

Abstract
Background:
While monoclonal antibodies against tumor necrosis factor-α (TNFα) are effective in treating Crohn’s disease (CD), approximately one-third of patients lose response. The mechanisms underlying this loss of response remain elusive.
Aim:
We sought to determine if novel biological pathways, including TNFα-independent inflammatory pathways, emerge in those with loss of response to anti-TNFα.
Methods:
Using RNA microarray technology in 28 patients with CD, we examined the colonic gene expression differences between those with active inflammation in the setting of loss of response to TNFα-antagonist therapy (“loss of responders”) compared to anti-TNFα naïve patients with active inflammation and those on anti-TNF therapy in disease remission. Pathway enrichment analyses were performed.
Results:
We found that colonic expression of chemokines known to drive inflammation (CXCL20, CXCL9, and CXCL10) were elevated in those with loss of response compared to those in remission. Expression of genes critical to modulating oxidative stress burden (DUOX2, DUOXA2, and NOS2) were also elevated. Additionally, MMP3, MMP1, and MMP12 were elevated in those with continued inflammation. Gene enrichment analysis revealed that loss of responders exhibited dysregulation in the cysteine and methionine metabolism pathway, suggesting alteration in oxidative stress burden. There were no differences in genes or pathways between loss of responders and those who were TNFα-naïve. However, loss of response occurred despite the ability of anti-TNFα therapy to normalize APO gene expression.
Conclusion:
Our analyses suggest that loss of response to anti-TNFα is not driven by the emergence of pathways that bypass the action or induce resistance to anti-TNFα therapy.
Keywords: Crohn’s disease, anti-TNF, loss-of-response, microarray
INTRODUCTION
Landmark clinical trials for antibodies that target anti-tumor necrosis factor-α (anti-TNFα) have demonstrated their efficacy in inducing and maintain remission in Crohn’s disease (CD) (1–3). However, up to a one-third of patients do not respond to these agents, and an additional one-third of patients experience secondary loss of response (4). Specifically, among primary responders to infliximab (IFX), 37% of patients eventually lose response at the rate of 13% per patient-years (4). In placebo-controlled trials for adalimumab (ADA), 17–20% of patients lose response by week 56 (5). In the placebo controlled trials for certolizumab (CZP), the rate of secondary loss of response at week 26 was 38% (6). Consequently, loss of response represents a significant clinical problem.
The mechanism for loss of response, nevertheless, remains elusive. Previous investigations into the mechanism of loss of response have focused on drug levels and antibody formation. However, development of anti-drug antibodies or subtherapeutic trough concentration explains loss of response in only a proportion of patients with CD, and many experience breakthrough inflammation despite therapeutic drug concentrations and no anti-drug antibodies (7). Furthermore, combined immunosuppression with immunomodulators that suppress antibody formation hasn’t been consistently shown to improve treatment durability or improve efficacy in all studies. Additionally, less immunogenic humanized anti-TNF therapies (ADA, CZP) have similar rates of loss of response as the chimeric IFX (8–9). Taken together, these data suggest that alternate TNFα-independent biological pathways independent of antibody formation and drug level may contribute mechanistically to loss of response.
Emerging evidence support a role for TNFα-independent pathways in perpetuating inflammation in CD. For example, certain patients exposed to anti-TNFα agents paradoxically experience inflammatory events. This includes a higher incidence of psoriasis in patients with rheumatoid arthritis receiving anti-TNFα therapy and new onset of IBD in patients with ankylosing spondylitis or juvenile arthritis being treated with IFX (10). In addition, prolonged anti-TNFα exposure has been shown to upregulate several inflammatory pathways, including type I interferon-mediated inflammatory pathways (11). Finally, it is well-appreciated that many TNFα -independent pathways promote intestinal inflammation in CD. Consequently, we performed this study with the following specific aims: (1) To define differentially expressed genes and pathways in patients experiencing loss of response to anti-TNF therapy compared to those with inflammation in a TNFα-naïve setting, and (2) to compare genes and pathways correlating with remission to anti-TNFα therapy.
