

Abstract
Overcoming the immunosuppressive tumor microenvironment (TME) is critical to realizing the potential of cancer immunotherapy strategies. Agonists of stimulator of interferon genes (STING), a cytosolic immune adaptor protein, have been shown to induce potent anti-tumor activity when delivered into the TME. However, the anionic properties of STING agonists make them poorly membrane permeable, and limit their ability to engage STING in the cytosol of responding cells. In this study, cationic liposomes with varying surface polyethylene glycol (PEG) levels were used to encapsulate cGAMP to facilitate its cytosolic delivery. In vitro studies with antigen-presenting cells (APCs) revealed that liposomal formulations substantially improved the cellular uptake of cGAMP and pro-inflammatory gene induction relative to free drug. Liposomal encapsulation allowed cGAMP delivery to metastatic melanoma tumors in the lung, leading to anti-tumor activity, whereas free drug produced no effect at the same dose. Injection of liposomal cGAMP into orthotopic melanoma tumors showed retention of cGAMP at the tumor site and co-localization with tumor-associated APCs. Liposomal delivery induced regression of injected tumors and produced immunological memory that protected previously treated mice from rechallenge with tumor cells. These results show that liposomal delivery improves STING agonist activity, and could improve their utility in clinical oncology.
Keywords: Liposomes, STING, cGAMP, cancer immunotherapy, adjuvants
Graphical abstract
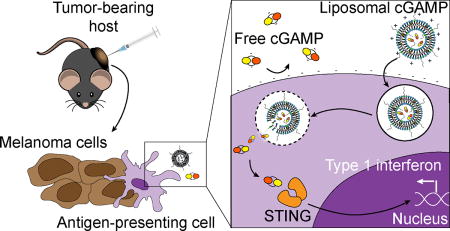
Immune activation is enhanced by STING agonist delivery via liposomes. Cationic liposomes show an improved ability to stimulate the STING pathway in immune cells in vitro relative to free drug. Liposomes allow increased activity of the drug in metastatic melanoma in the lungs, and enhance immune memory formation when injected into skin melanoma tumors.
1. Introduction
Cancer immunotherapy strategies have shown striking activity against a variety of tumor types. Immuno-oncology regimens involve leveraging immune cells, primarily cytotoxic T cells, to recognize and destroy tumor cells.[1] Current strategies such as checkpoint inhibitor antibodies,[2] adoptive T cell therapy,[3] chimeric antigen receptor T cell therapy,[4] and therapeutic cancer vaccines[5] have shown anti-tumor effects in the clinic. However, the majority of patients fail to achieve long-term disease control, making the development of complementary approaches to improve treatment efficacy a priority in oncology research.
Recently, interventions that act on the tumor microenvironment (TME) have gained increasing attention. The TME hinders the function of T cells as well as antigen-presenting cells (APCs) such as dendritic cells (DCs),[6] believed to be the primary APC that initiates anti-tumor T cell responses.[7] Tumor-resident DCs continuously collect tumor antigens and their subsequent exposure to immune danger signals can trigger their phenotypic maturation into potent T cell stimulators.[1] The stimulator of interferon genes (STING) pathway has emerged as critical for DC maturation in the TME.[8] STING is a cytosolic adaptor protein that is activated in certain microbial infections, autoimmunity, and cancer.[9] When cytosolic DNA is present in these settings, it is sensed by cyclic GMP-AMP synthase (cGAS) leading to production of the secondary messenger 2’3’-cyclic GMP-AMP (cGAMP), which binds to STING.[10,11] DNA shed by dying tumor cells triggers STING pathway activation in DCs leading to their phenotypic maturation, and the production of type I interferon and other cytokines.[12] Type I interferons, such as interferon β (IFN-β), are critical to the generation of anti-tumor T cell responses, and their intratumoral gene expression levels correlate with cytotoxic T cell infiltration into human melanoma metastases.[13] However, endogenous STING activation does not lead to tumor control and strategies to amplify STING signaling are being developed. It has been found that activating the STING pathway in the TME through exogenous delivery of STING agonists, like cGAMP, can augment anti-tumor immune responses. STING agonists applied to the TME produce innate and adaptive immune stimulation, leading to tumor control in mouse models of cancer.[14–16] Innate immune activation by STING agonists in the TME leads to vascular disruption and rapid tumor collapse, which is followed by T cell-mediated adaptive immunity against residual disease.[16] Intratumoral injection of an optimized STING agonist is currently being tested clinically in a phase I clinical trial in advanced cancer patients (NCT02675439). However, STING agonists such as cGAMP are hydrophilic and negatively charged, limiting their entry into the cytosol, a requirement for activation of the STING pathway. Additionally, systemic delivery of STING agonists for cancer therapy is currently not feasible, since they do not preferentially localize to tumor tissue and can induce off-target inflammation or autoimmunity. Ideally, STING agonists would be delivered efficiently into the cytosol of APCs in the target tissue to maximize drug efficacy. The development of delivery strategies that achieve this function may allow the full potential of these promising oncology drug candidates to be realized.
Biomaterials-based delivery strategies can be leveraged to improve the activity of immunologically active molecules by controlling their tissue and cell localization.[17] Liposomes, spherical lipid vesicle structures enclosing an aqueous core, are a class of nanomaterials that have been used extensively for nucleic acid delivery into cells.[18–20] Liposomes can be used to deliver immunomodulatory agents to immune cells and tumors,[21–24] and STING ligands have been delivered as vaccine adjuvants to the lymph nodes using liposomes.[25] Recent studies have explored STING agonist delivery via liposomes for anti-cancer therapy.[26,27] However, these studies did not compare the anti-tumor activity of the liposomal cGAMP formulation against free cGAMP, leaving the merit of liposomal delivery undefined. Additionally, these studies were performed with tumor models that were genetically modified to express proteins from foreign species, thus the efficacy of liposomal delivery of cGAMP into more clinically relevant, unmodified tumor models is still untested.
