

Abstract
β-Hydroxy difluoromethyl ketones represent the newest class of agonists of the GABAB receptor, and they are structurally distinct from all other known agonists at this receptor because they do not display the carboxylic acid or amino group of γ-aminobutyric acid (GABA). In this report, the design, synthesis, and biological evaluation of additional analogues of β-hydroxy difluoromethyl ketones characterized the critical nature of the substituted aromatic group on the lead compound. The importance of these new data is interpreted by docking studies using the X-ray structure of the GABAB receptor. Moreover, we also report that the synthesis and biological evaluation of β-amino difluoromethyl ketones provided the most potent compound across these two series.
Keywords: GABA, receptor, fluorine, agonist, GABAB, gamma-aminobutyric acid
Graphical abstract
The inhibitory neurotransmitter, γ-aminobutyric acid (GABA), reduces the excitability of neurons and assists in the regulation of other neurotransmitters, especially in the central nervous system. The two major types of GABA receptors are GABAA and GABAB and both are validated targets for drug discovery.1 The GABAA receptors are ion channels and can be controlled by three classes of pharmaceuticals, the barbiturates, the benzodiazepines, and the newer non-benzodiazepine sedatives, such as zaleplon and zopiclone.2 The GABAB receptors are metabotropic G-protein-coupled receptors, and serve as the target of the muscle relaxant, baclofen.3 Moreover, the GABAB receptors are the focus of drug discovery efforts in muscle spasticity disorders,3 schizophrenia,4 pain,5 and gastroesophageal reflux disease (GERD).6 Agonists,7 antagonists,8 positive allosteric modulators,9,10 and negative allosteric modulators11 of the GABAB receptor are known, and in 2013, the X-ray structures of this receptor in the ligand-free state and in the presence of agonists and antagonists were reported.12
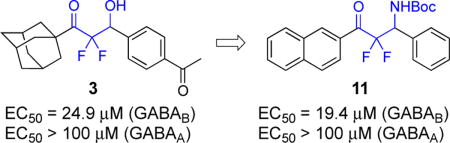
Nearly all of the known agonists of the GABAB receptor display the structure of GABA, and, for example, baclofen is the 3-para-chlorophenyl analogue of GABA (Figure 2).13 Although the pharmaceutical formulation of baclofen is a racemic mixture, the (R)-(−)-enantiomer is significantly more active. The 3-phenyl derivative is the agent, phenibut, and in a similar fashion, (R)-(−)-phenibut is the more active enantiomer.14 Also, the 2-chlorothienyl group is a surrogate for the para-chlorophenyl group of baclofen and is displayed in GABAB agonist 1.15 Other key analogues of baclofen, which are also agonists of the GABAB receptor, are the pyridinyl methoxy derivative 2,16 (R)-(−)-GABOB,17 and CGP44532.18 The only known agonist of the GABAB receptor that does not display the backbone of GABA or baclofen is the difluoromethyl ketone 3.19
Figure 2.