METHODS
Study approval
Human experimentation conformed to ethical standards, and was approved by the Institutional Review Board (IRB) at the Massachusetts General Hospital.
Patient selection
Patients 18 years and older with an established diagnosis of CD (ileocolonic or colonic), based on clinical, radiologic, endoscopic and pathologic criteria, were eligible for study inclusion. For inclusion as the primary study population of interest, patients had to demonstrate a secondary loss of response to anti-TNFα therapy after achieving an initial response. Secondary loss of response was defined by evidence of disease recurrence clinically (Harvey Bradshaw Index > 4) and endoscopically in those patients who had initial clinical and endoscopic remission after initiation of an anti-TNFα for at least one year. Patients were not on any concurrent therapy, including steroids or thiopurines. We did not have information on serum concentration of the anti-TNFα therapy in our patients. Exclusion criteria included lack of standard loading regimen for the anti-TNFα therapeutic, objective endoscopic evidence of active disease at the time of enrollment, isolated ileal Crohn’s disease, and primary non-response to anti-TNFα therapy.
We included two control groups into our study. To control for the effects of inflammation, we recruited patients naïve to anti-TNFα therapy who were undergoing colonoscopy for assessment of disease activity prior to initiation of anti-TNFα therapy. To control for the effects of anti-TNFα exposure, we also included patients who were on anti-TNFα therapy for at least one year and had no evidence for active inflammation at the time of colonoscopy (Figure 1).
Figure 1.
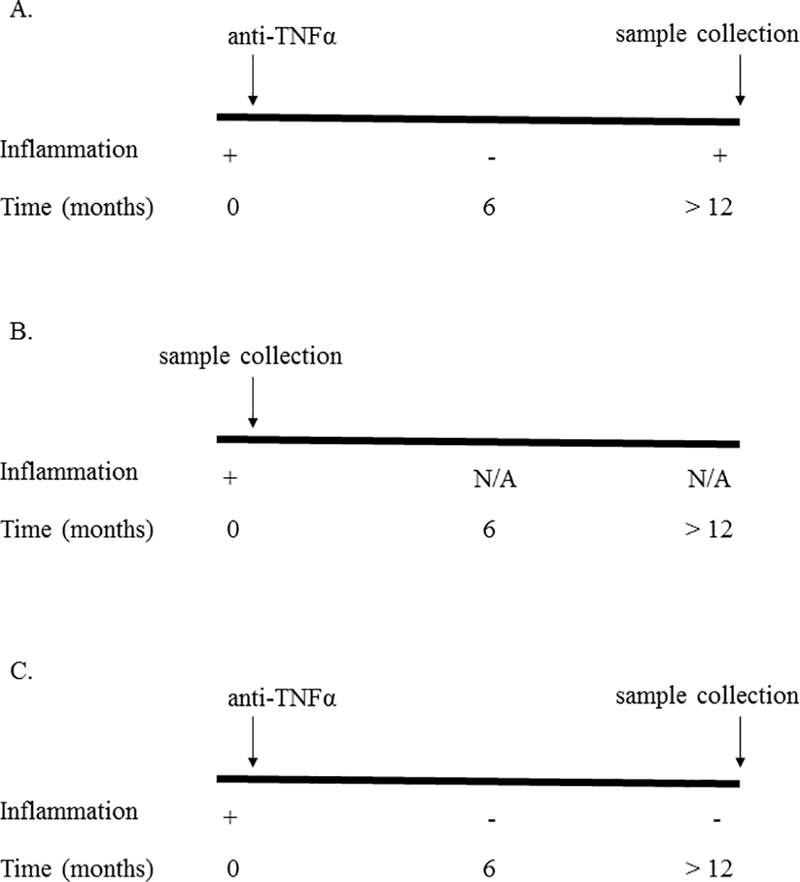
Study design. We compared the colonic gene expression of Crohn’s disease (CD) patients who developed (A) loss of response (N = 10 patients), (B) CD patients with active colonic inflammation who had never been exposed to anti-TNFα therapy (N = 10 patients), and (C) CD patients who were in remission while on anti-TNFα therapy (N = 8 patients).
