

Abstract
Hypothesis-driven research has led to many scientific advances, but hypotheses cannot be tested in isolation: rather, they require a framework of aggregated scientific knowledge to allow questions to be posed meaningfully. This framework is largely still lacking in microbiome studies, and the only way to create it is by discovery-, tool-, and standards-driven research projects. Here we illustrate these issues using several such non-hypothesis-driven projects from our own laboratories, including spatial mapping, the American Gut Project, the Earth Microbiome Project (which is an umbrella project integrating many smaller hypothesis-driven projects), and the knowledgebase-driven tools GNPS and Qiita. We argue that an investment of community resources in infrastructure tasks, and in the controls and standards that underpin them, will greatly enhance the investment in hypothesis-driven research programs.
Introduction
Microbiome research is making dramatic progress, with thousands of papers now published each year linking specific microbes and/or host-microbe co-metabolites to specific diseases, physiological properties, or environmental parameters. Much of this research is performed in a traditional, hypothesis-driven way, or at least presented as a rational reconstruction that fits this model, much as Darwin re-wrote much of his discovery-driven work as hypothesis driven to increase its respectability under the influence of contemporary philosophers of science such as William Whewell (1). However, it should be noted that hypothesis-driven science was not always so respectable -- Isaac Newton famously wrote “Hypotheses non fingo”, or “I feign no hypotheses”, in an essay appended to the second edition of the Principia (2) -- so the tradition of modifying how science is framed to meet respectability criteria dates back at least 300 years. What can be framed as a testable hypothesis suffers important limitations based on what we can measure and what we already know.
Ten years ago Chris Anderson, editor of Wired magazine, set off an international debate with his article “The End of Theory: The Data Deluge Makes the Scientific Method Obsolete” (3). The idea was that with enough data, hypotheses will emerge (“Let the data speak for itself”) has become widely discussed in the rapidly growing data science profession. A thoughtful review of this topic was written in EMBO Reports in 2015-”Could Big Data be the end of theory in science? A few remarks on the epistemology of data-driven science” (4). As the author points out:
“Francis Bacon, the “father of the scientific method” himself, in his Novum Organum (1620), argued that scientific knowledge should not be based on preconceived notions but on experimental data. Deductive reasoning, he argued, is eventually limited because setting a premise in advance of an experiment would constrain the reasoning so as to match that premise. Instead, he advocated a bottom-up approach: In contrast to deductive reasoning, which has dominated science since Aristotle, inductive reasoning should be based on facts to generalize their meaning, drawing inferences from observations and data.”
We recently reviewed experimental design considerations for traditional hypothesis-driven microbiome studies elsewhere (5, 6), and do not discuss these issues further in this review. Here we describe the danger of jumping too soon into hypothesis testing, and describe the need for four major categories of non-hypothesis-driven research: better spatial and abstract maps, better tools, and better standards. Given space constraints, we illustrate these primarily using the American Gut Project (7), the Earth Microbiome Project (8), and tools we developed in our laboratories.
The challenge of unknown unknowns
In microbiome research, a recurring challenge has been that factors intuitively suspected to drive differences in the microbiome are less important than other, more surprising factors. For example, sex has a small impact on microbiomes across the human body (9, 10) and has a much weaker effect than many other variables such as age (even within adults), or the time of year the sample was collected(11, 12). However, sex is far more frequently reported than time of year. Similarly, although long-term dietary habits are correlated with the overall composition of the human microbiome within and between populations (7, 13–16), and dietary changes over months can lead to changes in overall microbiome composition larger than the differences between arbitrarily chosen individuals (17, 18), but short-term changes have transient effects smaller than typical differences between individuals (14, 19). However, many studies focus on short-term rather than long-term diet. Perhaps even more surprisingly, factors such as temperature and pH have much smaller impacts on environmental microbiomes than salinity(8, 20), and even the saline vs. non-saline difference is much smaller than the host-associated vs free-living difference (8, 21). Samples from different parts of the same person’s body differ more from one another in their overall microbial communities than radically different free-living microbial communities, such as soils versus oceans (8). Differences of this magnitude can also occur within the gut of a single person, with sufficiently large perturbation (7).
Because factors of large effect are often unknown and unreported, studies testing hypotheses concerning intuitively obvious factors of small effect are often subject to important confounding variables, that, when uncovered, prompt complete reinterpretation of the study. For example, suppose an investigator is unaware that cage effects are important in the microbiome (22), and profiles microbiomes in two cages each of two different genotypes of mice. The results will likely be driven by which cages happens to resemble each other more closely. If the variable of cage is not measured, or not tested in an unsupervised model, this important confounding variable will likely remain undiscovered, and the interpretation of the experiment entirely incorrect
Similarly, a frequent practice is to discard unannotated microbes or unannotated molecules, focusing on the subset of microbes or molecules that can be matched to an existing database. Because databases of both microbes and molecules are heavily biased (microbes, by studies of known pathogens that come from only a few taxonomic groups, and molecules, by commercially available compounds), the entities that best discriminate among classes of samples may be lost in the analysis: often, only 60% of sequences and 2% of molecular features from an untargeted metabolomics experiment can be annotated by existing references (23, 24). However, a rational reconstruction of why the annotatable microbes or molecules are plausibly connected to a phenotype of interest can frequently be developed, especially given the characteristics of these highly multivariate datasets that can lead to high false discovery rates when the true number of implicitly tested hypotheses is considered (25).
The need for spatial maps
An important metaphor in science and information visualization is the idea of the map. As data volumes increase, it is frequent that the main research activity in a field moves from tests of hypotheses of differences in individual variables among sites, to tests of these hypotheses with replicates at each site, to spatially or temporally explicit sampling, to detailed spatial maps that reveal otherwise unsuspected patterns. This progression has occurred in 16S rRNA ampliconbased microbiome studies over the past decade (8, 26), and increasingly characterizes mass spectrometry-based metabolome studies over the past four years (27–32).
