

Abstract
Cleft palate is one of the most common craniofacial congenital defects in humans. It is associated with multiple genetic and environmental risk factors, including mutations in the genes encoding signaling molecules in the sonic hedgehog (Shh) pathway, which are risk factors for cleft palate in both humans and mice. However, the function of Shh signaling in the palatal epithelium during palatal fusion remains largely unknown. Although components of the Shh pathway are localized in the palatal epithelium, specific inhibition of Shh signaling in palatal epithelium does not affect palatogenesis. We therefore utilized a hedgehog (Hh) signaling gain-of-function mouse model, K14-Cre;R26SmoM2, to uncover the role of Shh signaling in the palatal epithelium during palatal fusion. In this study, we discovered that constitutive activation of Hh signaling in the palatal epithelium results in submucous cleft palate and persistence of the medial edge epithelium (MEE). Further investigation revealed that precise downregulation of Shh signaling is required at a specific time point in the MEE during palatal fusion. Upregulation of Hh signaling in the palatal epithelium maintains the proliferation of MEE cells. This may be due to a dysfunctional p63/Irf6 regulatory loop. The resistance of MEE cells to apoptosis is likely conferred by enhancement of a cell adhesion network through the maintenance of p63 expression. Collectively, our data illustrate that persistent Hh signaling in the palatal epithelium contributes to the etiology and pathogenesis of submucous cleft palate through its interaction with a p63/Irf6-dependent biological regulatory loop and through a p63-induced cell adhesion network.
Keywords: Shh signaling, submucous cleft palate, palatal fusion, palatal epithelial cell fate
Introduction
Cleft palate, one of the most common congenital craniofacial birth defects, has an incidence of 1:500 to 1:2500 (Schutte and Murray, 1999) and can be induced by the interaction of genetic and environmental risk factors. Many human cases involve submucous cleft palate, in which the cleft is covered by an intact mucosal membrane. Submucous clefts have a reported incidence of 1:1250 to 1:5000 (Reiter et al., 2012). Clinically, submucous cleft palate is characterized by a translucent zone (zona pellucida) along the midline of the posterior hard palate indicating a separation of the submucous palatal musculature, a palpable bony notch in the same area as the zona pellucida, and a bifid uvula (Kosowski et al., 2012). Patients with submucous cleft palate may require surgery or other interventions to assist them with speech and swallowing (Kosowski et al., 2012). Despite the high frequency of submucous cleft palate in humans, the molecular and developmental mechanisms involved are not well studied, partly due to a limited number of animal models mimicking this defect in humans.
Multiple developmental events must occur to achieve the complex process of normal palatogenesis. These events include the growth of the palatal primordia from the lateral edges of the maxillary process, reorientation and elevation of the palatal shelves, fusion of the palatal shelves at the midline, and disappearance of the medial edge epithelium (MEE). Disruption of these processes may result in the failure of palatal fusion, known as cleft palate. Disappearance of the MEE is a key event during the palatal fusion process. Persistence of the MEE has been proposed as a mechanism responsible for the occurrence of submucous cleft palate in mouse models with dysregulation of Tgf-βor Bmp signaling in the palatal epithelium (Iwata et al., 2013; Noda et al., 2016; Xu et al., 2006). Defects in palatine bone formation following proper fusion of palatal shelves also result in submucous cleft palate (Baek et al., 2011; Pauws et al., 2009). However, the mechanism that regulates the fate of the MEE remains largely unknown, in part due to a lack of animal models that exhibit persistence of the MEE.
Sonic hedgehog (Shh), a member of the hedgehog (Hh) family, is a secreted protein that is crucial to numerous developmental processes, including establishing the left-right axis; patterning the neural tube, limbs, and brain; and endoderm development (Ingham and McMahon, 2001; McMahon et al., 2003; Roessler and Muenke, 2003). Shh signaling involves binding to receptors from the patched family, namely Ptch1 and Ptch2. When Shh ligands are not present, Ptch1/2 inhibits Smoothened (Smo). Activation of Smo upon Shh signaling in turn leads to activation of downstream genes by transcription factors from the Gli family. Because Ptch1 and Gli1 are themselves Shh downstream targets, a regulatory feedback loop is established through this activation process (Ingham and McMahon, 2001; McMahon et al., 2003).
Shh signaling plays essential roles during palatogenesis in humans and mice. As the palate develops, Shh expression occurs only in the oral side of the palatal epithelium along the palatal rugae (Han et al., 2009). In contrast, the Shh ligand receptor Ptch1 and downstream target Gli1 are expressed in both the palatal epithelium and mesenchyme, indicating that Shh ligand secreted from palatal epithelial cells targets both epithelial and mesenchymal cells (Lan and Jiang, 2009). SHH gene mutation in humans results in holoprosencephaly, often presenting with cleft lip and palate (Nanni et al., 1999). Mutations in PTCH in humans cause nevoid basal cell carcinoma syndrome with cleft lip and palate (Sasaki et al., 2009). Utilizing mice with tissue-specific knockouts of Shh pathway-associated genes, Lan and Jiang found that specifically blocking Shh signaling activity in the palatal mesenchyme by deleting Smo results in cleft palate, which is also observed in mice with specific ablation of Shh in the palatal epithelium (Lan and Jiang, 2009). Interestingly, specifically blocking the downstream target of Shh in the palatal epithelium has no effect on palatal fusion (Rice et al., 2004). These studies suggest that Shh protein secreted from palatal epithelial cells exerts its effect on the adjacent mesenchyme through epithelial-mesenchymal interaction to control palatal outgrowth (Lan and Jiang, 2009; Rice et al., 2004). Because important components of the Shh signaling pathway, such as Shh, Ptch1, and Gli1, are all expressed in the palatal epithelium during development, we sought to investigate the function of Shh signaling in the palatal epithelium during the palatal fusion process.
In this study, we generated K14-Cre;R26SmoM2 mice with gain-of-function of Hh signaling in the palatal epithelium and found that these mice exhibit submucous cleft palate with 100% phenotype penetrance. Furthermore, we found that downregulation of Shh signaling in the MEE is specifically required during palatogenesis. We demonstrated that constitutively activating Hh signaling in the palatal epithelium affects the fate of MEE cells, possibly due to dysfunction of the regulatory loop between p63 and Irf6, as well as enhancement of a cell adhesion network induced by the maintenance of p63 expression in the MEE. Upregulation of cell adhesion-associated genes induced by the maintenance of p63 expression may serve as the common mechanism responsible for the survival of MEE cells in animal models with submucous cleft palate and persistent MEE.
Materials and methods
Generation of transgenic mice
The K14-Cre (a gift from Sarah E. Millar, University of Pennsylvania, Philadelphia, USA; (Andl et al., 2004)), R26SmoM2 (a gift from Andrew McMahon, University of Southern California, USA; (Jeong et al., 2004)), conditional Tgfbr2 floxed (a gift from Harold L. Moses, Vanderbilt University, USA; (Leveen et al., 2002)) and Gli1-LacZ knock-in/knock-out reporter (JAX#008211, The Jackson Laboratory; (Bai et al., 2002)) mouse lines have all been described previously. Gli1-LacZ knock-in/knock-out mice were used as heterozygotes. Mating K14-Cre with R26SmoM2 mice generated K14-Cre;R26SmoM2fl/+ embryos. K14-Cre;Gli1-LacZ+/− mice were crossed with R26SmoM2 mice to generate K14-Cre;R26SmoM2fl/+;Gli1-LacZ+/− embryos. To generate K14-Cre;Tgfbr2fl/fl embryos, we mated K14-Cre;Tgfbr2fl/+ with Tgfbr2fl/fl mice. In all studies involving animals, we used both male and female mice for our experiments. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Southern California.
X-gal staining and detection of β-galactosidase activity
Samples from embryonic day 14.5 (E14.5) mice were fixed in 0.2% glutaraldehyde in PBS with 2mM MgCl2 overnight at 4°C, dehydrated in 15% and 30% sucrose with 2mM MgCl2, embedded in OCT compound (Tissue-Tek, Sakura) and sectioned on a cryostat (Leica CM1850) at 10 μm prior to X-gal staining for LacZ expression. To detect β-galactosidase (β-gal) activity in tissue sections, cryosections were stained in X-gal staining solution (2mM MgCl2, 0.01% sodium deoxycholate, 0.005% nonidet P-40, 5mM potassium ferricyanide, 5mM potassium ferrocyanide, 20mM Tris pH7.3, and 1 mg/ml X-gal in PBS) for 3–4 hours at 37°C in the dark, followed by postfixation in 3.7% formaldehyde for 30 minutes at room temperature and counterstaining with nuclear fast red (Electron Microscopy Sciences, 2621203).
Histological analysis
Dissected samples were fixed in 4% paraformaldehyde (PFA) in diethyl pyrocarbonate (DEPC)-treated PBS overnight at 4°C, decalcified in 10% DEPC-treated EDTA (pH 7.4) for 1–5 days depending on the age of the sample, dehydrated through serial concentrations of ethanol for embedding in paraffin and sectioned at 6 μm using a microtome (Leica). Deparaffinized sections were stained with Hematoxylin and Eosin (H&E) using standard procedures for general morphology.
For cryosections, decalcified samples were dehydrated in 15% and 30% sucrose in DEPC-treated PBS overnight at 4°C, embedded in OCT compound (Tissue-Tek, Sakura) and sectioned at 8 μm using a cryostat (Leica CM1850).