This study tests the hypothesis that cationic liposomes can enhance STING activation by cGAMP and treat melanoma tumors after systemic or intratumoral delivery. Cationic liposomes have been used extensively for delivering encapsulated molecules into cells. Their positive surface charge allows binding to the negatively charged cell membrane, internalization, and release of their cargo into the cytosol. In this study, cGAMP was encapsulated in cationic liposomes with varying levels of polyethylene glycol (PEG) content and their stability, in vitro interactions with DCs, and in vitro gene expression were evaluated and compared to free cGAMP. The ability of liposomal cGAMP to induce pro-inflammatory gene expression and provide anti-tumor activity in vivo was assessed in aggressive lung metastatic and orthotopic melanoma tumor models.
cGAMP was encapsulated in cationic liposomes as a delivery vehicle to promote cellular internalization and STING pathway activation (Figure 1-A). 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP)/cholesterol liposomes were chosen since they have been utilized extensively for in vivo gene delivery[28] and vaccine[29] applications, and have been shown to be safe in patients with advanced cancer.[30] cGAMP containing phosphorothioate bonds was utilized since it shows increased resistance to esterase degradation and provides improved biological activity compared to native cGAMP.[31] The effect of incorporating PEG-containing lipids in the liposome formulation was explored since PEGylation can improve particle stability in the presence of serum, and enhance in vivo persistence.[32] Liposome formulations were tested that contained 5 and 10 mol% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG(2000)) content, at which surface PEG primarily exhibits a brush-like conformation providing high liposome stability.[33] It was hypothesized that cationic liposome delivery of cGAMP to the TME would enhance STING pathway activation in tumor-resident APCs relative to cGAMP delivered in the free form (Figure 1-B). cGAMP contains two negative charges, which results in low permeability through the negatively charged cell membrane and hydrophobic membrane interior. Cationic liposome-formulated cGAMP can bind the net anionic cell membrane, leading to enhanced cell-association, uptake into endosomes, and cytosolic delivery of cGAMP, and potentially providing enhanced stimulation of the STING pathway and increased type I interferon production in the TME.
Figure 1.

Schematic of liposomal cGAMP structure and therapeutic strategy. a) 2’3’-cGAMP is encapsulated in cationic liposomes formed from 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) and cholesterol using thin film rehydration, freeze thawing, and membrane extrusion. A polyethylene glycol(PEG)-containing lipid (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]; DSPE-PEG(2000)) is optionally included in the liposome preparation to create a PEG coating that improves liposome stability. b) In a therapeutic setting, melanoma tumor-bearing hosts are injected with free or liposomal cGAMP, where cells, for example antigen-presenting cells (APCs), in the tumor microenvironment take up liposomal cGAMP concurrent with melanoma cell antigens. (Inset) Free cGAMP has limited transport into the cytosol due to the presence of two negative charges that limit its permeability through the negatively charged cell membrane. cGAMP encapsulated in cationic liposomes shows improved cell membrane binding and uptake. Once internalized into the endosomal compartment, cationic liposomes facilitate the release of cGAMP into the cytosol, where cGAMP binds to the stimulator of interferon genes (STING) adaptor molecule, leading to type I interferon production by the APC.
2. Results and Discussion
2.1 Synthesis and characterization of liposomal cGAMP
Liposomes encapsulating cGAMP were formed using the thin film rehydration method, freeze thawing, and membrane extrusion. This loading strategy encapsulated ~20% of the input cGAMP in all liposome formulations tested (Table S1) and led to controlled cGAMP release over several days after entrapment (Figure S1). All liposome formulations contained particles of ~160 nm z-average diameter in phosphate-buffered saline (PBS), as measured by dynamic light scattering (DLS) measurements and observed by cryo-electron microscopy (cryoEM) (Figure 2 A,B), similar to unloaded liposomes (Figure S2). To assess particle stability in biological media, particle size measurements were performed in complete cell culture medium containing 10% fetal calf serum (FCS). Non-PEGylated liposomes (PEG0-cGAMP) showed a large diameter increase in complete cell culture medium, likely due to anionic serum protein binding on the cationic liposome surface causing liposome aggregation. Liposomes with 5 mol% DSPE-PEG(2000) (PEG5-cGAMP) and 10 mol% DSPE-PEG(2000) (PEG10-cGAMP) showed only a slight increase in liposome size suggesting PEGylation reduced protein binding and largely prevented aggregation of the liposomes in serum-containing media. Next, the zeta potential of all formulations was measured. More positive liposome zeta potential increases cellular binding and uptake of cationic liposomes.[34] PEGylation reduced the zeta potential of liposomal cGAMP formulations (Figure 2-C), as has been demonstrated in other DOTAP-containing liposome systems.[34] These results show that cationic liposomes encapsulating cGAMP could be formed with varying degrees of PEGylation, leading to differing serum stability and surface charge properties.
Figure 2.

Physical characterization of liposomal cGAMP formulations. a) Representative Gaussian-smoothed size intensity DLS histograms of liposomal cGAMP formulations containing 0 mol% (PEG0-cGAMP), 5 mol% PEG (PEG5-cGAMP), and 10 mol% PEG (PEG10-cGAMP) measured in PBS (solid lines) or cell culture medium containing 10% FCS (hatched lines). Inset shows a representative cryoEM micrograph of each formulation (scale bars: 50 nm) b) Z-average diameter of the same liposomal cGAMP formulations in PBS or cell culture media. c) Zeta potential measurements of liposomal cGAMP formulations. Data are shown as mean and standard error of three repeated measurements, and are representative of three independent particle preparations.
2.2 Cationic liposomes enhance cell-association of cGAMP
Since stimulation of APCs in the TME is critical to cGAMP activity, the association of cGAMP in free or liposomal form with bone marrow-derived dendritic cells (BMDC) was studied. Fluorescein-conjugated cGAMP was used to allow monitoring of total cell-associated cGAMP by flow cytometry, and its cellular localization by confocal microscopy over a 12-hour period. Treatment of BMDCs with free cGAMP showed only a low magnitude cell-associated signal that remained unchanged over time (Figure 3-A,B). However, cells treated with liposomal cGAMP showed an order of magnitude higher amount of cell-associated cGAMP signal at all time points studied. PEG0-cGAMP produced a higher total cell-associated cGAMP signal after 1 hour and 6 hour incubation relative to PEGylated formulations. This difference waned over time with all liposomal formulations showing a similar amount of signal after 12 hours. Confocal microscopy confirmed that liposomal cGAMP had a higher magnitude of total cell-associated fluorescent signal than free cGAMP at all time points tested (Figure 3-C). Interestingly, the cellular localization differed between PEG0-cGAMP and the PEGylated formulations after 12 hours of incubation (Figure 3-D). Whereas PEG0-cGAMP showed a punctate pattern of fluorescence throughout the cell, a large fraction of the cGAMP fluorescence in the PEGylated liposome formulations appeared to be membrane localized.
