

SUMMARY
Neuromodulation imposes powerful control over brain function, and cAMP-dependent protein kinase (PKA) is a central downstream mediator of multiple neuromodulators. Although genetically-encoded PKA sensors have been developed, single-cell imaging of PKA activity in living mice has not been established. Here, we used two-photon fluorescence lifetime imaging microscopy (2pFLIM) to visualize genetically-encoded PKA sensors in response to the neuromodulators norepinephrine and dopamine. We screened available PKA sensors for 2pFLIM and further developed a variant (named tAKARα) with increased sensitivity and a broadened dynamic range. This sensor allowed detection of PKA activation by norepinephrine at physiologically-relevant concentrations and kinetics and by optogenetically released dopamine. In vivo longitudinal 2pFLIM imaging of tAKARα tracked bidirectional PKA activities in individual neurons in awake mice, and revealed neuromodulatory PKA events that were associated with wakefulness, pharmacological manipulation, and locomotion. This new sensor combined with 2pFLIM will enable interrogation of neuromodulation-induced PKA signaling in awake animals.
ETOC BLURB
It remains challenging to visualize neuromodulation, a major mode of neuronal communication, in vivo. Ma et al. demonstrate that two-photon fluorescence lifetime imaging of an improved genetically-encoded sensor enables monitoring of neuromodulatory PKA signaling with single-cell resolution in behaving mice.
INTRODUCTION
Neuromodulation plays a critical role in many aspects of animal behavior, including arousal, stress, attention, and reward (Aston-Jones and Cohen, 2005; Greengard, 2001; Johansen et al., 2011; Petersen and Posner, 2012; Schultz, 2006; Sun et al., 2015). At the cellular level, many neuromodulators activate G protein-coupled receptors to trigger intracellular signaling events, thereby imposing powerful control over the excitability and plasticity of individual neurons. Despite their important functions, it remains challenging to monitor neuromodulators and their downstream signaling pathways with single-cell or subcellular resolution, especially in vivo.
Microdialysis (Zhang and Beyer, 2006) and fast-scan cyclic voltammetry (FSCV) (Rodeberg et al., 2017) have been applied in vivo to measure the extracellular concentrations of certain neuromodulators; but analysis at the single neuron level is not possible. At the same time, individual neurons may express distinct subtypes or levels of neuromodulator receptors and exhibit unique intracellular signaling dynamics in response to the same extracellular concentration of a neuromodulator. Monitoring subcellular signaling events on a cell-by-cell basis is essential for dissecting the function of neuromodulation in animal behavior.
Several key neuromodulators, including norepinephrine (NE), dopamine, serotonin, and acetylcholine, converge onto the intracellular cAMP/protein kinase A (PKA) pathway via activation of their corresponding G-protein coupled receptors (Beaulieu and Gainetdinov, 2011; Chen et al., 2017; Francis and Corbin, 1994; Greengard, 2001; Millan et al., 2008; Pierce et al., 2002). In turn, PKA regulates diverse functions in neurons, including gene transcription, protein trafficking, protein degradation, neuronal excitability, and synaptic plasticity (Brandon et al., 1997; Choi et al., 2002; Frey et al., 1993; Goldsmith and Abrams, 1992; Greenberg et al., 1987; Impey and Goodman, 2001; Kwon and Sabatini, 2011; Pedarzani and Storm, 1993; Weisskopf et al., 1994; Yasuda et al., 2003). Monitoring the activity of the PKA pathway in individual neurons, therefore, may serve as readout for neuromodulatory events in the same way that calcium imaging is used to report neuronal electrical activity.
A genetically-encoded sensor for PKA activity, namely A-kinase activity reporter (AKAR) that is based on Förster resonance energy transfer (FRET) was introduced in 2001 (Zhang et al., 2001), followed by a series of improved variants (Allen and Zhang, 2006; Depry et al., 2011; Komatsu et al., 2011; Zhang et al., 2005). The early sensors were developed for two-color ratiometric imaging methods using conventional microscopes. Recently, the use of two-photon fluorescence lifetime imaging microscopy (2pFLIM) has offered advantages for high-resolution imaging of FRET in light-scattering brain tissue (e.g., Figure S1) (Yasuda, 2006; Yasuda et al., 2006; Yellen and Mongeon, 2015). Several AKAR variants for 2pFLIM have also been generated (Chen et al., 2014; Tang and Yasuda, 2017; Tillo et al., 2017); however, it remains unknown how these recently developed sensors perform relative to one another. Moreover, although AKAR sensors have been used in vitro (e.g., (Chen et al., 2017; Dodge-Kafka et al., 2005; Dunn et al., 2006; Tang and Yasuda, 2017; Tillo et al., 2017; Yapo et al., 2017)), and in a few in vivo photometric experiments (Goto et al., 2015; Yamaguchi et al., 2015), there has been no report to date of successful imaging of PKA activity in individual neurons in awake mice.
Here, we compared several recently developed 2pFLIM AKARs in response to norepinephrine in cultured hippocampal slices. We further developed an AKAR variant, named targeted AKAR α (tAKARα), which has a 2.7-fold greater response amplitude to norepinephrine, by targeting the best available sensors to subcellular components where PKA is enriched (Scott and Pawson, 2009; Zhong et al., 2009). tAKARα reliably detected norepinephrine at concentrations as low as 10 nM with a sensitivity (K1/2 ~ 20 nM) and kinetics in alignment with noradrenergic regulation of the slow afterhyperpolarization (sAHP) in CA1 neurons (Madison and Nicoll, 1986a, b). tAKARα also readily detected optogenetically released endogenous dopamine in the striatum. Using this sensor, we found that PKA responses were cell type-specific in both the hippocampus and cortex. By integrating 2pFLIM capability with in vivo two-photon microscopy, we were able to longitudinally image bidirectional PKA activity in awake, head-fixed mice with single-neuron resolution. We found that cortical neurons exhibited wakefulness-dependent PKA activity associated with the noradrenergic pathway. Enforced locomotion elicited PKA activity in a cell-specific and brain region-specific manner. Overall, this study paves the way for interrogations of cellular responses to neuromodulation in vivo. In addition, the strategy of sensor localization may be employed to improve other signaling indicators.
RESULTS
FLIM-AKAR exhibits the largest response amplitude to norepinephrine among available sensors
2pFLIM measures the fluorescence lifetime of the donor fluorophore of a FRET pair and has advantages for imaging in brain tissue over conventional FRET measurements, such as the ratiometric method (i.e., comparing the relative fluorescence of the donor and acceptor of a FRET pair). For example, the fluorescence ratio of a single fluorophore split into two color channels changes when imaging deeper into brain tissue due to wavelength-dependent light scattering (Figure S1). In contrast, because 2pFLIM involves only one color channel, measurements are stable at different depths (Figure S1).
Several variants of AKARs for 2pFLIM have been reported, namely AKAR5, AKAR5’, FLIM-AKAR, and AKARet (Chen et al., 2014; Tang and Yasuda, 2017; Tillo et al., 2017). These sensors all share similar overall domain designs (Figure 1A) and all use EGFP as the donor fluorophore (Figure 1B). When the sensor is phosphorylated by PKA, the forkhead associated domain 1 (FHA1) binds to the phosphorylated substrate, bringing EGFP close to the acceptor fluorophore such that FRET increases and the lifetime of EGFP decreases. These AKAR variants differ in terms of their acceptor fluorophores (sREACh, a low-irradiant yellow fluorescent protein (Murakoshi et al., 2008), its circular-permutated variant (Chen et al., 2014; Tillo et al., 2017), or sREAChet, a sREACh variant with enhanced inter-fluorophore affinity (Tang and Yasuda, 2017)) and in terms of the linkers connecting the different functional sub-domains (Figure 1B). To compare these sensors, they were individually expressed along with a cytosolic marker, mCherry, in organotypic hippocampal slice cultures via biolistic transfection. 2pFLIM was then used to image the sensors in the apical dendrites of CA1 neurons in response to bath-applied norepinephrine (1 μM), followed by a mixture of the adenylate cyclase agonist forskolin (25 μM) and the phosphodiesterase inhibitor IBMX (50 μM). While we focused on the sensor response to the physiological neuromodulator norepinephrine, treatment with forskolin/IBMX (F + I) revealed the maximal response.
Figure 1. Schematics and comparisons of available AKARs.
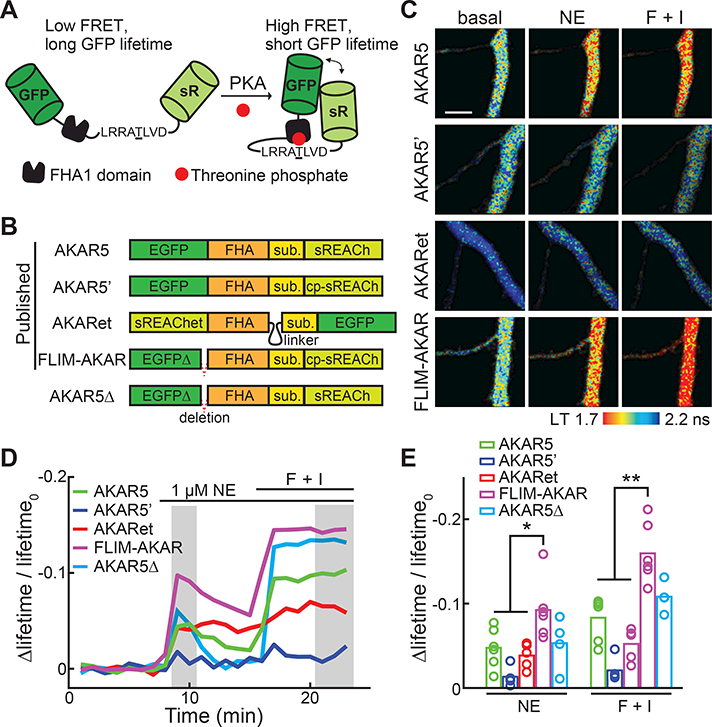
(A) Schematic of the general design of AKARs. sR: sREACh. FHA1: the forkhead associated domain 1 of the Rad53p protein. This panel is a modification from the original schematic published in (Zhang et al., 2001).
(B) Schematics of available 2pFLIM AKAR sensors illustrating their domain structures and differences. Sub: PKA substrate sequence.
(C) Representative lifetime images of current 2pFLIM AKAR sensors in the apical dendrites of CA1 neurons at rest, after stimulation with 1 μM norepinephrine (NE), followed by 25 μM forskolin/50 μM IBMX (F+I). All images are pseudo-colored according to the same lifetime scale to show the differences in baseline and response amplitudes across sensors.
(D, E) Representative timecourse traces (D) and amplitudes (E) of Δlifetime/lifetime0 of the indicated AKAR sensors. n = 6 neurons for AKAR5 and FLIM-AKAR; 5 for AKAR5’ and AKARet; and 4 for AKAR5Δ.
Error bars are s.e.m.
See also Figure S1 and S2.
Each sensor exhibited different resting fluorescence lifetimes and response amplitudes to the stimuli (Figures 1C and S2). To compare them quantitatively, we normalized the lifetime response amplitudes to their corresponding baselines (i.e., Δlifetime/lifetime0). This value is inversely related to the square root of the minimal number of photons required to achieve a certain signal-to-noise ratio (see STAR METHODS). Thus, a two-fold increase in Δlifetime/lifetime0 translates to a four-fold decrease in the required photon counts. For all current 2pFLIM sensors, norepinephrine triggered approximately 60% of the respective maximal responses induced by forskolin/IBMX (Figure 1D and 1E). Among these sensors, FLIM-AKAR exhibited the largest response (Δlifetime/lifetime0 = −0.092 ± 0.014 for norepinephrine; and −0.160 ± 0.014 for forskolin/IBMX), which was nearly two-fold as large as the next best sensor AKAR5 (Δlifetime/lifetime0, ne = −0.048 ± 0.010, p < 0.05 c.f. FLIM-AKAR; Figure 1E).
