

Abstract
Objective:
Cortical spreading depolarizations (CSDs) are intense and ubiquitous depolarization waves relevant for the pathophysiology of migraine and brain injury. CSDs disrupt the blood-brain barrier (BBB), but the mechanisms are unknown.
Methods:
A total of six cortical SDs (CSDs) were evoked over 1h by topical application of 300 mM KCl or optogenetically with 470 nm (blue) LED over the right hemisphere in anesthetized mice (C57BL/6J wild type, Thy1-ChR2-YFP line 18 and cav-1-/-). BBB disruption was assessed by Evans Blue (2% EB, 3ml/kg, intra-arterial) or Dextran (200 mg/kg, Fluorescein, 70,000 MW, intra-arterial) extravasation in parietotemporal cortex at 6–24h after CSD. Endothelial cell ultrastructure was examined using transmission electron microscopy 0–24h after the same CSD protocol in order to assess vesicular trafficking, endothelial tight junctions and pericyte integrity. Mice were treated with vehicle, isoform non-selective ROCK inhibitor fasudil (10 mg/kg, IP 30 min before CSD), or ROCK-2 selective inhibitor KD025 (200 mg/kg, PO bid for 5 doses before CSD).
Results:
We show that CSD-induced BBB opening to water and large molecules is mediated by increased endothelial transcytosis starting between 3 and 6 hours and lasting approximately 24 hours. Endothelial tight junctions, pericytes and basement membrane remain preserved after CSDs. Moreover, we show that CSD-induced BBB disruption is exclusively caveolin-1-dependent, and requires rho-kinase 2 activity. Importantly, CSD-induced BBB disruption is independent of tissue hypoxia, as hyperoxia failed to prevent CSD-induced BBB breakdown.
Interpretation:
Our data elucidate the mechanisms by which CSDs lead to transient BBB disruption, with diagnostic and therapeutic implications for migraine and brain injury.
Keywords: cortical spreading depression, blood-brain barrier, transcytosis, caveolin-1, rho-associated kinase, endothelium
Cortical spreading depolarizations (CSDs) are intense neuroglial pandepolarization waves that slowly propagate (millimeters per minute) within contiguous gray matter (Leao, 1944). Evolutionarily highly conserved from insect to man, CSDs are the electrophysiological substrate of migraine aura (Ayata, 2010), and numerous recurrent CSD waves occur spontaneously for hours and days after brain injury, worsening the outcome (Lauritzen et al., 2011; von Bornstadt et al., 2015). As such, CSD is central to the pathophysiology of many neurological disorders (Ayata and Lauritzen, 2015).
During CSD propagation, massive transmembrane ion and water shifts lead to elevated extracellular concentrations of K+ and virtually every neurotransmitter and neuromodulator molecule measured to date. These changes engulf cerebral vasculature like a tsunami wave, triggering blood flow responses larger than any other observed in the brain, disrupting the cerebrovascular reflexes (e.g. neurovascular coupling), and opening the blood-brain barrier (BBB) (Ayata and Lauritzen, 2015). Although MMP-9 upregulation has been implicated (Gursoy-Ozdemir et al., 2004), the effects of CSD on BBB constituents, and the underlying mechanisms, are unknown. Given the importance of both the BBB and CSDs in the pathophysiology of migraine and brain injury (Dreier, 2011; Lauritzen et al., 2011), we examined the ultrastructural features and molecular mechanisms underlying CSD-induced cerebrovascular hyperpermeability. Our data demonstrate for the first time that CSDs, without attendant tissue injury, open the BBB by activating caveolin-1 and rho-associated kinase (ROCK)-dependent endothelial transcytosis.
MATERIALS AND METHODS
Animals
A total of 199 C57BL/6J wild type (male, 22–36 g), 6 Thy1-ChR2-YFP line 18 (male, 25–30 g; B6.Cg-Tg(Thy1-COP4/EYFP)18Gfng/J; Jackson Laboratories, Bar Harbor, ME, USA), and 6 caveolin-1 knockout (cav-1-/-) and age-matched control mice (male, 25 g, JAX: 004585) were used. All experiments were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committees following the NIH Guide for Use and Care of Laboratory Animals.