Figure 3.

In vitro association of various cGAMP formulations with BMDC. a) Representative flow cytometry histograms showing timecourse of BMDC association with fluorescein-cGAMP delivered in free or liposomal form at 1 µg ml−1. b) Quantitative flow cytometry timecourse of cell-associated fluorescein-cGAMP signal. Triplicate sampels were used in (a) and (b). c) Confocal microscopy timecourse of BMDC association with fluorescein-cGAMP at 1 µg ml−1 (representative images from 3 slides/condition, scale bar: 20 µm). d) Magnified images of BMDC association with liposomal fluorescein-cGAMP (scale bar: 10 µm). Arrows indicate representative areas of membrane association. Data are shown as mean and standard deviation.
To further explore cellular localization of cGAMP, mild detergent treatment and phalloidin staining of the cytoskeleton were used to visualize cell morphology simultaneously with cGAMP signal (Figure 4-A). This staining protocol is expected to lead to disruption of extracellular liposomes and removal of cell membrane-bound cGAMP signal, allowing the assessment of internalized cGAMP. A higher internalized cGAMP signal was present in PEG0-cGAMP relative to the PEGylated formulations. Strong punctate cGAMP signal was present within BMDC after 1-hour incubation with PEG0-cGAMP, indicating rapid uptake into cells compared to PEGylated formulations. Z-stack confocal imaging at 12 hours showed punctate fluorescence signal throughout the cytoplasm in all liposome formulations, with PEG0-cGAMP showing the brightest signal intensity (Figure 4-B). Thus, even though all liposomal formulations showed a similar magnitude of total cell-associated signal (Figure 3A,B), PEGylation negatively affected internalization, which may hinder downstream STING pathway activation. This observation is consistent with reports showing that non-PEGylated cationic liposomes have a higher internalized to membrane-bound liposome ratio, whereas high amounts of PEGylation leads to a reversal of this ratio.[34] PEG-containing cationic liposomes are also known to have slower kinetics of uptake from the cell membrane.[35] Altogether, these data show that liposomal formulations enhance cell-association of cGAMP relative to free cGAMP, and that non-PEGylated liposomes provide faster cell-association kinetics and a higher level of internalization of cGAMP in vitro.
Figure 4.

In vitro uptake of various cGAMP formulations by BMDC. a) Confocal microscopy timecourse of BMDC uptake of fluorescein-cGAMP in free or liposomal form at 1 µg ml−1 (representative image from 3 slides/condition, scale bar: 20 µm). b) Z-stack maximum intensity projection image of BMDC uptake of liposomal fluorescein-cGAMP (scale bar: 20 µm). Main image shows view in z-plane and orthogonal side views are shown on top and right.
2.3 Liposomal formulation increases STING pathway stimulation by cGAMP
Next, the in vitro bioactivity of the various formulations was explored. To explore changes in potency of the liposomal formulations relative to free cGAMP in stimulating type I interferon production, the RAW-Blue ISG macrophage reporter cell line was used, since macrophages are a critical cell type for the anti-tumor activity of cGAMP in the TME.[16] These cells secrete a reporter enzyme in response to interferon regulatory factor (IRF) pathway activation by cGAMP directly stimulating the STING pathway. In addition, type I interferons produced as a result of STING pathway activation may induce further IRF pathway activation, increasing reporter enzyme secretion. Liposomal cGAMP induced IRF pathway reporter activity at an order of magnitude lower concentration of cGAMP compared to free form, indicating an increased overall potency of cGAMP when delivered in liposomal form (Figure 5-A). Unloaded liposomes did not produce a similar magnitude response in these reporter cells compared to cGAMP-loaded liposomes (Figure S3). Indeed, all cGAMP-loaded liposome formulations showed lower half maximal effective concentration (EC50) values relative to free cGAMP in this assay (Table S2). The maximal reporter induction decreased with increased PEGylation, with PEG10-cGAMP producing similar magnitude reporter cell responses as empty PEG0 liposomes at high levels. This may be due to slower kinetics and lower magnitude of internalization or cytosolic delivery of liposome contents with increased PEGylation of liposomes,[35] leading to reduced overall IRF pathway activation in these reporter cells over the culture period. The EC50 values were not increased with PEGylation as would be expected if internalization and cytosolic delivery were inhibited by PEGylation, leading to reduced STING pathway activation. This may be because this reporter cell assay is not simply a measure of direct activation of STING by cGAMP alone, as stated above. Further studies of the intracellular dynamics of cGAMP delivery with cationic liposomes are warranted.
Figure 5.

Comparison of biological activity of various cGAMP formulations. a) Dose-response curves in response to various cGAMP formulations by RAW-Blue ISG cells, which produce a reporter enzyme in response to stimulation of the interferon regulatory factor (IRF) pathway by cGAMP (3 wells/condition). Connecting lines are variable-slope dose-response curve fits. b) Representative flow cytometry histograms of BMDC CD86 expression after stimulation with various cGAMP formulations, and CpG as a positive control, at 1 µg ml−1 for 8 hours (3 wells/condition). Results with BMDCs isolated from both wild-type (WT) and STINGgt/gt mice, which lack STING, are shown. c) Gene expression analysis for interferon-β (Ifnb1), (C-X-C motif) ligand 9 (Cxcl9), (C-X-C motif) ligand 10 (Cxcl10), and tumor necrosis factor (Tnf). WT and STINGgt/gt mice treated with various cGAMP formulations at 1 µg ml−1 for 4 hours. PBS treatment was used as a negative control, while CpG treatment at 1 µg ml−1 for 2 hours was used as a positive control (4 wells/condition). Data are shown as mean and standard deviation.