FLIM-AKAR differs from AKAR5’ (the least sensitive sensor) by only an 11-residue deletion at the C terminus of EGFP (Figure 1A). However, the same deletion introduced into AKAR5 (construct named AKAR5Δ; Figure 1B) did not increase its signal amplitudes (Δlifetime/lifetime0, ne = −0.053 ± 0.016 for AKAR5Δ, p > 0.1 c.f. AKAR5). As these 11 residues likely function to link EGFP to the remaining sensor, these observations are consistent with the notion that linkers in a sensor play a highly sensitive role in determining critical response characteristics (Hires et al., 2008).
Targeting FLIM-AKAR to microtubules greatly enhances response amplitude
For FLIM-AKAR, a minimum of 120 photons are required to achieve a signal-to-noise ratio of 1 at saturating concentrations of norepinephrine (see STAR METHODS). A larger response amplitude would reduce the number of required photons while also making it easier to detect sub-saturating concentrations of norepinephrine, or to make repeated measurements within the challenging in vivo imaging environment. A hallmark of PKA signaling in neurons is that most PKA is anchored to specific subcellular locations at rest (Scott and Pawson, 2009; Theurkauf and Vallee, 1982). Once activated, a fraction of the catalytic subunit of PKA is liberated and translocates to the membrane (Tillo et al., 2017). We hypothesized that, by targeting FLIM-AKAR to the subcellular locations where PKA is enriched, the sensor response could be increased. A series of constructs was generated by fusing FLIM-AKAR with various targeting sequences or proteins (named targeted AKARs, or tAKARs). Each construct was designed for a different subcellular target: microtubule (tAKARα), membrane (tAKARγ), actin (tAKARδ), filamentous actin (tAKARε), or postsynaptic density (tAKARζ) (Figure 2A). To minimize sensor signals in the nucleus, which exhibited properties different from sensor signals in the cytosol (data not shown; see also (Chen et al., 2014)), we also generated a cytosol-only sensor by fusing it to a nuclear export signal (NES; sensor named tAKARß). All of these tAKARs were expressed well in CA1 neurons and exhibited the subcellular distributions expected for their respective targeting motifs (Figure 2B and S3A).
Figure 2. Microtubule-targeting enhances the performance of AKAR sensors.
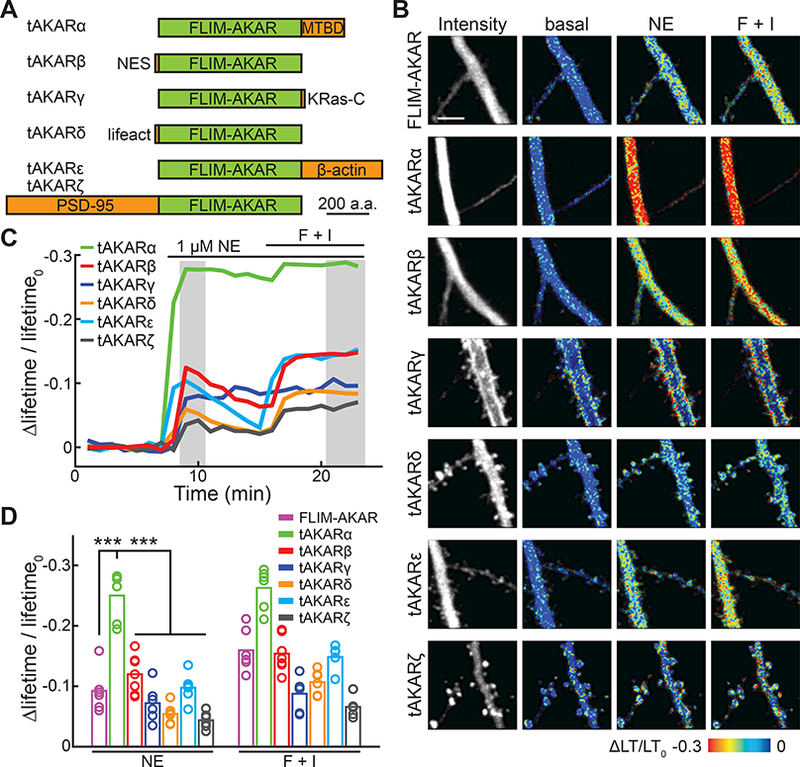
(A) Schematic of tAKAR variants.
(B) Representative basal fluorescence intensity and Δlifetime/lifetime0 images in the apical dendrite of CA1 neurons with the indicated simulations. LT: lifetime.
(C, D) Representative timecourse traces (C) and amplitudes (D) of Δlifetime/lifetime0 of tAKARs. n = 6 – 7 neurons for all sensors.
Error bars are s.e.m.
See also Figure S3.
The Δlifetime/lifetime0 response amplitudes of these tAKARs were compared. Tagging the sensor with NES (i.e., tAKARβ), which did prevent the sensor from entering the nucleus (Figure S3A), did not significantly alter the response amplitudes to norepinephrine or forskolin/IBMX (Figure 2C and 2D). In contrast, targeting to the microtubule (tAKARα) by fusing the sensor to the microtubule binding domain of MAP2c (Hedrick et al., 2016; Zhong et al., 2009) increased the response to norepinephrine by 2.7-fold (Δlifetime/lifetime0 = −0.250 ± 0.017 for tAKARα; p < 0.001, c.f. FLIM-AKAR or any other tAKARs) (Figure 2C and 2D). This increase reflects a combination of increased dynamic range (i.e., the maximal response amplitude; Δlifetime/lifetime0, F + I = −0.263 ± 0.014 for tAKARα; p < 0.001, c.f. FLIM-AKAR or any other tAKARs) and sensitivity (i.e., the saturating norepinephrine response shifting closer to the maximal response; norepinephrine to forskolin/IBMX response ratio = 95% for tAKARα, c.f. 58% for FLIM-AKAR). Notably, targeting AKAR4, a different AKAR designed for ratiometric FRET measurements (Depry et al., 2011), to the microtubule (construct named tAKAR4α), also enhanced the FRET response to norepinephrine by four-fold (ΔRatio/Ratio0, NE = 0.071 ± 0.016 for AKAR4 compared to 0.293 ± 0.051 for tAKAR4α, p < 0.01; Figure S3C, S3D, and S3E), suggesting that the subcellular targeting strategy can be generalized to other PKA sensors.
Targeting FLIM-AKAR to the actin cytoskeleton (tAKARδ and tAKARε) or to the postsynaptic density (tAKARζ) did not lead to larger response amplitudes. In addition, although we recently showed that PKA catalytic subunits will become enriched on the membrane upon activation (Tillo et al., 2017), targeting the sensor to the membrane (tAKARγ) did not result in a larger response amplitude. This may be because the sensor was already activated by basal PKA activity, as suggested by its notably lower lifetime at rest (Figure S3B).
tAKARα detects physiologically relevant PKA signaling events
tAKARα, with its enhanced sensitivity, may become the next generation sensor for visualizing PKA. We therefore examined whether its signal was physiologically relevant. The response of tAKARα to norepinephrine was completely eliminated when the PKA phosphorylation site was mutated (tAKARα-T391A; Figure 3A). Moreover, the response to norepinephrine was reversible (Figure 3B). It was completely blocked by the β-adrenergic receptor antagonist propranolol (1 μM), and was significantly reduced (74% reduction, p < 0.001) with the PKA antagonist H89 (20 μM) (Figure 3B). These results indicate that tAKARα reports PKA phosphorylation events triggered by norepinephrine via β-adrenergic receptors.
Figure 3. tAKARα response reflects physiologically relevant PKA signaling events.
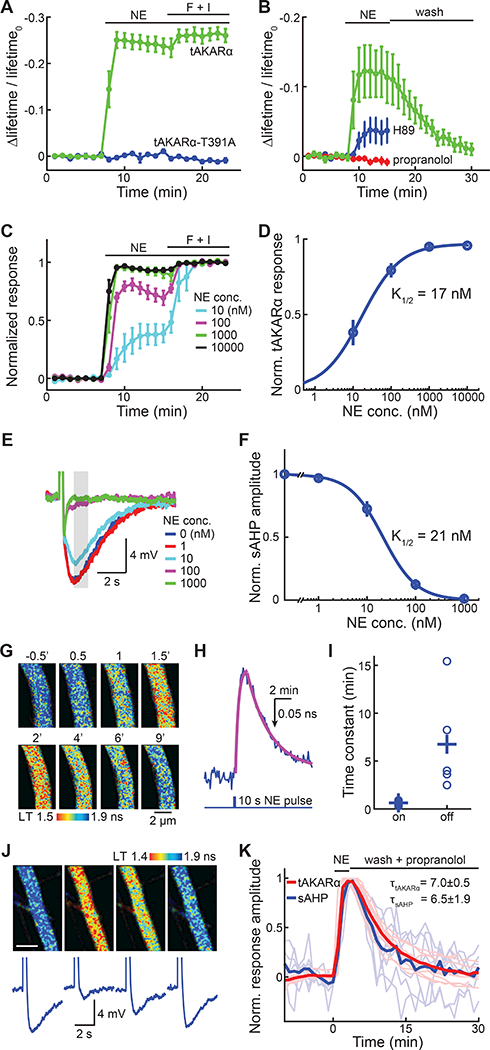
(A) Δlifetime/lifetime0 of tAKARα and its phosphorylation site mutant (tAKARα-T391A) responding to 1 μM norepinephrine (NE) stimulation followed by forskolin/ IBMX (F + I) application.
(B) Δlifetime/lifetime0 of tAKARα in response to 30 nM norepinephrine followed by washout, or in the presence of 20 μM H89 or 1 μM propranolol. n = 4 – 6 neurons.
(C) tAKARα responses to indicated concentrations of norepinephrine normalized to the maximal responses induced by F + I. n = 4 – 6 neurons.
(D) Dose-response curve for tAKARα responding to norepinephrine, fitted by the Hill equation.
(E, F) Representative traces (E) and dose response curve fitted by the Hill equation
(F) of sAHP attenuated by increasing concentrations of norepinephrine. n = 6 neurons.
(G, H, I) Representative images (G), a representative trace and its fitted curves (H), and collective kinetic time constants (I) of tAKARα responding to iontophoresis-released pulses of norepinephrine. n = 5 neurons.
(J, K) Representative tAKARα 2pFLIM images (J, upper panels), representative sAHP traces (J, lower panels), and collective normalized timecourses (K) of neurons that were imaged or recorded in response to a 3-min pulse of bath-applied norepinephrine (0.5 μM) followed by wash in the presence of propranolol. Both individual (thin and lighter colored) and averaged traces are shown. n = 11 neurons for tAKARα and 7 for sAHP.
Error bars are s.e.m.
See also Figure S4.
We next asked whether the sensitivity of tAKARα to norepinephrine was within a physiologically relevant range. First, a dose-response curve of tAKARα was constructed by normalizing the 2pFLIM response to different concentrations of norepinephrine to the maximal response induced by forskolin/IBMX (Figure 3C and 3D) in CA1 neurons. A concentration as low as 10 nM elicited a robust response of tAKARα (normalized response = 0.381 ± 0.085 at 10 nM) (Figure 3C and 3D), and the half maximal response concentration (K1/2) was determined to be 17 ± 10 nM (95% confidence level of fitting). Second, the sensitivity of noradrenergic regulation of a physiological function was measured. Norepinephrine activates PKA via β-adrenergic receptors in CA1 neurons to inhibit sAHP, which in turn influences neuronal excitability (Madison and Nicoll, 1986a, b). Whole-cell current-clamp recordings were used to measure the sAHP in CA1 neurons triggered by a 40 Hz train of 10 back-propagating action potentials. The sAHP was stable under control conditions, but decreased in a dose-dependent manner upon application of increasing concentrations of norepinephrine (Figure 3E and S4A). Fitting the dose-response curve gave a K1/2 value of 21 ± 6 nM (95% confidence level; Figure 3E and 3F), resembling the sensitivity of tAKARα (Figure 3D).