RNAscope in situ hybridization
Staining was carried out on cryosections using the RNAscope 2.5 HD Reagent Kit-RED assay (Advanced Cell Diagnostics, 322350) according to the manufacturer’s instructions. All probes used in our study were designed and synthesized by Advanced Cell Diagnostics (ACD), including Dsg3 (464301), Fgfr2 (443501), Gli1 (311001), Irf6 (462931), Itga3 (521011), Jag2 (417511), Lama5 (494911), Lgr5 (312171), Mmp13 (427601) and Tgfb3 (406211).
Immunostaining
Sections were immersed in a preheated antigen unmasking solution (Vector, H-3300) in a microwave oven at 95°C for 15–20 minutes, followed by cooling at room temperature for 20 minutes and incubation with a blocking reagent (PerkinElmer, FP1012) for 1 hour and then a primary antibody overnight at 4°C. The following primary antibodies were used in our study: active Caspase-3 (Casp3; Abcam, ab2302; 1:100), phospho-Histone H3 (pH3; Millipore, 06–570; 1:100), non-muscle myosin heavy chain IIA (NMHCIIA; Biolegend, 909801; 1:500), phospho-Smad2 (pSmad2; Cell Signaling, 3108; 1:500), phospho-Smad1/5/9 (pSmad1/5/9; Cell Signaling, 13820; 1:500), ΔNp63 (p63; Biolegend, 619002; 1:100), Sox2 (Abcam, ab97959; 1:1000), Sox2 (Santa Cruz, sc-17320; 1:100), and SSEA1 (Developmental Studies Hybridoma Bank, AB 528475; 1:10). After three washes in PBS, sections were incubated with the Alexa Fluor 568 or Alexa Fluor 488 (1:200, Invitrogen) secondary antibody for 2 hours at room temperature. For pSmad2 and pSmad1/5/9, HRP-labeled goat anti-rabbit IgG (PerkinElmer, NEF812001EA; 1:200) was used as secondary antibody and a TSA kit was used for signal detection (PerkinElmer, NEL741001KT). Sections were counterstained with DAPI (Sigma, D9542). Images were captured using a fluorescence microscope (Leica DMI 3000B) with filter settings for DAPI/FITC/TRITC.
In situ hybridization
After deparaffinization and serial hydration with ethanol, sections were treated with proteinase K (20 mg/ml) for 15 minutes at room temperature followed by a PBS rinse. Sections then were post-fixed in 4% paraformaldehyde in PBS for 20 minutes followed by a PBS rinse. After treatment with 0.1M triethanolamine and 0.25% acetic anhydride under vigorous stirring for 10 minutes, sections were washed with PBS for 5 minutes and dehydrated through serial concentrations of ethanol, followed by air drying for 30 minutes. After digoxigenin-labeled probes were diluted in hybridization solution (50% formamide, 0.3M NaCl, 20mM Tris-HCl pH7.4, 5mM EDTA pH 8.0, 10mM NaH2PO4-H2O pH8.0, 5% dextran sulfate, 1X Denhardt’s solution, 0.5 mg/ml yeast tRNA, 10mM dithiothreitol, 0.05% CHAPS, 10 mg/ml ssDNA in DEPC-treated H2O) in a 1.5 ml centrifuge tube, the tube was heated in a 100°C water bath for 5 minutes, followed by chilling on ice immediately. Then 100 to 150 μl of diluted probe was added per section and incubated in a humidified hybridization chamber at 55°C for 16 hours. After hybridization, cover slips were floated off in 2X SSC buffer, then sections were treated with 1 μg/ml RNase A in 2X SSC buffer for 30 minutes at 37°C, followed by 2X SSC buffer rinse. Sections were washed in pre-warmed 2X SSC buffer and 0.2X SSC buffer with 0.05% CHAPS 3 times (20 minutes each) at 65°C, followed by blocking with 20% sheep serum for 2–3 hours at room temperature and incubation with 1:2000 dilution of anti-digoxigenin-ap antibody (Roche, 11093274910) at 4°C overnight. Next, sections were washed with PBS with 0.1% tween 20 and 1mM Tetramisole hydrochloride 6 times (30 minutes each) at room temperature, followed by a wash with alkaline-phosphatase buffer (100mM NaCl, 100mM Tris-HCl pH9.5, 50mM MgCl2, and 0.1% tween 20 in H2O). Finally, sections were developed using a BCIP/NBT liquid substrate system (Sigma, B1911). Axin2 cDNA clones were kindly provided by Paul Sharpe (King’s College London, UK).
Western blot
For immunoblotting, E13.5 palatal shelves were lysed in a lysis buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.1% NP-40, 10% glycerol and protease inhibitor cocktail). After protein quantification using Bio-Rad protein assays (Bio-Rad Laboratories), 10–20 μg of protein were separated by SDS-PAGE and transferred to the 0.45 μm PVDF membrane. Membranes were blocked with 5% milk/TBST for 1 hour, followed by overnight incubation with an anti-ΔNp63 antibody (p63; Biolegend, 619002; 1:1000 dilution in 5% BSA/TBST) and 1 hour incubation with an HRP-conjugated secondary antibody (R&D, HAF008; 1:1000 dilution in 5% milk/TBST). Immunoreactive protein was detected using a LumiGLO Reagent (Cell Signaling, 7003) and Chemiluminescent Western Blot Imaging System (Azure, C300).
Quantitative RT-PCR
Total RNA was extracted from E14.5 mouse palatal shelves using RNeasy Micro Kit (Qiagen, 74004) according to the manufacturer’s instructions. The following PCR primers were used: Fgfr2, 5’-AATCTCCCAACCAGAAGCGTA-3’ and 5’-CTCCCCAATAAGCACTGTCCT-3’; Jag2, 5’-CAATGACACCACTCCAGATGAG-3’ and 5’-GGCCAAAGAAGTCGTTGCG-3’; Irf6, 5’-CTCTCCCCATGACTGACTTGG-3’ and 5’-CAGGTCCCCATAGAAGAGCC-3’; Dsg3, 5’-TGGCAGTCTGGAAGTCACC-3’ and 5’-CTGTAGAGGGTCAGGGATGG-3’; Itga3, 5’-CCTCTTCGGCTACTCGGTC-3’ and 5’-CCGGTTGGTATAGTCATCACCC-3’; Lama5, 5’-GCTGGCGGAGATCCCAATC-3’ and 5’-GTGTGACGTTGACCTCATTGT-3’.
Palatal shelf organ culture
Timed-pregnant mice were euthanized at E13.5. Palatal shelves were microdissected and cultured in a serum-free chemically defined medium as previously described (Ito et al., 2003). To test epithelial degeneration and fusion, palatal shelves were positioned contacting each other at the midline and cultured for 3 days along with 10 ng/ml bovine serum albumin (BSA) or 10 ng/ml TGFβ3 protein (R&D systems, 243-B3–002). After 3 days in culture, palatal shelves were fixed in 4% PFA and subsequently processed for H&E staining and immunostaining.
Statistical analysis
Two-tailed Student’s t-tests were applied for statistical analysis. P<0.05 was considered statistically significant.
Results
Downregulation of Shh signaling is specifically required in the MEE during palatal fusion
To explore the function of Shh signaling in the palatal epithelium during palatogenesis, we first examined Shh signaling activity during the entire palatal fusion process using Gli1-LacZ reporter mice. At E14.5, strong Gli1 expression was detectable in the epithelium at the tips of the palatal shelves before they made contact with one another (Figure 1A). However, the expression of Gli1 dramatically decreased in the palatal epithelium once the palatal shelves started to touch along the midline (Figure 1B) and completely disappeared in the MEE after they began to fuse (Figure 1C). This loss of expression suggests that there is no requirement for Shh signaling in the palatal epithelium during the fusion process, which explains why there is no palatal fusion defect after specific blockage of the Shh pathway in the palatal epithelium.
Figure 1. Downregulation of Shh signaling is specifically required in the MEE during palatal fusion.

(A-C) X-gal staining (blue) of frontal sections of the palatal shelves of E14.5 Gli1-LacZ reporter mice. Larger arrow indicates strong expression (A), smaller arrow indicates weak expression (B), and arrowhead indicates undetectable expression (C). (D-G) Macroscopic views of newborn (NB) control and K14-Cre;R26SmoM2 palates. Red boxed area in (E) is shown magnified in (F), highlighting the failure of fusion between the primary and secondary palate (arrow). Blue boxed area in (E) is shown magnified in (G), highlighting the translucent strip in the posterior part of the midline (arrow). Asterisk in (E) indicates cleft soft palate. (H-O) H&E staining of frontal sections of newborn (NB) control and K14-Cre;R26SmoM2 palate, from anterior to posterior (indicated by red lines). Arrows indicate palatal defects; asterisk indicates complete cleft in the soft palate region (O). NS, nasal septum; PP, primary palate; PS, palatal shelves; SP, secondary palate. Scale bars (A-C): 50 μm; (H-O): 200 μm.
