

Abstract
Multiple myeloma (MM) is amongst the most compelling examples of cancer in which research has markedly improved the length and quality of lives of those afflicted. Research efforts have led to 18 newly approved treatments over the last 12 years, including 7 in 2015. However, despite significant improvement in overall survival, MM remains incurable as most patients inevitably, yet unpredictably, develop refractory disease. Recent advances in high-throughput “omics” techniques afford us an unprecedented opportunity to (1) understand drug resistance at the genomic, transcriptomic, and proteomic level; (2) discover novel diagnostic, prognostic, and therapeutic biomarkers; (3) develop novel therapeutic targets and rational drug combinations; and (4) optimize risk-adapted strategies to circumvent drug resistance, thus bringing us closer to a cure for MM. In this review, we provide an overview of “omics” technologies in MM biomarker and drug discovery, highlighting recent insights into MM drug resistance gleaned from the use of “omics” techniques. Moving from the bench to bedside, we also highlight future trends in MM, with a focus on the potential use of “omics” technologies as diagnostic, prognostic, or response/relapse monitoring tools to guide therapeutic decisions anchored upon highly individualized, targeted, durable, and rationally informed combination therapies with curative potential.
Keywords: multiple myeloma, drug resistance; omics; genomics; proteomics; transcriptomics; metabolomics; immunomics; translational medicine; bench to bedside; immunotherapy
INTRODUCTION
Multiple Myeloma (MM) is a plasma cell neoplasm that accounts for 1.3% of all malignancies and 15% of hematological cancers, making MM the second most commonly diagnosed blood cancer (after non-Hodgkin lymphoma) [1]. Once considered an incurable disease with a short overall survival (OS), major progress in the understanding of MM biology and the development of highly active therapeutics has led to a distinct change in the natural history of MM. Indeed, MM is becoming a chronic illness for many patients, in which median OS has increased over 3 folds in the past 15 years. Therapeutic advancements have led to evolving treatment paradigms focusing on (1) autologous stem cell transplantation (ASCT), (2) therapies targeting MM in the context of the bone marrow (BM) microenvironment (e.g. proteasome inhibitors, immunomodulatory drugs, histone deacetylase inhibitors), and (3) immunotherapy (e.g. monoclonal antibodies, checkpoint inhibitors and T-cell immunotherapy) [2]. However, despite significant improvement in OS, MM remains incurable in the long-term as most patients inevitably, yet unpredictably, develop refractory disease (i.e. disease that fails to respond to induction or salvage therapy, or progresses within 60 days of last therapy) [3]. This is the product of genomic instability, clonal diversity, and MM’s unique relationship with the BM microenvironment [4]. The treatment of relapsed/refractory disease poses a special challenge due to significant heterogeneity in relapsed disease, clonal tiding, and the lack of clear biological-based recommendations on the choice of salvage therapies at different stages of disease progression [5]. A study by Kumar et al., has reported that patients who are double refractory to both proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs) do poorly, with a median OS and progression-free survival (PFS) of 9 and 5 months, respectively [6]. As such, there is an urgent need to decipher the underlying mechanisms of intrinsic and acquired drug resistance in MM.
“Omics” is the non-targeted, unbiased, and comprehensive study of genes (genomics), mRNA (transcriptomics), proteins (proteomics), lipids (lipidomics), and metabolites (metabolomics) in specific biological samples (Table 1) [7,8]. The success of genetic research in the discovery of therapeutic targets is exemplified by the use of the tyrosine kinase inhibitor Imatinib for the treatment of Philadelphia positive (Ph+) hematologic neoplasia [9]. In the decade following the discovery of the Philadelphia chromosome, the advent of next generation sequencing (NGS) has led to revolutionary advances in the diagnosis and management of hematologic (and solid) cancers with findings such as the BRAF V600E mutation in Hairy Cell Leukemia, the MYD88 265P mutation in Waldenstrom’s Macroglobulinemia, and the CALR mutation in JAK2 and MPL double negative myeloproliferative neoplasms [10–12]. In the field of MM, NGS has improved our understanding of the heterogeneous landscape of genetic alterations and facilitated the identification of multiple deregulated core signaling pathways and mutations of diagnostic and therapeutic significance [13]. A massive parallel sequencing study of samples from 203 patients diagnosed with MM reported frequent mutations in KRAS, NRAS, FAM46C, TP53, and DIS3 and less frequently in BRAF, TRAF3, CYLD, RB1, and PRDM1, which hold biological and therapeutic potential in MM [14]. NGS efforts have also been instrumental in uncovering clonal heterogeneity and evolution in MM patients [15,16]. Furthermore, the use of gene expression profiling has identified ≥ 20 different types of myeloma, each phenotypically different in treatment response and clinical behavior [17]. By using molecular profiles to understand disease mechanisms, predict drug response and patient relapse, “omics” data can be used to guide pre-clinical drug development and tailor personalized treatments for each individual patient and disease.
Table 1.
“Omics” technologies used in preclinical and clinical studies
| Omics Approach | Omics Technology | Source Material | Readout | Application | |
|---|---|---|---|---|---|
| Preclinical | Clinical | ||||
| WGS, WES | Genomics | Genomic DNA (Germline) | WGS: Sequence of entire chromosomal and
mitochondrial DNA WES: Sequence of all exomes in genome |
Discover new mechanisms of drug resistance, biomarker discovery | Patient risk stratification, predict response to therapy, guide therapeutic decisions |
| Cancer genome sequencing | Genomic DNA (Tumour) | Mutational profile of cancer | |||
| SNP array, CNV microarray | Genomic DNA (Germline or Tumour) |
Unbiased association of genotype and phenotype | Identification of genetic variation associated with response and/or adverse events to treatment | ||
| RNA Seq, RNA microarray | Transcriptomics | mRNA (cDNA) | Gene expression profile, disease associated genes, chemoresistance- associated genes | Monitor changes in mutational landscape of cancer, predict response to therapy, guide therapeutic decisions | |
| Protein analysis by LC-MS/MS, SILAC-MS, ITRAQ-MS | Proteomics | Proteins | Protein maps and predicted networks, disease-associated proteins, chemoresistance-associated proteins | Predict treatment response, guide therapeutic decisions, monitor treatment response and relapse | |
| Metabolome analysis by LC-MS/MS, NMR, ion-mobility spectrometry, Raman spectroscopy | Metabolomics | Metabolites | Metabolite profiles in cancer, tissues, and body fluids | ||
| Genome-wide DNA methylation assays, miRNA array, Histone modification assays | Epigenomics | DNA, proteins | DNA methylation, miRNAs, histone modifications | ||
| High-throughput screen
(knockdown/knockout shRNA/CRISPR-Cas9 screens, overexpression screens, drug screens) |
Genomics, Multi-omics |
Cells, proteins, embryo | Phenotype (i.e. survival, proliferation, chemoresistance), biochemical, etc… | NA | |
WGS: whole genome sequencing; WES: whole exome sequencing; SNP: single nucleotide polymorphism; CNV: copy number variant; LC-MS/MS: liquid chromatography-tandem mass spectrometry; miRNA: microRNA; NMR: nuclear magnetic resonance; shRNA: short hairpin RNA; CRISPR: clustered regularly interspaced short palindromic repeats
Indeed, recent advances in high-throughput “omics” techniques afford us an unprecedented opportunity to understand drug resistance at the genomic, transcriptomic, and proteomic level. The use of multi-”omics” has proven invaluable for investigating the genetic and molecular mechanisms of drug resistance in refractory MM in both clinical and pre-clinical studies. Specifically, a literature review of studies on “myeloma” “resistance”, published between 2010 and 2016, revealed 52, 9, and 3 papers that utilized genomics, proteomics, and metabolomics, respectively, to interrogate the mechanisms underlying drug resistance. In this review, we provide an overview of “omics” technologies in (1) developing MM clinical diagnostic and risk stratification tools, (2) understanding MM drug resistance in the era of conventional and targeted therapies, (3) developing new biomarkers and therapies in the era of targeted cancer immunotherapy. We also propose a model for the application of “omics” technologies in preclinical research (bench) and clinical practice (bedside) (Fig 1).
Figure 1. Application of “omics” technologies in preclinical research (bench) and clinical practice (bedside)Bench:
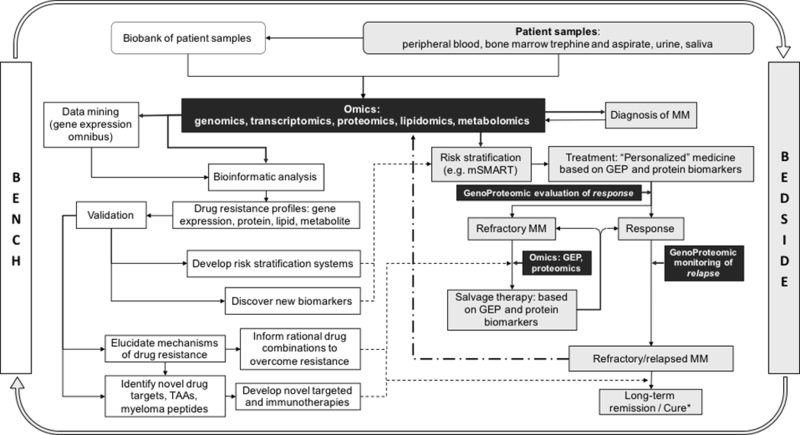
The use of “omics” technologies in pre-clinical research has and will continue to facilitate the development of (1) better risk stratification systems, (2) biomarker discovery, (3) rational drug combinations to overcome resistance, and (4) novel targeted and immunotherapies for use in the diagnostic workup and treatment of patients with MM. Bedside: Although at present, “omics” technologies are not ready for immediate clinical use as diagnostic, prognostic, or response/relapse monitoring tools, it can be envisaged that simple, rapid, robust, portable, and cost-effective clinical diagnosis, prognosis, and disease monitoring systems could be available in near future, which would not only improve clinical decisions but also guide the design of more clinically pertinent, bench to bedside research. Bench to bedside research, Beside to bench research, Application of “omics” technologies, TAA: tumor-associated antigens, GEP: gene expression profiling
CLINICAL APPLICATION OF “OMICS”: MOVING “OMICS”-BASED DIAGNOSTICS AND PROGNOSTICS FROM BENCH TO BEDSIDE
Potential of genomics in the identification of high-risk MM
Standard cytogenetics and FISH are key to risk stratify newly diagnosed multiple myeloma patients. However, there may be further heterogeneity even within groups with these genetic prognostic factors. From this perspective, standard karyotype and FISH only have limited value in guiding treatment decisions and aid in personalized therapeutic strategies [18–23].