We next examined whether the sensor response kinetics were physiologically relevant by using iontophoresis to apply a short pulse (10-s long) of norepinephrine to CA1 neuronal dendrites expressing tAKARα (Figure 3G). The rising and recovery phases of the sensor response were fit individually with single exponentials (Figure 3H and 3I). A 0.63 ± 0.11 min on kinetics and a 6.75 ± 2.4 min off kinetics were determined. These numbers represented the upper boundary of the sensor kinetics, as they included the time needed for norepinephrine to diffuse in and out of brain slices. To evaluate whether the tAKARα kinetics reflected physiological PKA signaling events, we compared tAKARα and sAHP responses under the same conditions to a short-duration (3 min) bath application of norepinephrine (0.5 μM), followed by washing in the presence of propranolol (10 μM). For both sAHP and tAKARα, the responses were kinetically similar (τoff = 6.5 ± 1.9 for sAHP and 7.0 ± 0.5 for tAKARα; p > 0.1; Figure 3J and 3K).
Importantly, neurons expressing tAKARα did not exhibit altered resting membrane potential (Figure S4B) or altered sAHP amplitude (Figure S4C). The action potential frequency elicited by current injection, another neuronal property known to be regulated by norepinephrine in a cAMP-dependent manner (Madison and Nicoll, 1986a, b), was also unchanged in neurons expressing tAKARα (Figure S4D). In addition, tAKARα expression did not alter the sensitivity of sAHP to a sub-saturating concentration (30 nM) of norepinephrine (Figure S4E). Thus, tAKARα expression does not appear to alter PKA regulation of neuronal function. Overall, these results indicate that tAKARα detects physiologically-relevant PKA activity triggered by norepinephrine activation of β-adrenergic receptors.
tAKARα excels in multiple neuronal subtypes
The above sensor development and characterizations were carried out in CA1 neurons. To test whether tAKARα also performs well in other neuronal subtypes, we compared tAKARα, tAKARβ, and tAKARγ in CA3 neurons in slice cultures. For both tAKARα and tAKARβ, the responses to norepinephrine were much smaller in CA3 neurons compared to CA1 neurons (Δlifetime/lifetime0,NE = −0.087 ± 0.025 for tAKARα, and −0.023 ± 0.017 for tAKARß, p < 0.001 c.f. the corresponding values in CA1 neurons) (Figure S5A), suggesting that norepinephrine triggers lower PKA activation in CA3 neurons. However, the response amplitude of tAKARα remained much greater than that of tAKARβ (3.7 fold; p < 0.001) (Figure 4A and 4B).
Figure 4. tAKARα reveals cell type-specific PKA dynamics.
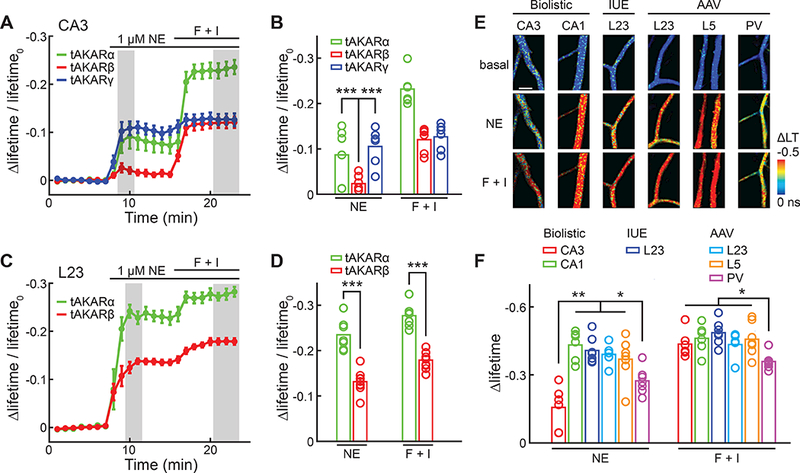
(A, B) The averaged timecourses (A) and amplitudes (B) of Δlifetime/lifetime0 of the indicated tAKARs in the apical dendrites of CA3 neurons in cultured hippocampal slices in response to the indicated pharmarcological manipulations. n = 6 neurons for all sensors.
(C, D) The average timecourses (C) and amplitudes (D) of Δlifetime/lifetime0 of tAKARα and tAKARβ in the apical dendrites of L23 pyramidal neurons in the somatosensory cortex in response to the indicated pharmarcological manipulations in acute brain slices. n = 7 neurons for tAKARα, and 9 for tAKARβ.
(E, F) Representative Δlifetime images (E) and collective quantifications (F) of neurons of the indicated type and transfection methods responding to the indicated simulations. n = 5 – 7 neurons. IUE: in utero electroporation; LT: lifetime.
Error bars are s.e.m.
See also Figure S5.
The response amplitude of tAKARγ to norepinephrine did not decrease in CA3 neurons compared to CA1 neurons (Δlifetime/lifetime0,NE = −0.105 ± 0.017 for tAKARγ in CA3; Figure S5A) and became 4.5-fold larger than that of tAKARβ in CA3 neurons (p < 0.001) (Figure 4A and 4B). This result confirms the earlier notion that in CA1 neurons, where overall PKA activity is higher, tAKARγ may have been largely phosphorylated at rest, resulting in a smaller dynamic range.
Next, tAKARα and tAKARβ were compared in layer 2/3 (L23) pyramidal neurons of the mouse somatosensory cortex in acute brain slices transfected by in utero electroporation. At postnatal day 18 – 22, both sensors were expressed robustly (Figure S5B and S5C). Consistent with the results in hippocampal slice cultures, tAKARα exhibited much larger response amplitudes to norepinephrine compared to tAKARß (Δlifetime/lifetime0, ne = −0.235 ± 0.013 for tAKARα, and −0.131 ± 0.008 for tAKARβ, p < 0.001; Figure 4C and 4D). Overall, these results indicate a much improved sensitivity of tAKARα for detecting norepinephrine across multiple neuronal subtypes.
Different neuronal subtypes exhibit distinct PKA responses to norepinephrine
Genetically-encoded PKA sensors hold the promise of dissecting cell type-specific subcellular response to a given neuromodulatory stimulus. Since norepinephrine induced much smaller PKA responses in CA3 neurons than in CA1 neurons (Figure 3A and 3B), it was of interest to also examine cell type-specific PKA responses in the cortex. Recombinant adeno-associated virus serotype 2/1 (Mao et al., 2011) was injected into the somatosensory cortex of PV-IRES-Cre;Ai9 double heterozygous mice (Hippenmeyer et al., 2005; Madisen et al., 2010) to express tAKARα under the control of the human synapsin promoter (AAV2/1-hSyn-tAKARα) in all neurons. The sensor was expressed well after approximately two weeks (Figure S5D, S5E and S5F). In addition, parvalbumin (PV)-positive interneurons in these mice were readily identified by their expression of the red fluorescent protein tdTomato as a result of the genetic background (Figure S5F).
The PKA responses in L23 and layer 5 (L5) pyramidal neurons and in PV-positive interneurons were measured in acute brain slices. Virally infected L23 neurons and L5 neurons exhibited similar response amplitudes compared to those of L23 neurons transfected by in utero electroporation in acute brain slices, or of CA1 neurons in cultured hippocampal slices (Figure 4E and 4F), suggesting that gene delivery methods do not affect the sensor’s ability to detect PKA dynamics. Interestingly, the response amplitudes of PV-positive interneurons to both norepinephrine and forskolin/IBMX were smaller than those of CA1, L23, and L5 pyramidal neurons (Figure 4F). These results suggest that, while PKA is activated by norepinephrine in all of the examined neuronal subtypes, the activation magnitude may be a cell type-specific property.
tAKARα readily detects PKA activation by dopamine in striatal neurons
Another major neuromodulator, dopamine, also couples to the PKA pathway. We asked whether tAKARα can detect dopamine in striatal neurons. tAKARα was expressed in the mouse striatum via AAV injection and was subsequently imaged in acute brain slices (2 – 4 weeks post injection). Fifty percent of striatal neurons (10 out of 20) responded to bath-applied dopamine (100 μM; Figure 5A, 5B and 5C), consistent with the notion that approximately half of striatal projection neurons express the D1-type dopamine receptor, which activates PKA. When analyzing only the responding cells, the Δlifetime/lifetime0 response of tAKARα was 0.17 ± 0.02 for dopamine and 0.22 ± 0.01 for forskolin/IBMX. The dopamine response was nearly completely abolished by the D1 antagonist SCH23390 (Δlifetime/lifetime0 = 0.03 ± 0.01 after drug; p < 0.001 c.f. paired control; Figure 5D), but not by the D2 dopamine receptor antagonist sulpiride (Δlifetime/lifetime0 = 0.20 ± 0.02 after drug; p = 0.1 c.f. paired control; Figure 5D).
Figure 5. tAKARα readily detects PKA activation by dopamine in striatum.
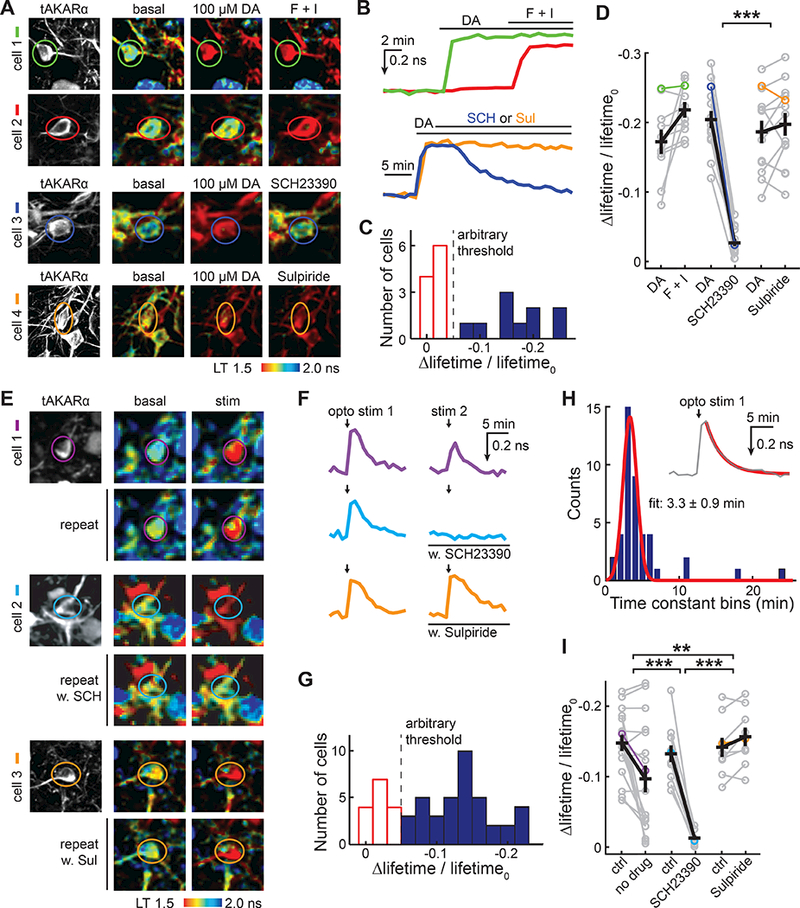
(A, B) Representative images (A) and corresponding color-coded traces (B) of striatal neurons in response to 100 μM dopamine (DA) followed by the application of forskolin/IBMX (F + I, cells 1 – 2), D1-antagonist SCH23390 (10 μM; SCH, cell 3), or D2-antagonist sulpiride (10 μM; Sul, cell 4). n = (neurons/mice, from left to right) = 10/4, 14/4, and 13/3.