To investigate whether an increase in Shh signaling may disrupt palatal fusion, we generated mouse models with gain-of-function of Hh signaling in the palatal epithelium. We generated K14-Cre;R26SmoM2 transgenic mice, in which Smo is constitutively expressed in all K14+ epithelial cells, including MEE cells, leading to constitutive activation of Hh signaling. We found that newborn (postnatal day 0.5) K14-Cre;R26SmoM2 mice died soon after birth and exhibited severe defects of the palate (Figure 1D-G): the primary palate failed to fuse with the secondary palatal shelves (Figure 1E, F), the posterior part of the midline contained a translucent strip similar to the zona pellucida seen in human patients with submucous cleft palate (Figure 1E, G), and the soft palate exhibited a complete cleft (Figure 1E). These phenotypes were identified in newborn K14-Cre;R26SmoM2 mice with 100% penetrance (n=20). Detailed histological analysis revealed additional palatal fusion defects in newborn K14-Cre;R26SmoM2 mice (Figure 1H-O), including failure of the nasal septum to fuse with the palatal shelves (Figure 1L), a persistent midline epithelial seam in the anterior part of the secondary palate (Figure 1M), and a thin epithelial bridge separating the palatine bone that prevented fusion along the midline (Figure 1N). In addition, the complete cleft in the soft palate region was also detectable histologically (Figure 1O). These data suggest that downregulation of Shh signaling is specifically required in the MEE during palatogenesis to ensure normal palatal fusion.
Constitutive activation of the Hh pathway in the MEE does not affect the Tgf-β, Smad-dependent Bmp, or canonical Wnt pathways
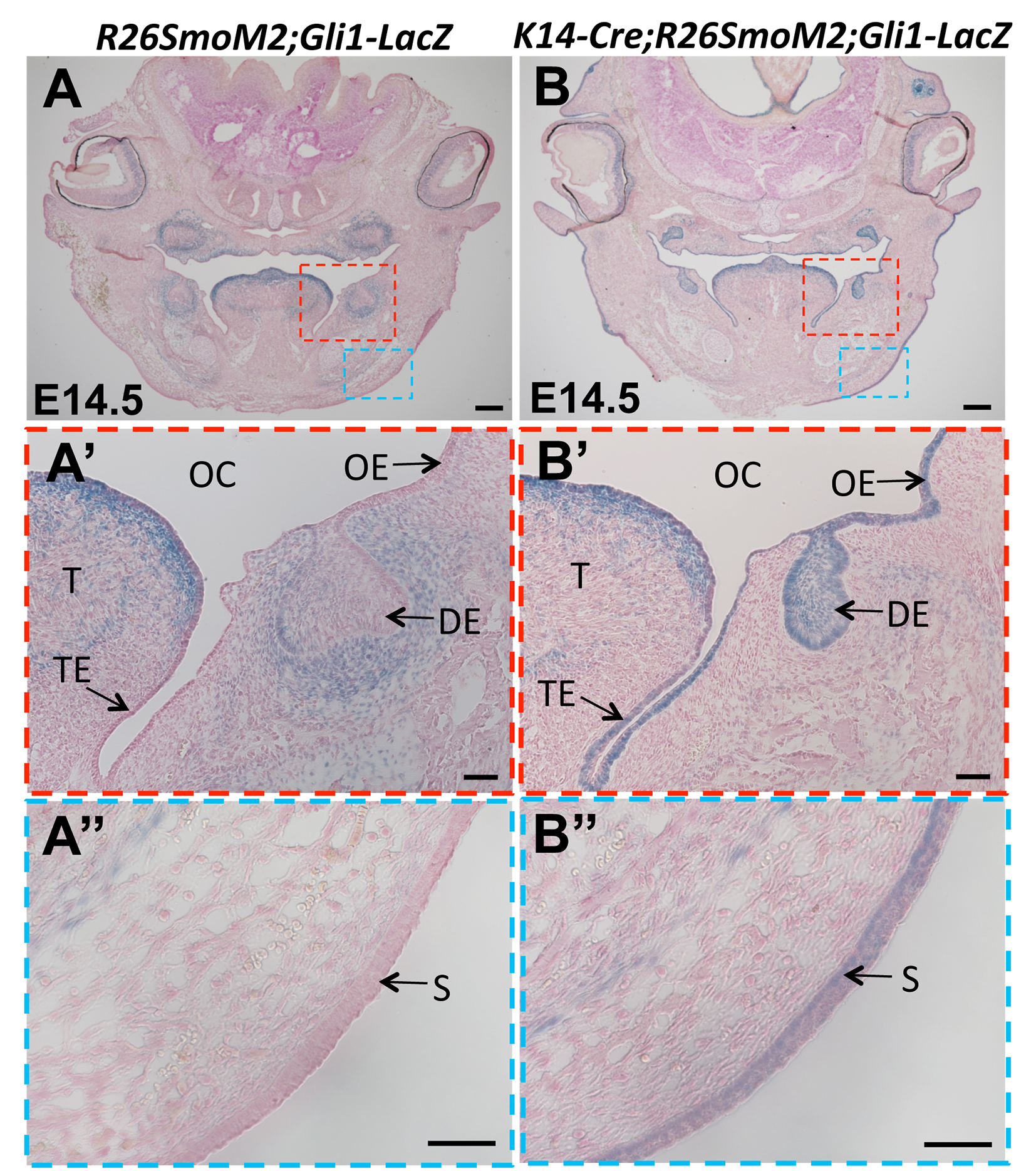
To verify that the Hh pathway is activated in the MEE of SmoM2 mutant mice, we generated K14-Cre;R26SmoM2;Gli1-LacZ mice and analyzed Gli1 expression by performing X-gal staining. At E14.5, strong Gli1 expression was detectable in the MEE of SmoM2 mutant mice (Figure 2B). In contrast, we failed to detect Gli1 expression in the MEE of control mice (Figure 2A). In addition to the MEE, constitutive Gli1 expression was also detectable in other K14+ epithelial structures of SmoM2 mutant mice at E14.5 (Figure S1A, B), including the epithelium of the tooth, tongue, oral cavity (Figure S1A’, B’), and skin (Figure S1A”, B”). These data confirm that the Hh pathway is constitutively activated in K14-Cre;R26SmoM2 epithelium, including the MEE.
Figure 2. The Hh pathway is constitutively activated in the MEE of SmoM2 mutant mice without affecting the Tgf-β, Smad-dependent Bmp, or canonical Wnt pathways.

(A-B) X-gal staining (blue) of frontal sections of E14.5 R26SmoM2;Gli1-LacZ (control) and K14-Cre;R26SmoM2;Gli1-LacZ palatal shelves. Arrow indicates activated Gli1 expression, whereas arrowhead indicates absence of Gli1 expression. (C-D) RNAscope in situ hybridization analysis of Tgfb3 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. (E-H) Immunofluorescence of phospho-Smad2 (pSmad2) and phospho-Smad1/5/9 (pSmad1/5/9) (red) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Arrows indicate positive signal; arrowheads indicate absence of signal. (I-J) In situ hybridization of Axin2 (dark blue) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Arrowheads indicate absence of Axin2 expression. Black and white dotted lines indicate the location of the MEE. Scale bars: 50 μm.
Previous studies have shown that dysregulation of the Tgf-β, Smad-dependent Bmp, or canonical Wnt pathways in the epithelium results in palate or lip fusion defects including persistence of the MEE (Iwata et al., 2013; Kurosaka et al., 2014; Noda et al., 2016; Xu et al., 2006). Therefore, we hypothesized that the Shh pathway regulates the fate of MEE cells during palatal fusion through interaction with the Tgf-β, Smad-dependent Bmp, or canonical Wnt pathways. To test our hypothesis, we analyzed the expression of important components of these pathways. We first examined Tgf-β signaling activity in the MEE of SmoM2 mutant mice. We found that the expression levels of Tgfb3 and pSmad2 were unaffected in K14-Cre;R26SmoM2 MEE (Figure 2C-F). We failed to detect changes in the activity of either Smad-dependent Bmp or canonical Wnt signaling in K14-Cre;R26SmoM2 MEE, indicated by the expression of pSmad1/5/9 (Figure 2G, H) and Axin2 (Figure 2I, J), respectively. These data suggest that constitutive activation of the Hh pathway results in the persistence of the MEE without affecting the Tgf-β, Smad-dependent Bmp, or canonical Wnt pathways.