The value of spatial maps is so self-evident that the results are often cursed by obviousness. For example, the finding that metabolomes cluster by individual, as revealed by principal coordinates analysis (PCoA), is interesting (Fig. 1A). However, the finding that a given molecule such as lauryl sulphate (m/z 355.219) is distributed across the body of one of the two individuals, but is absent from the other individual is obvious (Fig. 1B), especially when subject A, who is male, reports using the skin care product Nivea for Men, the source of the molecule(28). Similarly, the finding that samples from four individuals differ significantly in levels of specific purines between and within subjects might well prompt further investigation. However, a spatial map with dense sampling of the same individuals (Fig. 1C) makes it obvious that the molecule is something that is touched and consumed, and sometimes spilled, allowing one to guess that it is caffeine; similarly, the spatial map reveals that one person likely spends time in the ocean based on the distribution of Synechococcus spp. (Fig. 1D) (30).
Figure 1.
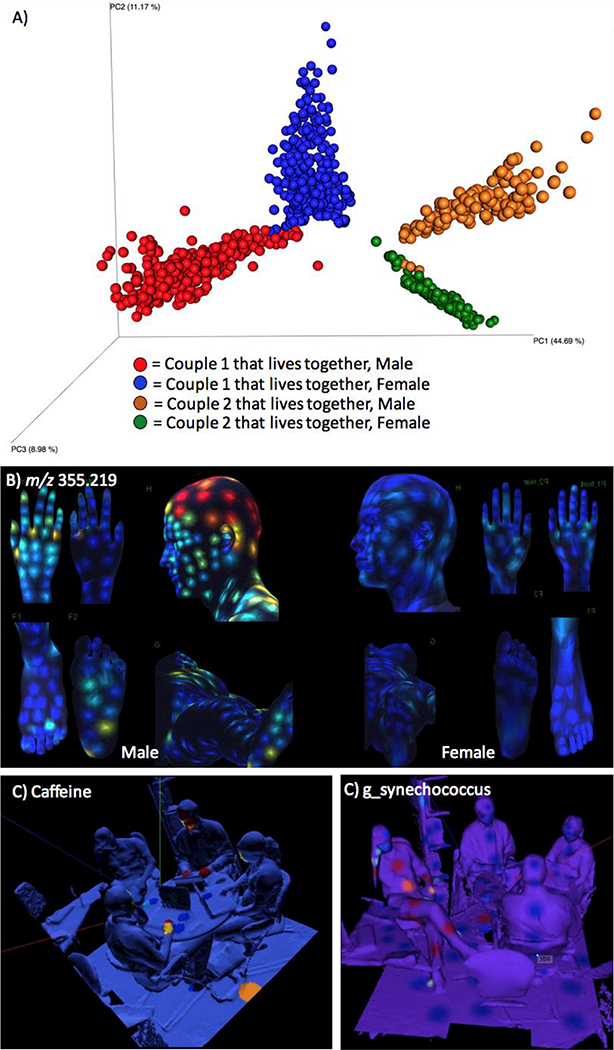
Spatial analysis based on metabolomics of skin samples and a human habitat. A) Principal coordinates analysis (Hellinger distance) of metabolomics data of skin swabs obtained from several hundreds locations on the human body of four volunteers. B) The detection of lauryl sulfate (m/z 355.219) from the shampoo Nivea for Men on a male volunteer. C) The distribution of caffeine (m/z 195.088) on four individuals and office environment. D) The distribution of Synechococcus spp. on within that same office environment.
We have no idea where most microbes and molecules occur in and on the human body, in natural environments, or in human-constructed and human-impacted environments. Spatial maps could make many of these distributional patterns obvious, just as John Snow’s map of cholera instantly led to the hypothesis that this disease was water-borne and stemmed from the Broad Street pump, reinforced by the map’s revelation that the block that drank alcohol rather than pump water was spared from cholera (33). In an analogous manner, systematically collected maps of microbes and molecules across different spatial scales will fundamentally transform the types of questions that can be asked of microbiome and metabolomics data.
The need for abstract maps
Despite the intuitive appeal of spatial maps, the value of abstract maps, including ordinations such as principal coordinates analysis (PCoA), non-metric multidimensional scaling (NMDS), tdistributed stochastic neighbor embedding (t-SNE), and network diagrams from object similarity (sequence or spectrum) or co-occurrence, is also considerable. The correct data frame and distance metric often immediately reveal the key result, without a specific hypothesis in mind. Consider the starting and ending time point of a fecal transplantation series (34) (Fig. 2A), where the different clusters are obvious, but the direction and meaning of this difference are not. Placing these samples in the context of the Human Microbiome Project data (10) reveals immediately that the difference between start and endpoint is much greater than the difference between healthy and diseased samples, and adding intermediate timepoints shows that the transition occurs rapidly. The map thus enables new hypotheses about how to move individuals along a desired trajectory in the abstract space. A major driving force behind both the Earth Microbiome Project (8) and the American Gut Project (7) has been to build out these abstract maps for additional sample types and populations.
Figure 2.
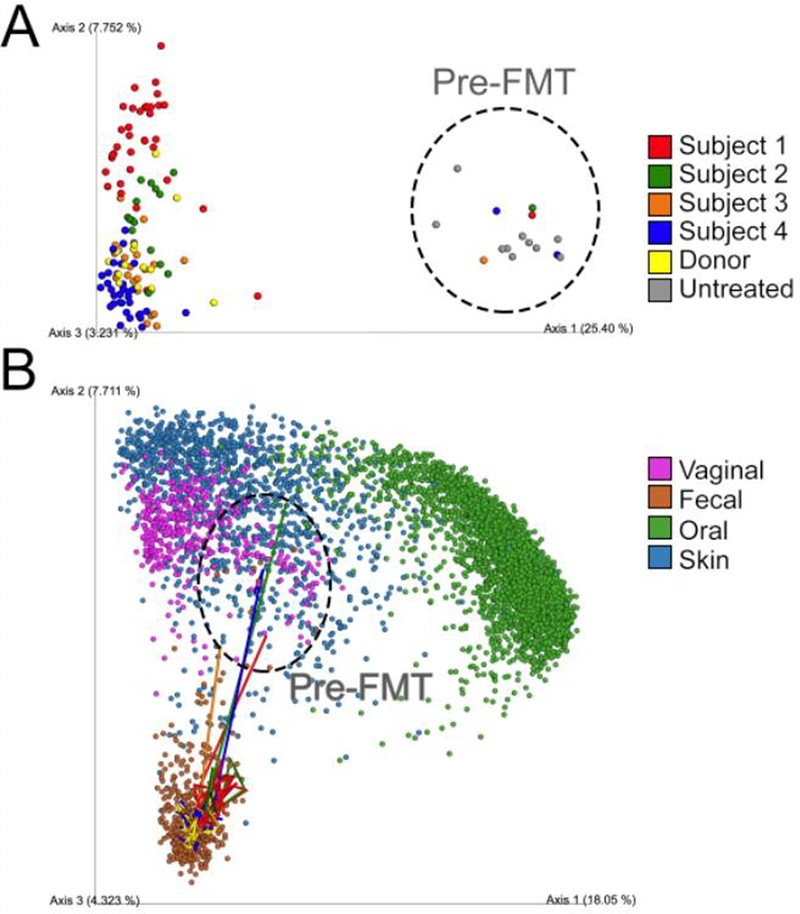
Untangling the meaning of complex microbial interactions through meta-analyses. (A) Principal coordinates analysis (unweighted UniFrac) of Clostridium difficile Infection subjects, before and after a fecal transplant, along with the fecal donor and 10 untreated subjects (34). (B) Principal coordinates analysis (unweighted UniFrac) of the Human Microbiome Project (HMP) (10) combined with the data in panel A, the longitudinal samples for subjects 1–4 are connected as lines displaying the temporal variability and the shift from a disjointed untreated state of the patients vs. the healthy frame of the HMP. A high-resolution version can be found athttps://www.dropbox.com/sh/paq9sdiqvzp5mog/AABTUuRkZlHzlPbsN0riWIICa?dl=0.