(C) Histogram of Δlifetime/lifetime0 response to 100 μM dopamine. An arbitrary Δlifetime/lifetime0 threshold of −0.05 (dashed line) was used to identify dopamine responding neurons (blue) versus non-responders (red).
(D) Quantification of dopamine-responding neurons and their responses to the addition of the indicated reagents. Data points from the representative images in panel A and traces in panel B are indicated using the corresponding colors.
(E, F, G) Representative images (E), corresponding color-coded traces (F), and a histogram of Δlifetime/lifetime0 (G) of striatal neurons in response to local photostimulation of dopaminergic axons in the DAT;Ai32 double heterozygous genetic background, with or without the presence of 10 μM SCH23390 or 10 μM sulpiride. n = (neurons/mice, from left to right) = 17/4, 11/3, and 9/3.
(H) Histogram of time constants during the decaying phase after photostimulation of dopaminergic axons in the DAT;Ai32 double heterozygous background. A single exponential was fitted (orange line) to the decaying phase after the photostimulation response (gray, inset).
(I) Quantification of neurons responding to optogenetically induced dopamine releases with or without the addition of the indicated reagents. Data points from the representative images in panel E and traces in panel F are indicated using the corresponding colors.
Error bars are s.e.m.
We next asked whether tAKARα is sensitive enough to detect endogenous dopamine release using optogenetic experiments. tAKARα-expressing AAV was injected into the dorsomedial striatum of DAT-Cre;Ai32 double heterozygous mice in which channelrhodopsin was expressed in dopaminergic neurons (Backman et al., 2006; Madisen et al., 2012). In acute slices, we stimulated dopaminergic axons in a manner that mimics the phasic activity of dopaminergic axons (Adamantidis et al., 2011; Grace, 1991) by using five trains (one train per second) of light stimuli, with each train consisting of five 1.5-ms pulses at 20 Hz. Robust dopaminergic responses were observed in a fraction of neurons (Δlifetime/lifetime0 = 0.15 ± 0.01 for the responders; Figure 5E, 5F, and 5G). The responses were reversible and exhibited an off kinetics of 3.3 ± 0.9 minutes (Figure 5F and 5H). The on kinetics were faster than a minute, but their precise determination was not possible due to the limitation of our setup, which required a manual switching step for this experiment. For responding neurons, a second stimulus resulted in a repeated but moderately smaller response (Δlifetime/lifetime0 = 0.10 ± 0.02 for the second stimulus; p < 0.001 c.f. the first response; Figure 5F and 5I). The reduced amplitude of the second stimulus was likely due to dopamine-dependent inhibition of dopaminergic axons via D2 dopamine receptors (Ford, 2014), as the reduction was eliminated by application of the D2 dopamine receptor antagonist sulpiride before the second stimulus (Δlifetime/lifetime0 = 0.14 ± 0.01 and 0.16 ± 0.01 for paired measurements before and after sulpiride, respectively). Application of the D1 dopamine receptor antagonist SCH23390 before the second stimulus completely abolished the response (Δlifetime/lifetime0 = 0.13 ± 0.01 and 0.013 ± 0.003 for paired measurements before and after SCH23390, respectively, p<0.001), indicating that the PKA response is mediated by D1 receptors. Together with our earlier results involving norepinephrine, these data demonstrate that tAKARα can detect multiple neuromodulatory pathways that are coupled to PKA.
In vivo imaging of tAKARα reveals tonic PKA activity in cortical neurons of awake mice mediated by β-adrenergic receptors
We asked whether tAKARα is sensitive enough for in vivo imaging of PKA. tAKARα was expressed in cortical neurons using either in utero electroporation or AAV injection, and 2pFLIM imaging was carried out on shallow layers of the barrel cortex through a window implemented via craniotomy (verified by intrinsic imaging; Figure S6) in head-fixed animals. Individual neurons and their dendrites can be clearly resolved (Movie S1 and S2). The same neurons could be repeatedly imaged over weeks (Figure 6A). We found that L23 neurons in awake mice showed significantly shorter EGFP lifetimes, i.e., higher PKA activity, compared to L23 neurons imaged in acute brain slices at the resting state (lifetime = 1.73 ± 0.03 for acute slices and 1.52 ± 0.01 for awake mice, p < 0.001; Figure 6B). This result suggests that a basal level of PKA activity exists in cortical neurons of awake mice.
Figure 6. In vivo imaging of tAKARα reveals basal tonic activity in cortical neurons of awake mice.
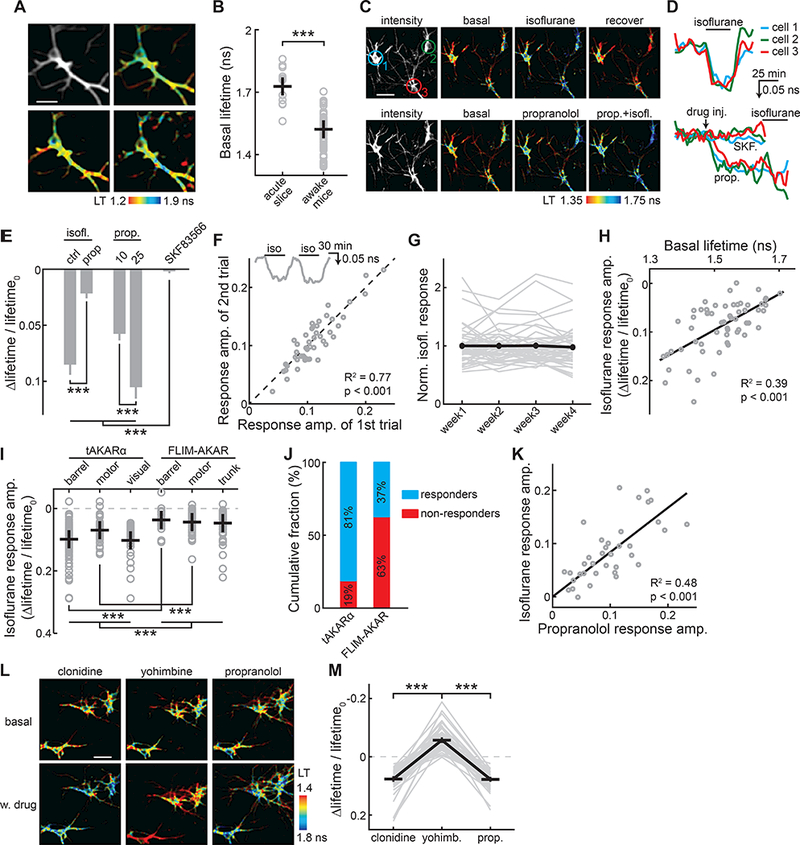
(A) Representative intensity and lifetime (LT) images of a neuron imaged over 35 days. The postnatal ages at the time of imaging are labeled.
(B) Comparison of basal tAKARα lifetimes between the apical dendrites of L23 neurons in acute brain slices and the neurons imaged in awake mice. n (neurons/mice) = 11/5 for acute slices, and 59/8 for awake mice.
(C, D) Representative lifetime images (C) and timecourse traces (D) of three example neurons in one field of view responding to the indicated drug administration.
(E) Collective results of PKA activity in the presence of the indicated pharmacological manipulation. Dosages are: 1.25% isoflurane with or without pretreatment with 25 mg/kg propranolol, 10 or 25 mg/kg propranolol, or 0.2 mg/kg SKF83566. n = 48 neurons from 5 mice.
(F) Isoflurane-induced responses of individual neurons from two consecutive trials. Inset shows a representative trace for the experiment. The line of equity (not from fitting) is also shown. n = 47 neurons from 3 mice.
(G) Isoflurane-induced responses across four weeks normalized to the averaged response of the first week. Both individual cell traces (gray lines) and the averaged response (black) are shown. n = 46 neruons from 3 mice.
(H) A linear fit of the isoflurane-induced PKA responses in individual neurons correlated with the basal lifetime in the corresponding neuron. n = 59 neurons from 8 mice.
(I) Comparison of isoflurane-induced PKA responses visualized by tAKARα and FLIM-AKAR across different cortical regions. Trunk: the trunk somatosensory cortex. n (neurons/mice, from left to right) = 91/5, 43/4, 31/2, 18/2, 49/2, and 43/4.
(J) Fraction of neurons imaged across brain areas that exhibited detectible responses to isoflurane using the indicated sensors. The threshold is 1. 5 times the standard deviation of the baseline reponses. N (neurons/mice) = 165/11 for tAKARα, and 110/8 for FLIM-AKAR.
(K) A linear fit of the isoflurane responses in individual neurons correlated with propranolol response in the same neurons. n = 48 neurons from 5 mice.
(L) Representative lifetime images of three example neurons in one field of view responding to the indicated pharmacological manipulations.
(M) Summary results of PKA responses in the presence of the indicated pharmarcological manipulations. Dosages are: 1 mg/kg clonidine, 5 mg/kg yohimbine, and 25 mg/kg propranolol. n = 63 neurons from 6 mice.
LT: lifetime; prop.: propranolol; isofl.: isoflurane; SKF.: SKF83566; yohimb.: yohimbine.
Error bars are s.e.m.
Wakefulness is known to be associated with increased activity in the noradrenergic pathways (Berridge et al., 2012; Brown et al., 2012; Mitchell and Weinshenker, 2010), which may result in tonic PKA activities. To test whether the observed basal PKA activity is associated with wakefulness, mice were lightly anesthetized with the general anesthetic isoflurane (1.25% via inhalation) (Figure 6C, 6D, and 6E). Isoflurane induced a decrease in PKA activity across neurons, as indicated by an increased sensor lifetime (Δlifetime/lifetime0 = 0.085 ± 0.010, p < 0.001 c.f. baseline; upper panels of Figure 6C and 6D, and the most left bar in Figure 6E). This effect was reversed when the isoflurane flow was stopped and the animal regained consciousness (upper panels of Figure 6C and 6D). The response amplitudes of individual neurons were stable between two consecutive trials (Figure 6F) and across repeated imaging sessions spanning over one month (Figure 6G). Importantly, the response amplitude correlated with the basal lifetime in the corresponding neurons, such that neurons with a shorter basal lifetime, i.e., higher basal PKA activities, exhibited larger isoflurane response amplitudes (Figure 6H). Neurons in other cortical regions also exhibited a similar reduction in PKA activity by isoflurane (Figure 6I). Consistent with the results in brain slices, tAKARα gave significantly larger response amplitudes than FLIM-AKAR in both barrel and motor cortices (Figure 6I). Results from the visual cortex for tAKARα and from the trunk somatosensory cortex for FLIM-AKAR, while they cannot be directly compared with each other, are also shown to provide a sense of the response amplitudes across multiple brain regions. Using an arbitrary threshold of 1.5 times the standard deviation of baseline, detectible responses were found in 81% of all imaged neurons when tAKARα was used (Figure 6J); in contrast, only 37% neurons responded when FLIM-AKAR was used, indicating the necessity of the enhanced signal amplitudes of tAKARα for comprehensive in vivo PKA imaging. Overall, these results indicate that a basal level of PKA activity is present in cortical neurons and is associated with wakefulness.