The enhancement of cGAMP potency by liposomes in stimulating BMDC maturation was tested next. STING pathway activation and IFN-β production in DCs are thought to be critical to generating endogenous anti-tumor immune responses.[12] In these studies, BMDCs from STINGgt/gt mice, which lack STING, were used as a control to verify STING-dependent DC maturation by the various cGAMP formulations. Upregulation of CD86, an important co-stimulatory receptor for induction of anti-tumor T cell responses, was assessed as a proxy for BMDC activation by the various cGAMP formulations. Liposomal cGAMP induced higher levels of CD86 expression on wild-type BMDCs compared to free cGAMP (Figure 5-B). This effect was STING pathway-dependent, since STINGgt/gt BMDCs failed to upregulate CD86 in response to cGAMP-containing formulations, but did upregulate CD86 expression in response to CpG oligonucleotide, which signals through the toll-like receptor 9 pathway. These results show that liposomal cGAMP formulations enhance DC maturation through the STING pathway relative to free cGAMP.
Next, the impact of cGAMP formulation on induction of pro-inflammatory gene expression in BMDC was tested. In addition to IFN-β, we assessed expression of (C-X-C motif) ligand 9 (CXCL9) and (C-X-C motif) ligand 10 (CXCL10), two critical chemokines for recruitment of effector T cells to tumors,[36] and tumor necrosis factor (TNF), a cytokine that induces rapid tumor vascular destruction after STING agonist delivery into tumors.[16] Liposomal formulation induced significantly higher expression of all of the pro-inflammatory genes tested in wild-type BMDCs relative to free cGAMP (Figure 5-C). Induction of these genes was not observed in STINGgt/gt BMDC, verifying STING pathway specificity. PEG0-cGAMP induced significantly higher levels of Ifnb1, Cxcl9, and TNF expression relative to the PEGylated formulations. Empty liposomes did not induce Ifnb1 expression, and free cGAMP in the presence of empty liposomes did not show similar enhancement of Ifnb1 induction (Figure S4), suggesting that cGAMP must be encapsulated in liposomes to produce the observed potentiation of inflammatory gene expression. These data indicate that liposome-encapsulated cGAMP induces superior pro-inflammatory gene induction via the STING pathway compared to free cGAMP.
2.4 Liposomal cGAMP has anti-tumor activity against metastatic melanoma
The potency of cGAMP delivered as a systemic therapy was next tested against metastatic lung tumors in the B16-F10 melanoma model. DOTAP/cholesterol liposomes passively target the lungs when delivered systemically and have been used for gene delivery to lung tumors.[33,37,38] We utilized PEG5-cGAMP in these studies to provide serum stability while maintaining suitable delivery efficiency, since DOTAP/cholesterol liposomes with 4 mol% PEG have been shown to have higher proportional delivery to the lung over other tissues relative to liposomes with 10 mol% PEG.[33] After intravenous injection of free cGAMP, gene expression analysis in the lungs of tumor-bearing mice revealed no significant increase in inflammatory gene expression relative to PBS injection (Figure 6-A). Strikingly, intravenously injected PEG5-cGAMP induced ~200× and ~1400× increases in Ifnb1 and Cxcl9 expression in the lung. Empty liposomes alone did not produce significant increases in the expression of these genes in the lungs of mice, indicating this inflammatory response was cGAMP-dependent (Figure S5). Next, the treatment efficacy of free cGAMP and PEG5-cGAMP against established lung tumors were compared (Figure 6-B). In this study, mice bearing B16-F10 tumors in the lung were concurrently treated with anti-CTLA-4 and anti-PD-1, since this regimen has recently been approved to treat BRAF V600 wild-type unresectable or metastatic melanoma in humans.[39] When lungs were assessed after treatment, neither free nor PEG5-cGAMP formulations reduced the number of tumor nodules in the lung relative to PBS-treated controls (Figure 6-C). The length of the longest axis of single metastatic foci was measured as an estimate of the size of individual tumor nodules in this multifocal tumor model. PEG5-cGAMP treated mice showed a significantly reduced median nodule length in the lung when compared to PBS-treated controls, whereas free drug-treated mice showed no significant reduction in individual tumor length (Figure 6-D, Figure S6, Table S3). This reduction in the size of individual tumor nodules on the lung is likely due to improved immune activity of cGAMP in the lung using liposomal delivery compared to free drug. Further optimization of particle characteristics, treatment regimen, or combination with complementary therapies such as radiation, may improve efficacy. These data show that liposomal cGAMP induces superior pro-inflammatory gene expression in the lung after systemic delivery and provides anti-tumor activity against metastatic melanoma in the lung, whereas free cGAMP does not.
Figure 6.

Impact of systemic delivery of cGAMP in a therapeutic lung metastatic melanoma model. a) Gene expression analysis for interferon-β (Ifnb1), and (C-X-C motif) ligand 9 (Cxcl9) in tumor-bearing lung tissue after intravenous injection of 0.35 µg of free or liposomal cGAMP, or PBS as a control (1-way ANOVA with Tukey’s post hoc test, n=4 mice/condition, ****p < 0.0001, n.s. = not significant). Mice were injected intravenously with B16-F10 cells to establish lung metastases three days prior to treatment. Lungs were harvested for gene expression analysis four hours after treatment. b) Treatment scheme for therapeutic B16-F10 melanoma lung metastatic model. Mice were injected in the tail vein with B16-F10 melanoma cells and treated twice with PBS, free cGAMP or PEG5-cGAMP intravenously on the indicated days. Anti-CTLA-4 and anti-PD-1 antibodies were given by intraperitoneal injection three times as indicated. c) Quantitation of total tumor nodules present on lung surface after study conclusion. Kruskal-Wallis analysis with Dunn’s multiple comparison test was performed and showed no significant differences. d) Quantitation of median tumor nodule length on the lung surface (Kruskal-Wallis analysis with Dunn’s multiple comparison test, **p < 0.01). Data are shown as mean and standard deviation.