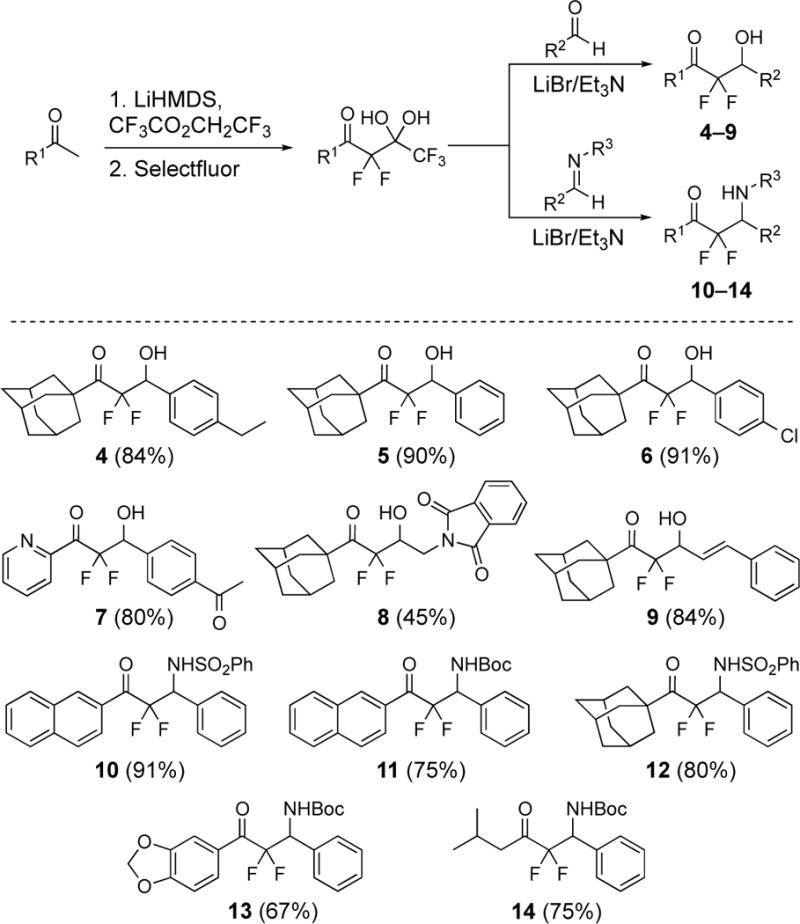
Preparation of β-hydroxy difluoromethyl ketones 4–9 and β-amino difluoromethyl ketones 10–14. Isolated yields for the aldol and imino-aldol reactions are given in parenthesis.
The β-hydroxy difluoromethyl ketones are a distinct group of ligands of the GABAB receptor and were first reported in 2013.19 The structure–activity relationships for the lead compound 3 in this series are that the fluorines and the β-hydroxy substituent are required for activity (Scheme 1).19 Specifically, the non-fluorinated analogue of 3 is inactive and methyl ether derivative created from methylation of the β-hydroxy substituent of 3 is also inactive. The bulky, lipophilic adamantyl group or naphthyl group is common in other active derivatives. On the other hand, the para-acetyl phenyl group of 3 tolerates many other structures such as alkyl, alkenyl, and aryl groups. Although these data are insightful, the agent 3 presents additional opportunities to identify new structure-activity relationships for agonist activity for the GABAB receptor. In the present study, we have prepared new β-hydroxy difluoromethyl ketones and characterized the β-amino difluoromethyl ketones as another complementary scaffold.
Scheme 1.
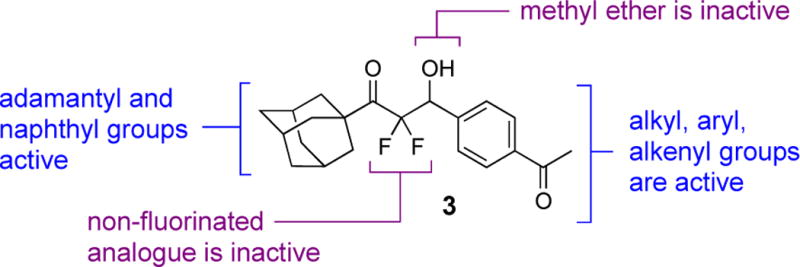
Structure–activity relationships for compd 3 and agonist activity at the GABAB receptor.
These unique compounds were discovered following the development of a new synthetic protocol that uses pentafluoro-gem-diols to produce difluoroenolates for aldol reactions20,21 and was later extended to imino-aldol reactions.22,23 The first objective was to define the role of substituents on the para-acetylphenyl group of 3. Compounds 4–6 were synthesized,24 and each displays a change to the para-acetyl group on the phenyl ring (Figure 3). Compound 7 was prepared to understand if a heteroaromatic group could replace the adamantyl group. The analogue 8 bears a larger isoindolinedione to replace the para-acetyl phenyl group. Next, the β-amino difluoromethyl ketones 10–14 were synthesized using the literature methods.22,23 The naphthyl and adamantyl groups were conserved (i.e., 10–11 and 12, respectively) and the N-phenylsulfonyl and N-tert-butylcarbamate groups were selected as substituents for the amine.
Figure 3.