On the other hand, gene expression profiling (GEP) has given rise to several genetic signatures that have been successfully implemented in MM to improve risk stratification [24–27]. The “Mayo stratification of myeloma and risk-adapted therapy model” (mSMART) put forward by the Mayo clinic Dysproteinemia group is an example of a model that integrates gene expression profiling (GEP) with conventional cytogenetics and fluorescent in-situ hybridization (FISH) [28]. Indeed, a study using GEP identified 70 genes that are associated with shorter durations of remission, event-free survival (EFS) and OS; 30% of which were found to be either upregulated or downregulated on chromosome 1. By utilizing a ratio of mean upregulated to downregulated gene expression, a high-risk score was formulated and shown to be an independent predictor of outcome in a multivariate analysis that included the International Staging System. The study further identified a 17-gene signature (subset of the original 70 genes) that could accurately define high-risk disease [24]. Another study identified 15 survival-associated genes through GEP. A risk score based on the expression level of these genes was calculated and used to stratify patients into a high-risk group (overexpression of cell cycle-related genes) and a low-risk group (heterogeneous GEP pattern with a hyperdiploid signature). This genetic signature was validated in three independent myeloma cohorts (n=853), with the low and high-risk groups reporting a three-year OS of 90.5% and 47.4%, respectively; hence attesting to the potential of GEP in myeloma risk stratification [29].
However, limitations to GEP do exist as the gene signatures used to stratify risk may not always be specific for a given clinical outcome, thus potentially leading to over- or under-treatment. Additionally, GEP cannot assay certain important prognostic factors, such as the presence of del(17p), and should therefore be combined with FISH analysis. Moreover, there still needs to be standardization in gene expression profiling methods, analysis techniques, and consensus and validation of the best genes to be used universally. Given these limitations, coupled with the fact that GEP is still largely experimental and not widely available, there are several issues that need to be addressed prior to clinical application [30]. Furthermore, while GEP has proven utility in tumor classification and survival risk prediction [29,31–40], gene expression alone may not be adequate in predicting complete response in MM, highlighting the need to adopt integrated omics approaches in the development of more accurate and comprehensive predictive models [41].
Treatment response and relapse monitoring using a genoproteomic approach
The early detection of MM relapse has been challenging due to a historical lack of sufficiently sensitive monitoring strategies [42]. Presently, however, a better understanding of disease biology coupled with progress in science and technology has enabled us to measure MRD in the bone marrow with sensitivities in the range of 10−5–10−6 cells through the development of cellular techniques such as next generation flow cytometry and “omics”-based approaches such as qASO-PCR (quantitative allele-specific oligonucleotide polymerase chain reaction) and next generation sequencing [43]. Specifically, NGS can be used to track clonal rearrangements in one or more of three Ig genes (IgH, IgLκ, IgLλ) unique to the malignant plasma cell over the course of disease and treatment to guide and further refine therapeutic decisions. Ultimately, the question today is no longer “can we detect MRD with sufficient sensitivity?” but rather “what is the practical value of minimal residual disease (MRD) monitoring and how can it be utilized to improve patient outcomes?”; a question that is thoroughly discussed in a current perspective piece by Anderson et al [44].
Researchers at the Mayo clinic have devised a new proteomics-based approach to monitor clonotypic peptides from M-protein heavy chain variable regions [45]. Compared with current analytical methods (e.g. protein electrophoresis/PEL, immune-fixation electrophoresis/IFE, and free light chain nephelometry/FLC), proteomics can detect clonotypic peptides in PEL-, IFE-, and FLC-negative samples. Thus, the use of proteomics to monitor myeloma progression and relapse has the capability to redefine clinical residual disease due to its superior sensitivity and specificity [45]. As tumor heterogeneity and evolution make myeloma a ‘moving’ molecular target, myeloma (M)-protein monitoring could be complimented by ongoing molecular profiling to evaluate how the genetic architecture of myeloma changes over time or in response to treatment. The proposed genoproteomic-based drug-repurposing program could be more effective than the current therapeutic approach (i.e. to treat empirically based on clinical trial evidence or to re-challenge with prior active agent or ASCT) adopted in patients who may be refractory to standard therapies [46].
PRE-CLINICAL APPLICATION OF “OMICS” IN THE ERA OF CONVENTIONAL AND TARGETED CHEMOTHERAPY: UNCOVERING AND OVERCOMING MECHANISMS OF MM DRUG RESISTANCE
As alluded to previously, a search of the published literature from 2010 to 2016 identified 62 studies (25 clinical; 37 pre-clinical) that utilized “omics” technologies to screen for genes, proteins, and metabolites dysregulated in drug-resistant MM (Table 2). These studies identified many deregulated pathways (e.g. survival, apoptosis, proliferation, cell-cycle regulation, DNA repair, epigenetic regulation, redox homeostasis, protein handling, drug-efflux, autophagy, inflammation, and plasma cell maturation) that could contribute to resistance to conventional chemotherapy, proteasome inhibitors, IMiDs, and small molecule inhibitors. In particular, decreased XBP1 splicing was recently found to be a marker of bortezomib resistance in MM [47]. By suppressing XBP1s, MM cells de-commit to plasma cell maturation and decrease immunoglobulin production, proteasome load, and ER stress, resulting in acquired resistance to PI [47,48]. Low cereblon (CRBN) expression on the other hand was discovered to be implicated in Lenalidomide and Pomalidomide resistance [49]. The next step would then be to validate and translate this data into (1) novel diagnostic, prognostic, and therapeutic biomarkers and (2) novel therapeutic targets and rational drug combinations, to optimize risk-adapted strategies to circumvent drug resistance and bring us closer to a potential cure for MM.
Table 2:
Literature review of “omics” studies on “myeloma” “resistance” published between 2010 and 2016
| CLINICAL STUDIES | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Resistant to | Primary (n) vs CL | Type of Cell Assayed | “Omic” Technology Used | Gene/Protein Expression Database | Pathways Involved in Resistance | Specific Findings | Country | Year | Reference (PMID) | |||||||||||
| Resistance to Proteasome Inhibitors | ||||||||||||||||||||
| 1 | BTZ | (544) | Primary MM cells | GEP | GSE2658 | Cell survival, proliferation, DNA repair | JUN and EGR-1 are critical components in BTZ-induced apoptosis in primary MM cells; knockdown of JUN or EGR-1 resulted in BTZ resistance in MM cell lines | USA | 2010 | (19837979) | ||||||||||
| 2 | BTZ, IMiD, CST | (1) | Drug-resistant extramedullary primary MM | WGS, WES, GEP | NA | 4 somatic insertions/deletions, 38 intrachromosomal rearrangements, 35 translocations, 271 nonsynonymous point mutations in KRAS, PIK3CA, ATM, NF-κB2; Truncating mutation of CRBN, point mutations in proteasome subunit G2 and glucocorticoid receptor | USA | 2013 | (23480694) | |||||||||||
| cGH | NA | Aneuploidy, deletions | del1(p13.2–34.2), monosomy 13, monosomy X | |||||||||||||||||
| 3 | BTZ, DEX | (345) | Primary MM cells | GEP | GSE2658 | Redox homeostasis | SCARA3 upregulation in MM cells results in BTZ and DEX resistance; SCARA3 protects against oxidative stress-induced cell killing | USA | 2013 | (23537707) | ||||||||||
| 4 | BTZ | (417) | Primary NPC (n=22); MGUS (n=44); MM (n=351) | GEP | GSE2658 | BTZ resistance is associated with tupregulation of antioxidant genes (CuZnSOD, GPx-1, GSH); the overexpression of SOD1 induced BTZ resistance while the inhibition of CuZnSOD increased BTZ sensitivity | USA | 2015 | (25485927) | |||||||||||
| 5 | BTZ | NA | Primary MM cells | GEP |
GSE4581 GSE6477 |
Hypoxia-induced and acquired BTZ resistance results in increased TrxR1 expression and NF-κB subunit p65 nuclear protein levels; inhibition of TrxR1 restored sensitivity of MM cells to BTZ and decreased hypoxia induced upregulation of p65 | AUS | 2016 | (26743692) | |||||||||||
| 36 | CFZ | NA | Primary MM cells (MRD) | GEP | GSE70399 | Redox homeostasis, protein handling, apoptosis, inflammation | CFZ resistance is associated with the upregulated expression of Nrf2 target genes which include autophagy related genes GABARAPL1 and SQSTM1/p62. CFZ resistance is also associated with the induction of the unfolded protein response (UPR) and PERK-eIF2α signalling which in turn results in NrF2 overexpression. EIF4E3, a downstream target and positive upstream regulator of NrF2, is upregulated in CFZ-resistant cells. Inhibition of the Nrf2-EIF4E3 axis or the PERK-eIF2α pathway, disruption of redox homeostasis, or the inhibition of fatty acid oxidation conferred sensitivity to CFZ | USA | 2016 | (27626179) | ||||||||||
| CL | CFZ-resistant MM cell line: LP-1/CFZ | GSE78069 | ||||||||||||||||||
| 7 | PAD | (77) | Primary RR MM (to THAL-based regimen) | LC-MS/MS | NA | Reduced response to PAD is associated with: (1) Upregulation of proteasome proteins and molecules related to protein folding; (2) Increased relative abundance of TXN, TXNDC5, thioredoxin-dependent peroxide reductase, and thioredoxin-like protein 1, upregulation of PRDX2/5/6, decreased levels of catalase, myeloperoxidase, and glutathione S-transferase P; (3) Annexin A1/A6 and vimentin downregulation; (4) upregulation of four programmed cell death proteins; (5) downregulation of proteins involved in inflammatory and defence response, apart from MHCII and macrophage migration inhibitory factor which were increased | POL | 2016 | (27527861) | |||||||||||
| 8 | BTZ | NA | Primary MM cells | GEP | NA | BTZ resistance is associated with del(8)(p21) which results in altered expression of genes such as TRAIL-R4, CCDC25, RHOBTB2, PTK2B, SCARA3, MYC, BCL2, and TP53 | SWE | 2015 | (26378933) | |||||||||||