An important question in building maps is often whether, given a fixed sequencing budget, it is better to have more points (samples), or more accurate or detailed characterization of each point. In our experience, for amplicon sequencing, having more samples outweighs the value of having more sequences per sample, down to surprisingly low thresholds. For example, Fig. 3 shows the Earth Microbiome Project dataset (8) sampled at 500,000 sequences per sample, 1000 sequences per sample, and just 200 sequences per sample. The overall patterns, e.g. the host/non-host split and the saline/non-saline split, are much clearer with more samples than with more precision about the location of each sample in PCoA space. Multinomial sampling considerations make it immediately clear why this is true: with 100 sequences per sample, the standard error in inferring the proportion of a taxon at 5% frequency is ~√(100*.95*.05) or 2.18%, or nearly 50% error in proportion; the standard error at a taxon at 1% frequency is ~√(100*.99*.01) or 0.99%, essentially 100% error. Consequently, even low-abundance taxa are sampled accurately enough to place a sample in a map with surprisingly few sequences. Logically, this must be true, or all ordination diagrams in microbial ecology before nextgeneration sequencing would have been useless.
Figure 3.
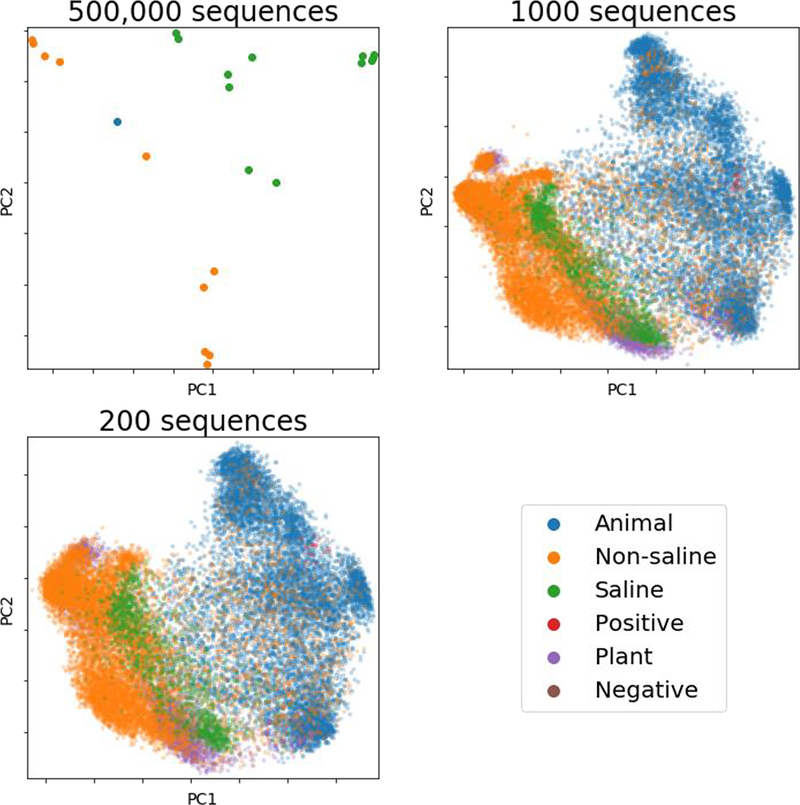
Broader sampling improves abstract maps of the microbial world in the Earth Microbiome Project, even with low resolution. All panels show principal coordinates analysis of unweighted UniFrac distances between samples. (A) Samples rarefied to 500,000 sequences, showing only those exceeding this threshold sampling depth. (B) Samples rarefied to 1000 sequences. (C) Samples rarefied to 200 sequences. Even with few observations per sample, the overall relationships among sample types are preserved; in contrast, the overall pattern is lost with too few samples no matter how exquisitely characterized.
The need for improved tools
Amplicon studies have been greatly enabled by improvements in processing pipelines, distance metrics, and reference databases, which we have recently reviewed elsewhere (35), greatly enabling hypothesis-driven studies about relative abundance of particular microbial taxa and their placement on abstract maps such as those produced by the Earth Microbiome Project (8) and the American Gut Project (7). As we extend these projects to other data types, notably shotgun metagenomics and metabolomics, we face new challenges that can best be solved by new tools. However, tool production is fundamentally itself more of an engineering than a hypothesis-driven activity, especially when the main advances are in user interfaces (32, 36) or in software engineering (37).
Although most 16S rRNA fragments can now be identified, in shotgun metagenomics only a small fraction of the sequences can typically be associated with known taxonomy or function. Genome assembly is especially valuable in identifying biosynthetic pathways, allowing taxonomic resolution at the species or strain level, and generating high-resolution single nucleotide polymorphism (SNP) profiles to characterize novel strains and confirm functional variants (38). Consequently, methods that can identify genetic variation from lower-coverage data, and that can estimate features of interest from less data or with efficient target capture, are needed for improved sample throughput. Shotgun metagenomics also often requires host DNA depletion because total DNA extracts from biological specimens can be dominated by host DNA (39).
