOI] [PubMed] [Google Scholar]
- Perdu S, Castellana B, Kim Y, Chan K, Deluca L, and Beristain AG (2016). Maternal obesity drives functional alterations in uterine NK cells. JCI Insight 1, e85560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Petitt M, Fong A, Tsuge M, Tabata T, Fang-hoover J, Maidji E, Zydek M, Zhou Y, Inoue N, et al. (2014). Intrauterine Growth Restriction Caused by Underlying Congenital Cytomegalovirus Infection. J. Infect. Dis 209, 1573–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M, Cnattingius S, Villamor E, Soderling J, Pasternak B, Stephansson O, and Neovius M (2017). Risk of major congenital malformations in relation to maternal overweight and obesity severity: cohort study of 1.2 million singletons. BMJ 357, j2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaks V, Birnberg T, Berkutzki T, Sela S, Benyashar A, Kalchenko V, Mor G, Keshet E, Dekel N, Neeman M, et al. (2008). Uterine DCs are crucial for decidua formation during embryo implantation in mice. J. Clin. Invest 118, 3954–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt DJ, Smith AM, Arora N, Diamond MS, Coyne CB, and Miner JJ (2018). Zika virus – related neurotropic flaviviruses infect human placental explants and cause fetal demise in mice. Sci. Transl. Med 10, eaao7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhudas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, Fisher S, Golos T, Matzuk M, Mccune JM, et al. (2015). Immune mechanisms at the maternal-fetal interface: perspectives and challenges. Nat. Immunol 16, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purisch SE, and Gyamfi-Bannerman C (2017). Perinatology Epidemiology of preterm birth. Semin. Perinatol 41, 387–391. [DOI] [PubMed] [Google Scholar]
- Racicot K, Cardenas I, Wünsche V, Aldo P, Guller S, Means RE, Romero R, and Mor G (2013). Viral infection of the pregnant cervix predisposes to ascending bacterial infection. J. Immunol 191, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racicot K, Kwon JY, Aldo P, Abrahams V, El-guindy A, Romero R, and Mor G (2016). Type I Interferon Regulates the Placental Inflammatory Response to Bacteria and is Targeted by Virus: Mechanism of Polymicrobial Infection-Induced Preterm Birth. Am. J. Reprod. Immunol 75, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racicot K, Aldo P, El-guindy A, Romero R, and Mor G (2017). Cutting Edge: Fetal/Placental Type I IFN Can Affect Maternal Survival and Fetal Viral Load during Viral Infection. J. Immunol 198, 3029–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, and Saltiel AR (2017). Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol 13, 633–643. [DOI] [PubMed] [Google Scholar]
- Rinaldi SF, Makieva S, Saunders PT, Rossi AG, and Norman JE (2017). Immune cell and transcriptomic analysis of the human decidua in term and preterm parturition. Mol. Hum. Reprod 23, 708–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JR, and Bakardjiev AI (2012). Pathogens and the placental fortress. Curr. Opin. Microbiol 15, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RM (2007). Interferon-tau, a Type 1 interferon involved in maternal recognition of pregnancy. Cytokine Growth Factor Rev. 18, 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, and Denison FC (2011). Placental structure and inflammation in pregnancies associated with obesity. Placenta 32, 247–254. [DOI] [PubMed] [Google Scholar]
- Robertson SA, Christiaens I, Dorian CL, Zaragoza DB, Care AS, Banks AM, and Olson DM (2010). Interleukin-6 Is an Essential Determinant of On-Time Parturition in the Mouse. Endocrinology 151, 3996–4006. [DOI] [PubMed] [Google Scholar]
- Robertson SA, Chin P, Femia JG, and Brown HM (2018). Embryotoxic cytokines — Potential roles in embryo loss and fetal programming. J. Reprod. Immunol 125, 80–88. [DOI] [PubMed] [Google Scholar]