2.5 Liposomal cGAMP stimulates immune memory against melanoma
Based on the anti-tumor activity of liposomal cGAMP in the metastatic B16-F10 model, an orthotopic skin B16-F10 melanoma model was used to study the direct effect of the liposomal cGAMP formulations in the TME. In previous studies, STING agonists have been intratumorally injected at doses of 10–100 µg to treat established orthotopic B16-F10 tumors.[14,15] To assess enhancement of cGAMP anti-tumor activity by liposomes, a lower dose of 1 µg was used in this study. Gene expression analysis from tumors injected with free or liposomal cGAMP showed increased mean pro-inflammatory gene expression with liposomal formulations relative to free cGAMP (Figure 7-A). Surprisingly, PEG10-cGAMP induced the highest mean increase in inflammatory gene expression and produced significantly increased Cxcl9 expression compared to free cGAMP. The anti-tumor efficacy of these formulations in treating established orthotopic melanoma tumors was then tested (Figure 7-B). Empty liposomes were also tested as DOTAP itself has been shown to have adjuvant activity,[40,41] which may result in anti-tumor effects in the TME. Control tumors treated with PBS injection or empty liposomes grew rapidly, whereas tumors treated with cGAMP-containing formulations showed delayed growth (Figure 7-C). Free cGAMP, PEG5-cGAMP and PEG10-cGAMP treatment led to complete regression of tumors in half of the mice. PEG0-cGAMP showed delayed tumor growth but incomplete tumor control as these tumors eventually grew out. Next, to assess the induction of immunological memory after regression of tumors by local cGAMP treatment, surviving mice from the free cGAMP, PEG5-cGAMP, and PEG10-cGAMP treatment groups were rechallenged with B16-F10 cells in the opposite flank 60 days after primary challenge. Naïve mice succumbed to disease within 25 days, whereas surviving mice previously treated with free cGAMP showed 50% survival, PEG5-cGAMP showed 75% survival, and PEG10-cGAMP showed 100% survival at 60 days. Resistance to secondary tumor cell inoculation confirms the induction of an adaptive immune response against tumor antigens after treatment of the primary tumor. Taken together, these results suggest that PEGylated liposomal cGAMP formulations provide improved direct tumor control compared to non-PEGylated cGAMP and may improve the induction of adaptive immune memory responses against tumor antigens compared to free cGAMP.
Figure 7.

Local therapeutic delivery of cGAMP in an orthotopic melanoma model. a) Gene expression analysis for interferon-β (Ifnb1), and (C-X-C motif) ligand 9 (Cxcl9) in orthotopic B16-F10 melanoma tumors treated with various cGAMP formulations at a dose of 1 µg administered intratumorally (1-way ANOVA with Tukey’s post hoc test, n=3 mice/condition, ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05). Mice were injected intradermally with B16-F10 cells to establish an orthotopic tumor seven days prior to treatment and gene expression analysis. b) Treatment scheme for orthotopic therapeutic B16-F10 melanoma model. B16-F10 cells were injected intradermally in the lateral flank of mice. Four doses of PBS, empty liposomes, free cGAMP, or liposomal cGAMP were injected directly into the tumors at the indicated days, and the tumor size and mouse survival were monitored. Mice that cleared their tumors were injected intradermally in the opposite side of the flank on day 60 with B16-F10 cells and monitored for survival. c) Individual tumor growth curves of mice in various treatment groups. Black arrows indicate the times of intratumoral cGAMP injection. Number of surviving mice in each group are indicated at the bottom right of the graph. d) Overall survival Kaplan-Meier curves for mice treated with the indicated formulations during primary challenge (n=4–8 mice/condition; p values of treatments compared to PBS control by Mantel Cox test shown in legend) e) Overall survival curves for mice previously treated with the indicated formulations during rechallenge (n=3–4 mice/condition; p values of treatments compared to naive control by Mantel Cox test shown in legend). Data in (a) are shown as mean and standard deviation.
Next, the tissue distribution of cGAMP and changes in immune cell localization after cGAMP treatment were studied as a means to assess differences in the performance of the various cGAMP formulations. Free cGAMP was compared to the relatively poor-performing P0-cGAMP, and the higher performing PEG-10 cGAMP formulation. Since PEG5- and PEG10-cGAMP showed similar behavior in anti-tumor efficacy, but PEG10-cGAMP provided higher inflammatory gene expression and protection against rechallenge, we tested only the higher performing PEG10-cGAMP formulation to reduce animal usage. Tumors injected with fluorescein-cGAMP formulations were harvested 24 hours after injection, sectioned, and imaged to visualize cGAMP distribution (Figure 8-A). cGAMP signal was not seen at the tumor site 24 hours after free cGAMP was delivered, whereas PEG0-cGAMP and PEG10-cGAMP conditions both showed regions of cGAMP accumulation within and adjacent to the tumor. PEG0-cGAMP seemed to be largely deposited in a small volume in the tumor, whereas PEG10-cGAMP showed a more uniform distribution within the tumor mass. This limited permeation of PEG0-cGAMP in the tumor may account for its inferior anti-tumor activity compared to the PEGylated formulations tested. Immune cell infiltration, composed primarily of myeloid cells, increased dramatically with all cGAMP-containing treatments compared to PBS-injected tumors (Figure S7), indicating a potent innate immune recruitment effect with cGAMP treatment at tumor sites. Infiltration of NK cells, which may play a role in the anti-tumor efficacy of liposomal STING agonists,[27] did not change substantially after intratumoral cGAMP treatment (Figure S8). Staining for major histocompatibility complex II (MHC II)-positive APCs showed MHC II-expressing cells co-localized with cGAMP signal in the PEG10-cGAMP condition (Figure 8-B). This association with APCs, which are known to be highly responsive to STING pathway activation,[14] may account for the increased type I interferon gene signature induction and more potent immune memory response generation after intratumoral injection of PEG10-cGAMP relative to the other formulations tested. Further studies of APC activation in the TME in response to the various cGAMP formulations are warranted. Together, these data show that liposomal cGAMP formulations allow cGAMP retention at the tumor site for at least 24 hours after injection, and that PEG10-cGAMP more broadly distributes within the tumor mass relative to PEG0-cGAMP, and associates with APCs at the tumor site.
Figure 8.