CSD induction and monitoring
Mice were anesthetized with isoflurane (2.5% induction, 1% maintenance in 70% N2O/30% O2), and rectal temperature was kept at 37 ± 0.5°C throughout the anesthesia. Femoral artery was catheterized for continuous measurement of blood pressure (PowerLab; ADInstruments, Colorado Springs, MO, USA) and injection of Evans blue or 70 KDa-FITC-dextran. Two burr holes were drilled (1.5 mm anterior, 1.5 mm posterior, 2 mm lateral to the bregma) for CSD monitoring by extracellular steady (DC) potential and electrocorticogram recordings using a differential amplifier (EX1; Dagan Corporation, Minneapolis, MN), glass capillary microelectrodes (tip diameter ~10 μm, filled with 1M NaCl) placed 200 μm deep into the cortex, and an analog to digital converter (PowerLab; ADInstruments, Colorado Springs, CO). A third burr hole over occipital cortex (<500 μm dural exposure) was used to briefly (< 1 min) apply a cotton ball soaked in 300 mM KCl. Cortex was immediately washed with normal saline as soon as a CSD was detected. In our experience, small area and brief time of exposure, as well as high K+ clearance capacity of the brain tissue, limit K+ transfer by convection in paravascular spaces, so that extracellular K+ concentrations always remain stable when measured by K+-selective electrodes >1 mm away from the KCl application site (data not shown). The amplitude and duration at half-amplitude of the first CSD in each hemisphere were measured. In a separate cohort we induced CSDs non-invasively through intact skull using transgenic mice expressing channelrhodopsin-2 (Thy1-ChR2-YFP) in neurons as previously described (Chung et al., 2018; Houben et al., 2016). Briefly, the skull was exposed by midline scalp incision and cleared of connective tissue. Care was taken to prevent bone drying with application of mineral oil in order to maintain skull translucency. A 400 μm diameter optical fiber was positioned over motor cortex (2 mm anterior and 2 mm lateral to bregma). CSDs were induced optogenetically with 470 nm (blue) LED (MF470F3, DC2100; Thorlabs, Newton, NJ, USA) delivered for 10 seconds at 3 mW total power. A microscope objective USB camera (AmScope, MU300, Irvine, CA, USA) coupled to a dissecting stereomicroscope illuminated with unfiltered white light was used to take time-lapse intrinsic signal images every 2 seconds. The intrinsic signal is a well-established method to detect blood volume changes caused by CSDs (Chung et al., 2018; Yuzawa et al., 2012). CSDs were detected in real time by subtraction of adjacent images using MATLAB (Mathworks, Natick, MA, USA). Every stimulus under these conditions resulted in a CSD. After CSD inductions, local anesthetic (lidocaine gel 2%) was applied and mice were allowed to recover from anesthesia until the time of sacrifice using 5% isoflurane inhalation.
Brain water content
Six or 48 hours after CSD, parietotemporal cortex from CSD and nCSD sides were removed, weighed, and dried at 99°C for 48 hours and reweighed. Water content (%) was calculated as [(wet weight- dry weight)/wet weight] × 100.
Evans blue leakage
Evans blue (2%) was injected (3 ml/kg, 60 mg/kg) via femoral artery right after the last CSD. At this dose level, vast majority (>98%) of EB is bound to plasma proteins, predominantly albumin (Saunders et al., 2015). Mice were transcardially perfused with cold 0.9% NaCl, and parietotemporal cortex from CSD and nCSD hemispheres were dissected, weighed and homogenized in a sevenfold volume of 50% trichloroacetic acid with sonication for 2 min. After centrifugation (3000 G for 20 min), the supernatants were diluted fourfold with ethanol. Fluorescence intensity was measured by a microplate fluorescence reader (620 nm excitation, 680 nm emission; Wallac, Perkin Elmer, Wellesley, Massachusetts, USA). Calculations were based on external standard readings, and extravasated dye was expressed as ng/mg tissue.