| 9 | BTZ | (20) | BTZ treated primary MM cells | DNA Seq | NA | Protein handling | BTZ resistance results from inactivating XBP1 mutations: XBP1-L167I mutation (mapped to Ire1 splice site on XBP1u); XBP1s-P326R (non-conservative missense mutation within trans-activation domain of Xbp1s) | CAN | 2015 | (24029229) | ||||||||||
| 10 | VMP +/− RD | (7) | Primary MM cells: MRD subclone after Induction therapy | GEP | GSE70399 | BTZ resistance associated with inhibition of pathways involved in protein export, protein processing in ER, N-glycan biosynthesis. Overexpression of cancer related genes: FERMT3, TSPO, ALOX5, NCF4, FLNA, PREX1, PYCARD, MY01G found to contribute to BTZ resistance. Downregulation of cancer related genes: SERPINI1, DUSP11, ALCAM, CCNC, COPZ1, FZD3, E2F5; ER related genes: PDIA5, EMC7; PI related genes: PSMD10, SMILE/TMTC3, PSME3, PSMB6, CAV1 also associated with resistant phenotype | ESP | 2016 | (26755711) | |||||||||||
| 11 | CFZ | CL | MM cell line: NCI-H929/CFZ | GEP | NA | Drug-efflux | Upregulated ABCB1 expression confers resistance to CFZ; ABCB1-hi cells associated with upregulation of ASPM, KIF14, TMPO, ADM, EPAS1, HIF-2α, CXCL12/SDF1, HBEGF | USA | 2013 | (23475625) | ||||||||||
| (6) | Primary MM cells | NA | ||||||||||||||||||
| (304) | Primary MM cells | MMRC reference collection dataset | ||||||||||||||||||
| 12 | BTZ, DOX | (9) | Primary MM cells | GEP | GSE19554 | ALDH1A1 upregulates survival proteins (Akt and BCL2) and contributes to drug resistance; overexpression of ALDH1A1 also led to upregulation of ALDH1A1-RXRα-NEK2 pathway resulting in activation of drug-efflux pump ABCB1 | USA | 2014 | (25230277) | |||||||||||
| 13 | VD or VAD | (24) | Matched diagnostic and relapse primary MM samples | DNA Copy Number + LOH | GSE37459 | Cell cycle | Relapse associated with increase in CNA (diagnosis: 15.8 vs relapse: 19.1); Relapse represented intraclonal evolution and diversification of myeloma; Upregulation of NF-κB genes at relapse; 1q21 gain, TP53 deletions | FRA | 2013 | (22874878) | ||||||||||
| 14 | BTZ | (264) | Primary relapsed MM | GEP | NA | CKS1B expression is increased in relapsed MM and confers drug resistance to BTZ | USA | 2015 | (26156395) | |||||||||||
| 15 | BTZ | (1) | Primary MM cells | DNA Seq | NA | Eight nonsynonymous somatic mutations and several copy number variants including CCND1 and RB1 | JPN | 2015 | (26491355) | |||||||||||
| 16 | BTZ | NA | Primary MM cells (CR vs PD) | GEP | GSE9782 | Plasma cell maturation | Expression of XBP1 target genes lower in PD (BTZ-resistant) tumors; suppression of XBP1s induces BTZ resistance via de-commitment to plasma cell maturation and Ig production resulting in diminished ER front-loading and cytotoxic susceptibility to PI-induced inhibition of ER-associated degradation | CAN | 2015 | (24029229) | ||||||||||
| 17 | VMP +/− RD | (12) | Primary MM cells: MRD subclone after Induction therapy | DNA Copy Number + LOH | GSE70399 | Copy number abnormalities | CNA: 66 CNA (56 losses and 10
gains) uniquely present in either dignostic or MRD clonal PC
population CNN-LOH: 3 patients’ MRD clonal PCs lacked CNN-LOH present at diagnosis, whereas 1 patient’s MRD clonal PCs had a CNN-LOH in chromosome 1 that was not detected at diagnosis |
ESP | 2016 | (26755711) | ||||||||||
| 18 | BTZ | (9) | Primary drug-resistant pre-plasma cells | GEP | NA | Epigenetic | Drug-resistant pre-PCs (CD19- CD138-) are enriched in epigenetic regulators (e.g. histone methyltranferases and demethylases, histone acetyltransferases and deacetylases), suggesting drug resistance is associated with a reversible bidirectional phenotypic transition of myeloma-propagating cells | GBR | 2013 | (23169779) | ||||||||||
| 19 | BTZ | (6) | Primary MM cells (BTZ-responders vs BTZ-resistant) | miRNA array | NA | Resistance to BTZ associated with downregulation of exosomal miR-16–5p, miR-15a-5p, mir-17–5p, and miR-20a-5p | CHN | 2016 | (27129167) | |||||||||||
| Resistance to Conventional Chemotherapy | ||||||||||||||||||||
| 20 | MPH | (357) | CD138+ primary MM cells | GEP | NA | Drug-efflux, protein handling | Resistance associated genes: SLC31A2 (membrane pump), FBXW7, USP6, UBE2J1, Wnt-5a (UPS), CSGAL-NACT1 (component of CD138) | DEN | 2013 | (24376673) | ||||||||||
| 21 | MPH | (12) | Primary MM cells | GEP | GSE2658 | Cell-cycle | Kruppel-like factor 4 (KLF4) expression increased resistance of MM cells to MPH resulting cell cycle blockade; KLF4 associated with induction of p21(Cip1) and p27(Kip1) in cell lines with intact p53 pathway and of p27(Kip1) only in those with impaired p53 pathway | FRA | 2013 | (23585530) | ||||||||||
| 22 | MPH, DEX | CL and Primary | MM cell line: ARP-1 and primary human macrophage cells | GEP | NA | Apoptosis, survival, proliferation | PSGL-1 (P-selectin glycoprotein ligand-1)/selectins and ICAM-1/CD18 contribute to macrophage-mediated myeloma cell drug resistance; interaction of these molecules activated Src and Erk1/2 kinases and c-myc pathways and suppressed caspase activation induced by chemotherapy | USA | 2013 | (22996336) | ||||||||||
| Resistance to Other Therapies | ||||||||||||||||||||
| 23 | Multidrug | (51) | Primary MM cells | GEP | NA | Survival, proliferation | CKS1B overexpression activates downstream STAT3 and MEK/ERK pathways resulting in drug resistance | USA | 2010 | (20930946) | ||||||||||
| 24 | Multidrug | (19) | Primary MM cells | GEP | GSE19554 | NEK2, a chromosome instability gene, is highly associated with drug resistance; overexpression of NEK2 activates Akt and Wnt signalling | USA | 2012 | (23328480) | |||||||||||
| 25 | NK cell lysis | (12) | Primary MM cells | GEP | NA | Antigen presentation | MHCI highly expressed in patient MM cells that were relatively resistant to NK-92 cell lysis | CHN | 2014 | (24850305) | ||||||||||
| PRE-CLINICAL STUDIES | ||||||||||||||||||||
| Resistant to | Type of Cell Assayed | “Omic” Technology Used | Gene/Protein Expression Database | Pathways Involved in Resistance | Specific Findings | Country | Year | Reference (PMID) | ||||||||||||
| Resistance to Proteasome Inhibitors | ||||||||||||||||||||
| 26 | BTZ | MM cell line: KMS-11 | siRNA screen | NA | Plasma cell maturation | Suppression of XBP1s induces BTZ resistance by de-commitment to plasma cell maturation and immunoglobulin production thereby decreasing ER front-loading and cytotoxic susceptibility to PI-induced ERAD | CAN | 2015 | (24029229) | |||||||||||
| IRE1/XBP1 knockdown MM cell line: OCI-MY5 | GEP | GSE44968 | Protein handling | IRE1 knockdown associated with BTZ resistance | ||||||||||||||||
| 27 | BTZ | BTZ-resistant MM cell line: U266/R6 | GEP | NA | HSP88 overexpressed in BTZ-resistant cells; HSPB8 plays important role in elimination of aggregates | FRA | 2014 | (25051369) | ||||||||||||
| 28 | BTZ | BTZ-resistant MM cell lines: KMS-11/BTZ, OPM-2/BTZ | DNA Seq | NA | Ubiquitin-proteasome system | Point mutation (G322A) in PSMB5 gene mediates BTZ resistance | JPN | 2010 | (20555361) | |||||||||||
| 29 | BTZ | Multidrug-resistant MM cell line: RPMI-R5 | GEP | NA | USP24 overexpressed in BTZ-resistant MM cells | ITA | 2011 | (20977926) | ||||||||||||
| 30 | PI | CML cell line: KBM7 | WGS | PRJNA281714 | Resistance to MG132 and bortezomib associated with mutations in 19S proteasome subunit. Cells harbouring PSMC5 or PSMD2 mutations maintained a significant level of 20S proteasome activity in the presence of BTZ treatment | USA | 2015 | PMC4551903 | ||||||||||||
| GEP | PRJNA281613 | Increased resistance to BTZ in cells with 19S subunit reduction associated with suppression of cell cycle (repressed genes involved in DNA replication and cell cycle control), which primes cells to enter a protected, quiescent state | ||||||||||||||||||
| 31 | CFZ | MM cell line: U266 | shRNA screen | NA | 19S proteasomal regulator knockdown induced resistance to CFZ | USA | 2015 | (26327694) | ||||||||||||
| LC-MS/MS | NA | PSMD12 knockdown resulted in selective upregulation of protein turnover pathways such as SQSTM1/p62, UFD1L, and VCP/p97 | ||||||||||||||||||
| 32 | BTZ | MM cell lines: XG1, MDN, SBN, U266, LP1, L363, JJN3, RPMI-8226, NCI-H929 | GEP | NA | Survival, proliferation | Resistance to BTZ associated with induction of REDD1, a negative regulator of mTORC1 activity | FRA | 2010 | (20100206) | |||||||||||
| 33 | BTZ | BTZ-resistant MM cell lines: ANBL-6/BTZ, OPM-2/BTZ, RPMI-8226/BTZ | GEP | NA | IGF-1/IGF-1R signalling contributes to acquired BTZ resistance; OSI-906 (dual IGF-1R and insulin receptor inhibitor) overcame BTZ resistance in cell lines and primary samples in vitro and in vivo | USA | 2012 | (22932796) | ||||||||||||
| 34 | MPH, DOX, BTZ, ETP, STA, tm | MM cell line | GEP | GSE38204 | Drug resistance associated with higher cMET phosphorylation resulting in constitutive activation of cMET-dependent signalling pathways | ITA | 2013 | (23804425) | ||||||||||||
| Drug-resistant MM cell lines: RPMI-8226/R5, MM.1R | Proteome profiler assay | NA | ||||||||||||||||||
| 35 | PI | PI-resistant MM cell lines: LP-1/cIAP2, ANBL-6/cIAP2 | GEP |
GSE4589 GSE5900 |
cIAP2 overexpression confers MM cells with resistance to proteasome inhibitors; 12 target genes of the NF-κB pathway were downregulated; other genes downregulated were IL1RN, TNFAIP2, MBP, IL23A, RelB, FTH1, CTSB, S100A6, ASS1 | SWE | 2015 | (26036313) | ||||||||||||
| 36 | BTZ | BTZ-resistant MM cell line: RPMI-8226/BTZ | GEP | GSE66910 | Autophagy | GRP78-encoding HSPA5 (and hence autophagy) significantly upregulated in BTZ-resistant cells; metformin suppressed GRP78 and enhanced anti-proliferative effect of BTZ | USA | 2014 | (25605012) | |||||||||||
| 37 | BTZ | BTZ-resistant MM cell line: PS-R (U266/BTZ) | GEP | NA | BCL2L11 (Bim) downregulation in BTZ-resistant MM cells resulting in increased cytoprotective autophagy | USA | 2014 | (25208888) | ||||||||||||
| 38 | BTZ | CAF from BTZ-resistant patients | LC-MS/MS | NA | BTZ-resistance parallels activation of oxidative stress and pro-survival autophagy (increase LC3-II and inhibits p62 and p-mTOR) | ITA | 2016 | (26487273) | ||||||||||||
| 39 | CFZ | CFZ-resistant MM cell lines: KMS-11/CFZ, KMS-34/CFZ | GEP | GSE13411 | Cell-cycle | CFZ-resistance associated with autophagy; Kruppel-like factor 4 (KLF4) expression and nuclear localisation were elevated in CFZ-resistant MM cells | USA | 2015 | (26109433) | |||||||||||