Distribution of cGAMP delivered into tumor sites. a) Fluorescence images of 10 µm tissue sections showing distribution of various cGAMP formulations 24 hours after injection into melanoma tumors. Mice were injected intradermally with B16-F10 cells to establish an orthotopic tumor seven days prior to treatment (representative image from n=3 mice/condition, scale bar = 500 µm, white dashed line indicated tumor margin). b) Immunofluorescence on tissue section from PEG10-cGAMP-treated tumor showing cGAMP signal co-localizing with major histocompatibility complex class II (MHC II)-expressing cells (representative image from n=3 mice/condition, scale bar = 50 µm). Arrows show examples of MHC II-expressing cells highly associated with cGAMP signal.
3. Conclusion
In this report, the utility of cationic liposomes for enhancing the activity of cGAMP for cancer immunotherapy was tested. Three DOTAP/cholesterol liposome formulations with 0, 5, and 10 mol% PEG were used to encapsulate cGAMP. Liposomal cGAMP showed increased cell-association and uptake relative to cGAMP in its free form, with non-PEGylated liposomes providing faster initial cell-association and internalization. Liposomal formulation enhanced the potency of cGAMP in an IRF pathway macrophage reporter cell assay by an order of magnitude compared to free cGAMP. Induction of IFN-β gene expression in BMDC by cGAMP treatment was enhanced by two orders of magnitude by using cationic liposomes as a delivery vehicle. Cationic liposomes showed anti-tumor activity against metastatic melanoma in the lung, whereas free drug produced no effect. PEGylated liposomal cGAMP was capable of clearing established orthotopic melanoma tumors and inducing adaptive immunity that resisted a second challenge with the same tumor cells. Increased retention within the TME and co-localization with tumor-associated APCs may underly the superior type I interferon induction and adaptive immune response generation of PEGylated cGAMP. These results position cationic liposomes as a promising delivery strategy for systemic and local STING agonist delivery to tumors.
4. Experimental Section
Mice
All work with C57BL/6J and C57BL/6J-Tmem173gt/J (STINGgt/gt) mice (The Jackson Laboratory) was performed in compliance with the National Institutes of Health and institutional guidelines.
Cell culture
Primary BMDC cultures were established from female C57Bl/6J or C57BL/6J-Tmem173gt/J (STINGgt/gt) mice as previously described.[42] Briefly, bone marrow was flushed from isolated femurs using PBS. The marrow was seeded in petri dishes in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% heat inactivated FCS, 1% penicillin-streptomycin, 50 µM beta-mercaptoethanol, and 40 ng ml−1 murine GM-CSF (Peprotech). One third of the volume of fresh medium was added on day 3, and one third of the volume of medium was exchanged for fresh medium on day 6. The non-adherent cell fraction of these cultures, which are typically >75% CD11c+ by flow cytometry, were used for experiments between days 7–9 of culture.
B16-F10 murine melanoma cells (ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FCS in an incubator maintained at 37 °C and 5% CO2 atmosphere. Cells were passaged every 2 days and used before passage 10 after acquisition from the vendor.
RAW-Blue ISG cells (Invivogen) were cultured according to the manufacturer’s instructions.
Liposome synthesis
DOTAP, cholesterol, and DSPE-PEG(2000) were purchased from Avanti Polar Lipids. Lipids were mixed at a ratio of 1:1:X DOTAP:cholesterol:DSPE-PEG(2000) where X is 0, 0.1, and 0.2 for PEG0, PEG5, and PEG10 conditions, respectively. Lipid mixtures were dried under a nitrogen stream in glass test tubes. The resulting lipid films were placed in a desiccation system overnight. Dry films were hydrated in a solution of 250 µg ml−1 of 2’3’-cGAM(PS)2 (Rp/Sp) (Invivogen) in PBS with 6 cycles of vortexing for 30 seconds every 5 minutes. Samples were then freeze-thawed 6 times between liquid nitrogen and a 37 °C water bath and extruded using 21 passes in a mini-extruder (Avanti) through a 0.2 µm pore size polycarbonate filter (Whatman). Samples were placed in 10K MWCO dialysis cartridges (Thermo Scientific) and dialyzed against PBS for 6 hours before use. The cGAMP content of the liposomes was determined by measuring the absorbance peak of the liposomes at 254 nm, corresponding to the maximum absorbance of cGAMP, subtracting the lipid contribution to absorbance at this wavelength, and comparing to a standard curve of various cGAMP concentrations. For studies with fluorescent cGAMP-loaded liposomes, fluorescein-conjugated 2’3’-cGAMP (BIOLOG Life Science Institute) was used for rehydration of lipid films and further processed as described above.
Liposome characterization
The hydrodynamic diameter and zeta potential of liposomes were evaluated using a Malvern Zetasizer Nano ZS. Measurements were taken in PBS or DMEM with 10% FCS. For cryoEM of liposomes, samples were deposited on plasma-treated holey carbon Quantifoil grids and plunge frozen in liquid ethane using a Cryoplunge 3 System (Gatan). Frozen samples were kept under liquid nitrogen, and imaged on an FEI Tecnai Arctica CryoTEM system operating at 200kV. For release studies, purified liposomes containing fluorescein-cGAMP were placed in D-Tube Dialyzer Midi, MWCO 6–8 kDa (Millipore) and dialyzed at 37 °C against PBS. Liposome samples were collected over time, lysed with 0.5% Triton X-100, and total fluorescence was measured using a plate reader. These values were compared to the original fluorescent signal at the start of the release study to determine percentage release.
Flow cytometry
For BMDC liposome association experiments by flow cytometry, BMDCs were plated overnight at 5×105 cells/well of a 6-well tissue culture plates and treated with various formulations of fluorescein-cGAMP at 1 µg ml−1 for the indicated time. Cells were recovered with a cell scraper and stained for viability with Fixable Viability Dye eFluor 780 (eBioscience) and nucleated cells with Hoescht 33342 (Life Technologies). Cells were analyzed for fluorescein signal on a BD LSRFortessa flow cytometer. For BMDC activation studies, cells were stimulated for 4 hours with 1 µg ml−1 equivalent cGAMP, collected and stained for viability and nucleated cells as above, and further stained with CD86-PE (clone: GL1; eBioscience) before flow cytometry analysis. Data analysis was performed in Flowjo 10.0.8.