To test whether the basal PKA activity in awake mice is dependent on the noradrenergic pathway, we applied the β-adrenergic receptor antagonist propranolol to the experimental mice via IP injections. A dose-dependent increase in tAKARα lifetime corresponding to a decrease in PKA activity was observed (Δlifetime/lifetime0 = 0.057 ± 0.006 for 10 mg/kg, and 0.105 ± 0.10 for 25 mg/kg, p < 0.001; lower panels of Figure 6C and 6D, and Figure 6E). This effect was likely specific because the D1 dopamine receptor antagonist SKF83566 did not alter the basal PKA activity (Δlifetime/lifetime0 = 0.002 ± 0.002, p > 0.1 c.f. baseline; lower panel of Figure 6D, and Figure 6E). After propranolol injection, isoflurane exposure resulted in little additional increase in lifetime (Δlifetime/lifetime0 = 0.022 ± 0.005 after propranolol, p < 0.001 c.f. before propranolol; lower panel of Figure 6D, and second bar from the left in Figure 6E), suggesting that the isoflurane-induced decrease in PKA activity was due to a decrease in the noradrenergic tone in the cortex. Consistent with this notion, the response amplitudes to isoflurane correlated with those induced by propranolol (Figure 6K). These results suggest that the isoflurane-sensitive basal PKA activity is mediated by β-adrenergic receptors, presumably via tonic norepinephrine release in the cortex of awake animals.
tAKARα tracks bidirectional PKA activity
We next asked whether tAKARα is capable of detecting a further increase in PKA activity in awake animals. It is known that cortical neurons also express α2-adrenergic receptors, which inhibit the cAMP/PKA pathway via Gi proteins (Langer, 2015). We hypothesized that α2- and β-adrenergic receptors form a push-pull system to tightly regulate PKA activities. Indeed, inhibition of α2-receptors by the antagonist yohimbine (5mg/kg) resulted in increased PKA activity; in contrast, activation of α2-receptors using clonidine (1mg/kg) decreased the PKA activity to a degree comparable to inhibiting β-adrenergic receptors (Figure 6L and 6M). Thus, tAKARα can track bidirectional manipulation of PKA activity by pharmacological reagents.
tAKARα detects PKA activity elicited by enforced locomotion
We asked whether tAKARα may be used to detect neuromodulation fluctuations during animal behavior. Locomotion has been shown previously to be associated with increased noradrenergic activity in the cortex (Paukert et al., 2014; Polack et al., 2013). A motorized treadmill was used to control mouse locomotion (Movie S3) under the head-fixed configuration during in vivo 2pFLIM imaging. In the motor cortex, a large number of neurons exhibited a decreased tAKARα lifetime, corresponding to increased PKA activity, when the animal was forced to run (Figure 7A, 7B, and 7C). The PKA responses to locomotion were reversible and repeatable (Figure 7B). Similar responses were also observed in neurons in the visual cortex, but the responses in the barrel cortex were significantly smaller (Figure 7C), indicating that this locomotion-induced response is brain region-specific.
Figure 7. tAKARα detects enforced locomotion-induced PKA activity.
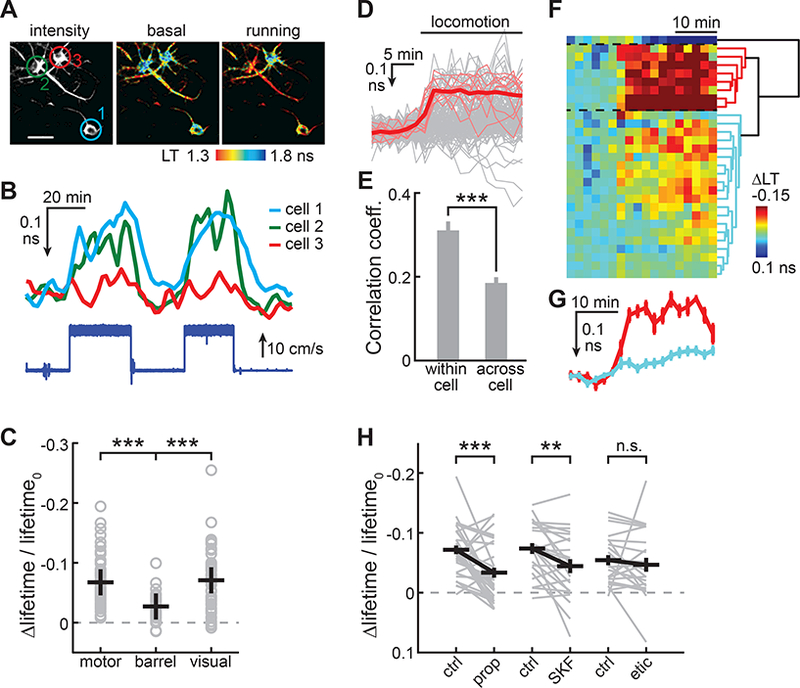
(A,B) Representative lifetime images (A) and timecourse Δlifetime traces (B) of example neurons responding to enforced running. Mouse running speed is indicated (bottom of panel B).
(C) Enforced locomotion-induced PKA responses at different cortical regions. n (neurons/mice) = 88/6, 32/2, and 50/2 for motor, barrel, and visual cortices, respectively.
(D) All individual trials of locomotion-induced Δlifetime responses of neurons with repeated trials (light gray), the individual trials of a representative neuron (thin red), and the averaged response of this neuron (thick red). 158 trials, 29 neurons, 5 mice.
(E) Correlation coefficient of individual trials of locomotion-induced PKA responses within a neuron or across neurons.
(F) The Δlifetime response timecourses averaged for each of 29 neurons (each row represents a neuron, and each column represents a time point) that are clustered into three groups (marked by dashed lines) according to the dendrogram shown on the right.
(G) The averaged response traces from the two color-coded groups in panel F.
(H) Both the β-adrenergic receptor antagonist propranolol and the D1 receptor antagonist SKF83566 attenuate locomotion-induced PKA responses, whereas the D2 receptor antagonist eticlopride does not. n (neurons/mice) = 33/5, 23/4, and 23/4 for prop., SKF. and etic., respectively
prop.: propranolol; SKF.: SKF83566 etic.: eticlopride.
Error bars are s.e.m.
See also Movie S3.
Different neurons in the same field of view of the motor cortex responded with different amplitudes and kinetic characteristics, although the responses among different trials of the same neurons appeared to be similar (Figure 7D). To characterize this objectively, for a subset of neurons with repeated trials (29 neurons, 2–8 trials per neuron with a mean ± s.d. of 5.4 ± 3.8 trials per neuron), we calculated the correlation coefficient within the neuron and across neurons (Figure 7E). On average, the correlation coefficients within a neuron were significantly higher than those across neurons (correlation coefficient = 0.31 ± 0.02 and 0.19 ± 0.01 for within and across neurons, respectively; p < 0.001). To categorize the neuronal responses across neurons, we averaged all trials for each neuron and performed a cluster analysis (Figure 7F). The 29 neurons were clustered into three groups based on the Euclidean distance across traces: 1) 27% (8 out of 29) neurons that had larger response amplitudes, 2) 69% (20) neurons with somewhat smaller response amplitudes, and 3) one neuron that responded with decreased PKA activity. By averaging all neurons within a group (Figure 7G), it appeared that, in addition to the amplitudes, the kinetics across the two positively responding groups (group 1 and 2) were also different. The large-responding group had fast on kinetics, whereas the small responders exhibited a response that continued to ramp up over time. Finally, we asked whether the locomotion-induced PKA activity is sensitive to blockade of neuromodulator receptors. We found that both the β-adrenergic receptor antagonist propranolol and the D1 receptor antagonist SKF83566 attenuated the response, although neither completely abolished the response, suggesting that both the noradrenergic and dopaminergic pathways may be involved. In contrast, the D2 receptor antagonist eticlopride did not significantly alter the response amplitude. Overall, these results indicate that tAKARα combined with 2pFLIM can detect PKA dynamics induced by animal behaviors.
DISCUSSION
Despite the importance and pervasiveness of neuromodulation throughout the nervous system, there is as yet no robust method for monitoring the intracellular consequences of neuromodulator release in vivo at single-cell resolution in rodents or other mammalian species. PKA is a central mediator of key neuromodulators in the brain, including norepinephrine, dopamine, acetylcholine, and serotonin. Thus, PKA imaging may serve as readout for neuromodulatory activity, in the same way that calcium imaging does for electrical activities. Applying high-resolution PKA imaging to mammalian brain tissue in vivo is challenging because of severe light scattering and light aberration (Denk and Svoboda, 1997; Zipfel et al., 2003). The “photon budget”, i.e., the number of collectible photons in a given sample, is typically greatly reduced in intact brain tissue. As a result, many imaging approaches that are effective in vitro encounter difficulties in vivo. Although AKARs and other PKA sensors (Nagai et al., 2000; Zaccolo and Pozzan, 2002; Zhang et al., 2018) have been developed, no in vivo application in rodents or other mammals with single-cell resolution has been established. The signal amplitude of tAKARα in response to norepinephrine is 2.7-fold larger than the previous sensors. Thus, tAKARα requires approximately seven-fold less photons (see Star Methods) to achieve the same signal-to-noise ratio. As demonstrated (Figure 6 and 7), this improved sensor is sensitive enough for in vivo imaging of bidirectional PKA activity with cellular resolution in awake mice. We found that different neurons or neuronal types may respond differently to the same neuromodulator stimuli (e.g., see Figure 4F and 5C). During animal behavior, seemingly similar neurons in the cortex may exhibit cell-specific responses (Figure 7). Thus, imaging PKA signaling events with single-cell resolution will enable a direct and in-depth dissection of the mechanism underlying neuromodulation.
We choose 2pFLIM for in-tissue and in vivo quantification of FRET. FLIM can be implemented onto most existing two-photon microscopes by the modular addition of several hardware components (Figure S7; see also STAR METHODS). In FLIM images, each pixel consists of a fluorescence decay curve of 64 to 256 points. The amount of data is therefore much larger than that of conventional two-photon images. Challenges associated with acquiring and handling such a large amount of data impose practical limits in data acquisition speed and in pixel resolution. On the other hand, 2pFLIM offers great advantages when measuring FRET in vivo. 2pFLIM is readily quantifiable (Yasuda, 2006; Yellen and Mongeon, 2015) and it does not suffer from wavelength-dependent light scattering (e.g., see Figure S1), making it possible to compare neurons across different depths, animals, and conditions. These properties enabled us to discover that cortical neurons in awake mice exhibit higher PKA activity than those in acute brain slices (Figure 6B). Another advantage is that a low-irradiating fluorescent protein can be used as the acceptor (in our case, cp-sREACh). This frees up one color channel compared to ratiometric sensors. This vacant color channel can then be used to simultaneously image orthogonal neuronal properties. In the current study, we simultaneously imaged the red cytosolic marker tdTomato (e.g., Figure S3A) to reveal neuronal morphology. In the future, multiplexed imaging of PKA with another subcellular signaling pathway, such as the calcium or PKC pathway (pending the availability of the appropriate sensors), would accelerate the investigation of signaling mechanisms underlying neuromodulation.