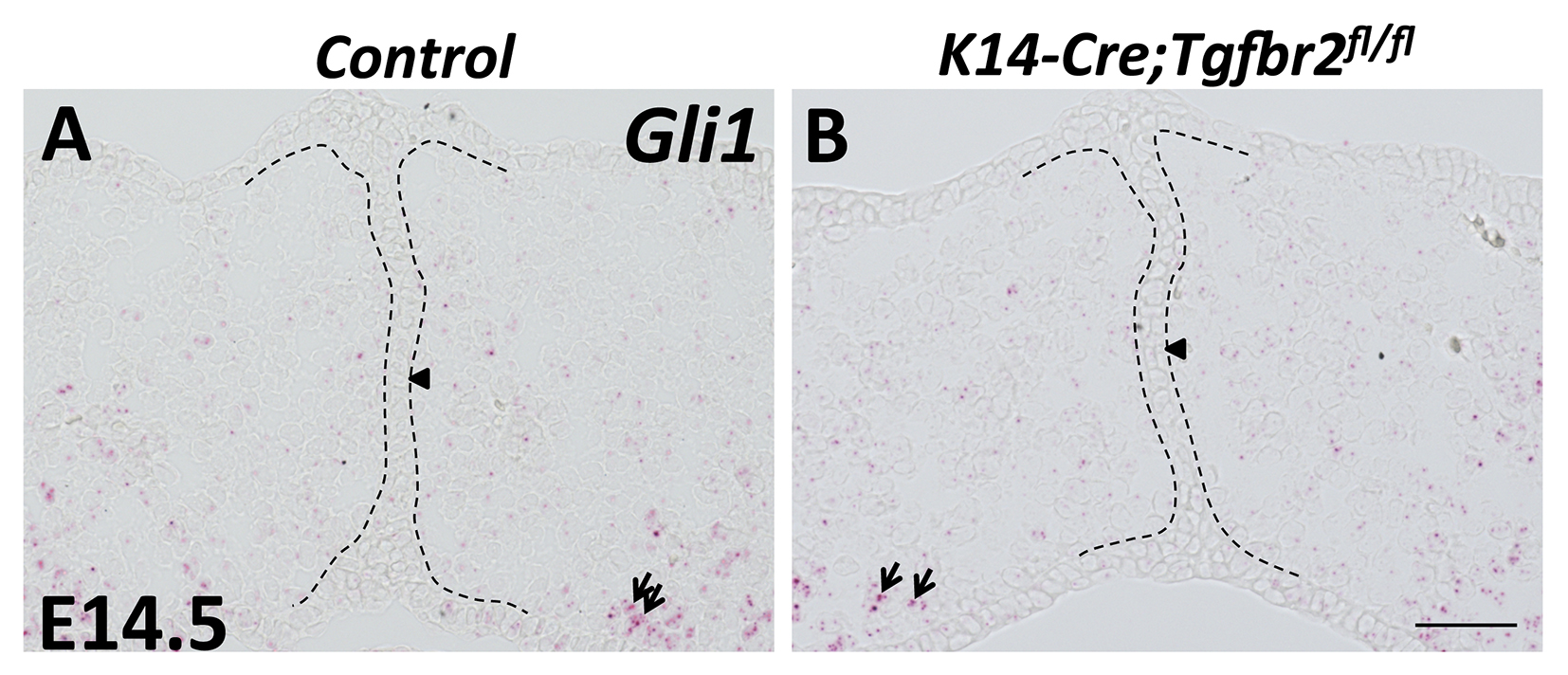
Tgf-β3 has a high level of expression specifically in the epithelial tips of the palatal shelves prior to fusion and in the MEE right after palatal fusion (Lane et al., 2014), which is complementary to the expression pattern of Gli1 in the MEE during palatal fusion. Additionally, loss-of-function of Tgf-β and gain-of-function of Hh signaling in the palatal epithelium result in similar MEE defects during palatal fusion. We therefore hypothesized that Tgf-β signaling induces the downregulation of Shh signaling in the MEE during palatal fusion. To test our hypothesis, we examined the expression of Gli1 in the MEE after specific deletion of Tgfbr2 in the palatal epithelium, and found that Gli1 expression was undetectable in the MEE of both control and K14-Cre;Tgfbr2fl/fl mice (Figure S2A, B), indicating that Tgf-β signaling is not required for the repression of Shh signaling in the MEE during palatal fusion.
Constitutive activation of the Hh pathway in the palatal epithelium alters MEE cell fate without affecting periderm disappearance or actomyosin-driven cell movement
Epithelial cell death is a crucial event common to the fusion of facial processes during development, including that of the palatal shelves (Cuervo and Covarrubias, 2004; Ray and Niswander, 2012). The presence of periderm initially prevents premature fusion during palatogenesis, but the periderm must be removed before the two palatal shelves come into contact to ensure proper palatal fusion (Lan et al., 2015). Previous studies also demonstrated that actomyosin-driven cell intercalation and extrusion serve as a general mechanism for tissue fusion in development, including palatal fusion (Kim et al., 2015). To determine why the MEE fails to disappear in SmoM2 mutant mice, we investigated whether apoptosis, proliferation or periderm disappearance was altered. In control mice at E14.5, apoptotic cells were detectable in the MEE, especially in the nasal and oral epithelial triangles (Figure 3A). In contrast, there were no detectable apoptotic cells in the MEE of SmoM2 mutant mice at E14.5 (Figure 3B). Next, we analyzed proliferation and found that the persistent MEE cells in SmoM2 mutant mice maintained active cell proliferation (Figure 3D), whereas no proliferative activity was detectable in the MEE of control mice at E14.5 (Figure 3C). Furthermore, we examined the expression of periderm marker SSEA1 and found that periderm disappearance was unaffected in SmoM2 mutant MEE (Figure 3E, F). Finally, we analyzed actomyosin contractility by examining the expression of non-muscle myosin heavy chain IIA (NMHCIIA) and found that it was indistinguishable in control and SmoM2 mutant MEE at E14.5 (Figure 3G, H). These data suggest that overactivation of the Hh pathway in the palatal epithelium changes MEE cell fate, resulting in the maintenance of proliferation rather than a switch to apoptosis.
Figure 3. Constitutive activation of the Hh pathway in the palatal epithelium alters MEE cell fate without affecting periderm disappearance or actomyosin-driven cell movement.

(A-H) Immunofluorescence of active caspase 3 (Casp3, green), phospho-histone H3 (pH3, red), SSEA1 (green), and non-muscle myosin heavy chain IIA (NMHCIIA, red) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Arrows indicate positive signal; arrowheads indicate absence of signal. White dotted lines indicate the location of the MEE. Scale bars: 50 μm.
Constitutively activated Hh signaling in the palatal epithelium results in maintenance of p63 expression in the MEE and upregulation of p63 target genes
Downregulation of the p63 isoform ΔNp63 in the MEE is required during normal palatal fusion (Moretti et al., 2010; Richardson et al., 2017; Thomason et al., 2010). Previous studies have demonstrated that both loss of Tgf-β signaling and constitutive activation of Bmp signaling in the palatal epithelium result in persistence of the MEE through increased ΔNp63 expression, and overactivation of ΔNp63 in the palatal epithelium also disrupts MEE disappearance (Iwata et al., 2013; Noda et al., 2016; Richardson et al., 2017; Xu et al., 2006). In addition, Shh signaling can regulate p63 expression during keratinocyte differentiation (Chari et al., 2013). Therefore, we hypothesized that constitutive activation of Hh signaling in the palatal epithelium may upregulate ΔNp63 expression, resulting in persistence of the MEE. We first examined the expression of ΔNp63 in the palatal epithelium at different developmental stages. Before palatal shelf elevation at E13.5, ΔNp63 expression in the basal layer of the entire palatal epithelium was indistinguishable in control and K14-Cre;R26SmoM2 mice (Figure 4A, B), confirmed by Western blotting analysis (Figure 4I). At E14, after the elevation of palatal shelves but before they touch along the midline, ΔNp63 expression persisted with the same pattern and comparable levels in control and SmoM2 mutant mice (Figure 4C, D). However, once the palatal shelves touched along the midline at E14.5, ΔNp63 expression was undetectable in the MEE cells of control mice, although it persisted in the basal layer of the nasal and oral sides of the palatal epithelium (Figure 4E). In contrast, ΔNp63 expression was maintained in the MEE of SmoM2 mutant mice at E14.5 (Figure 4F) and persisted in the MEE as late as the newborn stage (Figure 4G, H). Thus, constitutive activation of Hh signaling in the palatal epithelium results in the failure of ΔNp63 expression to disappear in the MEE when palatal fusion should be occurring, but does not affect ΔNp63 expression prior to palatal fusion.
Figure 4. Constitutively activated Hh signaling in the palatal epithelium results in maintenance of p63 expression in the MEE and upregulation of p63 target genes.

(A-H) Immunofluorescence of ΔNp63 (p63, red, indicated by arrows) in frontal sections of control and K14-Cre;R26SmoM2 palatal shelves at E13.5 (A, B), E14 (C, D), E14.5 (E, F) and newborn (NB; G, H) stages. Arrowheads indicate absence of ΔNp63 expression. (I) Western blotting analysis of ΔNp63 expression in E13.5 control and K14-Cre;R26SmoM2 (two samples: mu1 and mu2) palatal shelves. (J-O) RNAscope in situ hybridization of Fgfr2, Jag2 and Irf6 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. (P) qPCR analysis of Fgfr2, Jag2 and Irf6 in E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Values are expressed relative to control. *, p<0.05. Black and white dotted lines indicate the location of the MEE. Scale bars: 50 μm.
We also examined the expression of p63 direct target genes Fgfr2, Jag2, and Irf6 (Richardson et al., 2017), mutations in which result in cleft palate (Jiang et al., 1998; Kondo et al., 2002; Riley et al., 2007). Compared to the low-level expression of these three genes in control MEE (Figure 4J, L, N), Fgfr2, Jag2, and Irf6 expression levels were significantly elevated in the MEE of SmoM2 mutant mice at E14.5 (Figure 4K, M, O), as confirmed by q-PCR analysis (Figure 4P). Our data suggest that constitutive activation of Hh signaling in the palatal epithelium results in the maintenance of ΔNp63 expression in the MEE, which in turn upregulates the expression of p63 target genes.