Sample collection
Colon biopsy samples were obtained from the mid-descending colon at a site of active inflammation in the cases of secondary loss of responders and those naïve to anti-TNFα with inflammation. It should be noted that biopsies were taken while loss of responders were still on anti-TNFα therapy, as the decision to alter the medical regimen had not been determined prior to colonoscopy. In those with a sustained response to anti-TNFα, colonic biopsies were also obtained from the mid-descending colon, although no inflammation was present in these cases. Collected tissue was used to perform histological and mRNA analysis.
Microarray analysis
RNA was extracted from the biopsy specimens using the RNeasy Mini Kit (Qiagen, Valencia, California). The quantity and quality of RNA was assessed using the Nanodrop ND-1000 spectrophotometer. Furthermore, fragment size and distribution (RIN) was quantified by Agilent Bioanalyzer. Total RNA was reverse-transcribed into cDNA using the SuperScript Choice System (Invitrogen, Carlsbad, California), which includes both random hexamers and oligo(dT) primers. Nucleotides were hybridized overnight into the Affymetrix Human Genome U133 Plus 2.0 Array comprising 54,675 probe sets. The 28 samples were run on three different chips. Hybridization, washing, staining, and scanning were all normal. To ensure consistency, we also ran a cluster of all 28 samples and did not find any correlation between the cluster they were put in and the chip the samples were loaded on. GenomeStudio software was used to perform average normalization, and we exported these normalized signal intensities, with values on a linear scale, to perform pairwise differential analyses. Pairwise differential gene expression was assessed using the moderated (empirical Bayesian) t-test implemented in the limma package (version 3.14.4) (i.e., creating simple linear models with lmFit, followed by empirical Bayesian adjustment with eBayes). All microarray analyses were performed using the R environment for statistical computing (version 2.15.1). We compared the gene expression profile of patients with secondary loss of response to those anti-TNFα naïve patients with active colonic inflammation and to those with durable response to anti-TNFα therapy who were in endoscopic remission.
Gene set enrichment
Gene set enrichment analysis was performed using publicly available software from the Broad Institute (http://www.broadinstitute.org/gsea/index.jsp) (Version 6.0) to identify the pathways that are most perturbed between the two groups. Pathways were classified using the KEGG pathway database. The primary outcome of the analysis is the enrichment score (ES). The ES is an estimation of the degree to which a gene set is overrepresented at the top/bottom of the ranked gene list. The ES is then normalized (NES) for differences in gene set size and in correlations between gene sets and the expression dataset. A false discovery rate (FDR) of equal to or less than 25% was considered significant (12).
RESULTS
Patient demographics
Our study included twenty-eight patients with CD – ten patients each with secondary loss of response to anti-TNFα therapy and anti-TNFα naïve patients with active colonic inflammation, and eight patients with durable response to anti-TNFα therapy who were in endoscopic remission. The three groups were similar in gender, age, race, smoking history and distribution of disease (Table 1). Those with anti-TNFα loss of response were more likely to have stricturing or penetrating phenotype of their CD. No patients were on steroids or thiopurines in addition to anti-TNFα therapy. We did not have information on the serum concentration of anti-TNF in our patients.
Table 1.
Patient demographics
| Loss of anti-TNFα response (N = 10) |
Anti-TNFα naïve (N = 10) |
Anti-TNFα responder (N = 8) |
|
|---|---|---|---|
| Sex (% female) | 60 | 60 | 62.5 |
| Age at diagnosis | 19.2 | 24.9 | 24.3 |
| Race (% Caucasian) | 90 | 80 | 100 |
| Ileal involvement (%) | 100 | 100 | 100 |
| Disease behavior (% ) B1 B1P B2 B2P B3 B3P |
10 10 20 30 20 10 |
10 10 30 10 20 20 |
37.5 37.5 12.5 0 0 12.5 |
| Smoking (%) Never Prior Active |
70 30 0 |
60 40 0 |
75 25 0 |
Up-regulation in the expression of chemokines, genes involved in oxidative stress, and intestinal metalloproteinases in loss of responders compared to anti-TNFα responders
First, to identify genes associated with active inflammation, we compared colonic expression profiles from those who were in endoscopic remission on anti-TNFα therapy compared to those with secondary loss of response on this treatment. Several genes were over-expressed in those with secondary loss of response and active inflammation, the top ten are listed in Table 2. The most upregulated gene in those with secondary loss of response was CCL20 (fold change: 10.71, adjusted P-value: 0.01), a gene which influenced by TNFα for its production (13). Multiple other chemokines were also upregulated, including CXCL9 (fold change: 6.30, adjusted P-value: 0.06) and CXCL10 (fold change: 6.13, adjusted P-value: 0.05).