Dextran extravasation
Dextran (200 mg/kg, Fluorescein, 70,000 MW, Anionic, Lysine Fixable; Thermo Fisher Scientific, Waltham, MA, USA) was injected through femoral artery 6 or 48 hours after the last CSD. After 1 hour of dextran circulation, the brains were removed and freshly frozen in 2-methylbutane on dry ice. Cryosections (12 μm) were obtained and mounted on gelatinized slides. After air drying for 15 min, sections were post-fixed in 4% PFA at room temperature for 15 min, washed in PBS and co-stained with isolectin B4 to visualize blood vessels (1:500; I21411, Molecular Probes, CA, USA). Sections were cover-slipped with mounting medium containing DAPI (Vector, Burlingame, CA, USA), dried overnight and analyzed with a laser scanning confocal microscope. Quantification of vessel leakage was performed as previously described (Ben-Zvi et al., 2014). In brief, confocal images of 12 μm-thick brain sections from tracer-injected animals were co-stained with lectin and analyzed using ImageJ (NIH, MD, USA). The same acquisition parameters were applied to all images and the same threshold was used. Tracer-positive area outside a vessel (parenchyma) was used as a parameter for leakage. For each hemisphere, at least 10 sections of a fixed lateral cortical plate area were scored. Average representation of each leakage group was calculated for ipsilateral and contralateral hemispheres.
Transmission electron microscopy (TEM)
Tissue processing for TEM was performed as described previously (Ben-Zvi et al., 2014). For analysis of endothelial ultrastructure (i.e. TJs and vesicles) mice were perfused through the heart with PBS followed by fixative solutions. The brain was removed, post-fixed and washed. Coronal 50 μm-thick brain sections were processed free floating. Ultrathin sections were then cut, collected on grids, counterstained and examined under a 1200EX electron microscope (JEOL USA Inc., MA, USA) equipped with a 2k CCD digital camera (Advanced Microscopy Techniques Corp., MA, USA). All ultrastructural quantifications were performed using ImageJ (NIH, MD, USA). Measures of tight junction structure (length, width, thickness and angle) as well as basement membrane thickness were performed on 10 images (40,000x magnification) per hemisphere (nCSD vs. CSD; n=3 mice) at each time point post-CSD (0h, 3h, 6h, 12h, 24h, and 48h). Basement membrane thickness (nm) was measured manually using ImageJ; three measures were obtained per capillary profile and averaged. To quantify endothelial vesicle counts, each capillary profile was imaged by two electron micrographs (12,000x magnification), each covering half the capillary profile. Endothelial vesicles were manually counted in 10 capillary cross sectional profiles per hemisphere (nCSD vs. CSD; n=3 mice) at each time point post-CSD (0h, 3h, 6h, 12h, 24h, and 48h). (i.e. 20 micrographs/hemisphere/time point). Vesicle counts from 10 capillaries were averaged within each animal to obtain a single data point per animal. The coefficient of variation of vesicle counts within each animal was 54±2% (mean ± SEM) when averaged across a representative cohort. Data were expressed as number per endothelial cytoplasmic area (μm2) for “free” cytoplasmic vesicles, or per plasma membrane length (μm) for membrane-connected vesicles.
For analysis of transcytosis/leakage, Horseradish peroxidase (HRP) type II (10 mg/20 g in PBS; Sigma Aldrich, MO, USA) was injected through femoral artery catheter 6 and 24 hours after CSD induction. After 30 min of HRP circulation, brains were dissected and fixed in a sodium-cacodylate-buffered mixture followed by PFA and washes. Tissue sections (50 μm) were then processed free floating. To reveal HRP as an electron dense precipitate, sections were incubated in a Tris-HCl buffer containing 3–3’ diaminobenzidine and H2O2, post-fixed in osmium tetroxide, and dehydrated and embedded in epoxy resin (Ben-Zvi et al., 2014). Imaging and quantification of HRP-filled vesicles followed the same protocol as above.
Histology
Immunohistochemistry was performed as previously described (Ben-Zvi et al., 2014; Lacoste et al., 2014). Pericyte coverage of cortical vessels was quantified by analyzing the proportion of total CD31-positive endothelial length also positive for pericyte marker NG2. Pericyte coverage of brain microvascular networks was quantified on low magnification confocal images as the ratio between the total area of NG2-positive capillaries and the total area of CD31-positive capillaries.