- Romero R, Dey SK, and Fisher SJ (2014a). Preterm labor: One syndrome, many causes. Science (80-. ). 345, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Bieda J, Chaemsaithong P, Miranda J, Chaiworapongsa T, and Ravel J (2014b). The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers EF, Rosenkranz D, Roelen BAJ, Mahdipour M, Han C, He N, Chuva de Sousa Lopes SM, van der Westerlaken LAJ, Zischler H, Butter F, et al. (2015). Piwi Proteins and piRNAs in Mammalian Oocytes and Resource Piwi Proteins and piRNAs in Mammalian Oocytes and Early Embryos. Cell Rep. 10, 2069–2082. [DOI] [PubMed] [Google Scholar]
- Rowe HM, and Trono D (2011). Dynamic control of endogenous retroviruses during development. Virology 411, 273–287. [DOI] [PubMed] [Google Scholar]
- Rowe JH, Ertelt JM, Aguilera MN, Farrar MA, and Way SS (2011). Foxp3 + Regulatory T Cell Expansion Required for Sustaining Pregnancy Compromises Host Defense against Prenatal Bacterial Pathogens. Cell Host Microbe 10, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozner AE, Durning M, Kropp J, Wiepz GJ, and Golos TG (2016). Macrophages modulate the growth and differentiation of rhesus monkey embryonic trophoblasts. Am J Reprod Immunol 76, 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MD, Graham AM, Feczko E, Miranda-dominguez O, Rasmussen JM, Nardos R, Entringer S, Wadhwa PD, Buss C, and Fair DA (2018). Maternal IL-6 during pregnancy can be estimated from newborn brain connectivity and predicts future working memory in offspring. Nat. Neurosci 21, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone G, Berghella V, Maruotti GM, Ghi T, Rizzo G, Simonazzi G, Rizzo N, Facchinetti F, Asta AD, Visentin S, et al. (2017). Antiphospholipid antibody profile based obstetric outcomes of primary antiphospholipid syndrome: PREGNANTS study. Am. J. Obstet. Gynecol 216, 525.e1–525.e12. [DOI] [PubMed] [Google Scholar]
- Sadler AJ, and Williams BRG (2008). Interferon-inducible antiviral effectors. Nat. Rev. Immunol 8, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky DW, Adams KM, Gravett MG, Witkin SS, and Novy MJ (2006). Preterm labor is induced by intraamniotic infusions of interleukin-1 b and tumor necrosis factor- a but not by interleukin-6 or interleukin-8 in a nonhuman primate model. Am. J. Obstet. Gynecol 195, 1578–1589. [DOI] [PubMed] [Google Scholar]
- Santos S, Lorena A, Correa H, Carvalho D, Maria A, Valdrinez M, Lonardoni C, Galhardo I, Alves Q, and Neto DL (2017). Antiphospholipid syndrome and recurrent miscarriage: A systematic review and meta-analysis. J. Reprod. Immunol 123, 78–87. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorn AJ, Gutbrod MJ, Leblanc C, Martienssen R, Schorn AJ, Gutbrod MJ, Leblanc C, and Martienssen R (2017). LTR-Retrotransposon Control by tRNA- Derived Small RNAs. Cell 170, 61–71.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber K, Sciascia S, De Groot PG, Devreese K, Jacobsen S, Ruiz-Irastroza G, Salmon JE, Shoenfeld Y, Shovman O, and Hunt BJ (2018). Antiphospholipid syndrome. Nat. Rev. Dis. Prim 4, 18005. [DOI] [PubMed] [Google Scholar]
- Senegas A, Villard O, Neuville A, Marcellin L, Pfaff AW, and Steinmetz T (2009). Toxoplasma gondii- induced foetal resorption in mice involves interferon- gamma-induced apoptosis and spiral artery dilation at the maternofoetal interface. Int. J. Parasitol 39, 481–487. [DOI] [PubMed] [Google Scholar]
- Siewiera J, El Costa H, Tabiasco J, Berrebi A, Cartron G, Bouteiller P, and Jabrane-Ferrat N (2013). Human Cytomegalovirus Infection Elicits New Decidual Natural Killer Cell Effector Functions. Plos Pathog. 9, e1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes R, Buzzini R, Bernardo W, Cardoso F, Salomao A, and Cerri G (2016). Update on Zika virus infection in pregnancy. Rev. Assoc. Med. Bras 62, 106–107. [DOI] [PubMed] [Google Scholar]