Table 2.
Increased expression of genes from colonic biopsies in Crohn’s patients with loss of response to anti-TNFα therapy versus those who respond
| Gene | Fold Change | P-value | Adjusted P-value* | Rank** |
|---|---|---|---|---|
| CCL20 | 10.71 | 3.7 × 10−6 | 0.01 | 1 |
| DUOXA2 | 8.43 | 3.0 × 10−5 | 0.02 | 2 |
| NOS2 | 8.01 | 2.4 × 10−6 | 0.008 | 3 |
| SERPINA3 | 7.31 | 7.0 × 10−4 | 0.07 | 4 |
| UBD | 7.15 | 1.4 × 10−3 | 0.10 | 5 |
| MMP1 | 5.34 | 5.1 × 10−6 | 0.01 | 6 |
| CXCL9 | 6.30 | 5.0 × 10−4 | 0.06 | 7 |
| CXCL10 | 6.13 | 3.0 × 10−4 | 0.05 | 8 |
| DUOX2 | 6.02 | 1.0 × 10−4 | 0.05 | 9 |
| MMP3 | 5.57 | 7.0 × 10−4 | 0.04 | 10 |
= Boneferroni corrected
= calculated by weighing of fold change and P-value
Second, three of the top ten genes that were overexpressed in loss of responders are involved in modulating oxidative stress burden, including DUOX2 (fold change: 6.30, adjusted P-value: 0.06), NOS2 (fold change: 8.01, adjusted P-value: 0.008), and DUOXA2 (8.43, adjusted P-value: 0.02)
Finally, we found that multiple metalloproteinases, such as MMP3 (fold change: 5.57, adjusted P-value: 0.04), MMP1 (fold change: 5.33, adjusted P-value: 0.08), and MMP12 (fold change: 5.05, adjusted P-value: 0.04) were significantly upregulated in those with secondary loss of response, accounting for three of the top 10 overexpressed genes.
Although not in the top ten list of genes, we observed an association in genes that have been demonstrated to track with Crohn’s activity, including IL1-beta, IL-8, and IL-6. We did not observe a significant increase in oncostatin M (fold change: 1.7, adjusted P-value: 0.55), which has recently been shown to track with primary response (14).
Down-regulation of genes in those with loss of response compared to anti-TNFα responders
Several genes were under-expressed in those with loss of response, the top ten of which are listed in Table 3. The most down-regulated gene, based on fold change, was FAM151A (fold change: −5.18, adjusted P-value: 0.17), while the most significantly reduced gene, based on P-value, was CNTFR (fold change: −5.12, adjusted P-value: 7.6 × 10−5).
Table 3.