| 40 | PI | Multidrug-resistant MM cell line: RPMI-8226/R5 | ITRAQ-MS | NA | Overexpression of myristoylated alanine-rich C-kinase substrate (MARCKS) in drug-resistant R5 cells; pMARCKS promotes cell-cycle progression by facilitating SKP2 expression, suppressing p27(Kip1), and promoting Cyclin E/CDK2 activity to counteract drug-induced cell-cycle arrest | CAN | 2015 | (25179733) | ||||||||||||
| 41 | BTZ | BTZ-resistant Mouse MM cell lines | GEP | GSE41930 | Redox homeostasis | BTZ-sensitive (but not resistant) MM cells upregulate a cluster of NRF-2-mediated oxidative stress response genes: Hspb1, Dnajb1, Hspa1a, Hspa1b, and Ddit3 (CCATT/enhancer-binding protein homologous protein/CHOP); CMAP predicted response to HDACi | USA | 2013 | (23536725) | |||||||||||
| 42 | BTZ, DEX, MPH | MM cell line: MM1.S | GEP | GSE52315 | Hypoxia-induced drug resistance results from upregulation of HIF1A and its target gene LDHA; knockdown of LDHA restores sensitivity to BTZ | USA | 2015 | (25769724) | ||||||||||||
| BTZ | Metabolite Profiling | NA | ||||||||||||||||||
| 43 | BTZ | BTZ-resistant MM cell: ANBL-6/BTZ | Metabolite Profiling | NA | DNA metabolism | Significant alterations in anabolism/catabolism of purines, pyrimidines, and various CoAs between BTZ-resistant and sensitive MM cells | USA | 2014 | (24611431) | |||||||||||
| 44 | BTZ | Mouse MM cell line: 5T33MMvt | GEP | NA | Cytochrome p450 | Dll1/Notch activation contributes to BTZ resistance through downstream upregulation of CYP1A1 | BEL | 2012 | (23111325) | |||||||||||
| Resistance to Immunomodulatory Drugs | ||||||||||||||||||||
| 45 | LEN/POM | OPM2 MM cell line after either CRBN KD or LEN treatment | GEP | GSE31421 | Ubiquitin-proteasome system | Presence of CRBN critical for activity of LEN: NT control, LEN-sensitive cells showed 2-fold expression changes in ~1200 genes while CRBN-depleted, LEN-resistant OPM2 cells showed changes in ~180 genes after LEN treatment | USA | 2011 | (21860026) | |||||||||||
| LEN-resistant MM cell line: MM1.S/LEN | CGH + FISH | GSE31451 | MM1.S: 44 chromosomes, CRBN:
1 copy, MM1.S/LEN: 40-fold reduction of CRBN expression on MM1.S/LEN compared with MM1.S, under-expression of LAPTM4B, ASS1, DDIT4, C6orf190, MAG1, QKI, ANTXR2, COPZ2 |
|||||||||||||||||
| 46 | LEN | Cell Lines: 293FT, U937, MM1S, L363 | (LUC)-ORF Library | NA | LEN binds to cereblon to selectively enhance ubiquitination and proteasomal degradation of IKZF1 and IKZF3 | USA | 2014 | (24292623) | ||||||||||||
| 47 | LEN | MM cell line: MM1.S | SILAC-MS | NA | LEN binds DDB1 and CRBN (forms CRL4 E3 ubiquitin ligase together with CUL4 and ROC1) resulting in increased ubiquitination and proteasomal degradation of IKZF1 and IKZF3 | USA | 2014 | (24292625) | ||||||||||||
| 48 | LEN | LEN-resistant MM cell lines: (R10R) ANBL-6, KAS-6/1, U266, MM1.S | GEP | NA | Survival, proliferation | Activation of Wnt/β-catenin pathway mediates LEN-resistant MM cell lines and primary plasma cell samples while knockdown of β-catenin restores sensitivity to LEN | USA | 2011 | (21189262) | |||||||||||
| 49 | LEN/POM + DEX | In Vivo Murine Model (LEN + DEX or POM+ DEX-resistant MM.1S) | GEP | NA | Resistance to LEN/POM + DEX accompanied by upregulation of MEK/ERK pathway and addition of selumetinib (MEK inhibitor) resensitizes resistant cells | ESP | 2015 | (25102946) | ||||||||||||
| 50 | LEN | LEN-resistant MM cell lines: (R10R) ANBL-6, KAS-6/1, U266, MM1.S | GEP | NA | CAM-DR | Upregulation of CD44 confers CAM-DR in LEN resistant MM cells that could be overcome by Wnt/β-catenin suppression with FH535 and all-trans retinoic acid | USA | 2014 | (23760401) | |||||||||||
| Resistance to Conventional Chemotherapy | ||||||||||||||||||||
| 51 | PRD | PRD-resistant: MM cell line: U266/PRD, RPMI-8226/PRD | GEP | NA | Survival, proliferation, signaling | Upregulation of Rho family of GTPases; downregulation of ETS, NF-κB2; downregulation of TGFβ, SOCS2, SOCS4, WSB2 | TUR | 2012 | (22681910) | |||||||||||
| VICE | VICE-resistant: MM cell line: U266/VICE, RPMI-8226/VICE | Upregulation of TGFβ; Downregulation of Rho family of GTPases; downregulation of SOCS2, SOCS4, WSB2 | ||||||||||||||||||
| 52 | MPH | MPH-resistant MM cell lines: RPMI-8226/LR5, U266/LR6 | LC-MRM | NA | Survival, proliferation, DNA repair, apoptosis | NF-κB signaling, Bcl-2 family of apoptosis-regulating proteins, and Fanconi Anemia DNA repair components contribute to MPH resistance in MM cells | USA | 2011 | (21846842) | |||||||||||
| 53 | ATO | ATO-resistant cell line: RPMI-8226/ATOR05 | GEP | NA | Redox homeostasis | Upregulation of intracellular GSH associated with ATO resistance; cross resistance to MPH and DOX was observed as well | USA | 2012 | (23285138) | |||||||||||
| 54 | MPH | MPH-resistant MM cell line: RPMI-8226/MPH | GEP | GSE60970 | Redox homeostasis, warburg effect | MPH-resistance associated with metabolic switch conforming to Warburg effect (aerobic glycolysis), elevated oxidative stress response mediated by VEGF/IL8-signaling, upregulation of aldo-keto reductase levels of AKR1C family involved in prostaglandin synthesis | NOR | 2015 | (25769101) | |||||||||||
| SILAC-MS | PXD001276 | |||||||||||||||||||
| 55 | DOX | DOX-resistant MM cell line: U266/DOX, RPMI-8226/DOX | miRNA array | NA | Epigenetic | U266/DOX: 9 miRNAs
overexpressed; 16 miRNAs underexpressed RPMI-8226/DOX: 21 miRNAs overexpressed; 26 miRNAs underexpressed |
USA | 2010 | (20357429) | |||||||||||
| MPH | MPH-resistant MM cell line: U266/MPH, RPMI-8226/MPH | U266/MPH: 8 miRNAs
overexpressed; 10 miRNAs underexpressed RPMI-8226/MPH: 12 miRNAs overexpressed; 21 miRNAs underexpressed |
||||||||||||||||||
| 56 | DEX | DEX-resistant MM cell line: MM.1R | miRNA array | NA | 10 and 12 miRNAs upregulated and downregulated respectively in MM.1R cells vs MM.1S. DEX resistance associated with increased miR-221–222 expression and downregulation of PUMA | USA | 2015 | (26249174) | ||||||||||||
| 57 | ADM +
4-OHCY |
MM cell line: RPMI-8226 | GEP | GSE66466 | Chemotherapy increases H3K27me3 that is abrogated by CAM-DR via inactivating phosphorylation of transcription regulator EZH2, resulting in sustained expression of antiapoptotic genes: IGF1, BCL2, HIF1A; inhibition of IGF-1R/PI3K/Akt pathway reversed CAM-DR through EZH2 dephosphorylation and H3K27 hypermethylation | JPN | 2015 | (26517694) | ||||||||||||
| Immunoblot | NA | |||||||||||||||||||
| 58 | DEX | DEX-resistant MM cell line: MM.1R | GEP | GSE59805 | miR-150–5p, miR-26b, miR-125a-5p, miR146–5p, and miR-184 are DEX inducible in MM.1S but not in MM.1R | BEL | 2014 | (25474406) | ||||||||||||
| miRNA array | NA | Cell-cycle | DEX-resistant MM cells did not upregulate genes involved in cell cycle control, cell organisation, cell death, and immunological disease when treated with DEX while DEX-sensitive MM cells upregulated the aforementioned genes | |||||||||||||||||
| 59 | PRD | PRD-resistant: MM cell line: U266/PRD | GEP | NA | Cell-cycle, lipid metabolism | Dowregulation of CDK6, cyclin D3, TNFR, BNIP1, BNIP3, PDCD11, PDCD4, UGCG, ASAH1; overexpression of CDKN2B, CDKN2A, BIRC group of genes, LASS6, SMPD3, DEGS1 | TUR | 2014 | (25330516) | |||||||||||
| VICE | VICE-resistant MM cell line: U266/VICE | Overexpression of cyclin E2, E2F, E2F7, E2F8, TNFAIP6, TNFIP8, BIRC group of genes, BNIP1, BNIP3, PDCD11, PDCD4 | ||||||||||||||||||
| MPH | MPH-resistant MM cell line: U266/MPH | Downregulation of TNFR, CDKN1A, CDKN1C, LASS6, SMPD1 | ||||||||||||||||||
| Resistance to Other Therapies | ||||||||||||||||||||
| 60 | HDACi | Human myeloma cell lines (sensitive, intermediate, resistant) | GEP | NA |
Protein
handling, cytoskeleton |
35-gene signature significantly enriched for 2 pathways in resistant MM cells: (1) regulation of actin cytoskeleton, (2) protein processing in ER | AUS | 2014 | (24651437) | |||||||||||
| 61 | rmhTRAIL | MM cell lines: RPMI-8226 (TRAIL-sensitive); U266 (TRAIL-resistant) | LC-MS/MS | NA | Ubiquitin-proteasome system | Upregulation of small ubiquitin-related modifier 1 and ubiquitin proteasome pathway-related proteins contributes to TRAIL resistance | CHN | 2016 | (27284413) | |||||||||||
| 62 | Geftinib/ Afatinib | MM cell lines: L-363 (NRASmut), LP-1 (NRASwt) | Metabolite Profiling | NA | Pentose phosphate pathway | Significant increased metabolites from Pentose Phosphate Pathway (PPP) associated with Gefitinib resistance | CHN | 2015 | (25894462) | |||||||||||
NA: not available; CL: cell line; HDACi: histone deacetylase inhibitors; CST: corticosteroid; PRD: prednisolone; VICE: vincristine; ATO: arsenic trioxide; ETP: etoposide; STA: staturosporin; tm: tunicamycin; DOX: doxorubicin; MPH: melphalan; DEX: dexamethasone; IMiD: immunomodulatory drugs; THAL: thalidomide; LEN: lenalidomide; POM: pomalidomide, BTZ: bortezomib; CFZ: carfilzomib; VAD: vincristine, adriamycin, dexamethasone; CNA: copy number abnormalities; PI: proteasome inhibitor; CR: complete response; PD: progressive disease; MRD: minimal residual disease; VMP: bortezomib, melphalan, prednisone; RD: lenalidomide, dexamethasone; PAD: bortezomib, doxorubicin, dexamethasone; TXN: thioredoxin; TXNDC5: thioredoxin domain-containing protein 5; rmhTRAIL: recombinant human tumor necrosis factor-related apoptosis-inducing ligand; GSH: Glutathione; GPx-1: Glutathione prooxidant production; TrxR1: Thioredoxin reductase 1; CAF: cancer associated fibroblasts
Genomic identification of biomarkers predicting drug resistance
The use of gene expression profiling to identify novel biomarkers of drug response in MM has already been extensively reviewed [50–52]. As such, we will only briefly highlight a few studies that employed genomic evaluation to identify potential biomarkers associated with drug resistant MM. A recent study identified a 23-gene expression signature, by comparing the baseline gene expression of bortezomib-resistant (BzR) vs bortezomib-sensitive (BzS) mouse MM cell lines, that could significantly predict patient outcomes in the MMTT3 human drug trial [53]. Additionally, an RNAi screen identified 37 genes that could potentially be targeted to sensitize MM cells to proteasome inhibitors [54]. Current genes in the biomarker translational pipeline include CXCR4; a gene linked with bortezomib-resistance and a potential diagnostic biomarker that can predict patient response to borterzomib [55].