Constitutive activation of Hh signaling in the palatal epithelium upregulates cell adhesion-associated genes and epithelial progenitor cell-associated genes in the MEE
Previous studies have shown that p63 also regulates cell-cell and cell-matrix adhesion as well as cell survival in epithelial cells (Carroll et al., 2006). Loss of p63 induces cell detachment and apoptosis, and ectopic expression of p63 can protect cells from apoptosis caused by cell detachment, possibly due to p63-induced upregulation of adhesion proteins and enhanced integrin-mediated adhesion (Carroll et al., 2006). Therefore, we hypothesized that constitutive activation of Hh signaling in palatal epithelium strengthens cell adhesion through persistent p63 expression in the MEE. To test our hypothesis, we analyzed the expression of cell adhesion molecules in the MEE, such as Dsg3, Itga3, and Lama5, all previously recognized p63 target genes (Carroll et al., 2006; Ferone et al., 2013). Indeed, Dsg3, Itga3, and Lama5 were all elevated in the MEE of SmoM2 mutant mice (Figure 5A-F), confirmed by q-PCR (Figure 5G). Moreover, palatal fusion requires remodeling of the extracellular matrix (ECM), which is controlled in part by a family of matrix metalloproteinases (MMPs) (Brown et al., 2002). MMPs can regulate epithelial tissue architecture through cleavage of intercellular junctions and degeneration of basement membrane (Page-McCaw et al., 2007). Because epithelial cells must adhere to the ECM for survival, MMP-mediated disruption of subcellular matrices can induce apoptosis in anchorage-dependent cells (Sternlicht and Werb, 2001). Next, we investigated whether constitutively activated Hh signaling in the palatal epithelium affects ECM remodeling in the MEE through regulating the activity of MMPs. We found that Mmp13 was strongly expressed in the MEE of E14.5 control samples (Figure 5H). In contrast, Mmp13 was undetectable in the MEE of SmoM2 mutant mice (Figure 5I). Based on the previously reported transcriptional regulation of Mmp13 by p63 (Celardo et al., 2014), it is possible that downregulation of Mmp13 results from the deregulation of p63 expression in the MEE of SmoM2 mutant mice. These data indicate that constitutive activation of Hh signaling in palatal epithelium enhances cell adhesion and affects ECM remodeling in the MEE, possibly due to the maintenance of p63 expression, which may prevent apoptosis in MEE cells.
Figure 5. Constitutive activation of Hh signaling in palatal epithelium upregulates cell adhesion-associated genes and epithelial progenitor cell-associated genes in the MEE.

(A-F) RNAscope in situ hybridization of Dsg3, Itga3 and Lama5 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. (G) qPCR analysis of Dsg3, Itga3 and Lama5 in E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Values are expressed relative to control. *, p<0.05. (H-I) RNAscope in situ hybridization analysis of Mmp13 (red dots, indicated by arrow) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Arrowhead indicates absence of Mmp13 expression. (J-K) Immunofluorescence of ΔNp63 (p63, red) and Sox2 (green) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Boxed areas in (J) and (K) are shown magnified in the top right corners with red (p63), green (Sox2), blue (DAPI) and merged channels. Arrows indicate co-expression of ΔNp63 and Sox2. (L-M) RNAscope in situ hybridization analysis of Lgr5 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;R26SmoM2 palatal shelves. Black dotted lines indicate the location of the MEE. Scale bars: 50 μm.
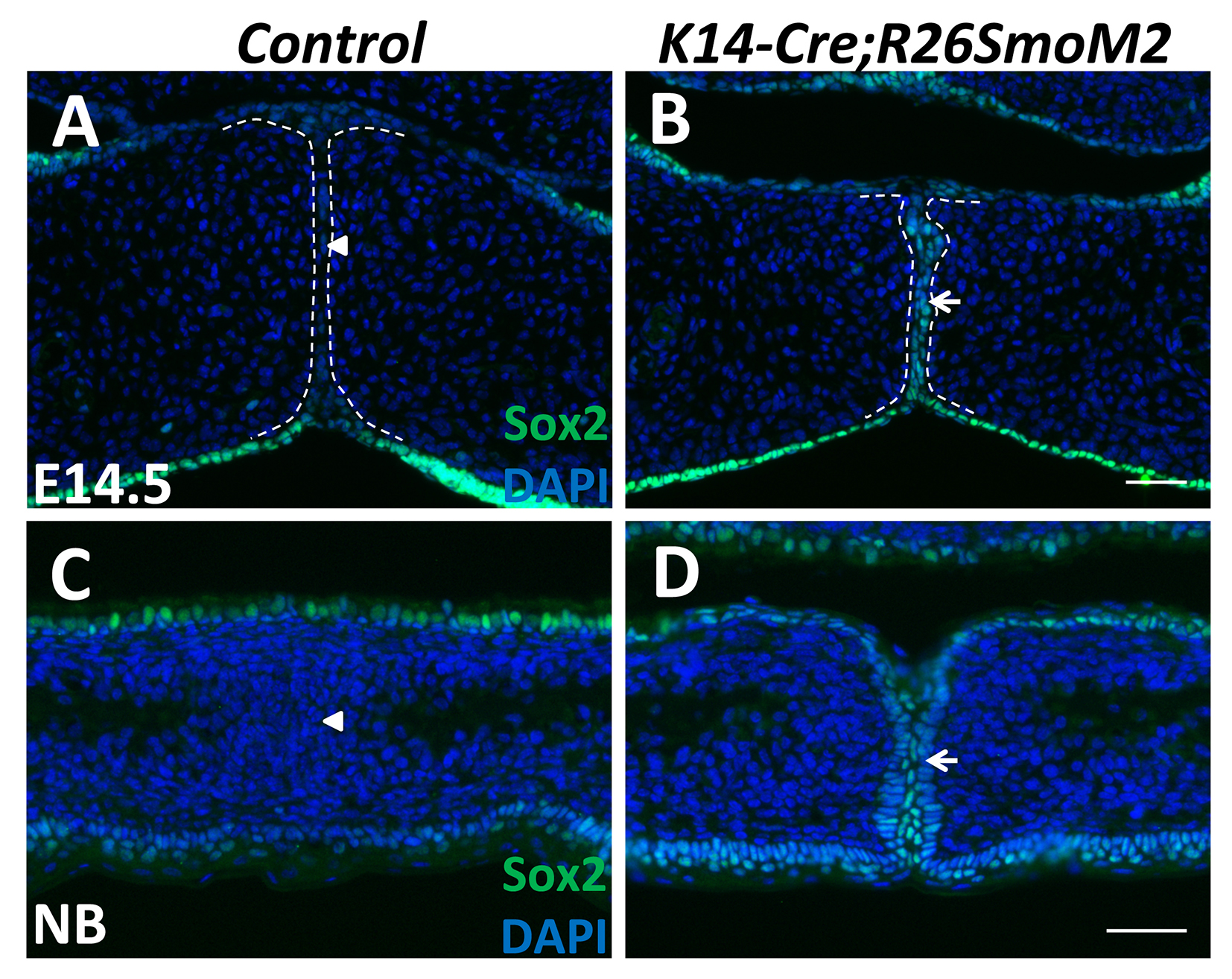
Because p63 is a marker of epithelial progenitor cells, we also investigated the expression of other epithelial progenitor cell markers, Sox2 and Lgr5, in the SmoM2 mutant MEE. We found that Sox2 has a similar expression pattern to that of p63 in the SmoM2 mutant MEE, persisting at E14.5 and as late as the newborn stage (Figure S3A-D). Moreover, the expression of Sox2 and p63 overlapped in the MEE cells of SmoM2 mutant mice (Figure 5J, K). Expression of Lgr5 was also significantly increased in SmoM2 mutant MEE cells compared to the control (Figure 5L, M). These data suggest that constitutive activation of Hh signaling in the palatal epithelium upregulates the expression of epithelial progenitor cell-associated genes in the MEE, which may contribute to the maintenance of proliferation of MEE cells.