Decreased expression of genes from colonic biopsies in Crohn’s patients with loss of response to anti-TNFα therapy versus those who respond
| Gene | Fold Change | P-value | Adjusted P-value* | Rank** |
|---|---|---|---|---|
| FAM151A | −5.18 | 0.005 | 0.17 | 1 |
| CNTFR | −5.13 | 1.6 × 10−9 | 7.6 × 10−9 | 2 |
| SULT2A1 | −4.81 | 0.006 | 0.19 | 3 |
| G6PC | −4.39 | 0.0004 | 0.05 | 4 |
| CDHR1 | −3.30 | 0.0001 | 0.03 | 5 |
| TNC22 | −2.96 | 0.0003 | 0.05 | 6 |
| ESRRG | −2.40 | 0.0004 | 0.05 | 7 |
| SLC25A34 | −2.23 | 0.0004 | 0.05 | 8 |
| FZD7 | −2.14 | 9.1 × 10−5 | 0.03 | 9 |
| MYOM3 | −2.10 | 6.2 × 10−5 | 0.03 | 10 |
= Boneferroni corrected
= calculated by weighing of fold change and P-value
Alteration in cysteine and methionine metabolism in loss of responders
We next performed gene set pathway enrichment analyses from genome-wide colonic expression data in those with loss of response versus those who responded to anti-TNFα to identify inflammatory pathways that associate with secondary loss of response. We found the cysteine and methionine metabolism pathway to be significantly altered in those with loss of response (NES: 1.64, FDR: 0.25) (Figures 2). Other pathways linked to CD pathogenesis and independent of TNFα, such as the JAK-SAT pathway, which is associated with a type I interferon response, were unchanged (NES: 1.11, FDR: 1.0).
Figure 2.
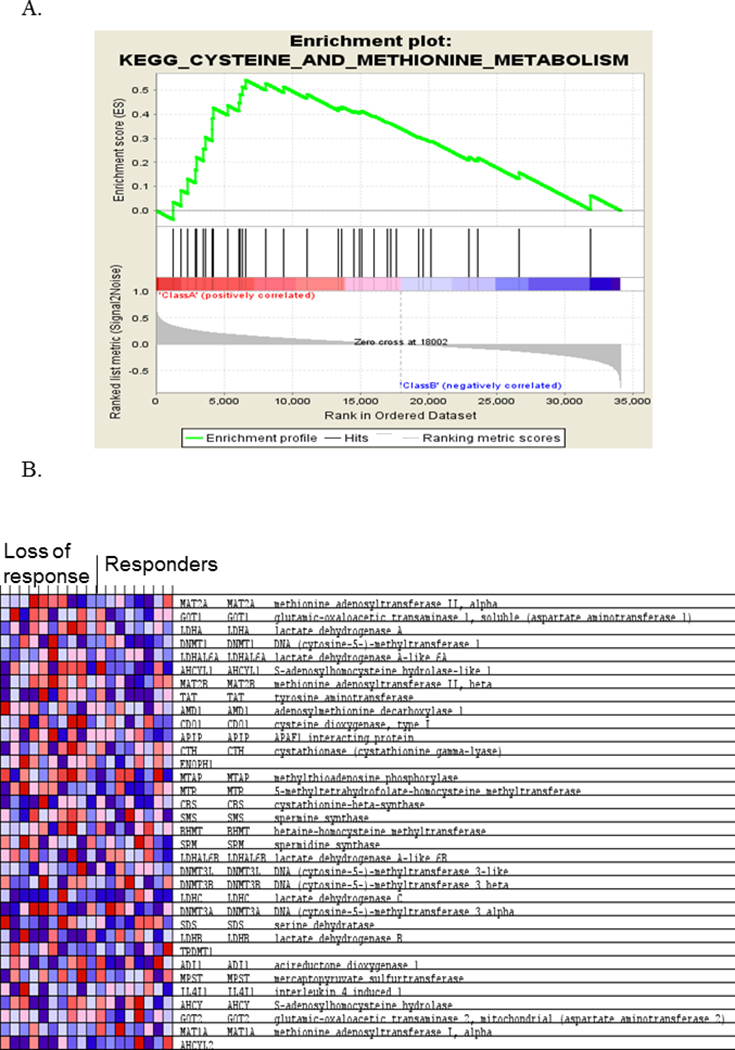
Loss of responders to anti-TNFα therapy exhibit dysregulation in the cysteine and methionine metabolism pathway. Gene set enrichment analysis was performed from the colonic gene expression data obtained from Crohn’s disease (CD) patients with loss of response compared to CD patients in remission while on anti-TNFα. (A) Genes in the cysteine and methionine pathway were most represented in the enrichment analysis, leading to an enrichment score (ES) of 0.59. (B) Heatmap of genes including in the cysteine and methionine pathway, per the KEGG database. Red color represents relative over-expression, while blue color represents under-expression.