Metabolomics poses different challenges (40). According to NHGRI, the cost per megabase of raw genome DNA sequence reduced in cost by almost six orders of magnitude since 2001(41), but mass spectrometry only decreased in cost by only two orders of magnitude during this period (40). However, the main limitation in metabolomics is the enormous chemical diversity, which hinders molecular identification and impacts the choice of extraction solvents, separation methods, instrumentation, and data analysis approaches. Because the multiplexing strategies that are successful in both amplicon- and shotgun-based sequencing approaches are not available in mass spectrometry, instrument time is directly proportional to the number of samples and limiting for large-scale projects. As with sequencing a decade ago, most molecular features found in a sample are unidentified, and many are likely technical artifacts, e.g. adducts formed in the gas phase, solvent artifacts (42) and multimers of the same compound (40). Better methods and incentives for aggregating community knowledge (24) (e.g. retention of knowledge of the large number of manual annotations performed by the community) and for automatically assigning unknown mass peaks and fragmentation spectra to molecules and have an estimation of error rates (43), as opposed to heuristics subject to personal interpretation rules (44), are urgently needed. Global Natural Products Social molecular networking (GNPS) (24) offers alternative solutions for computational mass spectrometry infrastructure. Spectral datasets can be publicly deposited with a unique identifier and transformed to “living data”, as they will be continuously searched against reference libraries to update users on new identifications. Furthermore, annotations can also be made by the scientific community within GNPS and propagated to all other data sets in the public domain with notifying subscribers on new annotations. Other expanded capabilities include automated species metabolome references (45) and the Molecular Explorer (24) for cross-searching annotated MS/MS spectra between datasets. Connections between several datasets, within the same knowledge base or between different spectral repositories such as Metabolights (46) and Metabolomics Workbench (47), can be made to highlight annotated compounds found in several data sets Such analysis is trivial in sequencing but still novel in mass spectrometry.
Integration of taxonomic, genomic and metabolomic data remains an important unsolved challenge. Although genome mining can successfully identify the sources of individual natural products (48), matching an overall taxonomic or functional microbiome profile to a molecular profile remains difficult because of procedural and analytical differences in data acquisition. In particular, the likelihood of time lags in chemical production or in genomic response to environmental changes, which may appear on different timescales, reduce the power of correlation approaches based on cross-sectional data (49). In cases where microbial and molecular composition is driven by a dominant effect (e.g. a dataset composed of soil and fecal samples will divide into two clusters driven by the difference between soil and feces), the molecular and metagenomic datasets will appear concordant by Procrustes analysis (50), but this is an artifact of the approach. An integrated systems biology approach that maps all data layers onto common pathways is likely needed, but cannot be performed today because most genes, pathways, and molecules are unknown and because even known system components lack coherent ontological conventions across databases.
The Need for Standards
Another branch of non-hypothesis-driven research critically important for framing precise hypotheses is standards development. In microbiome science these broadly take three tracks: analytical standards for determining the accuracy and fidelity of readouts, procedural standards for sample collection and handling, and annotation standards for integrating results across studies.
The lack of agreed-on standards stems from the origin of microbiome science in the discipline of ecology, where fundamental questions revolved around finding new kinds of organisms to fill out the phylogenetic tree of life (51), and finding statistically significant differences in microbial diversity or composition among samples within an individual study. Because the goal was to test whether any difference existed in the microbiome as a function of disease, physiological, or environmental state, biases (including missing taxa, or missing classes of molecules) were unimportant if a difference could be discovered. However, this situation diverges radically from the present, where physicians and engineers expect to be able to measure the correct, absolute abundance of all microbes or molecules in a given sample simultaneously. The realities of nucleic acid or organic extraction, detection methods for sequences and molecules, and downstream data processing do not support this important goal. Without consistent and welldefined measurements underpinned by a mechanistic causal model of error and bias, the state of microbiome-based predictions could be characterized as more like astrology than like astronomy, as pre-science rather than science.
To improve sample readout, we need known reference standards that can be spiked into samples at different stages, from original specimen to extracted DNA or compounds, that are agreed on, widely used, and have inexhaustible supply. Previous efforts, such as the HMP standards, have been limited by insufficient availability of materials, taxonomic complexity, or both. KatharoSeq in particular (52) benefits from having different spike-in standards at the level of primary sample and DNA, allowing different sources of contamination to be tracked down. Comparable development in mass spectrometry would be of tremendous value.
Sample collection and storage can bias specimen readout (53–55), but for most sample types the implications of different forms of degradation are unknown. Consequently, the conservative recommendation is always to expensively collect pristine samples (e.g. flash-frozen in liquid nitrogen), even though more practical methods would often suffice. For a few sample types, such as amplicon processing of stool, considerable data is now available on a range of conditions (55–58), and researchers can make informed decisions about which methods to use, as we did for citizen-scientists in the American Gut Project after exploring the limits of what can and cannot be usefully obtained for amplicon collection from stool shipped at room temperature (7). However, we know much less about the implications of sample degradation for most other types of biospecimens, and for the implications for reading out different molecular fractions with mass spectrometry (although see (59)).
Finally, integrating samples from different studies remains challenging because of differences in annotation (often called “metadata”). For example, different studies may refer to “stool”, “feces”, “gut”, or other synonyms, or rely on different units of measurement. Efforts such as the Genomic Standards Consortium MIxS family of standards (60), the Earth Microbiome Project Ontology (EMPO) (8), and other annotation schemes assist considerably in these tasks, but have been applied to relatively few datasets to date. The potential for natural language processing (NLP) and/or data-based methods for automatically applying annotations is considerable. These strategies were successful in Qiita for inferring EMPO annotations for tens of thousands of samples from the researcher-reported “sample_type.” However, further development is needed to enable researchers to “discover” variables and controlled vocabularies that can be generally applied.