Confocal microscopy
For confocal microscopy, BMDCs were seeded overnight on round 10 mm coverslips in a 12 well plate at 1×105 cells/well. Cells were treated with fluorescein-cGAMP in various formulations at 1 µg ml−1 for the indicated time. Coverslips were fixed with 4% paraformaldehyde (PFA) in PBS at room temperature for 10 minutes. For unpermeabilized samples, coverslips were stained with Hoechst 33342 for 20 minutes and mounted on microscope slides using ProLong Diamond Antifade Mountant (Life Technologies). For phalloidin staining, samples were permeabilized for 5 minutes in 0.1% Triton X-100, and further stained with Alexa-647 phalloidin (Life Technologies) and Hoechst 33342 for 20 minutes prior to mounting. Samples were imaged using an LSM 710 confocal microscope (Zeiss) with a 63× 1.46 NA oil immersion objective.
In vitro dose-response studies
RAW-Blue ISG cells were used to determine IRF pathway activation by the various liposome formulations. RAW-Blue ISG cells were plated with various cGAMP formulations in 96 well plates for 18 hours to allow production of secreted embryonic alkaline phosphatase (SEAP). 50 µl of cell supernatant was removed and added to 150 µl of QUANTI-Blue SEAP detection medium (Invivogen) and incubated for 2 hours at 37 °C. SEAP activity was assessed using absorbance at 630 nm on a plate reader.
Gene expression analysis
For in vitro gene expression studies, BMDCs were plated overnight at 5×105 cells/well of a 6-well tissue culture plates and treated with various formulations of cGAMP for 4 hours. Cells were lysed, and RNA was purified using an RNeasy Mini Kit (Qiagen), quantified using a Nanodrop (Thermo Scientific), and transcribed into cDNA using the iScript Reverse Transcription Supermix (Bio-Rad) according to the manufacturer’s instructions. Quantitative real-time PCR (qPCR) was performed in duplicate using validated PrimePCR primers and SsoAdvanced Universal SYBR green Supermix on a CFX96 Real-Time PCR detection system (Bio-Rad). Expression levels were analyzed using the delta-delta-Ct method with normalization to Gapdh and statistical analysis was performed on log2 transformed relative expression values as previously described.[43] Primer unique assay identifiers are listed below: Gapdh (qMmuCED0027497), Ifnb1 (qMmuCED0002606), Cxcl10 (qMmuCED0001068), Cxcl9 (qMmuCID0023784), and TNF (qMmuCED0004141).
For in vivo gene expression studies, samples were collected and placed immediately into RNAlater solution (Thermo Scientific) and stored at 4 °C overnight. The following day, samples were homogenized using 3.0 mm zirconium beads on a BeadBug Homogenizer (Benchmark Scientific). Samples were centrifuged to remove debris and RNA was purified and analyzed as above.
Tumor models, treatment, and analysis
For lung tumor metastasis studies, 200 µl of Hank’s Balanced Salt Solution (HBSS) containing 2×105 B16-F10 cells was injected through the lateral tail vein of 7–8 week old female C57Bl6/J mice. Mice were randomized and treated with 100 µl of freshly prepared cGAMP formulations at a concentration of 3.5 µg ml−1 (0.35 µg dose) via retro-orbital injection. Anti-CTLA-4 (clone 9H10; Bio X Cell) and anti-PD-1 (clone RMP1–14; Bio X Cell) were given at 100 µg and 250 µg doses, respectively, diluted in 200 µl of PBS by intraperitoneal injection. Mice were sacrificed and lungs were collected into Fekete’s solution to preserve and bleach the tissue as previously described.[44] The entire lung surface of blinded samples were digitally photographed using a Zeiss AxioZoom V16 microscope. Lung nodules were counted in a blinded manner and the length of the longest axis of each nodule was measured using NIH ImageJ version 2.00.
For the orthotopic melanoma model, 100 µl of HBSS containing 2.5×105 B16-F10 cells was injected intradermally into the shaved lateral flank of 6–8 week old female C57Bl6/J mice. On day 7 of tumor growth, tumors were measured and mice were randomized into groups having similar average tumor areas. For each treatment, freshly prepared cGAMP formulations were diluted to 20 µg ml−1 in PBS and 50 µl (1 µg dose) was injected using a 29G needle directly into the tumor mass. The length and width of the tumors were measured approximately three times per week and the tumor area was calculated as Area = π/4 × length × width. Animals surviving the primary challenge were rechallenged with 2.5×105 B16-F10 cells intradermally in the opposite flank and monitored for survival.
Sectioning and immunofluorescence staining
Seven days after injection with 2.5×105 B16-F10 cells intradermally, mice were injected with formulations containing 1 µg of fluorescein-cGAMP directly into the tumor mass. Tumors were harvested 24 hours later and fixed for two hours at room temperature in 4% PFA in PBS, equilibrated in a 30% sucrose in PBS solution overnight at 4 °C, placed in optimum cutting temperature formulation (O.C.T.; Sakura Finetek), and frozen in a cup of isopentane immersed in liquid nitrogen. Cryosectioning was performed to generate 10 µm frozen sections. To assess distribution of cGAMP, samples were briefly rehydrated in PBS, mounted using ProLong Diamond Antifade Mountant and tiled imaging was performed on a Zeiss Axio Observer Z1 epifluorescence microscope. For immune cell infiltration assessment, samples were cryosectioned and stained with allophycocyanin-conjugated antibodies (MHC II, CD45, CD11b, F4/80 (eBioscience) and NK-1.1 (Biolegend)) and Hoescht 33342 (Life Technologies). Slides were imaged using a Zeiss LSM 710 confocal microscope.
Supplementary Material
Acknowledgments
STK, LG, and DJM conceived the idea for the study. STK, ASC, LG, and DJM designed the studies. STK, ASC, LG, and ARG performed the experiments. STK, ASC, and LG analyzed the data. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. This work was supported by NIH grant R01 EB015498 to D.J.M. S.T.K. was supported by an HHMI ISRF. The authors thank V. Ramanan, O. Ali, C. Verbeke, and S. Perrault for scientific discussions, K. Vining for discussions and assistance with analyzing qPCR data, M. Pittet, C. Evavold, and C. Garris for discussions and experimental materials, and C. Marks for assistance with cryoEM studies. This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the National Science Foundation under NSF award no. ECS-0335765. CNS is part of Harvard University.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Sandeep T. Koshy, Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA.