Proteomic identification of biomarkers predicting drug resistance
Apart from gene expression profiling, mass spectrometric (MS) exploration of early biomarkers of bortezomib resistance has yielded some promising results. Apolipoprotein C-I and C-I’ were recently found to be significantly increased in the serum of treatment-refractory patients compared to treatment-responsive patients 24-hours post bortezomib administration [56]. In a separate study, an ‘isobaric tags for relative and absolute quantification’ (ITRAQ)-based approach implicated drug-resistance in the BzR RPMI-8226/R5 MM cell line with the overexpression of the MARCKs protein [57]. MS profiling of dexamethasone (dex)-sensitive MM.1S revealed FKBP5 overexpression following dex treatment which was not seen in the dex-resistant MM.1R cell line [58].
Membrane proteins play a significant role in chemoresistance [59–61]. Membrane proteomics represents a highly efficient way of identifying membrane proteins with unusual properties that can potentially lead to the discovery of novel therapeutic targets as well as important modulators of drug resistance. However, despite constituting 30% of the total genome, membrane proteins are under-represented in many proteome profiles. The under-representation of membrane proteins from proteome studies is attributed mainly due to the heterogeneous, hydrophobic, and low abundance nature of these proteins. Lately there have been significant developments made in the areas of membrane protein analysis due to the availability of superior solubilisation methods and the production of new mass spectrometers that can detect and quantify low abundant proteins such as those found in or associated with the membrane.
Apart from the regulation of protein expression, post-translational modifications (e.g. phosphorylation, glycosylation, ubiquitination, methylation, acetylation) provide an additional layer of control over protein function. Emerging evidence is showing that cancer progression is largely regulated by epigenetic alterations such as post-translational modifications (PTMs). PTMs play critical roles in gene regulation, cellular functions, tissue development, diseases, malignant progression and drug resistance. Mass spectrometry is now sensitive enough to reliably identify PTMs thus allowing us to further interrogate how PTMs, and not simply expression, of proteins underlie drug-resistance. Chemoresistance in MM has been associated with aberrant activation of FGFR3, through tyrosine phosphorylation, in 15–20% of MM due to a t(4;14)(p16.3;q32) translocation [62–64]. Phosphoproteomic profiling of proteins associated with FGFR3 expression, ligand activation, and drug inhibition was recently performed and several phosphotyrosine sites downstream of FGFR3 activation that could potentially serve as biomarkers of drug resistance were identified and quantified [65]. While the function of phosphorylation has been extensively studied over the last 20 years and is now relatively well-characterized, much less is understood about the role other PTMs (e.g. ubiquitination) play in MM drug resistance. Interestingly, multi-monoubiquitination can mark transmembrane proteins (for example, receptors) for removal from membranes (internalization) and fulfill several signaling roles within the cell. When cell-surface transmembrane molecules are tagged with ubiquitin, the subcellular localization of the protein is altered, often targeting the protein for destruction in the lysosomes. The conditions in the bone marrow microenvironment in MM and, in particular, the presence of growth factors (interleukin 6, insulin-like growth factor-1, and vascular endothelial growth factor) and their interaction with corresponding membrane receptors, can promote drug-resistance and plasma cell survival. Understanding how ubiquitination contributes to this phenotype, especially in presenting and internalizing membrane proteins may present opportunities to develop novel targeted therapies and biomarkers for monitoring patients.
MS analysis is a powerful and proven research tool to explore MM biology. However, its clinical implementation has several limitations. Firstly, high-abundant proteins such as albumin can mask low-abundant proteins while sample purification may result in the loss of low-abundant proteins through interactions with high-abundant proteins. Thus, careful analyses need to be performed at every purification step [57]. Furthermore, variables such as age differences, gender, ethnicity, menopause, and nutrition could confound biomarker discovery [57].
“Omics” identification of novel drug targets in MM
Significant sequencing efforts in MM have identified driver mutations (e.g. KRAS, NRAS, BRAF, FAM46C, TP53, DIS3, SP140, LTB, ROBO1) that can guide the development of novel targeted therapies exploiting oncogene addiction [14,66,67]. A recent study showed that whole genome sequencing (WGS) could detect BRAF mutations otherwise missed by FISH, thereby identifying a subset of patients that might benefit from BRAF inhibition [68]. However, the caveat here is that not only might some of these mutations only be present in a fraction of cells but, in addition, fluctuations of MM subclonal architecture make it difficult to predict the clinical efficacy of such a strategy. Nonetheless, “omics” approaches have led to the clinical development of potential drugs against drug resistant MM, some of which are highlighted in Table 3.
Table 3.
MM drugs in clinical development against pathways identified in “omics” studies
| Drug Name | Sponsor | Mechanism of Action | Status | Study Design | Identifier |
|---|---|---|---|---|---|
| Histone Deacetylase Inhibitors | |||||
| HDAC6-selective inhibitor | |||||
| ACY-241 | Acetylon (USA) |
Downregulation of MYC and IRF4 | Phase Ia/Ib | ACY-241 + POM + DEX vs ACY-241 alone in R/R MM | NCT02400242 |
| Ricolinostat | Acetylon (USA) |
Caspase 8/9 mediated apoptosis; Terminal UPR induction; PolyUb protein accumulation; Aggresome disruption | Phase I/II | Ricolinostat + LEN + DEX in R/R MM | NCT01583283 |
| pan-HDAC inhibitor | |||||
| Vorinostat | Merck (USA) |
p21 and p53 upregulation; Rb dephosphorylation; BID cleavage; Calpain activation | Phase III | Vorinostat + BTZ vs BTZ alone in R/R MM | NCT00773747 |
| Panobinostat | Novartis (CHE) |
Apoptosis; Cell cycle arrest | FDA approved | Panobinostat + BTZ + DEX or BTZ + DEX in Relapsed MM | NCT01023308 |
| Receptor Tyrosine Kinases | |||||
| MET inhibitors | |||||
| Tivantinib | NCI (USA) |
Perturbation of microtubule dynamics; G2/M arrest; Apoptosis | Phase II | Tivantinib alone in R/R MM | NCT01447914 |
| Cabozantinib | MGH (USA) |
Tumour growth inhibition; Anti-angiogenic | Phase I/II | Cabozantinib alone in R/R MM | NCT01866293 |
| MAPK Inhibitors | |||||
| MEK inhibitors | |||||
| Trametinib | GlaxoSmithKline (GBR) |
Tumour growth inhibition | Phase I/II | Trametinib + GSK2110183 in Solid Tumors or MM | NCT01476137 |
| Selumetinib | NCI (USA) |
Tumour growth inhibition; DNA damage | Phase II | Selumetinib alone in R/R MM | NCT01085214 |
| RAF inhibitors | |||||
| Encorafenib | University of Heidelberg
Medical Center (GER) |
Induction of senescence and autophagy; Cell cycle arrest | Phase II | Encorafenib + Binimetinib in R/R MM with BRAFV600E/K mutation | NCT02834364 |
| Binimetinib | Caspase 9 mediated apoptosis | ||||
| Sorafenib | Mayo Clinic (USA) |
Tumour growth inhibition; anti-angiogenic | Phase II | Sorafenib alone in Refractory MM | NCT00474929 |
| Dual RAF/MEK inhibitors | |||||
| RO5126766 | NHS (GBR) |
Apoptosis; Cell cycle arrest; Tumour growth inhibition | Phase I | RO5126766 alone in Solid Tumours or MM | NCT02407509 |
| PI3K-AKT Inhibitors | |||||
| GSK2141795 | NCI (USA) |
Inhibition of IL-6 pro-MM effect; Cell cycle arrest; UPR induction; Apoptosis | Phase II | Trametinib + GSK2141795 in R/R MM | NCT01989598 |
| CUDC-907 | Curis (USA) |
Tumour growth inhibition; Caspase 3/7 mediated apoptosis; Cell cycle arrest | Phase I | CUDC-907 alone in Lymphoma or MM | NCT01742988 |
| Nelfinavir | Swiss Group for Clinical Cancer Research
(CHE) |
Induction of ER stress; Inhibition of proteasome | Phase I/II | Nelfinavir + LEN + DEX in Progressive MM | NCT01555281 |
| Cell Cycle | |||||
| Selinexor | Karyopharm Therapeutics (USA) |
Apoptosis; Inhibition of MYC, MCL-1, and NF-κB; Cell cycle arrest | Phase II | Selinexor + DEX in R/R MM | NCT02336815 |
| Dinaciclib | NCI (USA) |
Apoptosis; Inhibition of XBP1s nuclear localization; Accumulation of p53; Downregulation of MCL-1 | Phase II | Dinaciclib alone in RRMM | NCT01096342 |
| Filanesib | PETHEMA Foundation (ESP) |
Mitotic arrest; Apoptosis | Phase 1/II | Filanesib + POM + DEX in R/R MM | NCT02384083 |
| Epigenetic Modulators | |||||
| Demethylating agents | |||||
| Azacitidine | Case Comprehensive Cancer Center
(USA) |
Apoptosis | Phase I/II | Azacitidine + LEN + DEX in R/R MM | NCT01155583 |
| BET bromodomain inhibitors | |||||
| GSK525762 | GlaxoSmithKline (GBR) |
MYC downregulation; Cell cycle arrest; Cell senescence | Phase I | GSK525762 in R/R haematological malignancies | NCT01943851 |
| CPI-0610 | Constellation
Pharmaceuticals (USA) |
CPI-0610 alone in R/R MM | NCT02157636 | ||
| Matrix Metalloproteinases | |||||
| Neovastat | Aeterna Zentaris (CAN) |
Anti-angiogenic | Phase II | Neovastat alone in R/R MM | NCT00022282 |
Targeted genome editing technologies (RNAi and CRISPR/Cas9) can also be harnessed to screen for novel “druggable” targets to overcome drug resistance. As a proof of concept, a CRISPR/Cas9 screen of protein domains in murine acute myeloma leukemia (AML) cells revealed six known drug targets and 19 additional dependencies [69]. Cell-based drug screening assays have also been used to screen compounds for their effects on cell viability in BzR MM cells [70]. Cancer researchers at the University of Helsinki’s Institute for Molecular Medicine Finland, in collaboration with the pharmaceutical company Pfizer, have developed cutting edge high-throughput systems biological platforms to functionally profile patient cells to develop new targeted cancer drugs in personalized and precision medicine projects [71]. A pilot screen using the NCI Diversity Set II (NCI Developmental Therapeutics Program) of ~1600 small molecules identified 4 compounds that either had greater single-agent activity against BzR cells or restored sensitivity to bortezomib in BzR cells co-treated with bortezomib [70]. These compounds were then validated and further downstream mechanistic studies were performed using next-generation “omics” approaches (e.g. gene expression profiling, chemical genomics) [70]. Therefore, the use of HTS approaches has utility not only in drug discovery, but also in helping us understand the molecular mechanisms for targeting drug-resistant MM.