p63 target genes and cell adhesion-associated genes are upregulated in K14-Cre;Tgfbr2fl/fl mice
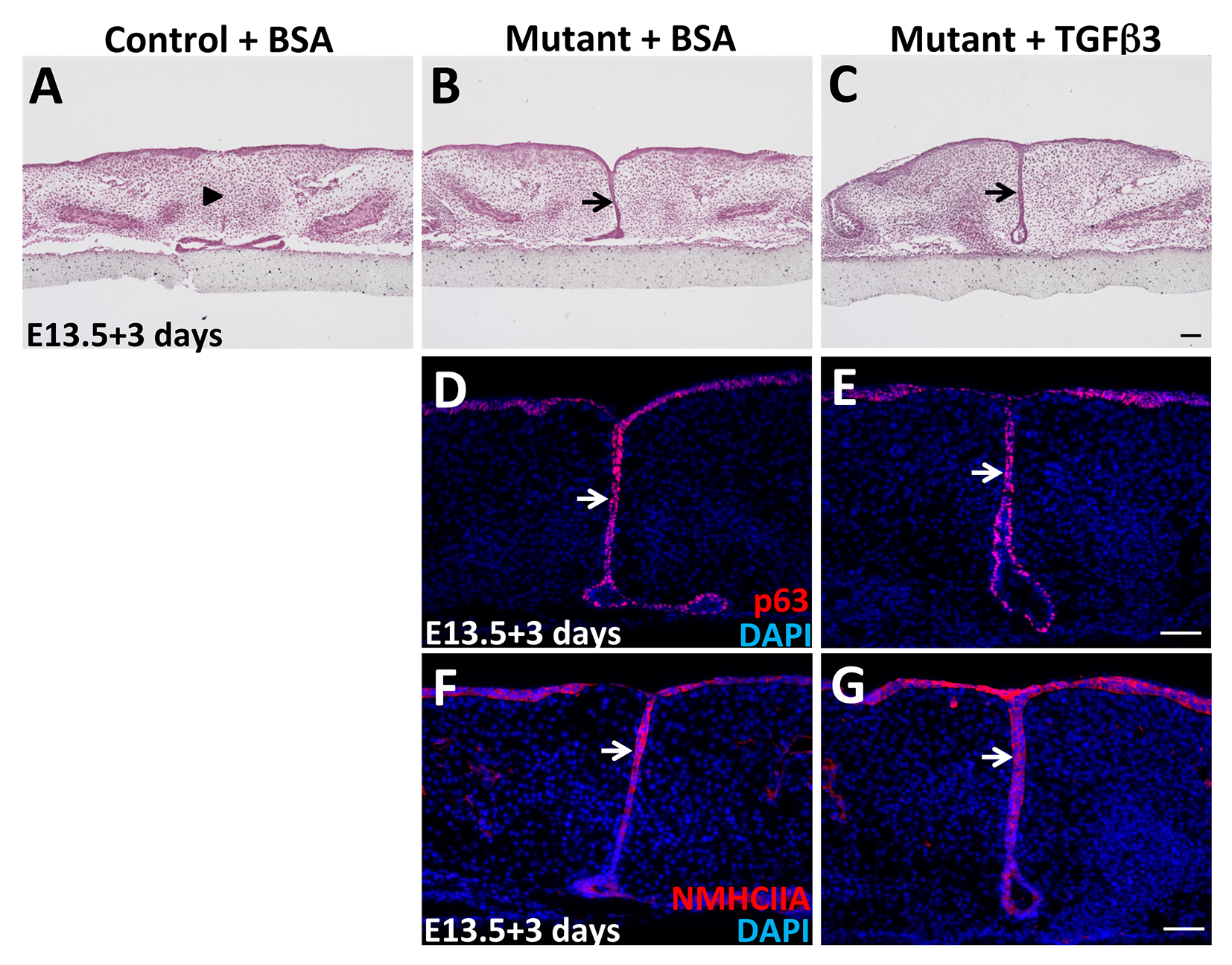
Previous studies have revealed that both loss of Tgf-β signaling and constitutive activation of Bmp signaling in the palatal epithelium result in persistence of the MEE through increased ΔNp63 expression (Iwata et al., 2013; Noda et al., 2016; Xu et al., 2006), consistent with our findings in K14-Cre;R26SmoM2 mice. Therefore, we hypothesized that enhanced expression of cell adhesion-associated genes induced by persistent p63 expression may serve as a converging point, responsible for the aberrant survival of MEE cells in multiple mouse models showing persistence of the MEE. To test our hypothesis, we examined the expression of genes upregulated in K14-Cre;R26SmoM2 mice in K14-Cre;Tgfbr2fl/fl mice, with loss of Tgf-β in the palatal epithelium (Iwata et al., 2013; Xu et al., 2006). We found that expression of Fgfr2 and Jag2, p63 direct target genes, were elevated in the MEE of K14-Cre;Tgfbr2fl/fl mice (Figure 6A-D). Cell adhesion-associated genes Dsg3, Itga3, and Lama5 were also increased in Tgfbr2 mutant MEE (Figure 6E-J), consistent with K14-Cre;R26SmoM2 mice. To confirm that Hh and Tgf-β signaling work in parallel and converge at p63 in the regulation of MEE cell fate during palatal fusion, we cultured SmoM2 mutant palatal shelves with or without the addition of TGFβ3 protein. As expected, exogenous TGFβ3 protein treatment had no effect on the expression level of p63 and NMHCIIA in SmoM2 mutant MEE and failed to induce the complete fusion of SmoM2 mutant palatal shelves (Figure S4A-G). These data suggest that enhancement of the cell adhesion network induced by the maintenance of p63 expression may serve as the common mechanism responsible for the survival of MEE cells in submucous cleft palate-associated animal models with persistent MEE, including both K14-Cre;R26SmoM2 and K14-Cre;Tgfbr2fl/fl mice.
Figure 6. p63 target genes and cell adhesion-associated genes are upregulated in K14-Cre;Tgfbr2fl/fl mice.

(A-J) RNAscope in situ hybridization of Fgfr2, Jag2, Dsg3, Itga3 and Lama5 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;Tgfbr2fl/fl palatal shelves. Black dotted lines indicate the location of the MEE. Scale bars: 50 μm.
Discussion
We have found that gain-of-function of Hh signaling in the palatal epithelium has unexpected effects on palatal fusion: it results in persistence of the MEE that prevents the fusion of palatal shelves and separates the palatine bone and the submucous palatal musculature, mimicking the characteristics of submucous cleft palate in human patients. More importantly, our study offers a new animal model for investigating the molecular mechanism of submucous cleft palate in humans, and provides new insights into the biological function of Shh signaling in the palatal epithelium during palatogenesis.
Shh signaling pathway plays a crucial role during craniofacial development, especially during palatogenesis. Both loss-of-function and gain-of-function of SHH signaling result in cleft palate in humans (Nanni et al., 1999; Sasaki et al., 2009). In our study, we first systematically analyzed the dynamic changes in Shh signaling activity in the palatal epithelium during palatal fusion and found that a dramatic downregulation of Shh signaling occurs in the MEE right after the palatal shelves touch, with Shh signaling becoming undetectable in the MEE following palatal fusion. Our data provide a clear explanation for the lack of a requirement for Shh signaling in the MEE during palatal fusion, based on normal palatal fusion after specific loss-of-function of Shh signaling in the palatal epithelium. Our study highlights the importance of precise temporal control of Shh signaling in the palatal epithelium to ensure palatal fusion, suggesting that turning off the downstream target genes of Shh signaling is required in the MEE during palatogenesis. From a developmental perspective, our discovery is consistent with the broader notion that tightly controlled Shh signaling is critical during embryogenesis, because Shh signaling regulates cell differentiation and organ formation in a concentration-dependent manner (Pasca di Magliano and Hebrok, 2003).
In humans, p63 mutations cause autosomal dominant ectodermal dysplasias, including ectrodactyly-ectodermal dysplasia-clefting (EEC) and ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndromes. Both of these syndromes are associated with cleft lip and/or cleft palate (CL/P) and defects in ectodermal tissues including the epidermis, hair, teeth, and glands (Rinne et al., 2007; van Bokhoven et al., 2001). Ectodermal abnormalities including CL/P are also observed subsequent to mutations in interferon regulatory factor 6 (IRF6), which causes Van der Woude syndrome and popliteal pterygium syndrome (Kondo et al., 2002). Considering that IRF6 and p63 are expressed in ectoderm-derived epithelia, and that there are obvious similarities between phenotypes observed in humans and mice with mutations in these two genes, several studies investigated the genetic interaction between Irf6 and p63. It has been determined that this interaction allows epithelial cells to exit the cell cycle during fusion processes: ΔNp63 directly activates Irf6 transcription, which in turn promotes the downregulation of ΔNp63, establishing a cell-autonomous negative regulatory loop (Moretti et al., 2010; Thomason et al., 2010). However, in our study, we found that constitutive activation of Hh signaling in the palatal epithelium results in high levels of both ΔNp63 and Irf6 expression, which is not consistent with a negative feedback loop between p63 and Irf6. We propose that constitutive activation of Hh signaling in the palatal epithelium results in the failure of Irf6-induced downregulation of p63 in the MEE. Upregulated Irf6 expression may be due to the combination of normal Tgf-β signaling and abnormal maintenance of p63 expression in the MEE after constitutive activation of Hh signaling in the palatal epithelium, because Irf6 can be directly regulated by TGF-β pathway as well as by ΔNp63 (Iwata et al., 2013; Thomason et al., 2010). Therefore, our study suggests that constitutive activation of Hh signaling in the palatal epithelium affects the regulatory loop between p63 and Irf6, causing the failure of p63/Irf6-dependent cell cycle exit, which may be part of the reason for the persistence of the MEE after constitutive activation of Hh signaling in the palatal epithelium (see Figure 7). In addition, previous studies have demonstrated that ΔNp63 activates transcription of Irf6, which, in turn, induces proteasome-mediated ΔNp63 degradation in keratinocytes (Moretti et al., 2010). Moreover, microRNAs also play critical roles during palatal fusion. For example, overexpression of miR-200b results in a failure of palatal fusion due to persistence of the MEE (Shin et al., 2012a; Shin et al., 2012b), and both miR-203 and members of the miR-302/367 cluster target isoforms of p63, loss of which leads to cleft lip and palate (Warner et al., 2014). Therefore, it would be very interesting to investigate whether constitutive activation of Hh signaling in the palatal epithelium affects the p63/Irf6 feedback loop through post-transcriptional regulation of p63, such as through ubiquitination or microRNAs, in our future work.
Figure 7. Schematic diagram of the mechanism underlying the persistence of the MEE in K14-Cre;R26SmoM2 mice.

Constitutive activation of Hh signaling in the palatal epithelium affects the regulatory loop between p63 and Irf6, causing the failure of p63/Irf6-dependent cell cycle exit, which may result in the persistence of the MEE in K14-Cre;R26SmoM2 mice. Maintenance of p63 expression in SmoM2 mutant MEE may prevent apoptosis in MEE cells through enhanced cell-cell and cell-matrix adhesion, which may also contribute to the persistence of the MEE in K14-Cre;R26SmoM2 mice.