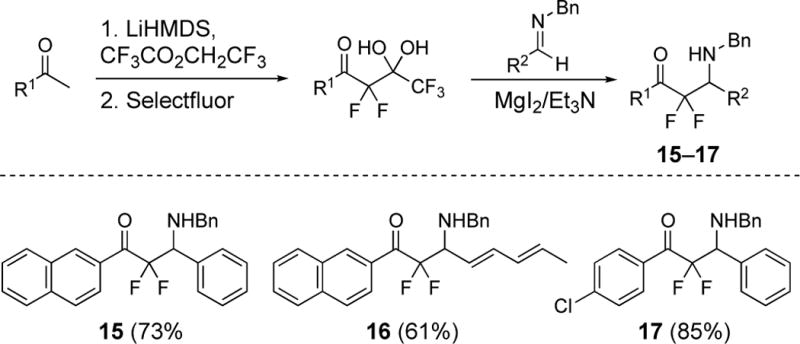
Synthesis of β-amino difluoromethyl ketones 15, 16, and 17. Isolated yields for the imino-aldol reactions are given in parenthesis. See ref. 25.
In 2018, we reported a procedure in which unactivated imines bearing N-benzyl, aryl, or alkyl group reacted with difluoroenolates generated from pentafluoro-gem-diols in the presence of magnesium salts.25 This process enables the creation of additional β-amino difluoromethyl ketones displaying a N-benzyl group to complement those bearing N-phenylsulfonyl and N-tert-butylcarbamate group (Figure 4). The naphthyl derivative 15 completes the series from 10 and 11, and compound 16 and 17 were prepared to obtained additional structure-activity data.25
Figure 4.
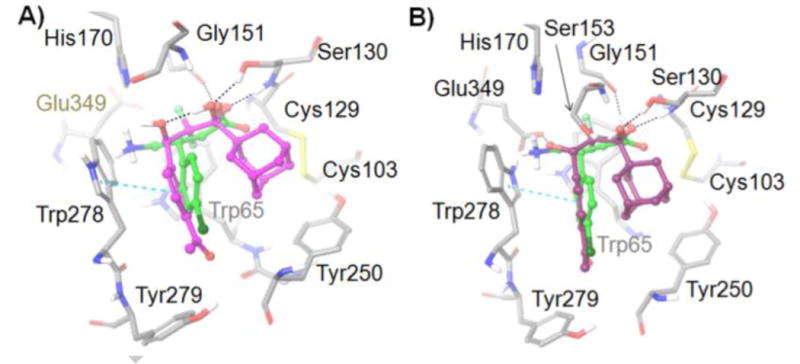
Active site residues (gray) of the GABAB receptor from docking studies with: A) (R)-3 (GlideScore = −10.080) (magenta) and B) (S)-3 (GlideScore = −9.606) (purple). (R)-Baclofen (GlideScore = −11.063) is also shown in each image (green). Amino acids are depicted as tubes and ligands are represented as ball-and-stick. H-bonds and aromatic π–π stacking are shown as black and cyan dashed lines, respectively.