- Simon A, and Laufer N (2012). Assessment and treatment of repeated implantation failure (RIF). J Assist Reprod Genet 29, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, and Aravin AA (2011). PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol 12, 246–258. [DOI] [PubMed] [Google Scholar]
- Smith SEP, Li J, Garbett K, Mirnics K, and Patterson PH (2007). Maternal Immune Activation Alters Fetal Brain Development through Interleukin-6. J. Neruoscience 27, 10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Kontgen F, and Abbondanzo SJ (1992). Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 359, 76–79. [DOI] [PubMed] [Google Scholar]
- Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, and Liggett LA (2016). Trisomy 21 consistently activates the interferon response. Elife 5, e16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaba FM, Tighe M, Kummer LW, Lanzer KG, Ward JM, Lanthier P, Kim IJ, Kuki A, Blackman MA, Thomas SJ, et al. (2018). Zika virus infection in immunocompetent pregnant mice causes fetal damage and placental pathology in the absence of fetal infection. PLOS Pathog. 14, e1006994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, Harris E, and Pereira L (2016). Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 20, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantbirojn P, Crum CP, and Parast MM (2008). Pathophysiology of Placenta Creta: The Role of Decidua and Extravillous Trophoblast. Placenta 29, 639–645. [DOI] [PubMed] [Google Scholar]
- Tessier DR, Yockell-Lelievre J, and Gruslin A (2014). Uterine Spiral Artery Remodeling : The Role of Uterine Natural Killer Cells and Extravillous Trophoblasts in Normal and High-Risk Human Pregnancies. Am. J. Reprod. Immunol 74, 1–11. [DOI] [PubMed] [Google Scholar]
- Thaxton JE, Romero R, and Sharma S (2009). TLR-9 Activation Coupled To IL-10 Deficiency Induces Adverse Pregnancy Outcomes. J Immunol 183, 1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton JE, Nevers T, Lippe EO, Blois SM, Saito S, and Sharma S (2013). NKG2D Blockade Inhibits Poly(I:C)-Triggered Fetal Loss in Wild Type but Not IL-10–/– Mice Jessica. J Immunol 190, 3639–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M, Viall CA, and Chamley LW (2015). Antiphospholipid antibodies and the placenta: A systematic review of their In vitro effects and modulation by treatment. Hum. Reprod Update 21, 97–118. [DOI] [PubMed] [Google Scholar]
- Tribe RM, Moriarty P, Dalrymple A, Hassoni AA, and Poston L (2003). Interleukin-1-beta Induces Calcium Transients and Enhances Basal and Store Operated Calcium Entry in Human Myometrial Smooth Muscle. Biol. Reprod 68, 1842–1849. [DOI] [PubMed] [Google Scholar]
- Vinet É, Pineau CA, Scott S, Clarke AE, Platt RW, and Bernatsky S (2014). Increased congenital heart defects in children born to women with systemic lupus erythematosus results from the offspring of systemic lupus erythematosus mothers registry study. Circulation 131, 149–156. [DOI] [PubMed] [Google Scholar]
- Vinet É, Pineau CA, Clarke AE, Scott S, Fombonne É, Joseph L, Platt RW, and Bernatsky S (2015). Increased Risk of Autism Spectrum Disorders in Children Born to Women With Systemic Lupus Erythematosus: Results From a Large Population-Based Cohort. Arthritis Rheumatol. 67, 3201–3208. [DOI] [PubMed] [Google Scholar]
- Wahid HH, Dorian CL, Chin PY, Hutchinson MR, Rice KC, Olson DM, Moldenhauer LM, and Robertson SA (2015). Toll-Like Receptor 4 Is an Essential Upstream Regulator of On-Time Parturition and Perinatal Viability in Mice. Endocrinology 156, 3828–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pham-mitchell N, Schindler C, and Campbell IL (2003). Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN- α - stimulated STAT2-independent production of IFN- γ in the brain. 112, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lin W, Popko B, and Campbell IL (2004). Inducible production of interferon-gamma in the developing brain causes cerebellar dysplasia with activation of the Sonic hedgehog pathway. Mol. Cell. Neurosci 27, 489–496. [DOI] [PubMed] [Google Scholar]