The strategy we employed to improve AKAR sensors is to target the sensor to microtubules where PKA activity is enriched. Although sensor targeting has been used to direct the subcellular localization of sensors (e.g., (Mao et al., 2008; Tang and Yasuda, 2017)), we show here that it can also be used to enhance the sensitivity and dynamic range of the sensor. This strategy is generalizable to AKAR4 (Figure S3C, S3D, and S3E), an AKAR for ratiometric FRET measurements, and should also be applicable to forthcoming generations of PKA activity sensors. Conceptually similar strategies may be applied to sensors for other subcellular signaling events, such as those mediated by PKC. From another perspective, our AKAR targeting results also have their own biological implications with regard to PKA signaling mechanisms in neurons. The enhanced performance of tAKARα supports our previous finding that the abundant microtubule-binding protein MAP2 is the dominant A-kinase anchoring protein (AKAP) in neurons (Zhong et al., 2009). The results associated with the membrane-targeted tAKARγ, which is largely phosphorylated even at the resting state, are also consistent with the recent finding that once PKA is activated, the catalytic subunit of PKA becomes enriched on the plasma membrane (Tillo et al., 2017).
Although we favor tAKARα as a general sensor because of its large signal amplitudes, other tAKARs may be useful under certain conditions. One limitation of tAKARα is that it does not label dendritic spines because microtubules are normally absent from spines. Current evidence suggests that PKA activity is not compartmentalized at the level of individual spines (Chen et al, 2014, Tang and Yasuda, 2017); therefore, sensors situated at the dendrite should still readily detect PKA signaling events triggered in the adjacent spine. In the event that PKA activity needs to be studied specifically in spines, other spine-localized tAKARs, including tAKARβ-ζ can be used (Figure 2). In addition, MAP2 is primarily expressed in neurons (Dehmelt and Halpain, 2005). It is not clear whether microtubule targeting would also improve PKA sensitivity in non-neuronal cells. In these cases, tAKARβ may serve as an alternative, although it generates smaller signal amplitudes. Improved amplitudes may be achieved by targeting the AKAR sensor to the primary PKA anchoring site in the corresponding cell type.
Although tAKARα has a relatively fast on kinetics, its off kinetics is on the order of a few minutes (Figure 3H, 3K and 5H). These kinetics are approximately two orders of magnitude slower than those reported for the extracellular concentration fluctuation of synaptically released neuromodulators (Courtney and Ford, 2014; Ford et al., 2009). However, these kinetics may in fact represent the endogenous dynamic properties of PKA, as they match the biochemically measured cAMP dissociation rate from PKA regulatory subunits (Kd ~ 0.15 min−1; (Francis and Corbin, 1994)), and they closely follow the PKA regulation kinetics of sAHP in response to norepinephrine (Figure 3K). Slow subcellular signaling kinetics may allow the seemingly fast neuromodulator releases to interact or integrate over a previously unanticipated long separation (several minutes). Such long-term interactions may have a significant impact on neuronal physiology and animal behavior, and may become an interesting future research direction.
Using tAKARα combined with 2pFLIM, we found a tonic level of PKA activity present in most cortical neurons of awake mice (Figure 6). This basal PKA activity is dependent on wakefulness, is mediated by β-adrenergic receptors, and is stable over weeks. Wakefulness is thought to be associated with increased activity in the norepinephrine pathway originating from the locus coeruleus (LC) (Berridge et al., 2012; Brown et al., 2012; Carter et al., 2010; Mitchell and Weinshenker, 2010). A tonic level of norepinephrine may be present in the awake brain (Bellesi et al., 2016; Constantinople and Bruno, 2011; Kalen et al., 1989). Our results reveal the cellular consequence of this tonic norepinephrine: it results in a significant level of subcellular cAMP/PKA signaling activity. This may contribute to the differences in brain function between sleep and wakefulness considering that increased PKA activity can enhance neuronal excitability, plasticity, and learning and memory. In addition, we found that the activation and blockade of α2 receptors using clonidine and yohimbine, respectively, have profound impacts on PKA activities in the cortex. Both of these drugs are widely used in humans. In vivo PKA imaging may contribute to a better understanding of the functions and side effects of these compounds.
Multiple neuromodulators converge onto PKA. Monitoring PKA responses under behavioral paradigms involving different pathways may therefore be used to read out the respective neuromodulatory activities. Mechanistic dissection of the contribution of individual neuromodulatory pathways may be achieved by pharmacological experiments and optogenetic manipulation of specific pathways. For example, we showed that basal PKA activity in cortical neurons of awake mice is sensitive to a blockade of the β-adrenergic receptors, but not to a blockade of the D1 dopamine receptors (Figure 6E), suggesting that norepinephrine is the primary tonic neuromodulator under these conditions. In contrast, locomotion-induced PKA activity is sensitive to the blockade of both β-adrenergic receptors and D1 dopamine receptors (Figure 7H), suggesting the involvement of both pathways.
Overall, neuromodulation exerts profound impacts on brain function, from the cellular to the behavioral level. Our improved genetically-encoded PKA sensor presented here may be used to dissect fundamental mechanisms related to neuromodulatory activities in vitro and in vivo, in a manner analogous to the use of calcium imaging in detecting neuronal electrical activities.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Haining Zhong (zhong@ohsu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal handling and experimental protocols were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals, written by the National Research Council (US) Institute for Laboratory Animal Research, and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Oregon Health & Science University (#IP00000420).
Hippocampal Slice Culture and Transfection
Hippocampi were dissected from P6 - P7 rat pups (both sexes) and sectioned to generate 400 μm slices in dissection medium containing (in mM) 1 CaCl2, 5 MgCl2, 10 glucose, 4 KCl, 26 NaHCO3, and 248 sucrose, with the addition of 0.00025% phenol red. The slices were then seeded onto a cell culture insert (Millipore, # PICM0RG50) and cultured at 35°C with 5% CO2 in medium containing 7.4 g/L MEM (ThermoFisher #11700–077) with the following supplements (in mM unless labeled otherwise): 16.2 NaCl, 2.5 L-Glutamax, 0.58 CaCl2, 2 MgSO4, 12.9 D-glucose, 5.2 NaHCO3, 30 HEPES, 0.075% ascorbic acid, 1 μg/ml insulin, and 20% heat-inactivated horse serum.
For sensor expression, mammalian expression plasmids (with the CMV promoter) were coated onto 1.6 μm gold particles (Bio-Rad #165–2264; ~ 1 μg DNA/mg gold) and were transfected to cultured slices at 2 – 3 weeks in vitro using the biolistic method (Bio-Rad Helios gene gun). Cultured slices were imaged or recorded at 2 – 4 days post-transfection in a chamber perfused with gassed (with 95% O2 / 5% CO2) artificial cerebral spinal fluid (ACSF) containing (in mM) 127 NaCl, 25 NaHCO3, 25 D-glucose, 2.5 KCl, 1.25 NaH2PO4, 4 CaCl2, and 4 MgCl2.
For the experiments described in Figure 3J and 3K, the CA1 regions of hippocampal slices were microinjected at one week in vitro with approximately 10 nl of 1:20 diluted adeno-associated virus serotype 2/1 (AAV2/1) in phosphate-buffered saline (PBS). Recordings were performed at 2 – 3 weeks post-injection.
Acute Brain Slices
Mice (P18 - P50) with neurons transfected using in utero electroporation or AAV injection were transcardially perfused with ice-cold, gassed ACSF containing 2 mM CaCl2 and 1 mM MgCl2. The brain was then dissected and coronal slices were prepared using a vibratome (Leica VT1200S) in a choline cutting solution (gassed with 95% O2 / 5% CO2) containing (in mM) 110 choline chloride, 25 NaHCO3, 25 D-glucose, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 11.5 sodium ascorbate, and 3 sodium pyruvate. The slices were then incubated in gassed ACSF at 35°C for 30 minutes and subsequently kept at room temperature for up to 8 hours. For experiments involving the striatum (Figure 5), brain slices were prepared in gassed warm ACSF (37°C); mice were perfused with ACSF before the brain was dissected and sectioned in ACSF. The slice was incubated in 37°C ACSF for 30 minutes and subsequently kept at room temperature for up to 6 hours.
Animals, Transfections, and Surgeries
C57BL/6 mice (Charles River, or home-bred within 5 generations from Charles River breeders) were used. Mice were either transfected via in utero electroporation or by stereotaxic viral injection. In utero electroporation was performed at E16.5 by injecting plasmid (1 μl/embryo, concentration ~3 – 4 μg/μl, mixed with a 0.2% final concentration of fast green for visualization) into the lateral ventricle of mouse embryos, which were then electroporated with five 100-ms pulses (38V) using an electroporator (BEX #CUY21). Stereotaxic viral injection was performed as previously described (Hunnicutt et al., 2014) at ~P21 using a custom-modified David Kopf system. The coordinates were: for the somatosensory cortex, 1 mm posterior to the bregma, 2.6 mm lateral to the midline, and 0.2 mm below the pia; for the motor cortex, 1.2 mm anterior to the bregma, 1.2 mm lateral to the midline, and 0.2 mm below the pia; and for the dorsal medial striatum, 0.95 mm anterior to the bregma, 1.7 mm lateral to the midline, and 3 mm below the bregma. Typically, 20 – 50 nl of AAV was injected. With the appropriate parameters (for in utero electroporation, the DNA concentration and the number of pulses used, and for viral infection, the volume injected and the dilution of the virus) and surgery, both transfection techniques can achieve a balanced labeling efficiency that is sparse enough for visualizing individual neurons while still having enough cell density for population imaging.
When in vivo imaging was involved, a glass window was installed on the skull of the mouse at postnatal days 30 – 60. Briefly, the open skull surgery was performed under isoflurane anesthesia (4% for induction, and 1.5 – 2% for maintenance). After exposure, the skull was cleaned using a Q-tip, and an aluminum plate (for head fixation) was attached to the skull using dental acrylic. A circular (~5.5 mm diameter) craniotomy was then made above the cortical region of interest. Imaging was performed at least two weeks post-surgery under a head-fixed configuration while the animals were awake. The animals were allowed to rest or to move at will on a one-dimensional treadmill, except for during the experiments in Figure 7. In those experiments, a motorized treadmill was used to control the animal’s locomotion. Isoflurane was administered via inhalation, and other pharmacological reagents were administered by intraperitoneal injections.
METHOD DETAILS
Plasmid Constructs
Constructs were made using standard mutagenesis and subcloning methods, or by gene synthesis (Genewiz). All previously unpublished constructs, including viral plasmids, and their sequences will be deposited to Addgene.
FLIM-AKAR was generated independently from the published sequence (Chen et al., 2014) by deleting the nucleotides encoding the last 11 residues (G228-K238) of monomeric EGFP in AKAR5’. We verified that the corresponding protein sequence was identical to that of the FLIM-AKAR sequence deposited at Addgene (#63058); however, the nucleotide sequence was slightly different. For tAKARα and tAKAR4α, the microtubule-binding domain of MAP2c (Zhong et al., 2009) was fused to the C terminus of FLIM-AKAR and AKAR4, respectively, with a peptide linker of SGVYKGT. tAKARβ was constructed by adding the nuclear export signal (NES) from protein kinase inhibitor peptide (PKI; with the sequence being MLQNELALKLAGLDINKTG) (Xu et al., 2012) to the N terminus of AKAR5.1 with a four-residue linker (PVAT). tAKARγ was generated by fusing the membrane-targeting farnesylation sequence from KRas (GKKKKKKSKTKCVIM) (Hancock et al., 1989) with a three-residue linker (GEF) to the C terminus of FLIM-AKAR. The lifeact peptide sequence (MGVADLIKKFESISKEE) (Riedl et al., 2008) and rat PSD-95 were fused to the N terminus of FLIM-AKAR to yield tAKARδ and tAKARζ with the linker sequences of GDPPVAT and VPRARDPPVAT, respectively. Human β-actin was fused to the C terminus of FLIM-AKAR with the linker SGLRSRA to yield tAKARε.