PRE-CLINICAL APPLICATION OF “OMICS” IN THE ERA OF CANCER IMMUNOTHERAPY: EXPLOITING IMMUNOMICS AND “CHO”-OMICS
It has become apparent in recent years that conventional and targeted chemotherapy, while highly effective in lowering tumor burden, unfortunately lacks long-term durability as MM will evolve, recur, and become refractory to any conventional or targeted therapies. The observation that selected MM cases can be de facto cured with allogeneic hematopoietic stem cell transplant (HSCT) suggests that active cancer immunotherapy plays a fundamental role in inducing lasting disease remission due to its ability to target the malignant phenotype of MM cells rather than specific dysregulated pathways [72]. However, HSCT-related mortality remains an obstacle to the widespread implementation of this therapeutic avenue for most MM patients, necessitating the exploration of other immunotherapeutic strategies. Currently, three broad approaches exist to enhance anti-myeloma immunity and stimulate a “host-versus-myeloma” effect: (1) immunomodulation using IMiDs, checkpoint inhibitors, and cytokines; (2) stimulation of myeloma specific T cell immunity using MM vaccines (dendritic cell based, peptide based) and adoptive T cell transfer (CAR T cells); and (3) monoclonal antibodies (anti-CD38 daratumumab, isatuximab, and MOR202, and anti-SLAMF7/CS1 elotuzumab) [72]. Immunomics aims to characterize the tumor-host interface through integration of immunology, genomics, proteomics, transcriptomics, and bioinformatics [73]. The relevance of “omics” in IMiDs research has already been covered in Table 2. Herein, we will discuss the enormous potential of immunomics in the development of highly effective and and specific anti-myeloma immunotherapeutic strategies.
An immunomics approach to vaccine and CAR-T cell development
Enhancing myeloma-specific T cell immunity through vaccination against cancer-specific antigens holds great promise, particularly in the clinical setting of early-stage or minimal residual disease. A multi-peptide vaccine (PVX-410) consisting of a cocktail of four HLA-A2-specific peptides (XBP1u, XBP1s, CD138, SLAMF7) is currently being evaluated in a phase I/IIa trial in patients with smoldering MM with the goal of delaying their progression to active disease (NCT01718899) [74]. Chimeric antigen receptor (CAR)-T cells are engineered by cloning antigen-specific T cell receptors onto T cells collected from patients. After ex vivo engineering, these cancer-specific CAR-T cells are expanded and then infused back into the patient in a process known as adoptive cell transfer. Encouraged by the remarkable results of CD19-directed CAR-T cell therapy in relapsed and refractory chronic lymphocytic leukemia, non-Hodgkin lymphoma, and acute lymphoblastic leukemia, researchers are now looking to develop CAR-T cells against myeloma-specific antigens [75,76]. Specifically, CD138 and BCMA-directed CAR-T cells are currently undergoing phase I clinical trial (NCT01886976, NCT02215967) while CD38 and SLAMF7-directed CAR-T cells are still in preclinical development [77].
The success of MM vaccination and CAR-T cell development hinges on the identification of MHC class I-restricted myeloma peptides that can generate highly avid, myeloma-specific memory cytotoxic T lymphocytes (CTLs) to provide a long-lasting immune response. The main challenges to this approach are the complex tumor-host interaction and the molecular and phenotypic heterogeneity of MM [73]. Immunomics provides a systematic framework for the identification of cancer-specific antigens and epitopes that interact with the host immune system. Gene expression analysis and reverse vaccinology has led to the discovery of a wide array of myeloma-associated T-cell antigens (e.g. CD138, XBP1, SLAMF7, WT1, RHAMM, hTERT, Survivin) [78–94]. Tumor exome sequencing and cDNA libraries can be used to screen for myeloma-specific mutated proteins in patient tumors. These neoepitopes (positive hits) can then be further characterized using an MHC binding algorithm to identify candidate mutated T cell epitopes. Next generation immunosequencing can be used to profile T-cell receptor sequences to determine the mature T-cell repertoire of MM-specific T cells which can then guide development of CAR-T cells and ImmTACs (immune mobilizing monoclonal TCRs Against Cancer). Mass spectrometric analysis of the HLA-presented peptidome can also be used to the screen for novel, non-mutated, myeloma-specific T-cell epitopes [95].
A promising strategy that utilizes next-generation platforms to discover novel T-cell epitopes has been described [96]. Firstly, MHC-prediction algorithms are used to identify possible myeloma peptides with high binding affinity to the MHC I-complex [96]. Each candidate peptide-MHC tetramer is then labelled with a distinct three-metal staining code; each metal selected from a pool of 10 different metal tags. This system, which utilizes only 10 of the ~40 currently available cytometry by time of flight (CyTOF) heavy-isotope channels for three-dimensional antigen-specificity encoding (assignment of a distinct three metal tag to each antigen specificity), provides us with 120 unique combinations of three metals to label up to 120 different tetramers simultaneously [96]. CD8+ T cell-enriched MM patient samples are then stained with the metal-labelled tetramers and sorted using magnetized columns to further enrich for “tetramer-positive” T cells. Subsequent analysis of both sorted and pre-sorted samples using mass cytometry gives an objective readout of the frequency of pre-sorted antigen-specific T-cell in each donor sample, by fusing a back-calculation approach [96]. Metal-tagged antibodies specific for phenotypic markers of interest (e.g. cell surface markers, memory cell markers, functional markers, co-stimulatory/inhibitory markers) can also be added to further characterize the peptide-specific CD8+ T cells. Multi-parameter analyses would then enable a more stringent selection of peptides that preferentially induce the expansion of highly functional memory T cells against MM [96].
“Omics” approaches to streamline monoclonal antibody development and production
Monoclonal antibodies (mAbs) bind against specific antigens expressed on the surface of cells. They can then induce cell death through a number of mechanisms: (1) antibody-dependent cell-mediated cytotoxicity, (2) complement-dependent cytotoxicity, (3) antibody-dependent cellular phagocytosis, and (4) direct cytotoxicity via alterations in intracellular signaling, inhibition of function of growth factor receptors and adhesion molecules or induction of apoptosis by crosslinking receptors [72,97]. Two FDA approved mAbs, daratumumab and elotuzumab, have proven to be highly efficacious in MM. In particular, three-drug regimens incorporating daratumumab, with either bortezomib and dexamethasone or lenalidomide and dexamethasone showed unprecedented results in phase III trials in RR MM with circa 60% reduction in risk of death or progression compared to the control arm [98,99]. Antibody-drug conjugates (ADCs) utilize mAbs to selectively deliver cytotoxins to target cells, with the goal of increasing specificity and limiting side effects. Indatuximab ravtansine (chimeric anti-CD138-conjugated maytansinoid DM4) and J6M0-mcMMAF (humanized and afucosylated anti-BCMA-conjugated monomethyl auristatin F) are examples of ADCs undergoing clinical trials for use in RR MM [100]. Another area of mAb research focuses on bispecific T cell engagers (BiTEs). These molecules bind on one arm to a specific antigen and on the other to CD3, thus redirecting the activity of cytotoxic T cells against a specific target cell. BI 836909 is a novel BiTE in phase I clinical development that targets BCMA; a highly expressed protein in most MM cells (NCT02514239).
The design of novel cytotoxic mAb therapies (mAbs, ADCs, BiTEs) is challenging due to the limited availability of suitable tumor-associated antigens (TAA) that are: (1) specifically overexpressed on MM and not on normal tissue surfaces (to allow for the effective induction of anti-tumor immunity with as little side effects as possible), (2) involved in oncogenesis or MM survival, (to limit the chances of downregulation upon treatment pressure), and (3) highly immunogenic [101]. The use of genome-wide microarray analysis which gives us unbiased and comprehensive gene expression profiles of both normal and cancer tissues can be used to guide the selection of ideal TAAs [101]. Proteomic-based approaches can also be utilized to screen and identify potential TAAs. One such study utilized a polyclonal antibody, generated by immunizing rabbits with ARH-77 MM cells, to probe for potential TAAs, which were then identified by mass spectrometric analysis [102].
Commercial production wise, Chinese Hamster Ovarian (CHO) cells are used in large scale mAb manufacturing [103]. The process involves the transfection and expression of the mAb transgene followed by subsequent purification of recombinant mAb from the CHO cell culture supernatant. While improvements in recombinant DNA technology have significantly enhanced production yield by more than 100-fold over the last 20 years, there still exists considerable, and unpredictable, variation in yield between different production cell lines as the factors controlling protein (and gene) expression have yet to be uncovered [104]. Application of “omics” techniques have enabled us to decode the CHO cell genome, transcriptome, proteome, glycome, and metabolome, allowing us to better understand and exploit the molecular basis of high productivity [103,104]. For example, “omics” can be used to identify key markers of good production lines and optimize CHO-cell engineering [104]. Complete sequencing of the CHO cell provides us with numerous opportunities and possibilities for strategies to increase production yield and consistency, and reduce both costs and process-development time to ultimately expedite delivery of products into the clinic.