- Weldon Taubeneck M, Daston GP, Rogers JM, Gershwin ME, Ansari A, and Keen CL (1995). Tumor necrosis factor-alpha alters embryonic zinc metabolism and is developmentally toxic in mice Nutrient. J. Nutr 125, 908–919. [DOI] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, O’Connor JF, Baird DD, Schlatterer JP, Canfield RE, Armstrong GE, and Nisula BC (1988). Incidence of Early Loss of Pregnancy. N. Engl. J. Med 319, 189–194. [DOI] [PubMed] [Google Scholar]
- Wilson RM, Marshall NE, Jeske DR, Purnell JQ, Thornburg K, and Messaoudi I (2015). Maternal obesity alters immune cell frequencies and responses in umbilical cord blood samples. Pediatr. Allergy Immunol. 26, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, Jakobsson J, Szulc J, Liao S, Quazzola A, Beerman F, Aebischer P, and Trono D (2007). The Kruppel-associated Box Repressor Domain Can Trigger de Novo Promoter Methylation during Mouse Early Embryogenesis. J. Biol. Chem 282, 34535–34541. [DOI] [PubMed] [Google Scholar]
- Woidacki K, Popovic M, Metz M, Schumacher A, Linzke N, Teles A, Poirier F, Fest S, Jensen F, Rabinovich GA, et al. (2013). Mast cells rescue implantation defects caused by c-kit deficiency. Cell Death Dis. 4, e462–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, and Goff SP (2009). Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Thi VLD, Huang Y, Billerbeck E, Saha D, Hoffmann H, Wang Yy., Silva LAV, Sarbanes S, Sun T, et al. (2017). Stem Cells Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 172, 423–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellon SM (2017). Contributions to the dynamics of cervix remodeling prior to term and preterm birth. Biol. Reprod 96, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YS, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, Kim JY, Kim S, Kim H, Waisman A, et al. (2017). Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, Szigeti-Buck K, Van den Pol A, Lindenbach BD, Horvath TL, et al. (2016). Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 166, 1247–1256.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, Hastings AK, Fikrig E, Kong Y, Horvath TL, et al. (2018). Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol 3, eaao1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldovich VB, Clausen CH, Bradford E, Fletcher DA, Maltepe E, Robbins JR, and Bakardjiev AI (2013). Placental Syncytium Forms a Biophysical Barrier against Pathogen Invasion. Plos Pathog. 9, e1003821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenclussen AC, and Hammerling GJ (2015). Cellular regulation of the uterine microenvironment that enables embryo implantation. Front. Immunol. 6, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Chen Y, Wang H, Ji Y, Ning H, Wang S, Zhang C, Lu J, Duan Z, and Al ZET (2008). Reactive Oxygen Species Contribute to Lipopolysaccharide- Induced Teratogenesis in Mice. Toxicol. Sci 103, 149–157. [DOI] [PubMed] [Google Scholar]
- Zhao M, Chen Y, Dong X, Zhou J, Chen X, Wang H, and Wu S (2013). Folic Acid Protects against Lipopolysaccharide-Induced Preterm Delivery and Intrauterine Growth Restriction through Its Anti-Inflammatory Effect in Mice. PLoS One 8, e82713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zwan A, Bi K, Norwitz ER, Crespo ÂC, Claas FHJ, Strominger JL, and Tilburgs T (2018). Mixed signature of activation and dysfunction allows human decidual CD8 + T cells to provide both tolerance and immunity. Proc Natl Acad Sci U S A 115, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]