Comparison of anti-TNF secondary non-responders to anti-TNF naïve inflammation
Next, we compared the colonic expression profile in patients with secondary loss of response to those with CD naïve to anti-TNFα who exhibited colonic inflammation to identify unique genes and pathways that are associated with secondary loss of response in the setting of anti-TNF use. There were no significantly differentially expressed gene between the two groups, using an adjusted P-value of 0.05 or less. The top 10 genes are listed in Table 4. The three genes most associated with loss of response to anti-TNFα therapy were APOC3 (fold change: 10.71, unadjusted P-value: 5.0 × 10−4), APOA1 (fold change: 8.43, unadjusted P-value: 7.0 × 10−4), and APOA4 (fold change: 8.01, unadjusted P-value: 0.01). (Table 4). All three of these genes were up-regulated in the colonic mucosa of CD patients who lose response to anti-TNFα therapy compared to those with active inflammation naïve to anti-TNFα therapy.
Table 4.
Differentially expressed genes from colonic biopsies in patients with loss of response compared to anti-TNFα-naïve Crohn’s patients with inflammation
| Gene | Fold Change | P-value | Adjusted P-value* | Rank** |
|---|---|---|---|---|
| APOC3 | 10.71 | 5.0 × 10−4 | 0.853 | 1 |
| APOA1 | 8.43 | 7.0 × 10−3 | 0.853 | 2 |
| APOA4 | 8.01 | 0.01 | 0.853 | 3 |
| CCL25 | 7.31 | 0.01 | 0.853 | 4 |
| REG3G | 1.15 | 0.04 | 0.858 | 5 |
| HLA-DRB5 | 6.98 | 0.19 | 0.890 | 6 |
| GSTA1 | 6.30 | 0.02 | 0.853 | 7 |
| NTS | 6.13 | 0.01 | 0.853 | 8 |
| MS4A10 | 6.02 | 0.01 | 0.853 | 9 |
| ITLN2 | 5.57 | 0.005 | 0.853 | 10 |
= Boneferroni corrected
= calculated by weighing of fold change and P-value
We also performed gene set enrichment analyses comparing the expression profiles between these two groups. There were no differences in the examined pathways between the groups, including the cysteine and methionine pathways.
DISCUSSION
Secondary loss of response is an important clinical problem in Crohn’s disease and is associated with significant morbidity. While sub-therapeutic dosing and development of anti-drug antibodies explain this mechanism in some, many patients with loss of response have breakthrough of their inflammation despite adequate drug levels suggesting alternative mechanisms including upregulation of non-TNF dependent pathways may contribute to this phenomenon. In this study, we demonstrate that loss of response to anti-TNF therapy is associated with upregulation of several inflammatory genes that may partially depend on TNFα for their production, drive inflammation, or cleave anti-TNFα, compared to those with sustained responders. However, there were no pathways that were uniquely different between those with anti-TNF loss of response and anti-TNF naïve inflammation, suggesting that emergence of non-TNF dependent pathways may not be the mechanism underlying loss of response.
Loss of response to anti-TNFα represents a significant clinical problem, with estimates suggesting one-third of patients receiving anti-TNFα therapy will lose response after achieving an initial response. Although the mechanism for loss of response is likely multifactorial, the preponderance of work to-date has focused on correlating serum levels of anti-TNFα drugs or the presence of antibodies directed against the medications to loss of response. However, clinical trials have identified patients who exhibit loss of response despite adequate drug levels and no antibodies (7), suggesting other pathways may be involved. One hypothesis offered by our findings is that loss of response to anti-TNF therapy may be due to oxidative stress. In our analysis, the cysteine and methionine pathway was dysregulated in those with anti-TNF loss of response compared to those with sustained response. This pathway plays an important role in the production of critical mediators of oxidative stress, including nicotinamide adenine dinucleotide phosphate (NADPH) and s-adenosylmethionine (SAM). These proteins contribute to the production of free oxygen radical scavengers, which defend the host against oxidative stress. Its dysregulation suggests an increased oxidative stress burden in loss of responders. We also found expression of MMP3 to be higher in responders. Notably, higher levels of oxidative stress have been demonstrated to induce expression of MMP3 (15). MMP3, in-turn cleaves anti-TNFα, making the agent less able to neutralize TNFα and protect against inflammation (16). Consistent with this, we found that the colonic expression of MMP3 to be significantly elevated in loss of responders. It is feasible, therefore, that as oxidative stress drives more of the inflammation seen in CD, this induces MMP3 production, breakdown of anti-TNFα therapy, and resulting loss of response. Another potential explanation is that the increased oxidative stress burden alters the intestinal microbiome (17), leading to over expression of other inflammatory pathways.