CONCLUSION AND FUTURE DIRECTIONS: PROPOSED MODEL FOR THE APPLICATION OF “OMICS” TECHNOLOGIES IN PRECLINICAL RESEARCH (BENCH) AND CLINICAL PRACTICE (BEDSIDE)
Remarkable progress in our understanding of MM biology has led to significant refinements in how we diagnose, prognosticate, treat, and monitor MM. The expanding repertoire of novel therapeutics, designed to exploit MM’s three Achilles’ heels, fall largely into three hierarchical categories: drugs that target the (1) molecular aberrations of MM (e.g. MAPK and PI3K-Akt pathway inhibitors), (2) unique phenotype of MM resulting from these molecular aberrations and ongoing DNA damage (e.g. blocking stress responses, immunotherapy), and (3) mechanisms underlying genomic instability in plasma cells (e.g. APOBEC, APEX1). 20 years ago, there were not enough therapeutic options available to our patients. Today, clinicians face a different but welcomed challenge: one that involves having to figure out the right drugs to use, in the appropriate combination, at the correct time, and in the right sequence [105]. Bearing in mind that every cancer is as unique as the person fighting it, the goal would likewise be to have treatment regimens specifically tailored to the individual patient. Integrative personal “omics” profiling (iPOP) provides clinicians with a powerful tool to meet this challenge as we move forward into the era of precision medicine [106].
Rapid advances in science and technology offer huge potential for innovation at the crossroads of medicine, biotechnology, and Big Data. Indeed, the use of “omics” technologies has significantly advanced our understanding of the molecular biology of MM which has greatly advanced preclinical drug development. However, as Einstein famously puts it: “the more (we) learn, the more (we) realize how much (we do not) know”; the same can be said of our ongoing battle to decode MM. As we push the boundaries of science, it is important not to become lost in the multitude of data but to instead focus on making the research count for the patients. The use of “omics” technologies in pre-clinical research has and will continue to facilitate the development of (1) better risk stratification systems, (2) biomarker discovery, (3) rational drug combinations to overcome resistance, and (4) novel targeted and immunotherapies for use in the diagnostic workup and treatment of patients with MM (Fig 1). Although at present, “omics” technologies are not ready for immediate clinical use as diagnostic, prognostic, or response/relapse monitoring tools, they can be envisaged as simple, rapid, robust, portable, and cost-effective clinical diagnosis, prognosis, and disease monitoring systems that could be available soon, which would not only improve clinical decisions but also guide the design of more clinically pertinent, bench to bedside research.
REFERENCES
- 1.Palumbo A, Anderson K. Multiple Myeloma. The New England Journal of Medicine 2011;364:1046–1060. [DOI] [PubMed] [Google Scholar]
- 2.San Miguel JF. Introduction to a series of reviews on multiple myeloma. Blood 2015. [DOI] [PubMed] [Google Scholar]
- 3.Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdi J, Chen G, Chang H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget 2013;4:2186–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nooka AK, Kastritis E, Dimopoulos MA, Lonial S. Treatment options for relapsed and refractory multiple myeloma. Blood 2015;125:3085–3099. [DOI] [PubMed] [Google Scholar]
- 6.Kumar SK, Lee JH, Lahuerta JJ, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2011;26:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horgan RP, Kenny LC. ‘Omic’ technologies: genomics, transcriptomics, proteomics and metabolomics. The Obstetrician & Gynaecologist 2011;13:189–195. [Google Scholar]
- 8.Meyer UA, Zanger UM, Schwab M. Omics and drug response. Annual review of pharmacology and toxicology 2013;53:475–502. [DOI] [PubMed] [Google Scholar]
- 9.Sawyers CL. Chronic myeloid leukemia. The New England Journal of Medicine 1999;340:1330–1340. [DOI] [PubMed] [Google Scholar]
- 10.Tiacci E, Trifonov V, Schiavoni G, et al. BRAF Mutations in Hairy-Cell Leukemia. The New England Journal of Medicine 2011;364:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. The New England Journal of Medicine 2012;367:826–833. [DOI] [PubMed] [Google Scholar]
- 12.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. The New England Journal of Medicine 2013;369:2379–2390. [DOI] [PubMed] [Google Scholar]
- 13.Bhutani M, Landgren O, Usmani SZ. Multiple myeloma: is it time for biomarker-driven therapy? American Society of Clinical Oncology educational book / ASCO . American Society of Clinical Oncology. Meeting 2015:503. [DOI] [PubMed] [Google Scholar]
- 14.Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer cell 2014;25:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nature Reviews Cancer 2012;12:335–348. [DOI] [PubMed] [Google Scholar]
- 16.Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–928. [DOI] [PubMed] [Google Scholar]
- 17.Richardson PG. The Future of Myeloma Therapy: One Size Does Not Fit All. Journal of oncology practice / American Society of Clinical Oncology 2016;12:295–296. [DOI] [PubMed] [Google Scholar]
- 18.Sagaster V, Ludwig H, Kaufmann H, et al. Bortezomib in relapsed multiple myeloma: response rates and duration of response are independent of a chromosome 13q-deletion. Leukemia 2007;21:164–168. [DOI] [PubMed] [Google Scholar]
- 19.Jagannath S, Richardson PG, Sonneveld P, et al. Bortezomib appears to overcome the poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials. Leukemia 2007;21:151–157. [DOI] [PubMed] [Google Scholar]
- 20.Reece D, Song KW, Fu T, et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: adverse effect of deletion 17p13. Blood 2009;114:522–525. [DOI] [PubMed] [Google Scholar]
- 21.Dimopoulos MA, Kastritis E, Christoulas D, et al. Treatment of patients with relapsed/refractory multiple myeloma with lenalidomide and dexamethasone with or without bortezomib: prospective evaluation of the impact of cytogenetic abnormalities and of previous therapies. Leukemia 2010;24:1769–1778. [DOI] [PubMed] [Google Scholar]
- 22.Avet-Loiseau H, Soulier J, Fermand JPP, et al. Impact of high-risk cytogenetics and prior therapy on outcomes in patients with advanced relapsed or refractory multiple myeloma treated with lenalidomide plus dexaméthasone. Leukemia 2010;24:623–628. [DOI] [PubMed] [Google Scholar]
- 23.Chang H, Jiang A, Qi C, Trieu Y, Chen C, Reece D. Impact of genomic aberrations including chromosome 1 abnormalities on the outcome of patients with relapsed or refractory multiple myeloma treated with lenalidomide and dexamethasone. Leukemia & lymphoma 2010;51:2084–2091. [DOI] [PubMed] [Google Scholar]
- 24.Shaughnessy JD, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007;109:2276–2284. [DOI] [PubMed] [Google Scholar]
- 25.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 2005;106:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chng WJ, Braggio E, Mulligan G, et al. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood 2008;111:1603–1609. [DOI] [PubMed] [Google Scholar]
- 27.Fonseca R, Miguel J. Prognostic Factors and Staging in Multiple Myeloma. Hematology/Oncology Clinics of North America 2007;21:1115–1140. [DOI] [PubMed] [Google Scholar]
- 28.Mikhael JR, Dingli D, Roy V, et al. Management of Newly Diagnosed Symptomatic Multiple Myeloma: Updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Guidelines 2013. Mayo Clinic Proceedings 2013;88:360–376. [DOI] [PubMed] [Google Scholar]
- 29.Decaux O, Lodé L, Magrangeas F, et al. Prediction of Survival in Multiple Myeloma Based on Gene Expression Profiles Reveals Cell Cycle and Chromosomal Instability Signatures in High-Risk Patients and Hyperdiploid Signatures in Low-Risk Patients: A Study of the Intergroupe Francophone du Myélome . Journal of Clinical Oncology 2008;26:4798–4805. [DOI] [PubMed] [Google Scholar]
- 30.Prideaux SM, O’Brien E, Chevassut TJ. The RAG Model: A New Paradigm for Genetic Risk Stratification in Multiple Myeloma. Bone Marrow Research 2014;2014:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghadimi BM, Grade M. Cancer gene profiling for response prediction. Methods in molecular biology (Clifton, N.J.) 2010;576:327–339. [DOI] [PubMed] [Google Scholar]
- 32.Jensen EH, McLoughlin JM, Yeatman TJ. Microarrays in gastrointestinal cancer: is personalized prediction of response to chemotherapy at hand? Current opinion in oncology 2006;18:374–380. [DOI] [PubMed] [Google Scholar]
- 33.Mariadason JM, Arango D, Shi Q, et al. Gene expression profiling-based prediction of response of colon carcinoma cells to 5-fluorouracil and camptothecin. Cancer research 2003;63:8791–8812. [PubMed] [Google Scholar]
- 34.Nagasaki K, Miki Y. Molecular prediction of the therapeutic response to neoadjuvant chemotherapy in breast cancer. Breast Cancer 2008;15:117–120. [DOI] [PubMed] [Google Scholar]
- 35.Schauer M, Janssen K-PP, Rimkus C, et al. Microarray-based response prediction in esophageal adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2010;16:330–337. [DOI] [PubMed] [Google Scholar]
- 36.Anguiano A, Tuchman SA, Acharya C, et al. Gene expression profiles of tumor biology provide a novel approach to prognosis and may guide the selection of therapeutic targets in multiple myeloma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009;27:4197–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreaux J, Klein B, Bataille R, et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica 2011;96:574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood 2006;108:2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhan F, Barlogie B, Arzoumanian V, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007;109:1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhan F, Barlogie B, Mulligan G, Shaughnessy JD, Bryant B. High-risk myeloma: a gene expression based risk-stratification model for newly diagnosed multiple myeloma treated with high-dose therapy is predictive of outcome in relapsed disease treated with single-agent bortezomib or high-dose dexamethasone. Blood 2008;111:968–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amin SB, Yip WKK, Minvielle S, et al. Gene expression profile alone is inadequate in predicting complete response in multiple myeloma. Leukemia 2014;28:2229–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnan SR, Jaiswal R, Brown RD, Luk F, Bebawy M. Multiple myeloma and persistence of drug resistance in the age of novel drugs (Review). International journal of oncology 2016;49:33–50. [DOI] [PubMed] [Google Scholar]
- 43.Munshi NC, Avet-Loiseau H, Rawstron AC, et al. Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA Oncology 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson KC, Auclair D, Kelloff GJ, et al. The Role of Minimal Residual Disease Testing in Myeloma Treatment Selection and Drug Development: Current Value and Future Applications. Clin Cancer Res. [DOI] [PubMed] [Google Scholar]
- 45.Barnidge DR, Tschumper RC, Theis JD, et al. Monitoring M-Proteins in Patients with Multiple Myeloma Using Heavy-Chain Variable Region Clonotypic Peptides and LC–MS/MS. Journal of Proteome Research 2014;13:1905–1910. [DOI] [PubMed] [Google Scholar]
- 46.Kidd BA, Readhead BP, Eden C, Parekh S, Dudley JT. Integrative network modeling approaches to personalized cancer medicine. Personalized Medicine 2015;12:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leung-Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer cell 2013;24:289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bianchi G, Oliva L, Cascio P, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009;113:3040–3049. [DOI] [PubMed] [Google Scholar]