Compounds 4–17 were tested for agonist activity at the GABAA and GABAB receptors according to the previously reported procedures.19 The screen for agonist activity at the GABAB receptor is conducted in a HEK293/Cre-luc cell line expressing both subunits of the human GABAB receptor. Activation of the GABAB receptor accesses an endogenous signaling pathway and results in the inhibition of cAMP production. In this assay, forskolin is applied to stimulate cAMP production, and inhibition of the forskolin-stimulated cAMP production is measured (assays are performed in triplicate). All of the compounds that display activity at the GABAB receptor are also screened for agonist activity at the GABAA receptor using a whole-cell voltage-clamp method with HEK293 cells transfected with the α1β2γ2 subunits of the rat GABAA receptor. The pharmaceutical, baclofen, is a racemic mixture and selective agonist for the GABAB receptor; however, most of its pharmacological activity results from the (−)-baclofen enantiomer.19 In a similar fashion, the lead compound 3 also has more agonist activity from a single enantiomer (i.e., the (+)-3-enantiomer); however, an absolute stereochemical assignment of secondary alcohol was not reported.19 The fluorinated compounds 4–17 were all tested as racemic mixtures. The β-hydroxy difluoromethyl ketones 4–8 do not display any observable activity, even at concentrations up to 100 μM. These results demonstrate the low tolerance for changes to the para-acetyl phenyl group, especially as the replacement of the acetyl with an ethyl group produces an inactive compound (i.e., compound 4). On the other hand, some structural variation is allowed because our prior studies validated that the styrene 9 displays activity of 40 μM compared to the lead compound 3 at 24.9 μM at the GABAB receptor.19 The β-amino difluoromethyl ketone 11 was characterized as the most active agent across the entire difluoromethyl ketone series. The β-amino difluoromethyl ketone 11 displays EC50 = 19.4 μM, and, notably, when the N-Boc group in 11 is replaced with N-SO2Ph in 10 or with N-Bn in 15, agonist activity decreases to EC50 = 32.7 μM or EC50 = 91 μM, respectively. All three of these molecules display no activity at the GABAA receptor, which correlates well with the β-hydroxy difluoromethyl ketones, which also preferential for the GABAB receptor (over the GABAA receptor). The other β-amino analogues 12–14 and 16 were inactive at the GABAB receptor. The para-chlorophenyl derivative 17 displays moderate activity at the GABAB receptor.
Additional biological characterizations of compounds 3 and 11 were performed using a radio-ligand binding assay screen with cloned neurotransmitter receptors and transporters (Table 2).26 Difluoromethyl ketones are known in the literature to display activity at many targets,27,28 so these screens provide an important perspective on the relative role of these ligand for the GABAB receptor over other neurotransmitter receptors. Adrenoreceptors and biogenic amine, dopamine, opioid, histamine, muscarinic, serotonin, and sigma receptors were examined, and interestingly, both 3 and 11 display less than 30% inhibition and are considered inactive in all of these assays. These data strengthen the knowledge of the preference of this class of compounds (i.e., β-hydroxy difluoromethyl ketone and β-amino difluoromethyl ketone) for the GABAB receptor.
Table 2.
Radio-ligand binding assay screen for compds 3 and 11 in cloned receptors and transporters. Both 3 and 11 are inactive in all assays.
| Adrenoreceptors | α1A, α1B, α1Δ, α2A, β1, β2, β3 |
| Biogenic amine transporters | DAT, NET, SERT |
| Dopamine receptors | D1, D2, D3, D4, D5 |
| Opioid receptors | DOR, KOR, MOR |
| Histamine receptors | H1, H2, H3, H4 |
| Muscarinic receptors | M1, M2, M3, M4, MS |
| Serotonin receptors | 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT5A, 5-HT6, 5-HT7 |
| Sigma receptors | rSigma1, rSigma2 |
In order to integrate the new structure–activity relationships with the prior data, both enantiomers of the lead compound 3 were docked into the (R)-baclofen-bound GABAB receptor complex (PDB ID: 4MS4),12 using the Induced Fit Docking protocol of the Schrӧdinger small molecule drug discovery suite.24 The difluoromethyl ketone 3 is hydrated in the model, because difluoromethyl ketones are known to exist in the hydrated (gem-diol) form at physiological pH.19 The respective gem-diol, in turn, occupies a similar site as the carboxylate of baclofen. Hydrogen-bonding is conserved with Ser130 and Gly171 (as observed with GABA-bound X-ray structure, PDB-ID:4MS3) and the gem-diol maintains a close proximity to the Ser153 (Figure 5). The β-hydroxy substituent in both the (R)-3 and (S)-3 alcohols cannot occupy the same site as the amino group of baclofen due to the shorter intramolecular distance from the difluoromethyl ketone group. However, the para-acetylphenyl group of both the (R)-3 and (S)-3 compounds employs a similar site as the para-chlorophenyl group of baclofen and maintains key interactions with the Trp278 residue. Lastly, docking studies present potential docking poses for 3 that accommodate the bulky adamantyl group within the ligand binding site of the GABAB receptor.
Figure 5.