Conclusions
Although hypothesis-driven science has immense value, it depends to a considerable degree on a framework of maps, tools, and standards whose own development often does not fit meaningfully into a hypothesis-driven framework. However, without these developments, hypotheses more explicit than “differences in the microbiome” or “elevation or depletion of specific pre-defineid taxa or molecules” cannot be tested, and completely new ideas about how to read out or control the microbiome will not be developed.
Extraordinary advances in data collection technologies leave us in a world where we regularly make make millions of observations of organisms about which we know virtually nothing -- as exemplified by the recent ‘discovery’ of the most abundant phage in the human gut via metagenome mining (61). To bring about a future of precision medicine and precision ecological remediation, where we can specify precise microbiome changes and bring them about through defined interventions, a vast amount of non-hypothesis-driven research, often dismissed as “technical work” or “fishing expeditions”, remains to be done.
Acknowledgements
This work was supported in part by National Institute of Justice Award 2015-DN-BX-K047, the Alfred P. Sloan Foundation, and the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ruse M The Darwinian Revolution: Science Red in Tooth and Claw Chicago: University of Chicago Press; 1999. • This is the definitive treatment of Darwinism, and the revolution in biology triggered by Darwinian thinking. It contains fascinating material from Darwin’s correspondence about how hard he worked to make his new theories acceptable given trends in philosophy of science at the time. [Google Scholar]
- 2.Cohen IB. The First English Version of Newton’s Hypotheses non fingo. Isis. 1962;53(3):379–88. [Google Scholar]
- 3.Anderson C THE END OF THEORY: THE DATA DELUGE MAKES THE SCIENTIFIC METHOD OBSOLETE. Wired. 2008. 6/23/2008. [Google Scholar]
- 4.Mazzocchi F Could Big Data be the end of theory in science? A few remarks on the epistemology of data-driven science. EMBO Rep. 2015;16(10):1250–5. doi: 10.15252/embr.201541001. PubMed PMID: ; PMCID: PMC4766450. • This influential though controversial review proposes that the combination of vast datasets and machine learning techniques may make much of the traditional quest for scientific theory obsolete. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debelius J, Song SJ, Vazquez-Baeza Y, Xu ZZ, Gonzalez A, Knight R. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol. 2016;17(1):217. doi: 10.1186/S13059-016-1086-X. PubMed PMID: ; PMCID: PMC5072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, Knight R, Ley RE. Conducting a microbiome study. Cell. 2014;158(2):250–62. doi: 10.1016/j.cell.2014.06.037. PubMed PMID: ; PMCID: PMC5074386. • This excellent review covers aspects of bench and computational work essential for framing and testing traditional hypothesis-driven questions in microbiome science, with an emphasis on animal model research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, DeRight Goldasich L, Dorrestein PC, Dunn RR, Fahimipour AK, Gaffney J, Gilbert JA, Gogul G, Green JL, Hugenholtz P, Humphrey G, Huttenhower C, Jackson MA, Janssen S, Jeste DV, Jiang L, Kelley ST, Knights D, Kosciolek T, Ladau J, Leach J, Marotz C, Meleshko D, Melnik AV, Metcalf JL, Mohimani H, Montassier E, Navas-Molina J, Nguyen TT, Peddada S, Pevzner P, Pollard KS, Rahnavard G, Robbins-Pianka A, Sangwan N, Shorenstein J, Smarr L, Song SJ, Spector T, Swafford AD, Thackray VG, Thompson LR, Tripathi A, Vazquez-Baeza Y, Vrbanac A, Wischmeyer P, Wolfe E, Zhu Q, American Gut C, Knight R. American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems. 2018;3(3). doi: 10.1128/mSystems.00031-18. PubMed PMID: ; PMCID: PMC5954204. •• This massive citizen-science effort represents the microbiome study with the largest number of participants to date, considerably expanding our understanding of the types of microbiome configurations that are out there in the general population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vazquez-Baeza Y, Gonzalez A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, Earth Microbiome Project C. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551(7681):457–63. doi: 10.1038/nature24621. PubMed PMID: •• This open community effort spanning tens of thousands of biospecimens provides the first comprehensive map relating different types of microbial communities across the planet, removing methodological variability by using standardized methodologies for DNA extraction, PCR amplification, DNA sequencing, computational analysis, and metadata capture. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon Jl, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960): 1694–7. Epub 2009/11/07. doi: 10.1126/science. 1177486. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Human Microbiome Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. doi: 10.1038/nature11234. PubMed PMID: ; PMCID: PMC3564958.•• This large-scale NIH-funded project provided an unprecedented resource for human microbiome studies by sampling microbes across the human body, reporting resuts for 242 participants, and combining 16S rRNA and shotgun metagenomic profiling. It has been widely re-used as a data frame for other studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davenport ER, Mizrahi-Man O, Michelini K, Barreiro LB, Ober C, Gilad Y. Seasonal variation in human gut microbiome composition. PLoS One. 2014;9(3):e90731. doi: 10.1371/journal.pone.0090731. PubMed PMID: ; PMCID: PMC3949691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, Dominguez-Bello MG, Sonnenburg JL. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 2017;357(6353):802–6. doi: 10.1126/science.aan4834. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, Tito RY, Chaffron S, Rymenans L, Verspecht C, De Sutter L, Lima-Mendez G, D’Hoe K, Jonckheere K, Homola D, Garcia R, Tigchelaar EF, Eeckhaudt L, Fu J, Henckaerts L, Zhernakova A, Wijmenga C, Raes J. Population-level analysis of gut microbiome variation. Science. 2016;352(6285):560–4. doi: 10.1126/science.aad3503. PubMed PMID: •• This exciting study of the Flemish Gut population provided one of the first two population-level studies (with Zhernakova et al., below) relating overall composition of the human gut microbiome to dozens of covariates, measured in a consistent way in each subject, [DOI] [PubMed] [Google Scholar]