- 49.Zhu YX, Braggio E, Shi C-XX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011;118:4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fall DJ, Stessman H, Patel SS, et al. Utilization of translational bioinformatics to identify novel biomarkers of bortezomib resistance in multiple myeloma. Journal of Cancer 2014;5:720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Egan P, Drain S, Conway C, Bjourson AJ, Alexander DH. Towards Stratified Medicine in Plasma Cell Myeloma. International Journal of Molecular Sciences 2016;17:1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agnelli L, Tassone P, Neri A. Molecular profiling of multiple myeloma: from gene expression analysis to next-generation sequencing. Expert opinion on biological therapy 2013;13 Suppl 1:68. [DOI] [PubMed] [Google Scholar]
- 53.Stessman HA, Baughn LB, Sarver A, et al. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Molecular cancer therapeutics 2013;12:1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu YX, Tiedemann R, Shi C-XX, et al. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood 2011;117:3847–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stessman HA, Mansoor A, Zhan F, et al. Reduced CXCR4 expression is associated with extramedullary disease in a mouse model of myeloma and predicts poor survival in multiple myeloma patients treated with bortezomib. Leukemia 2013;27:2075–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsieh FY, Tengstrand E, Pekol TM, Guerciolini R, Miwa G. Elucidation of potential bortezomib response markers in mutliple myeloma patients. Journal of pharmaceutical and biomedical analysis 2009;49:115–122. [DOI] [PubMed] [Google Scholar]
- 57.Micallef J, Dharsee M, Chen J, et al. Applying mass spectrometry based proteomic technology to advance the understanding of multiple myeloma. Journal of Hematology & Oncology 2010;3:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rees-Unwin KS, Craven RA, Davenport E, et al. Proteomic evaluation of pathways associated with dexamethasone-mediated apoptosis and resistance in multiple myeloma. British journal of haematology 2007;139:559–567. [DOI] [PubMed] [Google Scholar]
- 59.Abdullah L, Chow E. Mechanisms of chemoresistance in cancer stem cells. Clinical and translational medicine 2013;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abraham J, Salama NN, Azab AK. The role of P-glycoprotein in drug resistance in multiple myeloma. Leukemia & lymphoma 2014:1–8. [DOI] [PubMed] [Google Scholar]
- 61.Micallef J, Taccone M, Mukherjee J, et al. Epidermal Growth Factor Receptor Variant III–Induced Glioma Invasion Is Mediated through Myristoylated Alanine-Rich Protein Kinase C Substrate Overexpression. Cancer research 2009;69:7548–7556. [DOI] [PubMed] [Google Scholar]
- 62.Trudel S, Stewart AK, Rom E, et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma cells. Blood 2006;107:4039–4046. [DOI] [PubMed] [Google Scholar]
- 63.Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998;92:3025–3034. [PubMed] [Google Scholar]
- 64.Pollett JB, Trudel S, Stern D, Li ZH, Stewart AK. Overexpression of the myeloma-associated oncogene fibroblast growth factor receptor 3 confers dexamethasone resistance. Blood 2002;100:3819–3821. [DOI] [PubMed] [Google Scholar]
- 65.St-Germain JR, Taylor P, Tong J, et al. Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proceedings of the National Academy of Sciences of the United States of America 2009;106:20127–20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Melchor L, Brioli A, Wardell CP, et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014;28:1705–1715. [DOI] [PubMed] [Google Scholar]
- 67.Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nature Communications 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011;471:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nature Biotechnology 2015;33:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stessman HA, Lulla A, Xia T, et al. High-throughput drug screening identifies compounds and molecular strategies for targeting proteasome inhibitor-resistant multiple myeloma. Leukemia 2014;28:2263–2267. [DOI] [PubMed] [Google Scholar]
- 71.Pietiainen V, Saarela J, von Schantz C, Turunen L, Ostling P, Wennerberg K. The high throughput biomedicine unit at the institute for molecular medicine Finland: high throughput screening meets precision medicine. Combinatorial chemistry & high throughput screening 2014;17:377–386. [DOI] [PubMed] [Google Scholar]
- 72.Kocoglu M, Badros A. The Role of Immunotherapy in Multiple Myeloma. Pharmaceuticals 2016;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Sousa KP, Doolan DL. Immunomics: a 21st century approach to vaccine development for complex pathogens. Parasitology 2016;143:236–244. [DOI] [PubMed] [Google Scholar]
- 74.Bae J, Prabhala R, Voskertchian A, et al. A multiepitope of XBP1, CD138 and CS1 peptides induces myeloma-specific cytotoxic T lymphocytes in T cells of smoldering myeloma patients. Leukemia 2015;29:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England Journal of Medicine 2014;371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. The New England Journal of Medicine 2011;365:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maus MV, June CH. Zoom Zoom: racing CARs for multiple myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19:1917–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou F-LL, Meng S, Zhang W- GG, et al. Peptide-based immunotherapy for multiple myeloma: current approaches. Vaccine 2010;28:5939–5946. [DOI] [PubMed] [Google Scholar]
- 79.Hundemer M, Schmidt S, Condomines M, et al. Identification of a new HLA-A2-restricted T-cell epitope within HM1.24 as immunotherapy target for multiple myeloma. Experimental hematology 2006;34:486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jalili A, Ozaki S, Hara T, et al. Induction of HM1.24 peptide-specific cytotoxic T lymphocytes by using peripheral-blood stem-cell harvests in patients with multiple myeloma. Blood 2005;106:3538–3545. [DOI] [PubMed] [Google Scholar]
- 81.Chiriva-Internati M, Liu Y, Weidanz JA, et al. Testing recombinant adeno-associated virus-gene loading of dendritic cells for generating potent cytotoxic T lymphocytes against a prototype self-antigen, multiple myeloma HM1.24. Blood 2003;102:3100–3107. [DOI] [PubMed] [Google Scholar]
- 82.Rew SB, Peggs K, Sanjuan I, et al. Generation of potent antitumor CTL from patients with multiple myeloma directed against HM1.24. Clinical cancer research : an official journal of the American Association for Cancer Research 2005;11:3377–3384. [DOI] [PubMed] [Google Scholar]
- 83.van Rhee F, Szmania SM, Zhan F, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood 2005;105:3939–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schuberth PC, Jakka G, Jensen SM, et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene therapy 2013;20:386–395. [DOI] [PubMed] [Google Scholar]
- 85.Bae J, Tai Y-TT, Anderson KC, Munshi NC. Novel epitope evoking CD138 antigen-specific cytotoxic T lymphocytes targeting multiple myeloma and other plasma cell disorders. British journal of haematology 2011;155:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bae J, Carrasco R, Lee AHH, et al. Identification of novel myeloma-specific XBP1 peptides able to generate cytotoxic T lymphocytes: a potential therapeutic application in multiple myeloma. Leukemia 2011;25:1610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bae J, Smith R, Daley J, et al. Myeloma-specific multiple peptides able to generate cytotoxic T lymphocytes: a potential therapeutic application in multiple myeloma and other plasma cell disorders. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18:4850–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brossart P, Schneider A, Dill P, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer research 2001;61:6846–6850. [PubMed] [Google Scholar]
- 89.Oka Y, Tsuboi A, Fujiki F, et al. WT1 peptide vaccine as a paradigm for “cancer antigen-derived peptide”-based immunotherapy for malignancies: successful induction of anti-cancer effect by vaccination with a single kind of WT1 peptide. Anti-cancer agents in medicinal chemistry 2009;9:787–797. [DOI] [PubMed] [Google Scholar]
- 90.Kuball J, de Boer K, Wagner E, et al. Pitfalls of vaccinations with WT1-, Proteinase3- and MUC1-derived peptides in combination with MontanideISA51 and CpG7909. Cancer immunology, immunotherapy : CII 2011;60:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Greiner J, Schmitt A, Giannopoulos K, et al. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica 2010;95:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmitt M, Schmitt A, Rojewski MT, et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 2008;111:1357–1365. [DOI] [PubMed] [Google Scholar]
- 93.Rapoport AP, Aqui NA, Stadtmauer EA, et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood 2011;117:788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hobo W, Strobbe L, Maas F, et al. Immunogenicity of dendritic cells pulsed with MAGE3, Survivin and B-cell maturation antigen mRNA for vaccination of multiple myeloma patients. Cancer immunology, immunotherapy : CII 2013;62:1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walz S, Stickel JS, Kowalewski D, et al. The antigenic landscape of multiple myeloma: mass spectrometry (re)defines targets for T-cell–based immunotherapy. Blood 2015;126:1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, Davis MM. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nature Biotechnology 2013;31:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weiner GJ. Building better monoclonal antibody-based therapeutics. Nature Reviews Cancer 2015;15:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. The New England Journal of Medicine 2016;375:754–766. [DOI] [PubMed] [Google Scholar]
- 99.Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. The New England Journal of Medicine 2016;375:1319–1331. [DOI] [PubMed] [Google Scholar]
- 100.Bianchi G, Richardson PG, Anderson KC. Promising therapies in multiple myeloma. Blood 2015;126:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nishimura Y, Tomita Y, Yuno A, Yoshitake Y, Shinohara M. Cancer immunotherapy using novel tumor‐associated antigenic peptides identified by genome‐wide cDNA microarray analyses. Cancer science 2015;106:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mu B, Zhang H, Cai X, et al. Screening of Multiple Myeloma by Polyclonal Rabbit Anti-Human Plasmacytoma Cell Immunoglobulin. PLoS One 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Datta P, Linhardt RJ, Sharfstein ST. An ‘omics approach towards CHO cell engineering. Biotechnology and bioengineering 2013;110:1255–1271. [DOI] [PubMed] [Google Scholar]
- 104.Orellana CA, Marcellin E, Schulz BL, Nouwens AS, Gray PP, Nielsen LK. High-antibody-producing Chinese hamster ovary cells up-regulate intracellular protein transport and glutathione synthesis. Journal of Proteome Research 2015;14:609–618. [DOI] [PubMed] [Google Scholar]
- 105.Anderson KC. Progress and Paradigms in Multiple Myeloma. Clinical Cancer Research 2016;22:5419–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen R, Mias GI, Li-Pook-Than J, et al. Personal Omics Profiling Reveals Dynamic Molecular and Medical Phenotypes. Cell 2012;148:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]