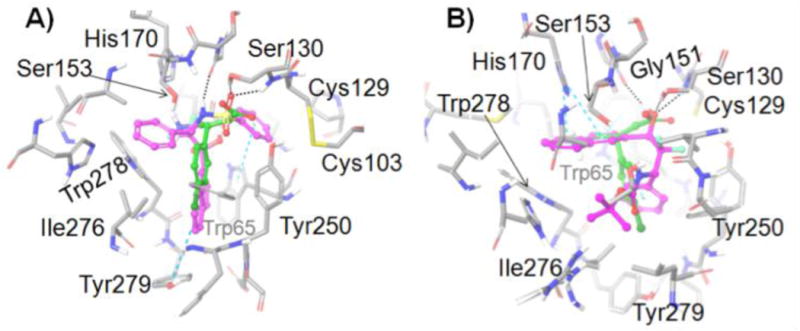
Active site residues (gray) of the GABAB receptor from docking studies with: A) (S)-10 (GlideScore = −11.037) (magenta) and B) (S)-11 (GlideScore = −10.592) (magenta). (R)-Baclofen is also shown in each image (green). Amino acids are depicted as tubes and ligands are represented as ball-and-stick. H-bonds and aromatic π–π stacking are shown as black and cyan dashed lines, respectively.
We attempted to dock both enantiomers of 10 and 11 into the GABAB receptor using both SP and XP Induced Fit Docking, and the methods gave similar results, so we report the XP ones. The sulfonamide group of (S)-10 is engaged in hydrogen-bonding with Ser130 whereas the gem-diol of (S)-11 is hydrogen-bonded to the Ser130 and Gly151, and both these functional groups lie in close proximity to Ser153 (Figure 5). The phenyl group attached to the stereogenic carbon in (S)-10 occupies the same site as the amino group of baclofen. By contrast, the naphthalene ring of (S)-11 occupies this site. Moreover, the naphthyl group of (S)-10 is in the site occupied by the para-chlorophenyl group of baclofen, whereas for (S)-11 this site is occupied by its phenyl group (Figure 6). (R)-10 failed to dock into the binding site in several trials with different grid settings. The docked pose of (R)-11 (GlideScore = −8.500) is provided in the supplementary information. Hence, we predict that the (S)-enantiomers are the more active for 10 and 11.
The difluoromethyl ketones represent the newest class of agonists of the GABAB receptor, and further investigations through synthesis, biological evaluation, and computational analysis have not only revealed that enhancements in potency are possible but also that the structure–activity relationships are distinct from other classes of agonists that display the structure of GABA. The critical nature of the substituent on the aromatic group of the β-hydroxy difluoromethyl ketone was characterized. Also, the identification of the β-amino difluoromethyl ketones as potent agonists of the GABAB receptor is another significant advance.
Supplementary Material
Figure 1.
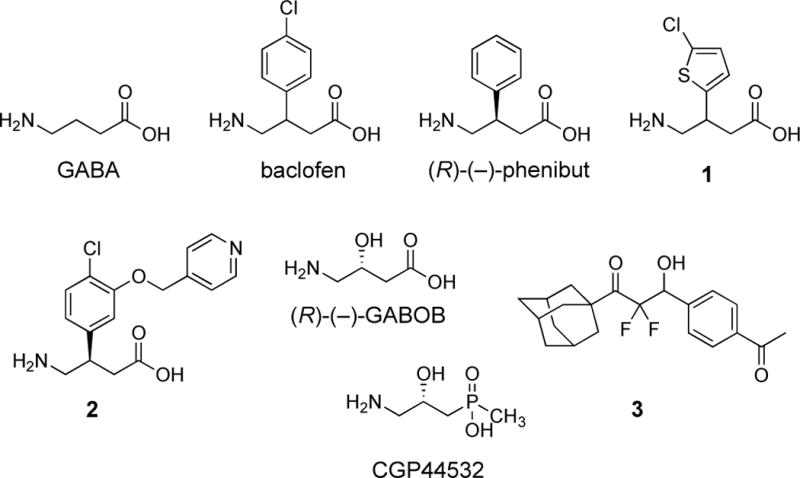
Structures of some known agonists of the GABAB receptor.