Additionally, we found that the ability of anti-TNFα therapy to normalize APOA4 levels did not correlate with improved outcome in loss of responders. In fact, we found that those with loss of response had elevated levels of certain APO genes, although not statistically significant. This is contrast to previous data suggesting higher levels of APO4 associate with improved outcomes in patients with IBD (18). The explanation for this observation is not clear; however, it is well-documented that APOA1 and APOA4 are lipoproteins with antioxidant properties. Therefore, in line with our previous findings, the upregulation of these antioxidant lipoproteins may be in response to the increased oxidative stress burden in those with loss of response. Accordingly, this upregulation may be a result of increased oxidative stress in the tissue as opposed to a direct of anti-TNFα therapy.
The etiology for secondary loss of response is likely multifactorial. For one, the presence of anti-TNF antibodies may influence the efficacy of therapy. In similar fashion, there are data suggesting auto-antibodies, such as antinuclear antibodies (ANA) and double-stranded DNA (anti-dsDNA), contribute to loss of response. In a study of patients with psoriasis, non-responders to anti-TNF therapy displayed higher levels of these autoantibodies and concentrations of these antibodies, suggesting a potential interaction with these antibodies and treatment success (19). Furthermore, Brandse et al. demonstrated that loss of drug in the stool associates with drug levels and likelihood for continued response (20). Although this study looked at primary non-response, it is possible that loss of drug in the stool may also contribute to secondary loss of response. Additionally, studies have associated the presence of obesity and smoking with loss of response (21–22).
The limitations of our study must be noted. First, our sample size may not be sufficient for identification of biologically relevant genes and pathways. Larger cohorts are necessary to more robustly define mechanistic basis of loss of response. Second, we did not routinely obtain serum or fecal anti-TNFα levels, both of which have been associated with loss of response. Finally, the presence of antibodies to anti-TNFα were not available in our data set and consequently we were unable to compare loss of response in those with sub-therapeutic dosing compared to those who have breakthrough in spite of adequate circulating drug. Future studies investigating for the mechanism of loss of response to anti-TNFα should address these limitations, and specifically examine if the dominant pathways are different with loss of response occurring in the setting of sufficient circulating anti-TNFα levels.
There are a few strengths of our study. Most prior studies have examined gene expression at baseline in the context of primary response to anti-TNFα therapy; few have examined mechanisms that pertain to loss of response. This is an important question as drug pharmacokinetic factors alone are insufficient in their ability to predict loss of response in many. In particular, whether anti-TNFα exposure induces emergence of other non-TNFα dependent inflammatory pathways as has been hypothesized remains to be robustly established but is critically important with the emergence of therapies with distinct mechanisms of action, including anti-integrin and anti-IL23 therapies.
In conclusion, our data suggests that increasing oxidative stress may be one mechanisms for loss of response to anti-TNFα therapy, potentially through the ability of oxidative stress to upregulate genes that promote breakdown of anti-TNFα therapy. However, no pathways were uniquely different between those with anti-TNFα loss of response and anti-TNFα naïve inflammation, suggesting the loss of response may not be due to upregulation of TNFα-independent inflammatory pathways. Further study is needed to understand the mechanism by which patients develop loss of response to anti-TNFα therapy.
Acknowledgments
Financial Support: 5K23DK103119 to MKG, Crohn’s and Colitis Foundation senior research award and AGA Elsevier pilot award to ANA, P30 DK043351 to RJX
Footnotes
Conflicts of Interest:
SJP have equity interest in Heprotech Inc. MG has equity interest in New Amsterdam Genomics. Ananthakrishnan has served on the scientific advisory boards for Gilead, Takeda, and Abbvie. All other authors have no conflicts to declare.