Integrins promote cell attachment to the ECM, which is important for the structural organization of tissues as well as for the proliferation, migration, differentiation, and survival of epithelial cells. When epithelial cells physically detach from the ECM, apoptosis or anoikis typically occurs (Frisch and Francis, 1994; Frisch and Ruoslahti, 1997; Reginato et al., 2003). This is true of the MEE cells in the palatal epithelium, which partly explains the disappearance of the MEE during palatal fusion (Kim et al., 2015). Previous studies have demonstrated that p63 is a key regulator of cellular adhesion and survival in epithelial cells, protecting cells from detachment-induced apoptosis (Carroll et al., 2006). It has been demonstrated both in vivo and in vitro that gain or loss of p63 function can directly or indirectly affect an array of genes involved in cell-cell adhesion (e.g., desmosome genes) and cell-matrix adhesion (e.g., genes for integrins and ECM components) (Carroll et al., 2006; Richardson et al., 2017). Increased expression of p63 in epithelial cells in vitro has been shown to reverse the effect induced by knockdown of endogenous p63, upregulating genes encoding key cell adhesion molecules and thereby protecting cells from apoptosis following detachment by increasing cellular adhesion to ECM through enhanced integrin signaling (Carroll et al., 2006). In our study, we found that constitutive activation of Hh signaling in the palatal epithelium alters the fate of MEE cells. Their failure to switch to apoptosis results in persistence of the MEE and ultimately in submucous cleft palate. Furthermore, we revealed that the maintenance of p63 expression in the MEE may be caused by dysfunction of a p63/Irf6 regulatory loop, which in turn upregulates multiple key cell adhesion-related genes in the MEE. Based on the well-documented functions of p63 in regulating epithelial cell adhesion and survival, we postulate that the maintenance of p63 expression after constitutive activation of Hh signaling in the palatal epithelium may confer MEE cells with resistance to apoptosis through enhanced cell-cell and cell-matrix adhesion, which may be part of the reason for the persistence of the MEE (see Figure 7). Although crosstalk between Hh and Tgf-β signaling is critical during development (Guo and Wang, 2009), our results suggest that the maintenance of p63 expression in the MEE is induced by different mechanisms in K14-Cre;Tgfbr2fl/fl and K14-Cre;R26SmoM2 mice. However, we found that these two models share multiple cellular and molecular characteristics, including the persistence of the MEE with an absence of apoptosis and strengthening of the cell adhesion network in the MEE. Therefore, we further propose that enhancement of this cell adhesion network, induced by the maintenance of p63 expression, may serve as the common mechanism responsible for the survival of MEE cells in submucous cleft palate-associated animal models with a persistent MEE. From a clinical perspective, our study may help illuminate the etiology of submucous cleft palate with a persistence of the MEE and suggest targets for future therapeutic strategies.
Using palatal shelves as a tissue fusion model, previous studies have demonstrated that actomyosin-driven cell convergence and extrusion are required for normal fusion of the mammalian secondary palate, and loss of function of the actomyosin contractility pathway results in persistence of the MEE during palatal fusion (Kim et al., 2015). Given the common phenotype of a persistent MEE during palatal fusion in mouse models with dysregulation of the Tgf-β, Bmp, and in our case, Hh signaling pathways in palatal epithelium, it would be very interesting to study whether these pathways control MEE cell fate during palatal fusion through the regulation of actomyosin activity. In our study, we failed to detect any change in non-muscle myosin II (NMII)-mediated actomyosin activity in SmoM2 mutant MEE, indicating that overactivation of Hh signaling in the palatal epithelium does not affect actomyosin-driven cell movement in the midline epithelium during palatal fusion. However, future work may provide additional understanding of this process.
Still another important question needs to be answered: what molecule or signaling pathway exerts temporal control over Shh signaling during palatogenesis, precisely inducing the downregulation of Shh signaling in the MEE right after the palatal shelves touch? Given the spatially and temporally complementary expression patterns of Tgf-β3 and Gli1, and considering the similarities in the MEE defects between animal models with loss-of-function of Tgf-β and gain-of-function of Hh signaling in palatal epithelium, we initially suspected that Tgf-β signaling may function upstream of Shh signaling and promote its downregulation in the MEE during palatal fusion. However, Shh signaling did not appear to be upregulated in the MEE following blockage of Tgf-β signaling in the palatal epithelium, indicating that some other molecule or signaling pathway may serve as a repressor upstream of Shh signaling in the MEE to ensure proper palatal fusion. We will pursue investigation of this upstream mechanism in future work.
In summary, we utilized a mouse model with gain-of-function of Hh signaling to investigate the biological function of Shh signaling in the palatal epithelium during palatogenesis. We found that constitutive activation of Hh signaling in the palatal epithelium results in submucous cleft palate with persistence of the MEE, providing a unique animal model for investigating the etiology and pathogenesis of submucous cleft palate in humans, which shares many similar features. We further demonstrated that downregulation of Shh signaling is required in the MEE during a specific temporal window in palatogenesis. Upregulation of Hh signaling in the palatal epithelium affects the fate of MEE cells, maintaining their proliferation when they should instead undergo apoptosis. We suggest this is due to dysfunction of the p63/Irf6 regulatory loop, and the observed resistance to apoptosis is likely induced by enhancement of the cell adhesion network through the maintenance of p63 expression. Our study provides new insights into the functional significance of tightly controlled Shh signaling during organogenesis and might also offer therapeutic targets that could potentially be manipulated to benefit individuals with altered Shh signaling and submucous cleft palate.
Supplementary Material
Figure S1. Analysis of Gli1 expression in heads of E14.5 K14-Cre;R26SmoM2;Gli1-LacZ mice
(A-B) X-gal staining (blue) in frontal sections of E14.5 R26SmoM2;Gli1-LacZ (control) and K14-Cre;R26SmoM2;Gli1-LacZ heads. Boxed areas in (A) and (B) are shown magnified in (A’, A”) and (B’, B”), respectively. DE, dental epithelium; OC, oral cavity; OE, oral epithelium; S, skin; T, tongue; TE, tongue epithelium. Scale bars (A-B): 200 μm; (A’, A”, B’, B”): 50 μm.
{kind=link}
Figure S2. Analysis of Gli1 expression in E14.5 K14-Cre;Tgfbr2fl/fl palatal shelves
(A-B) RNAscope in situ hybridization of Gli1 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;Tgfbr2fl/fl palatal shelves. Arrowheads indicate absence of Gli1 expression. Black dotted lines indicate the location of the MEE. Scale bars: 50 μm.
{kind=link}
Figure S3. Analysis of Sox2 expression in K14-Cre;R26SmoM2 palatal shelves
(A-D) Immunofluorescence of Sox2 (green, indicated by arrows) in frontal sections of control and K14-Cre;R26SmoM2 palatal shelves at E14.5 (A-B) and newborn (NB; C-D) stage. Arrowheads indicate absence of Sox2 expression. White dotted lines indicate the location of the MEE. Scale bars: 50 μm.
{kind=link}
Figure S4. Exogenous TGFβ3 protein treatment fails to induce the complete fusion of SmoM2 mutant palatal shelves in vitro
(A-C) H&E staining of E13.5 palate explants from control and K14-Cre;R26SmoM2 (mutant) mice treated with TGFβ3 protein (C) or BSA (A, B) for 3 days. Arrows indicate the persistence of the MEE, whereas arrowhead indicates the disappearance of the MEE. (D-G) Immunofluorescence of ΔNp63 (p63, red) and non-muscle myosin heavy chain IIA (NMHCIIA, red) in E13.5 palate explants from K14-Cre;R26SmoM2 (mutant) mice treated with TGFβ3 protein (E, G) or BSA (D, F) for 3 days. Arrows indicate positive signal in the MEE. Scale bars: 50 μm.
{kind=link}
Acknowledgements
We thank Sarah E. Millar for K14-Cre mice, Andrew McMahon for R26SmoM2 mice, Harold L. Moses for Tgfbr2fl/fl mice, and Bridget Samuels and Julie Mayo for critical reading of the manuscript.
This work was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health, USA (R37 DE012711 and U01 DE024421 to Yang Chai).