Virus Preparation
To generate AAV particles for expressing tAKARα, the tAKARα cDNA preceded by the Kozak sequence was cloned into a recombinant AAV vector containing the human synapsin promoter (Borghuis et al., 2011), a woodchuck post-transcriptional regulatory element (WPRE), an SV40 polyadenylation sequence, and two inverted terminal repeats. Viruses (rAAV 2/1, rep/cap) were assembled using a helper-free system (Stratagene) and purified on sequential cesium gradients according to published methods (Grieger et al., 2006). Titers were measured using a payload-independent qPCR technique (Aurnhammer et al., 2012). Typical titers were > 1010 genomes/microliter. Plasmids for making virus will be deposited to Addgene, and viral aliquots will be provided upon request.
Electrophysiology
Whole-cell voltage-clamp recordings were performed at room temperature using an Axopatch 200B amplifier (Molecular Devices). Electrophysiological signals were filtered at 2 kHz, and digitized and acquired at 20 kHz with custom software written in MATLAB. Slices were perfused with gassed ACSF containing 4 mM CaCl2 and 4 mM MgCl2. For the sAHP experiments described in Figure 3E and 3F, 10 μM NBQX was added to suppress recurring activities in culture slices. The internal solution contained (in mM) 140 KMeSO4, 10 KCl, 10 HEPES, 10 Tris-phosphocreatine, 2 Mg-ATP, and 0.4 Na-GTP with an osmolarity adjusted to 300 mOsmol/kg. The junction potential (ca. −11 mV) was not corrected. Small currents (≤ 50 pA) were injected constantly to hold neurons at a resting potential of −55 ± 1 mV under the current-clamp mode. sAHP was elicited by injecting ten 2-ms square pulses of 1.5 nA currents at 40 Hz, and was measured as the baseline-subtracted potential averaged between 1 – 2 seconds after the onset of stimulation. The serial resistance was carefully maintained to be 30 – 60 MΩ to reduce washout of the cell by moving the recording pipet slightly backwards, and by adjusting the pressure to the pipet. Under this condition, sAHP recording is stable for over 30 minutes or longer (Figure S5A). For Figures 3J and 3K, amphotericin B-based perforated patch recording was employed. This was carried out by diluting amphotericin B to 0.25 mg/ml from a fresh 25 mg/ml stock solution in DMSO in the whole cell recording internal solution. After forming Giga-seal under voltage clamp, it typically took 15 – 30 minutes for the access resistance to decrease below 60 MΩ. The cell was recorded under the current-clamp mode while being held at −55 mV. For these experiments, 1 μM tetrodotoxin (TTX) was used instead of NBQX. sAHP was elicited in 4 mM CaCl2 and 1 mM MgCl2 by injecting twenty 4-ms square pulses of 1.5 nA currents at 60 Hz, and was measured as the baseline-subtracted potential averaged between 1.5 – 1.6 seconds after the onset of stimulation. For Figure S4E, the cells were held at −70 mV under voltage clamp and sAHP currents were elicited by a voltage jump to 20 Mv for 500 ms and then back to −50 mV for 14 s. sAHP currents were measured at 1.6 – 1.7 seconds after the offset of stimulation.
Norepinephrine Iontophoresis
A glass electrode pipette filled with 6 μl of a 1 M norepinephrine solution was placed close to a tAKARα-expressing dendrite located near the slice surface. A −10 nA holding current was applied to prevent uncontrolled norepinephrine leakage. FLIM images of the dendrite at a single z plane were acquired every 10 seconds. After at least 6 minutes of baseline imaging, norepinephrine was released by injecting a single 10 second 150 nA current pulse controlled by Axopatch-2D using custom software written in MATLAB.
Intrinsic Imaging
Approximately two weeks after the glass window was implanted, intrinsic signals were measured from the cortical surface of lightly anesthetized mice while stimulating their whiskers as described previously (Masino et al., 1993). Anesthesia was achieved using chlorprothixene hydrochloride (0.4 mg/kg, administered intramuscularly; Sigma #C1671) and a low level of isoflurane (0.5 – 1%). Intrinsic signals were acquired through a custom-built video acquisition system. A 12-bit CCD camera (QImaging Retiga-2000) was focused onto the surface of the cortex via a zoom lens (Thorlabs #MVL6X12Z). The cortex was illuminated with light generated by a red (627 nm) LED (Luxeon Star LEDs, # LXM2-PD01–0050). Whiskers were stimulated with a piezo actuator (Piezo Systems #PSI-5A4E) coupled to a large vinyl surface that could stimulate the majority of the mouse’s whiskers. Video frames were recorded at a frame rate of 1 Hz with 16 frames for baseline and 4 frames for whisker-stimulated conditions. This protocol was repeated 30 times. The baseline and stimulated images were then averaged, respectively, and subtracted to yield the final intrinsic imaging image.
Two-photon and 2pFLIM Imaging
Two-photon imaging setups were custom-built and controlled by the ScanImage software (Pologruto et al., 2003). In particular, the in vivo two-photon microscope was built based on the open-access design of the Modular In vivo Multiphoton Microscopy System (MIMMS) from Howard Hughes Medical Institute Janelia Research Campus (https://www.janelia.org/open-science/mimms). We used 960 nm light to excite the donor fluorophore (monomeric EGFP). The fluorescence emission from EGFP and other fluorophores, such as mCherry, at different wavelengths were unmixed using a dichroic mirror (Chroma 565DCXR) and band-pass filters (Chroma ET500/40m barrier filter for green and Semrock FF01–630/92 for red). Fluorescence lifetime imaging was carried out in the time domain by integrating four additional hardware components onto the two-photon setup. Specifically, lifetime measurements were achieved by comparing the arrival of the laser pulses, as detected by a photodiode (Thorlabs FDS010), to the arrival of donor-emitted photons, as detected by a fast, cooled photomultiplier tube (Hamamatsu H7422PA-40 or H10769PA-40), using a TCSPC-730 or SPC-150 (Becker and Hickl) time-correlated single photon counting board, as described previously (Yasuda et al., 2006). A Becker and Hickl frequency-to-analog converter (HPM-CON-02) was used to split the signal into one output with unaltered waveform for lifetime measurements and another slower output (~1 – 2 μs) for simultaneous conventional two-photon imaging. Further details of the setup will be provided upon request. Data acquisitions were controlled by custom software (called FLIMimage) in MATLAB kindly provided by Dr. Ryohei Yasuda with modification. Slice imaging was carried out at room temperature in gassed ACSF containing 4 mM CaCl2 and 4 mM MgCl2 for cultures, and 2 mM CaCl2 and 1 mM MgCl2 for acute slices. Optogenetic photostimulation of dopaminergic axons in the striatum was delivered using a custom-built 470nm LED illumination through the 60× imaging objective. PMTs were manually shut down for ~ 10 seconds during the delivery of the photostimulation (5 trains at 1Hz, with each train containing five 1.5 ms light pulses at 20 Hz).
Enforced Locomotion on a Treadmill
A motorized spherical treadmill (20 cm diameter) was used to control head-fixed mice to run or rest under the 2pFLIM microscope, as shown in Movie S3. During running sessions, the movement speed was approximately 20 cm/s and was kept stable throughout each trial (~25 min). The indicated drugs were administered via intraperitoneal injection 10 min after the offset of a first, control trial. The test trial started approximately 15 minutes after drug administration.
Image Analysis
Data analyses were performed using custom software written in MATLAB. In particular for 2pFLIM analysis, we developed a software suite called FLIMview written in MATLAB. The software will be distributed freely upon request. Where appropriate, regions of interest were drawn to minimize the potential contamination of background photons. In all analyses, the mean photon emission time τ was reported as an approximation of the fluorescence lifetime (Harvey et al., 2008):
where t is the emission time of individual photons in the measurement window and t0 reflects the timing of laser pulses. t0 is measured separately under ideal imaging conditions and is a fixed property of a given hardware configuration. Because the measurement window (tw) is finite (≤ 10 ns passing t0 due to laser pulse repetition and single photon counting hardware properties), the measured τ (τapparent) is in fact slightly smaller than the real value:
For a typical τ of 2.0 ns and a measurement window of 9 ns, the measured τ is 1.90, or 95% of the real value.
Use of Δlifetime/lifetime
Approximating the fluorescence lifetime decay as a single exponential, the error in determining τ (δτ) is related to the number of collected photons (N) by (Yasuda et al., 2006):
For a given signal Δτ, and a desired signal-to-noise ratio (SNR), δτ has to be equal to or smaller than δτ/SNR, and the minimum number of photon required is:
Therefore, (Δτ/τ determines the minimal number of photons for a given SNR. Every twofold increase in (Δτ/τ will reduce the demand for photons by four-fold.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical tests were performed using custom software written in MATLAB. Averaged data are presented as mean ± s.e.m., unless noted otherwise. Throughout the paper, “n” indicates the number of neurons, unless noted otherwise. In slice experiments, most (> 90%) of the neurons came from different slices. p values were obtained from one-way ANOVA tests, unless noted otherwise. In all figures, *: p ≤ 0.05, **: p ≤ 0.01, and ***: p ≤ 0.001. Throughout the figures, responses are presented in reversed axes because a decrease in lifetime indicates increased PKA activity.
DATA AND SOFTWARE AVAILABILITY
We have developed a software suite called FLIMview written in MATLAB for 2pFLIM data analysis. This software will be made available without cost upon reasonable request to the Lead Contact.
Supplementary Material
Movie S1. Related to Figure 6. Representative intensity z-stack of in vivo 2pFLIM imaging.
Several cortical neurons and their neurites are shown. Field of view is 140 μM. The animal was anesthetized to minimize potential movement artifacts that may blur neurites.
Movie S2. Related to Figure 6. The lifetime z-stack of Movie S1.
The lifetime scale is same as that in Figure 6A.
Movie S3. Related to Figure 7. Representative video of mouse resting and running on motorized treadmill.
HIGHLIGHTS.
An improved PKA sensor (called tAKARα) with enhanced sensitivity and dynamic range
tAKARα tracks physiologically relevant PKA signaling activated by neuromodulators
2pFLIM of tAKARα enables single-cell imaging of PKA activity in behaving mice
Wakefulness and locomotion are associated with cell-specific PKA dynamics
ACKNOWLEDGEMENTS
We thank Dr. John Williams for critical discussions throughout the project; Ms. Yang Chen for comments on the manuscript; Dr. Ryohei Yasuda at Max Planck Florida for 2pFLIM acquisition software and AKARet cDNA; Dr. Jin Zhang at University of California, San Diego for AKAR4 cDNA; Drs. Wenzhi Sun, Na Ji, and Vijay Iyer, and Mr. Daniel Flickinger at Howard Hughes Medical Institute Janelia Research Campus, Drs. Joseph Wekselblatt and Cris Niell at University of Oregon, Dr. Vivek Unni at Oregon Health & Science University, and Dr. Christopher Harvey at Harvard for help with surgeries, hardware, and software for in vivo two-photon imaging; and Dr. Daniel O’Connor at Johns Hopkins University for intrinsic imaging. This work was supported by two NIH BRAIN Initiative awards U01NS094247 (H.Z. and T.M.) and R01NS104944 (H.Z. and T.M.), an NINDS R01 grant R01NS081071 (T.M.) and a NINDS R21 grant R21NS097856 (H.Z.).