- 14.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052): 105–8. doi: 10.1126/science. 1208344. PubMed PMID: ; PMCID: PMC3368382. • This important paper contrasts the large effect of long-term diet with the much smaller effect of a short-term inpatient dietary intervention in structuring the human gut microbiome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon Jl. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–7. doi: 10.1038/nature 11053. PubMed PMID: ; PMCID: PMC3376388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S, Wang J, Imhann F, Brandsma E, Jankipersadsing SA, Joossens M, Cenit MC, Deelen P, Swertz MA, LifeLines cohort s, Weersma RK, Feskens EJ, Netea MG, Gevers D, Jonkers D, Franke L, Aulchenko YS, Huttenhower C, Raes J, Hofker MH, Xavier RJ, Wijmenga C, Fu J. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352(6285):565–9. doi: 10.1126/science.aad3369. PubMed PMID: ; PMCID: PMC5240844. •• This exciting study of the Dutch LLDeep cohort provided one of the first two population-level studies (with Falony et al., above) relating overall composition of the human gut microbiome to dozens of covariates, measured in a consistent way in each subject, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ley RE, Turnbaugh PJ, Klein S, Gordon Jl. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122): 1022–3. doi: 10.1038/4441022a. PubMed PMID: • This study provided the first evidence that human gut microbes were linked to obesity, showing large changes in microbiome within an individual linked to changes in long-term diet. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Yin A, Li H, Wang R, Wu G, Shen J, Zhang M, Wang L, Hou Y, Ouyang H, Zhang Y, Zheng Y, Wang J, Lv X, Wang Y, Zhang F, Zeng B, Li W, Yan F, Zhao Y, Pang X, Zhang X, Fu H, Chen F, Zhao N, Hamaker BR, Bridgewater LC, Weinkove D, Clement K, Dore J, Holmes E, Xiao H, Zhao G, Yang S, Bork P, Nicholson JK, Wei H, Tang H, Zhang X, Zhao L. Dietary Modulation of Gut Microbiota Contributes to Alleviation of Both Genetic and Simple Obesity in Children. EBioMedicine. 2015;2(8):968–84. doi: 10.1016/j.ebiom.2015.07.007. PubMed PMID: ; PMCID: PMC4563136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. doi: 10.1038/nature 12820. PubMed PMID: ; PMCID: PMC3957428. • This widely cited study demonstrates that extreme short-term dietary interventions can result in changes that are comparable in magnitude across different subjects, and that some of the taxonomic shifts are reproducible. However, it does not establish that the direction as well as the magnitude of overall microbial community change are similar across individuals, and numerous studies now suggest that the direction of short-term diet-induced change differs in different individuals (as is also the case for antibiotic intervention). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lozupone CA, Knight R. Global patterns in bacterial diversity. Proc Natl Acad Sci USA. 2007; 104(27): 11436–40. doi: 10.1073/pnas.0611525104. PubMed PMID: ; PMCID: PMC2040916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon Jl. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6(10):776–88. doi: 10.1038/nrmicro1978. PubMed PMID: ; PMCID: PMC2664199. • This review and meta-analysis represented the first large-scale attempt to integrate hostassociated and free-living microbial communities into a single abstract map using common analysis techniques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCafferty J, Muhlbauer M, Gharaibeh RZ, Arthur JC, Perez-Chanona E, Sha W, Jobin C, Fodor AA. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. The ISME journal. 2013;7(11):2116–25. doi: 10.1038/ismej.2013.106. PubMed PMID: ; PMCID: PMC3806260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Silva RR, Dorrestein PC, Quinn RA. Illuminating the dark matter in metabolomics. Proc Natl Acad Sci USA. 2015;112(41):12549–50. doi: 10.1073/pnas.1516878112. PubMed PMID: ; PMCID: PMC4611607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu WT, Crusemann M, Boudreau PD, Esquenazi E, Sandoval-Calderon M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu CC, Floras DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw CC, Yang YL, Humpf HU, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, P CAB, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJN, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodriguez AMC, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard PM, Phapale P, Nothias LF, Alexandrov T, Litaudon M, Wolfender JL, Kyle JE, Metz TO, Peryea T, Nguyen DT, VanLeer D, Shinn P, Jadhav A, Muller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BO, Pogliano K, Linington RG, Gutierrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC, Bandeira N. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol. 2016;34(8):828–37. doi: 10.1038/nbt.3597. PubMed PMID: ; PMCID: PMC5321674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.loannidis JP. Why most published research findings are false. PLoS Med. 2005;2(8):e124. doi: 10.1371/journal.pmed.0020124. PubMed PMID: ; PMCID: PMC1182327. •• This important article introduces the first resource for online analysis and comparison of mass spectrometry datasets, introducing many concepts such as living data and crowdsourced annotation to the mass spectrometry field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez A, Knight R. Advancing analytical algorithms and pipelines for billions of microbial sequences. Curr Opin Biotechnol. 2012;23(1):64–71. doi: 10.1016/j.copbio.2011.11.028. PubMed PMID: ; PMCID: PMC3273654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouslimani A, Melnik AV, Xu Z, Amir A, da Silva RR, Wang M, Bandeira N, Alexandrov T, Knight R, Dorrestein PC. Lifestyle chemistries from phones for individual profiling. Proc Natl Acad Sci USA. 2016;113(48):E7645-E54. doi: 10.1073/pnas.1610019113. PubMed PMID: ; PMCID: PMC5137711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouslimani A, Porto C, Rath CM, Wang M, Guo Y, Gonzalez A, Berg-Lyon D, Ackermann G, Moeller Christensen GJ, Nakatsuji T, Zhang L, Borkowski AW, Meehan MJ, Dorrestein K, Gallo RL, Bandeira N, Knight R, Alexandrov T, Dorrestein PC. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci USA. 2015;112(17):E2120-9. doi: 10.1073/pnas. 1424409112. PubMed PMID: ; PMCID: PMC4418856. • This paper describes the first high-resolution (hundreds of sites) spatial map of microbes across the skin surface of the human body. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garg N, Wang M, Hyde E, da Silva RR, Melnik AV, Protsyuk I, Bouslimani A, Lim YW, Wong R, Humphrey G, Ackermann G, Spivey T, Brouha SS, Bandeira N, Lin GY, Rohwer F, Conrad DJ, Alexandrov T, Knight R, Dorrestein PC. Three-Dimensional Microbiome and Metabolome Cartography of a Diseased Human Lung. Cell Host Microbe. 2017;22(5):705–16 e4. doi: 10.1016/j.chom.2017.10.001. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapono CA, Morton JT, Bouslimani A, Melnik AV, Orlinsky K, Knaan TL, Garg N, Vazquez-Baeza Y, Protsyuk I, Janssen S, Zhu Q, Alexandrov T, Smarr L, Knight R, Dorrestein PC. Creating a 3D microbial and chemical snapshot of a human habitat. Sci Rep. 2018;8(1):3669. doi: 10.1038/s41598-018-21541-4. PubMed PMID: ; PMCID: PMC5829137. • This paper describes the first 3D visualization of humans in their built environment, with many patterns immediately revealed by the 3D map that would have been difficult or impossible to understand in the data if analyzed non-spatially. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petras D, Nothias LF, Quinn RA, Alexandrov T, Bandeira N, Bouslimani A, Castro-Falcon G, Chen L, Dang T, Floras DJ, Hook V, Garg N, Hoffner N, Jiang Y, Kapono CA, Koester I, Knight R, Leber CA, Ling TJ, Luzzatto-Knaan T, McCall LI, McGrath AP, Meehan MJ, Merritt JK, Mills RH, Morton J, Podvin S, Protsyuk I, Purdy T, Satterfield K, Searles S, Shah S, Shires S, Steffen D, White M, Todoric J, Tuttle R, Wojnicz A, Sapp V, Vargas F, Yang J, Zhang C, Dorrestein PC. Mass Spectrometry-Based Visualization of Molecules Associated with Human Habitats. Anal Chem. 2016;88(22): 10775–84. doi: 10.1021/acs.analchem.6b03456. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Protsyuk I, Melnik AV, Nothias LF, Rappez L, Phapale P, Aksenov AA, Bouslimani A, Ryazanov S, Dorrestein PC, Alexandrov T. 3D molecular cartography using LC-MS facilitated by Optimus and ‘ili software. Nat Protoc. 2018;13(1):134–54. doi: 10.1038/nprot.2017.122. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 33.Paneth N, Vinten-Johansen P, Brody H, Rip M. A rivalry of foulness: official and unofficial investigations of the London cholera epidemic of 1854. Am J Public Health. 1998;88( 10): 1545–53. PubMed PMID: ; PMCID: PMC1508470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weingarden A, Gonzalez A, Vazquez-Baeza Y, Weiss S, Humphry G, Berg-Lyons D, Knights D, Unno T, Bobr A, Kang J, Khoruts A, Knight R, Sadowsky MJ. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome. 2015;3:10. doi: 10.1186/s40168-015-0070-0. PubMed PMID: ; PMCID: PMC4378022. •• This paper uses the Human Microbiome Project data as an abstract data frame to enable interpretation of a second dataset, in this case the dynamics of re-colonization of the gut after fecal transplantation, enabling straightforward analysis of the ecological situation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, Gonzalez A, Kosciolek T, McCall LI, McDonald D, Melnik AV, Morton JT, Navas J, Quinn RA, Sanders JG, Swafford AD, Thompson LR, Tripathi A, Xu ZZ, Zaneveld JR, Zhu Q, Caporaso JG, Dorrestein PC. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16(7):410–22. doi: 10.1038/S41579-018-0029-9. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 36.Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2(1):16. doi: 10.1186/2047-217X-2-16. PubMed PMID: ; PMCID: PMC4076506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon Jl, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME Allows Analysis of High-throughput Community Sequencing Data. Nat Methods. 2010;7(5):335–6. PubMed PMID: •• This paper describes one of the most widely used tools in microbial ecology, QIIME, which owes its success in large part to software construction techniques such as unit testing, modularity, and adoption of open standards and formats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017;27(4):626–38. doi: 10.1101/gr.216242.116. PubMed PMID: ; PMCID: PMC5378180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. doi: 10.1186/S40168-018-0426-3. PubMed PMID: ; PMCID: PMC5827986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aksenov AA, da Silva R, Knight R, Lopes NP, Dorrestein PC. Global chemical analysis of biology by mass spectrometry. Nat Rev Chem. 2017; 1:0054. [Google Scholar]
- 41.Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) 2018. [cited 2018 July 1]. Available from: https://www.genome.gov/27541954/dna-sequencing-costs-data/. • This useful resource describes changes in sequencing costs at NHGRI and their underlying basis, and provides the information in frequently updated and useful graphical form.
- 42.Sauerschnig C, Doppler M, Bueschl C, Schuhmacher R. Methanol Generates Numerous Artifacts during Sample Extraction and Storage of Extracts in Metabolomics Research. Metabolites. 2017;8(1). doi: 10.3390/metabo8010001. PubMed PMID: ; PMCID: PMC5875991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheubert K, Hufsky F, Petras D, Wang M, Nothias LF, Duhrkop K, Bandeira N, Dorrestein PC, Bocker S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat Commun. 2017;8(1):1494. doi: 10.1038/s41467-017-01318-5. PubMed PMID: ; PMCID: PMC5684233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Members MSIB, Sansone SA, Fan T, Goodacre R, Griffin JL, Hardy NW, Kaddurah- Daouk R, Kristal BS, Lindon J, Mendes P, Morrison N, Nikolau B, Robertson D, Sumner LW, Taylor C, van der Werf M, van Ommen B, Fiehn O. The metabolomics standards initiative. Nat Biotechnol. 2007;25(8):846–8. doi: 10.1038/nbt0807-846b. PubMed PMID: •• This important consortium effort describes an initiative to define standards for the field of metabolomics. [DOI] [PubMed] [Google Scholar]