Table 1.
GABAA and GABAB assay data for baclofen and compds 3–17.
| compd | GABAB EC50 (μM)a | GABAA EC50 (μM)a |
|---|---|---|
| GABAc | 0.53 ± 0.33 | 2.30 ± 0.59 |
| (±)-baclofenc | 1.7 ± 0.10 | >100 |
| (±)-3b | 24.9 ± 1.30 | >100 |
| (−)-3b | 37.8 ± 0.78 | ndc |
| (+)-3b | 15.9 ± 1.84 | nd |
| 4 | >100d | nd |
| 5 | >100d | nd |
| 6 | >100d | nd |
| 7 | >100 | nd |
| 8 | >100 | nd |
| 9c | 40 ± 3.59 | >100 |
| 10 | 32.7 ± 1.44 | >100 |
| 11 | 19.4 ± 7.69 | >100 |
| 12 | >100 | nd |
| 13 | >100 | nd |
| 14 | >100 | nd |
| 15 | 91 ± 5.7 | >100 |
| 16 | >100 | nd |
| 17 | 57.4 ± 2.2 | >100 |
Values are given with the standard error.
Ref 19.
nd is not determined.
Cytotoxic in assay at 50 μM and 100 μM doses.
Highlights.
β-Hydroxy difluoromethyl ketones have distinct SAR data at the GABAB receptor
β-Amino difluoromethyl ketones are now characterized as GABAB agonists
Docking studies at the GABAB receptor suggest similar binding modes to baclofen
Acknowledgments
The authors acknowledge funding from the University of Mississippi, the Ralph W. and Grace M. Showalter Research Trust, and the National Institute of General Medical Sciences (Grant P20GM104932). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health (NIH). Receptor binding profiles was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth, MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Experimental procedures; molecular modeling procedures and supporting results; and 1H, 19F, and 13C NMR spectra. Supplementary data associated with this article can be found in the online version.
References and notes
- 1.Froestl W. Future Med Chem. 2011;3:163–175. doi: 10.4155/fmc.10.285. [DOI] [PubMed] [Google Scholar]
- 2.Hanson SM, Morlock EV, Satyshur KA, Czajkowski CJ. Med Chem. 2008;51:7243–7252. doi: 10.1021/jm800889m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Froestl W. Adv Pharmacol. 2010;58:19–62. doi: 10.1016/S1054-3589(10)58002-5. [DOI] [PubMed] [Google Scholar]
- 4.Mizukami K, Sasaki M, Ishikawa M, Iwakiri M, Hidaka S, Shiraishi H, Iritani S. Neurosci Lett. 2000;283:101–104. doi: 10.1016/s0304-3940(00)00939-3. [DOI] [PubMed] [Google Scholar]
- 5.Balerio GN, Rubio MC. Pharmacol Res. 2002;46:281–286. doi: 10.1016/s1043-6618(02)00147-0. [DOI] [PubMed] [Google Scholar]
- 6.Alstermar C, Amin K, Dinn SR, Elebrin T, Fjellström O, Fitzpatrick K, Geiss WB, Gottfries J, Guzzo PR, Harding JP, Holmén A, Kothare M, Lehmann A, Mattsson JP, Nilsson K, Sundén G, Swanson M, von Unge S, Woo AM, Wyle MJ, Zheng X. J Med Chem. 2008;51:4315–4320. doi: 10.1021/jm701425k. [DOI] [PubMed] [Google Scholar]
- 7.Froestl W, Mickel SJ, Hall RG, von Sprecher G, Strub D, Baumann PA, Brugger F, Gentsch C, Jaekel J. J Med Chem. 1995;38:3297–3312. doi: 10.1021/jm00017a015. [DOI] [PubMed] [Google Scholar]
- 8.Froestl W, Mickel SJ, von Sprecher G, Diel PJ, Hall RG, Maier L, Strub D, Melillo V, Baumann PA. J Med Chem. 1995;38:3313–3331. doi: 10.1021/jm00017a016. [DOI] [PubMed] [Google Scholar]