References
- Andl T, Ahn K, Kairo A, Chu EY, Wine-Lee L, Reddy ST, Croft NJ, Cebra-Thomas JA, Metzger D, Chambon P, Lyons KM, Mishina Y, Seykora JT, Crenshaw EB 3rd, Millar SE, 2004. Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development (Cambridge, England) 131, 2257–2268. [DOI] [PubMed] [Google Scholar]
- Baek JA, Lan Y, Liu H, Maltby KM, Mishina Y, Jiang R, 2011. Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Developmental biology 350, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL, 2002. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development (Cambridge, England) 129, 4753–4761. [DOI] [PubMed] [Google Scholar]
- Brown NL, Yarram SJ, Mansell JP, Sandy JR, 2002. Matrix metalloproteinases have a role in palatogenesis. Journal of dental research 81, 826–830. [DOI] [PubMed] [Google Scholar]
- Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, Brugge JS, Ellisen LW, 2006. p63 regulates an adhesion programme and cell survival in epithelial cells. Nature cell biology 8, 551–561. [DOI] [PubMed] [Google Scholar]
- Celardo I, Antonov A, Amelio I, Annicchiarico-Petruzzelli M, Melino G, 2014. p63 transcriptionally regulates the expression of matrix metallopeptidase 13. Oncotarget 5, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari NS, Romano RA, Koster MI, Jaks V, Roop D, Flores ER, Teglund S, Sinha S, Gruber W, Aberger F, Medeiros LJ, Toftgard R, McDonnell TJ, 2013. Interaction between the TP63 and SHH pathways is an important determinant of epidermal homeostasis. Cell death and differentiation 20, 1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo R, Covarrubias L, 2004. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development (Cambridge, England) 131, 15–24. [DOI] [PubMed] [Google Scholar]
- Ferone G, Mollo MR, Thomason HA, Antonini D, Zhou H, Ambrosio R, De Rosa L, Salvatore D, Getsios S, van Bokhoven H, Dixon J, Missero C, 2013. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum Mol Genet 22, 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Francis H, 1994. Disruption of epithelial cell-matrix interactions induces apoptosis. The Journal of cell biology 124, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Ruoslahti E, 1997. Integrins and anoikis. Current opinion in cell biology 9, 701–706. [DOI] [PubMed] [Google Scholar]
- Guo X, Wang XF, 2009. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell research 19, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Mayo J, Xu X, Li J, Bringas P Jr., Maas RL, Rubenstein JL, Chai Y, 2009. Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development (Cambridge, England) 136, 4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP, 2001. Hedgehog signaling in animal development: paradigms and principles. Genes & development 15, 3059–3087. [DOI] [PubMed] [Google Scholar]
- Ito Y, Yeo JY, Chytil A, Han J, Bringas P Jr., Nakajima A, Shuler CF, Moses HL, Chai Y, 2003. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development (Cambridge, England) 130, 5269–5280. [DOI] [PubMed] [Google Scholar]
- Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Urata M, Dixon MJ, Chai Y, 2013. Smad4-Irf6 genetic interaction and TGFbeta-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development (Cambridge, England) 140, 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP, 2004. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes & development 18, 937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serreze DV, Weinmaster G, Gridley T, 1998. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes & development 12, 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lewis AE, Singh V, Ma X, Adelstein R, Bush JO, 2015. Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS biology 13, e1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC, 2002. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nature genetics 32, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosowski TR, Weathers WM, Wolfswinkel EM, Ridgway EB, 2012. Cleft palate. Seminars in plastic surgery 26, 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka H, Iulianella A, Williams T, Trainor PA, 2014. Disrupting hedgehog and WNT signaling interactions promotes cleft lip pathogenesis. The Journal of clinical investigation 124, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Jiang R, 2009. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development (Cambridge, England) 136, 1387–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Xu J, Jiang R, 2015. Cellular and Molecular Mechanisms of Palatogenesis. Current topics in developmental biology 115, 59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J, Yumoto K, Pisano J, Azhar M, Thomas PS, Kaartinen V, 2014. Control elements targeting Tgfb3 expression to the palatal epithelium are located intergenically and in introns of the upstream Ift43 gene. Frontiers in physiology 5, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S, 2002. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood 100, 560–568. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ, 2003. Developmental roles and clinical significance of hedgehog signaling. Current topics in developmental biology 53, 1–114. [DOI] [PubMed] [Google Scholar]
- Moretti F, Marinari B, Lo Iacono N, Botti E, Giunta A, Spallone G, Garaffo G, Vernersson-Lindahl E, Merlo G, Mills AA, Ballaro C, Alema S, Chimenti S, Guerrini L, Costanzo A, 2010. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. The Journal of clinical investigation 120, 1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, de Die-Smulders C, Giannotti A, Imaizumi K, Jones KL, Del Campo M, Martin RA, Meinecke P, Pierpont MEM, Robin NH, Young ID, Roessler E, Muenke M, 1999. The Mutational Spectrum of the Sonic Hedgehog Gene in Holoprosencephaly: SHH Mutations Cause a Significant Proportion of Autosomal Dominant Holoprosencephaly. Human Molecular Genetics 8, 2479–2488. [DOI] [PubMed] [Google Scholar]
- Noda K, Mishina Y, Komatsu Y, 2016. Constitutively active mutation of ACVR1 in oral epithelium causes submucous cleft palate in mice. Developmental biology 415, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, Werb Z, 2007. Matrix metalloproteinases and the regulation of tissue remodelling. Nature reviews. Molecular cell biology 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano M, Hebrok M, 2003. Hedgehog signalling in cancer formation and maintenance. Nature reviews. Cancer 3, 903–911. [DOI] [PubMed] [Google Scholar]
- Pauws E, Hoshino A, Bentley L, Prajapati S, Keller C, Hammond P, Martinez-Barbera JP, Moore GE, Stanier P, 2009. Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum Mol Genet 18, 4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray HJ, Niswander L, 2012. Mechanisms of tissue fusion during development. Development (Cambridge, England) 139, 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS, 2003. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nature cell biology 5, 733–740. [DOI] [PubMed] [Google Scholar]
- Reiter R, Brosch S, Ludeke M, Fischbein E, Haase S, Pickhard A, Assum G, Schwandt A, Vogel W, Hogel J, Maier C, 2012. Genetic and environmental risk factors for submucous cleft palate. European journal of oral sciences 120, 97–103. [DOI] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP, 2004. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. The Journal of clinical investigation 113, 1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R, Mitchell K, Hammond NL, Mollo MR, Kouwenhoven EN, Wyatt ND, Donaldson IJ, Zeef L, Burgis T, Blance R, van Heeringen SJ, 2017. p63 exerts spatio-temporal control of palatal epithelial cell fate to prevent cleft palate. PLoS Genet 13, e1006828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, Marazita ML, Murray JC, 2007. Impaired FGF signaling contributes to cleft lip and palate. Proceedings of the National Academy of Sciences of the United States of America 104, 4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne T, Brunner HG, van Bokhoven H, 2007. p63-associated disorders. Cell cycle (Georgetown, Tex.) 6, 262–268. [DOI] [PubMed] [Google Scholar]
- Roessler E, Muenke M, 2003. How a Hedgehog might see holoprosencephaly. Hum Mol Genet 12 Spec No 1, R15–25. [DOI] [PubMed] [Google Scholar]
- Sasaki R, Saito K, Watanabe Y, Takayama Y, Fujii K, Agawa K, Miyashita T, Ando T, Akizuki T, 2009. Nevoid basal cell carcinoma syndrome with cleft lip and palate associated with the novel PTCH gene mutations. Journal Of Human Genetics 54, 398. [DOI] [PubMed] [Google Scholar]
- Schutte BC, Murray JC, 1999. The Many Faces and Factors of Orofacial Clefts. Human Molecular Genetics 8, 1853–1859. [DOI] [PubMed] [Google Scholar]
- Shin JO, Lee JM, Cho KW, Kwak S, Kwon HJ, Lee MJ, Cho SW, Kim KS, Jung HS, 2012a. MiR-200b is involved in Tgf-beta signaling to regulate mammalian palate development. Histochemistry and cell biology 137, 67–78. [DOI] [PubMed] [Google Scholar]
- Shin JO, Nakagawa E, Kim EJ, Cho KW, Lee JM, Cho SW, Jung HS, 2012b. miR-200b regulates cell migration via Zeb family during mouse palate development. Histochemistry and cell biology 137, 459–470. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z, 2001. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17, 463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason HA, Zhou H, Kouwenhoven EN, Dotto GP, Restivo G, Nguyen BC, Little H, Dixon MJ, van Bokhoven H, Dixon J, 2010. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. The Journal of clinical investigation 120, 1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG, 2001. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. American journal of human genetics 69, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DR, Mukhopadhyay P, Brock G, Webb CL, Michele Pisano M, Greene RM, 2014. MicroRNA expression profiling of the developing murine upper lip. Development, growth & differentiation 56, 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P Jr., Urata MM, Chai Y, 2006. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Developmental biology 297, 238–248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of Gli1 expression in heads of E14.5 K14-Cre;R26SmoM2;Gli1-LacZ mice
(A-B) X-gal staining (blue) in frontal sections of E14.5 R26SmoM2;Gli1-LacZ (control) and K14-Cre;R26SmoM2;Gli1-LacZ heads. Boxed areas in (A) and (B) are shown magnified in (A’, A”) and (B’, B”), respectively. DE, dental epithelium; OC, oral cavity; OE, oral epithelium; S, skin; T, tongue; TE, tongue epithelium. Scale bars (A-B): 200 μm; (A’, A”, B’, B”): 50 μm.
Figure S2. Analysis of Gli1 expression in E14.5 K14-Cre;Tgfbr2fl/fl palatal shelves
(A-B) RNAscope in situ hybridization of Gli1 (red dots, indicated by arrows) in frontal sections of E14.5 control and K14-Cre;Tgfbr2fl/fl palatal shelves. Arrowheads indicate absence of Gli1 expression. Black dotted lines indicate the location of the MEE. Scale bars: 50 μm.
Figure S3. Analysis of Sox2 expression in K14-Cre;R26SmoM2 palatal shelves
(A-D) Immunofluorescence of Sox2 (green, indicated by arrows) in frontal sections of control and K14-Cre;R26SmoM2 palatal shelves at E14.5 (A-B) and newborn (NB; C-D) stage. Arrowheads indicate absence of Sox2 expression. White dotted lines indicate the location of the MEE. Scale bars: 50 μm.
Figure S4. Exogenous TGFβ3 protein treatment fails to induce the complete fusion of SmoM2 mutant palatal shelves in vitro
(A-C) H&E staining of E13.5 palate explants from control and K14-Cre;R26SmoM2 (mutant) mice treated with TGFβ3 protein (C) or BSA (A, B) for 3 days. Arrows indicate the persistence of the MEE, whereas arrowhead indicates the disappearance of the MEE. (D-G) Immunofluorescence of ΔNp63 (p63, red) and non-muscle myosin heavy chain IIA (NMHCIIA, red) in E13.5 palate explants from K14-Cre;R26SmoM2 (mutant) mice treated with TGFβ3 protein (E, G) or BSA (D, F) for 3 days. Arrows indicate positive signal in the MEE. Scale bars: 50 μm.