REFERENCES
- 1.Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359(9317): 1541–1549. [DOI] [PubMed] [Google Scholar]
- 2.Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132(1): 52–65. [DOI] [PubMed] [Google Scholar]
- 3.Sandborn WJ, Feagan BG, Stoinov S, et al. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357(3): 228–238. [DOI] [PubMed] [Google Scholar]
- 4.Gisbert JP, Panes J. Loss of response and requirement of infliximab dose intensification in Crohn’s disease: a review. Am J Gastroenterol 2009; 104(3): 760–767. [DOI] [PubMed] [Google Scholar]
- 5.Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56(9): 1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandborn WJ, Feagan BG, Stoinov S, et al. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357(3): 228–238. [DOI] [PubMed] [Google Scholar]
- 7.Roblin X, Rinaudo M, Del Tedesco E et al. Development of an algorithm incorporating pharmacokinetics of adalimumab in inflammatory bowel diseases. Am J Gastroenterol 2014; 109: 1250–1256. [DOI] [PubMed] [Google Scholar]
- 8.M, Karmiris K, Louis E, Van Assche G, Ben-Horin S. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: Definitions, frequency and pharmacological aspects. Journal of Crohns and Colitis. 2010; 4: 355–366. [DOI] [PubMed] [Google Scholar]
- 9.Chowers A, Sturm A, Sans M, Papadakis K. Report of the ECCO workshop on anti-TNF therapy failures in inflammatory bowel diseases: Biological roles and effects of TNF and TNF antagonists. Journal of Crohns and Colitis 2010; 4: 367–376. [DOI] [PubMed] [Google Scholar]
- 10.Harrison MJ, Dixon WG, Watson KD, et al. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68(2): 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci U S A 2005; 102(9): 3372–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102(43): 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marafini I, Monteleone I, Dinallo V, et al. CCL20 Is Negatively Regulated by TGF-β1 in Intestinal Epithelial Cells and Reduced in Crohn’s Disease Patients With a Successful Response to Mongersen, a Smad7 Antisense Oligonucleotide. J Crohns Colitis 2017; 11(5): 603–609. [DOI] [PubMed] [Google Scholar]
- 14.West NR, Hegazy AN, Owens BMJ, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med 2017; 23(5): 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alge-Priglinger CS, Kreutzer T, Obholzer K, et al. Oxidative stress-mediated induction of MMP-1 and MMP-3 in human RPE cells. Invest Ophthalmol Vis Sci 2009; 50(11): 5495–503. [DOI] [PubMed] [Google Scholar]
- 16.Biancheri P, Brezski RJ, Di Sabatino A, et al. Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology 2015; 149: 1564–1574. [DOI] [PubMed] [Google Scholar]
- 17.Gyuraszova M, Kovalcikova A, Gardlik R. Association between oxidative status and the composition of intestinal microbiota along the gastrointestinal tract. Med Hypotheses 2017; 103: 81–85. [DOI] [PubMed] [Google Scholar]
- 18.Haberman Y, Tickle TL, Dexheimer PJ, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest 2014; 124(8): 3617–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pink AE, Fonia A, Allen MH, et al. Antinuclear antibodies associate with loss of response to antitumour necrosis factor-alpha therapy in psoriasis: a retrospective, observational study. Br J Dermatol 2010; 162: 780–785. [DOI] [PubMed] [Google Scholar]
- 20.Brandse JF, van den Brink GR, et al. Loss of Infliximab into feces Is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology 2015; 149(2): 350–5. [DOI] [PubMed] [Google Scholar]
- 21.Kong JY, Bundell C, Pawlik J, et al. Low trough serum infliximab and antibodies to infliximab in smokers. Inflamm Bowel Dis 2013; 19: E35–E36. [DOI] [PubMed] [Google Scholar]
- 22.Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease 2014. Autoimmun Rev 13: 24–30. [DOI] [PubMed] [Google Scholar]