- 9.Guery S, Floersheim P, Kaupmann K, Froestl W. Bioorg Med Chem Lett. 2007;17:6206–6211. doi: 10.1016/j.bmcl.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mugnaini C, Pedani V, Casu A, Lobina C, Casti A, Maccioni P, Porcu A, Giunta D, Lamponi S, Solinas M, Dragoni S, Valoti M, Colombo G, Castelli MP, Gessa GL, Corelli F. J Med Chem. 2013;56:3620–3635. doi: 10.1021/jm400144w. [DOI] [PubMed] [Google Scholar]
- 11.Chen L-H, Sun B, Zhang Y, Xu T-J, Xia Z-X, Liu J-F, Nan F-J. ACS Med Chem Lett. 2014;5:742–747. doi: 10.1021/ml500162z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng Y, Bush M, Mosyak L, Wang F, Fan QR. Nature. 2013;504:254–259. doi: 10.1038/nature12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown KM, Roy KK, Hockerman GH, Doerksen RJ, Colby DA. J Med Chem. 2015;58:6336–6347. doi: 10.1021/jm5018913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dambrova M, Zvejniece L, Liepinsh E, Cirule H, Zharkova O, Veinberg G, Kalvinsh I. Eur J Pharmacol. 2008;583:128–134. doi: 10.1016/j.ejphar.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Pirard B, Carrupt P-A, Testa B, Tsai R-S, Berthelot P, Vaccher C, Debaert M, Durant F. Bioorg Med Chem. 1995;3:1537–1545. doi: 10.1016/0968-0896(95)00144-6. [DOI] [PubMed] [Google Scholar]
- 16.Xu F, Peng G, Phan T, Dilip U, Chen JL, Chernov-Rogan T, Zhang X, Grindstaff K, Annamalai T, Koller K, Gallop MA, Wustrow DJ. Bioorg Med Chem Lett. 2011;21:6582–6585. doi: 10.1016/j.bmcl.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Hinton T, Chebib M, Johnston GAR. Bioorg Med Chem Lett. 2008;18:402–404. doi: 10.1016/j.bmcl.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Ong J, Bexis S, Marino V, Parker DAS, Kerr DIB, Froestl W. Eur J Pharmacol. 2001;412:27–37. doi: 10.1016/s0014-2999(00)00945-6. [DOI] [PubMed] [Google Scholar]
- 19.Han C, Salyer AE, Kim EH, Jiang X, Jarrard RE, Powers MS, Kirchhoff AM, Salvador TK, Chester JA, Hockerman GH, Colby DA. J Med Chem. 2013;56:2456–2465. doi: 10.1021/jm301805e. [DOI] [PubMed] [Google Scholar]
- 20.Han C, Kim EH, Colby DA. J Am Chem Soc. 2011;133:5802–5805. doi: 10.1021/ja202213f. [DOI] [PubMed] [Google Scholar]
- 21.Han C, Kim EH, Colby DA. Synlett. 2012;23:1559–1563. [Google Scholar]
- 22.Xie C, Wu L, Mei H, Soloshonok VA, Han J, Pan Y. Tetrahedron Lett. 2014;55:5908–5910. [Google Scholar]
- 23.Xie C, Wu L, Zhou J, Mei H, Soloshonok VA, Han J, Pan YJ. Fluorine Chem. 2015;172:13–21. [Google Scholar]
- 24.See Supplementary Material for details
- 25.Nguyen AL, Khatri HR, Woods JR, Baldwin CS, Fronczek FR, Colby DA. J Org Chem. 2018;83:3109–3118. doi: 10.1021/acs.joc.7b03014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Nature. 2012;492:212–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fä C, Hardegger LA, Baitsch L, Schweizer WB, Meyer S, Bur D, Diederich F. Org Biomol Chem. 2009;7:3947–3957. doi: 10.1039/b908489d. [DOI] [PubMed] [Google Scholar]
- 28.Moore CL, Leatherwood DD, Diehl TS, Selkoe DJ, Wolfe MS. J Med Chem. 2000;43:3434–3442. doi: 10.1021/jm000100f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.