- 45.Salek RM, Conesa P, Cochrane K, Haug K, Williams M, Kale N, Moreno P, Jayaseelan KV, Macias JR, Nainala VC, Hall RD, Reed LK, Viant MR, O’Donovan C, Steinbeck C. Automated assembly of species metabolomes through data submission into a public repository. Gigascience. 2017;6(8):1–4. doi: 10.1093/gigascience/gix062. PubMed PMID: ; PMCID: PMC5737527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haug K, Salek RM, Conesa P, Hastings J, de Matos P, Rijnbeek M, Mahendraker T, Williams M, Neumann S, Rocca-Serra P, Maguire E, Gonzalez-Beltran A, Sansone SA, Griffin JL, Steinbeck C. MetaboLights--an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013;41(Database issue):D781-6. doi: 10.1093/nar/gks1004. PubMed PMID: ; PMCID: PMC3531110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS, Sumner S, Subramaniam S. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016;44(D1):D463-70. doi: 10.1093/nar/gkv1042. PubMed PMID: ; PMCID: PMC4702780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158(6):1402–14. doi: 10.1016/j.cell.2014.08.032. PubMed PMID: ; PMCID: PMC4164201. •• This fascinating and important article uses genome mining to discover gene cassettes capable of synthesizing novel antibiotics within the human gut itself. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicholson JK, Lindon JC. Systems biology: Metabonomics. Nature. 2008;455(7216): 1054–6. doi: 10.1038/4551054a. PubMed PMID: •• This important perspective introduced the concept of using chemical characterization to read out metabolic state as an analog to the use of genomics to read out genotype. [DOI] [PubMed] [Google Scholar]
- 50.Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, Henrissat B, Knight R, Gordon Jl. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332(6032):970–4. doi: 10.1126/science. 1198719. PubMed PMID: ; PMCID: PMC3303602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276(5313):734–40. PubMed PMID: •• This groundbreaking and influential review redefined concepts of microbial life and its phylogenetic and geographic distribution for a generation of microbial ecologists. [DOI] [PubMed] [Google Scholar]
- 52.Minich JJ, Zhu Q, Janssen S, Hendrickson R, Amir A, Vetter R, Hyde J, Doty MM, Stillwell K, Benardini J, Kim JH, Allen EE, Venkateswaran K, Knight R. KatharoSeq Enables High-Throughput Microbiome Analysis from Low-Biomass Samples. mSystems. 2018;3(3). doi: 10.1128/mSystems.00218-17. PubMed PMID: ; PMCID: PMC5864415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gika HG, Theodoridis GA, Wilson ID. Liquid chromatography and ultra-performance liquid chromatography-mass spectrometry fingerprinting of human urine: sample stability under different handling and storage conditions for metabonomics studies. J Chromatogr A. 2008;1189(1–2):314–22. doi: 10.1016/j.chroma.2007.10.066. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 54.Lou JJ, Mirsadraei L, Sanchez DE, Wilson RW, Shabihkhani M, Lucey GM, Wei B, Singer EJ, Mareninov S, Yong WH. A review of room temperature storage of biospecimen tissue and nucleic acids for anatomic pathology laboratories and biorepositories. Clin Biochem. 2014;47(4–5):267–73. doi: 10.1016/j.clinbiochem.2013.12.011. PubMed PMID: ; PMCID: PMC3976177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song SJ, Amir A, Metcalf JL, Amato KR, Xu ZZ, Humphrey G, Knight R. Preservation Methods Differ in Fecal Microbiome Stability, Affecting Suitability for Field Studies. mSystems. 2016;1(3). doi: 10.1128/mSystems.00021-16. PubMed PMID: ; PMCID: PMC5069758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choo JM, Leong LE, Rogers GB. Sample storage conditions significantly influence faecal microbiome profiles. Sci Rep. 2015;5:16350. doi: 10.1038/srep16350. PubMed PMID: ; PMCID: PMC4648095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hale VL, Tan CL, Niu K, Yang Y, Cui D, Zhao H, Knight R, Amato KR. Effects of field conditions on fecal microbiota. J Microbiol Methods. 2016;130:180–8. doi: 10.1016/j.mimet.2016.09.017. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 58.Vogtmann E, Chen J, Amir A, Shi J, Abnet CC, Nelson H, Knight R, Chia N, Sinha R. Comparison of Collection Methods for Fecal Samples in Microbiome Studies. Am J Epidemiol. 2017; 185(2): 115–23. doi: 10.1093/aje/kww177. PubMed PMID: ; PMCID: PMC5253972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loftfield E, Vogtmann E, Sampson JN, Moore SC, Nelson H, Knight R, Chia N, Sinha R. Comparison of Collection Methods for Fecal Samples for Discovery Metabolomics in Epidemiologic Studies. Cancer Epidemiol Biomarkers Prev. 2016;25(11):1483–90. doi: 10.1158/1055-9965.EPI-16-0409. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, Gilbert JA, Karsch- Mizrachi I, Johnston A, Cochrane G, Vaughan R, Hunter C, Park J, Morrison N, Rocca-Serra P, Sterk P, Arumugam M, Bailey M, Baumgartner L, Birren BW, Blaser MJ, Bonazzi V, Booth T, Bork P, Bushman FD, Buttigieg PL, Chain PS, Charlson E, Costello EK, Huot-Creasy H, Dawyndt P, DeSantis T, Fierer N, Fuhrman JA, Gallery RE, Gevers D, Gibbs RA, San Gil I, Gonzalez A, Gordon Jl, Guralnick R, Hankeln W, Highlander S, Hugenholtz P, Jansson J, Kau AL, Kelley ST, Kennedy J, Knights D, Koren O, Kuczynski J, Kyrpides N, Larsen R, Lauber CL, Legg T, Ley RE, Lozupone CA, Ludwig W, Lyons D, Maguire E, Methe BA, Meyer F, Muegge B, Nakielny S, Nelson KE, Nemergut D, Neufeld JD, Newbold LK, Oliver AE, Pace NR, Palanisamy G, Peplies J, Petrosino J, Proctor L, Pruesse E, Quast C, Raes J, Ratnasingham S, Ravel J, Reiman DA, Assunta-Sansone S, Schloss PD, Schriml L, Sinha R, Smith Ml, Sodergren E, Spo A, Stombaugh J, Tiedje JM, Ward DV, Weinstock GM, Wendel D, White O, Whiteley A, Wilke A, Wortman JR, Yatsunenko T, Glockner FO. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 2011;29(5):415–20. doi: 10.1038/nbt.1823. PubMed PMID: ; PMCID: PMC3367316. •• This important community effort led by the Genomic Standards Consortium defines standards for annotating amplicon and metagenomic sequences with sample annotations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yutin N, Makarova KS, Gussow AB, Krupovic M, Segall A, Edwards RA, Koonin EV. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol. 2018;3(1):38–46. doi: 10.1038/s41564-017-0053-y. PubMed PMID: ; PMCID: PMC5736458. •• This fascinating study reveals that even the most abundant viruses in the gut are still poorly characterized and that there is much to discover before hypotheses about the human virome can even be adequately framed. The same is likely true for other components of the human microbiome besides bacteria, which have received the most attention to date. [DOI] [PMC free article] [PubMed] [Google Scholar]