Footnotes
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and three movies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adamantidis AR, Tsai HC, Boutrel B, Zhang F, Stuber GD, Budygin EA, Tourino C, Bonci A, Deisseroth K, and de Lecea L (2011). Optogenetic interrogation of dopaminergic modulation of the multiple phases of reward-seeking behavior. J Neurosci 31, 10829–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MD, and Zhang J (2006). Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun 348, 716–721. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, and Cohen JD (2005). An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28, 403–450. [DOI] [PubMed] [Google Scholar]
- Aurnhammer C, Haase M, Muether N, Hausl M, Rauschhuber C, Huber I, Nitschko H, Busch U, Sing A, Ehrhardt A, et al. (2012). Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 23, 18–28. [DOI] [PubMed] [Google Scholar]
- Backman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, and Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, and Gainetdinov RR (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63, 182–217. [DOI] [PubMed] [Google Scholar]
- Bellesi M, Tononi G, Cirelli C, and Serra PA (2016). Region-Specific Dissociation between Cortical Noradrenaline Levels and the Sleep/Wake Cycle. Sleep 39, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Schmeichel BE, and Espana RA (2012). Noradrenergic modulation of wakefulness/arousal. Sleep Med Rev 16, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghuis BG, Tian L, Xu Y, Nikonov SS, Vardi N, Zemelman BV, and Looger LL (2011). Imaging light responses of targeted neuron populations in the rodent retina. J Neurosci 31, 2855–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, and McKnight GS (1997). PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol 7, 397–403. [DOI] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, and McCarley RW (2012). Control of sleep and wakefulness. Physiol Rev 92, 1087–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, and de Lecea L (2010). Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci 13, 1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Granger AJ, Tran T, Saulnier JL, Kirkwood A, and Sabatini BL (2017). Endogenous Galphaq-Coupled Neuromodulator Receptors Activate Protein Kinase A. Neuron 96, 1070–1083 e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Saulnier JL, Yellen G, and Sabatini BL (2014). A PKA activity sensor for quantitative analysis of endogenous GPCR signaling via 2-photon FRET-FLIM imaging. Front Pharmacol 5, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Ko J, Park E, Lee JR, Yoon J, Lim S, and Kim E (2002). Phosphorylation of stargazin by protein kinase A regulates its interaction with PSD-95. J Biol Chem 277, 12359–12363. [DOI] [PubMed] [Google Scholar]
- Constantinople CM, and Bruno RM (2011). Effects and mechanisms of wakefulness on local cortical networks. Neuron 69, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney NA, and Ford CP (2014). The timing of dopamine- and noradrenaline-mediated transmission reflects underlying differences in the extent of spillover and pooling. J Neurosci 34, 7645–7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehmelt L, and Halpain S (2005). The MAP2/Tau family of microtubule-associated proteins. Genome Biol 6, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, and Svoboda K (1997). Photon upmanship: why multiphoton imaging is more than a gimmick. Neuron 18, 351–357. [DOI] [PubMed] [Google Scholar]
- Depry C, Allen MD, and Zhang J (2011). Visualization of PKA activity in plasma membrane microdomains. Mol Biosyst 7, 52–58. [DOI] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, and Scott JD (2005). The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437, 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn TA, Wang CT, Colicos MA, Zaccolo M, DiPilato LM, Zhang J, Tsien RY, and Feller MB (2006). Imaging of cAMP levels and protein kinase A activity reveals that retinal waves drive oscillations in second-messenger cascades. J Neurosci 26, 12807–12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP (2014). The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, and Williams JT (2009). The time course of dopamine transmission in the ventral tegmental area. J Neurosci 29, 13344–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, and Corbin JD (1994). Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol 56, 237–272. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, and Kandel ER (1993). Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260, 1661–1664. [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, and Abrams TW (1992). cAMP modulates multiple K+ currents, increasing spike duration and excitability in Aplysia sensory neurons. Proc Natl Acad Sci U S A 89, 11481–11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto A, Nakahara I, Yamaguchi T, Kamioka Y, Sumiyama K, Matsuda M, Nakanishi S, and Funabiki K (2015). Circuit-dependent striatal PKA and ERK signaling underlies rapid behavioral shift in mating reaction of male mice. Proc Natl Acad Sci U S A 112, 6718–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA (1991). Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41, 1–24. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Castellucci VF, Bayley H, and Schwartz JH (1987). A molecular mechanism for long-term sensitization in Aplysia. Nature 329, 62–65. [DOI] [PubMed] [Google Scholar]
- Greengard P (2001). The neurobiology of slow synaptic transmission. Science 294, 1024–1030. [DOI] [PubMed] [Google Scholar]
- Grieger JC, Choi VW, and Samulski RJ (2006). Production and characterization of adeno-associated viral vectors. Nat Protoc 1, 1412–1428. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Magee AI, Childs JE, and Marshall CJ (1989). All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, and Svoboda K (2008). A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A 105, 19264–19269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, and Yasuda R (2016). Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, and Arber S (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hires SA, Zhu Y, and Tsien RY (2008). Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc Natl Acad Sci U S A 105, 4411–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunnicutt BJ, Long BR, Kusefoglu D, Gertz KJ, Zhong H, and Mao T (2014). A comprehensive thalamocortical projection map at the mesoscopic level. Nat Neurosci 17, 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, and Goodman RH (2001). CREB signaling--timing is everything. Sci STKE 2001, PE1. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, and LeDoux JE (2011). Molecular mechanisms of fear learning and memory. Cell 147, 509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalen P, Rosegren E, Lindvall O, and Bjorklund A (1989). Hippocampal Noradrenaline and Serotonin Release over 24 Hours as Measured by the Dialysis Technique in Freely Moving Rats: Correlation to Behavioural Activity State, Effect of Handling and Tail-Pinch. Eur J Neurosci 1, 181–188. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, and Matsuda M (2011). Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell 22, 4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, and Sabatini BL (2011). Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer SZ (2015). alpha2-Adrenoceptors in the treatment of major neuropsychiatric disorders. Trends Pharmacol Sci 36, 196–202. [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ 3rd, Gu X, Zanella S, et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, and Nicoll RA (1986a). Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol 372, 221–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, and Nicoll RA (1986b). Cyclic adenosine 3’,5’-monophosphate mediates beta-receptor actions of noradrenaline in rat hippocampal pyramidal cells. J Physiol 372, 245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, Kusefoglu D, Hooks BM, Huber D, Petreanu L, and Svoboda K (2011). Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron 72, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, O’Connor DH, Scheuss V, Nakai J, and Svoboda K (2008). Characterization and subcellular targeting of GCaMP-type genetically-encoded calcium indicators. PLoS One 3, e1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Kwon MC, Dory Y, and Frostig RD (1993). Characterization of functional organization within rat barrel cortex using intrinsic signal optical imaging through a thinned skull. Proc Natl Acad Sci U S A 90, 9998–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Marin P, Bockaert J, and Mannoury la Cour C (2008). Signaling at G-protein-coupled serotonin receptors: recent advances and future research directions. Trends Pharmacol Sci 29, 454–464. [DOI] [PubMed] [Google Scholar]
- Mitchell HA, and Weinshenker D (2010). Good night and good luck: norepinephrine in sleep pharmacology. Biochem Pharmacol 79, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Lee SJ, and Yasuda R (2008). Highly sensitive and quantitative FRET-FLIM imaging in single dendritic spines using improved non-radiative YFP. Brain Cell Biol 36, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Miyazaki M, Aoki R, Zama T, Inouye S, Hirose K, Iino M, and Hagiwara M (2000). A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nat Biotechnol 18, 313–316. [DOI] [PubMed] [Google Scholar]
- Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, and Bergles DE (2014). Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, and Storm JF (1993). PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Petersen SE, and Posner MI (2012). The attention system of the human brain: 20 years after. Annu Rev Neurosci 35, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, and Lefkowitz RJ (2002). Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3, 639–650. [DOI] [PubMed] [Google Scholar]
- Polack PO, Friedman J, and Golshani P (2013). Cellular mechanisms of brain state-dependent gain modulation in visual cortex. Nat Neurosci 16, 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, and Svoboda K (2003). ScanImage: Flexible software for operating laser-scanning microscopes. BioMedical Engineering OnLine 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat Methods 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeberg NT, Sandberg SG, Johnson JA, Phillips PE, and Wightman RM (2017). Hitchhiker’s Guide to Voltammetry: Acute and Chronic Electrodes for in Vivo Fast-Scan Cyclic Voltammetry. ACS Chem Neurosci 8, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W (2006). Behavioral theories and the neurophysiology of reward. Annu Rev Psychol 57, 87–115. [DOI] [PubMed] [Google Scholar]
- Scott JD, and Pawson T (2009). Cell signaling in space and time: where proteins come together and when they’re apart. Science 326, 1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Hunt S, and Sah P (2015). Norepinephrine and Corticotropin-Releasing Hormone: Partners in the Neural Circuits that Underpin Stress and Anxiety. Neuron 87, 468–470. [DOI] [PubMed] [Google Scholar]
- Tang S, and Yasuda R (2017). Imaging ERK and PKA Activation in Single Dendritic Spines during Structural Plasticity. Neuron 93, 1315–1324 e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurkauf WE, and Vallee RB (1982). Molecular characterization of the cAMP-dependent protein kinase bound to microtubule-associated protein 2. J Biol Chem 257, 3284–3290. [PubMed] [Google Scholar]
- Tillo SE, Xiong WH, Takahashi M, Miao S, Andrade AL, Fortin DA, Yang G, Qin M, Smoody BF, Stork PJS, et al. (2017). Liberated PKA Catalytic Subunits Associate with the Membrane via Myristoylation to Preferentially Phosphorylate Membrane Substrates. Cell Rep 19, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, and Nicoll RA (1994). Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science 265, 1878–1882. [DOI] [PubMed] [Google Scholar]
- Xu D, Farmer A, Collett G, Grishin NV, and Chook YM (2012). Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell 23, 3677–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Goto A, Nakahara I, Yawata S, Hikida T, Matsuda M, Funabiki K, and Nakanishi S (2015). Role of PKA signaling in D2 receptor-expressing neurons in the core of the nucleus accumbens in aversive learning. Proc Natl Acad Sci U S A 112, 11383–11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapo C, Nair AG, Clement L, Castro LR, Hellgren Kotaleski J, and Vincent P (2017). Detection of phasic dopamine by D1 and D2 striatal medium spiny neurons. J Physiol 595, 7451–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, and Malenka RC (2003). A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 6, 15–16. [DOI] [PubMed] [Google Scholar]
- Yasuda R (2006). Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Curr Opin Neurobiol 16, 551–561. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, and Svoboda K (2006). Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci 9, 283–291. [DOI] [PubMed] [Google Scholar]
- Yellen G, and Mongeon R (2015). Quantitative two-photon imaging of fluorescent biosensors. Curr Opin Chem Biol 27, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, and Pozzan T (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295, 1711–1715. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, and Tsien RY (2005). Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature 437, 569–573. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ma Y, Taylor SS, and Tsien RY (2001). Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A 98, 14997–15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MY, and Beyer CE (2006). Measurement of neurotransmitters from extracellular fluid in brain by in vivo microdialysis and chromatography-mass spectrometry. J Pharm Biomed Anal 40, 492–499. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Huang H, Zhang L, Wu R, Chung CI, Zhang SQ, Torra J, Schepis A, Coughlin SR, Kornberg TB, et al. (2018). Visualizing Dynamics of Cell Signaling In Vivo with a Phase Separation-Based Kinase Reporter. Mol Cell 69, 334–346 e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Sia GM, Sato TR, Gray NW, Mao T, Khuchua Z, Huganir RL, and Svoboda K (2009). Subcellular dynamics of type II PKA in neurons. Neuron 62, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, and Webb WW (2003). Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21, 1369–1377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 6. Representative intensity z-stack of in vivo 2pFLIM imaging.
Several cortical neurons and their neurites are shown. Field of view is 140 μM. The animal was anesthetized to minimize potential movement artifacts that may blur neurites.
Movie S2. Related to Figure 6. The lifetime z-stack of Movie S1.
The lifetime scale is same as that in Figure 6A.
Movie S3. Related to Figure 7. Representative video of mouse resting and running on motorized treadmill.