Alexander S. Cheung, Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA.
Luo Gu, Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA.
Amanda R. Graveline, Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA
David J. Mooney, Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA.
References
- 1.Mellman I, Coukos G, Dranoff G. Nature. 2011;480:480. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Drake CG, Pardoll DM. Cancer Cell. 2015;27:1. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Restifo NP. Science. 2015;348:62. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Annu. Rev. Med. 2014;65:333. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. J. Clin. Invest. 2015;125:3401. doi: 10.1172/JCI80009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joyce JA, Fearon DT. Science. 2015;348:74. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 7.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Veer LJV, Sperling AI, Wolf DM, Krummel MF. Cancer Cell. 2014;26:1. [Google Scholar]
- 8.Woo S-R, Corrales L, Gajewski TF. Trends Immunol. 2015;36:1. doi: 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barber GN. Nat. Rev. Immunol. 2015;15:760. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, Wu J, Du F, Chen X, Chen ZJ. Science. 2013;339:786. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Science. 2013;339:826. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre M-L, Gajewski TF. Immunity. 2014;41:830. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corrales L, Gajewski TF. Clin. Cancer Res. 2015;21:4774. doi: 10.1158/1078-0432.CCR-15-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo S-R, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Gajewski TF. Cell Rep. 2015;11:1018. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M. Proc. Natl. Acad. Sci. U.S.A. 2015;112:15408. doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baird JR, Friedman D, Cottam B, Dubensky TW, Kanne DB, Bambina S, Bahjat K, Crittenden MR, Gough MJ. Cancer Res. 2016;76:50. doi: 10.1158/0008-5472.CAN-14-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koshy ST, Mooney DJ. Curr. Opin. Biotechnol. 2016;40:1. doi: 10.1016/j.copbio.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen TM, Cullis PR. Adv. Drug Delivery Rev. 2013;65:36. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 19.Kanasty R, Dorkin JR, Vegas A, Anderson D. Nat. Mater. 2013;12:967. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 20.Tseng Y-C, Mozumdar S, Huang L. Adv. Drug Delivery Rev. 2009;61:721. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. ACS Nano. 2015;9:16. doi: 10.1021/nn5062029. [DOI] [PubMed] [Google Scholar]
- 22.Kwong B, Liu H, Irvine DJ. Biomaterials. 2011;32:5134. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ. Cancer Res. 2013;73:1547. doi: 10.1158/0008-5472.CAN-12-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meraz IM, Savage DJ, Segura-Ibarra V, Li J, Rhudy J, Gu J, Serda RE. Mol. Pharmaceutics. 2014;11:3484. doi: 10.1021/mp5002697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ. J. Clin. Invest. 2015;125:2532. doi: 10.1172/JCI79915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H. J. Controlled Release. 2014;184:20. doi: 10.1016/j.jconrel.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, Harashima H. J. Controlled Release. 2015;216:149. doi: 10.1016/j.jconrel.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 28.Ueno NT, Bartholomeusz C, Xia W, Anklesaria P, Bruckheimer EM, Mebel E, Paul R, Li S, Yo GH, Huang L, Hung M-C. Cancer Res. 2002;62:6712. [PubMed] [Google Scholar]
- 29.Zhuang Y, Ma Y, Wang C, Hai L, Yan C, Zhang Y, Liu F, Cai L. J. Controlled Release. 2012;159:135. doi: 10.1016/j.jconrel.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, Nunez MI, Wistuba II, Erasmus JJ, Hicks ME, Grimm EA, Reuben JM, Baladandayuthapani V, Templeton NS, McMannis JD, Roth JA. PLoS ONE. 2012;7:e34833. doi: 10.1371/journal.pone.0034833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Yin Q, Kuss P, Maliga Z, Millán JL, Wu H, Mitchison TJ. Nat. Chem. Biol. 2014;10:1043. doi: 10.1038/nchembio.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Immordino ML, Dosio F, Cattel L. Int. J. Nanomed. 2006;1:297. [PMC free article] [PubMed] [Google Scholar]
- 33.Gjetting T, Arildsen NS, Christensen CL, Poulsen TT, Roth JA, Handlos VN, Poulsen HS. Int. J. Nanomed. 2010;5:371. doi: 10.2147/ijn.s10462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Wang J, Gao Y, Zhu J, Wientjes MG, Au JLS. AAPS J. 2011;13:585. doi: 10.1208/s12248-011-9298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan C-L, Majzoub RN, Shirazi RS, Ewert KK, Chen Y-J, Liang KS, Safinya CR. Biomaterials. 2012;33:4928. doi: 10.1016/j.biomaterials.2012.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gajewski TF, Schreiber H, Fu Y-X. Nat. Immunol. 2013;14:1014. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramesh R, Saeki T, Templeton NS, Ji L, Stephens LC, Ito I, Wilson DR, Wu Z, Branch CD, Minna JD, Roth JA. Mol. Ther. 2001;3:337. doi: 10.1006/mthe.2001.0266. [DOI] [PubMed] [Google Scholar]
- 38.Ito I, Ji L, Tanaka F, Saito Y, Gopalan B, Branch CD, Xu K, Atkinson EN, Bekele BN, Stephens LC, Minna JD, Roth JA, Ramesh R. Cancer Gene Ther. 2004;11:733. doi: 10.1038/sj.cgt.7700756. [DOI] [PubMed] [Google Scholar]
- 39.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS. N. Engl. J. Med. 2015;372:2006. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen W, Yan W, Huang L. Cancer Immunol. Immunother. 2007;57:517. doi: 10.1007/s00262-007-0390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan W, Chen W, Huang L. Mol. Immunol. 2007;44:3672. doi: 10.1016/j.molimm.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Boudreau J, Koshy S, Cummings D, Wan Y. J. Visualized Exp. 2008:e769. doi: 10.3791/769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rieu I, Powers SJ. Plant Cell. 2009;21:1031. doi: 10.1105/tpc.109.066001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ya Z, Hailemichael Y, Overwijk W, Restifo NP. Curr. Protoc. Immunol. 2015;108 doi: 10.1002/0471142735.im2001s108. 20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.