Pharmacological studies
Mice were randomized to receive vehicle (saline, 0.1 ml), or isoform-nonselective ROCK inhibitor fasudil (10 mg/kg; Tocris Bioscience, Bristol, UK) via intraperitoneal injection 1 hour before CSD induction. The ROCK2-selective inhibitor KD025 (formerly SLx-2119) (200 mg/kg; Kadmon Corp. LLC, NY, USA) or its vehicle (0.4% methylcellulose) was administered every 12h via orogastric gavage for 48 hours before to 24 hours after CSD induction. These doses were chosen based on prior reports in mice from our laboratory (Lee et al., 2014; Shin et al., 2007).
RT-qPCR
Total RNA was isolated from CSD and non-CSD brain tissue using illustra RNAspin Mini Kit (GE Healthcare Life Sciences, Pittsburg, PA, USA) and reverse transcription was performed using SuperScript III first-strand synthesis system (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. The following primers were used: ROCK1 5’-CTGCTGAAGTCGTGCTTGCA-3’ (forward primer) and 5’-AGCATGTTATCGGGCTTCACA-3’ (reverse primer); ROCK2, 5’-CTGAA-TGAAATGCAGGCTCAA-3’ (forward primer) and 5’-CCCTGGTCCACTGCCTATAC-3’ (reverse primer). The PCR products were quantified by ABI Prism® 7500 Sequence Detection System.
Study design and statistical analysis
Non-CSD hemispheres served as the control for CSD hemispheres within each animal. All experiments testing an intervention (ROCK inhibition, cav-1-/- knockout) were carried out and analyzed in a blinded fashion. Analyses were made by Prism 6 (GraphPad Software, Inc., CA, USA). Data are presented as mean ± S.E.M or as whisker-box plots. Whenever appropriate, differences between groups were determined by t-test (two-group comparisons), or one-way or two-way ANOVA (multigroup comparisons) followed post-hoc analysis. In the absence of pilot data, initial sample sizes were selected empirically and the data from these were used to calculate final sample sizes to achieve 80% power to detect 50% effect size (α=0.05). Sample sizes in EM cohorts (n=3 mice, 20 replicates per mouse) are typical for this method (Ben-Zvi et al., 2014). P value less than 0.05 was considered statistically significant.
RESULTS
Initial experiments aimed to characterize BBB opening after CSD and its time course. Brief topical (epidural) KCl application (300 mM for <1 minute followed by saline wash), the most frequently used depolarizing stimulus (Ayata, 2013; Karatas et al., 2013), reliably triggered a CSD in occipital cortex, which propagated across the ipsilateral hemisphere as detected electrophysiologically by two serially placed glass microelectrodes (Figure 1A). Using this approach in mice, we triggered 6 CSDs over an hour (Eikermann-Haerter et al., 2012). Then, using three independent but complementary methods, we examined BBB integrity in the parietotemporal cortex, away from the KCl application site. The contralateral hemisphere served as an internal control, since CSD does not propagate between hemispheres under these experimental conditions (Ayata, 2013). First, we measured the parenchymal water content and found a marked increase at 6 hours post-CSD, and to a lesser extent at 24 hours, when compared with the contralateral hemisphere (Figure 1B). We next examined cerebral extravasation of Evans blue (EB) dye administered intravascularly immediately after the last CSD. At the selected dose, vast majority of EB binds to serum albumin (~65 kDa), and the complex (~70 kDa) does not cross an intact BBB (Saunders et al., 2015). As with cortical water content, EB extravasation significantly increased within 6 hours after CSDs compared to the contralateral hemisphere, and progressed for at least 24 hours (Figure 1C, left). In a separate group of mice, we found that even a single CSD was able to open the BBB, although its magnitude was smaller compared to 6 CSDs (Figure 1C, middle). Sham-operated mice showed very little EB leakage, and we found no histological evidence of cortical injury due to our CSD induction paradigm (Data not shown). Nevertheless, because epidural KCl application is somewhat invasive (i.e. requires craniotomy), we also triggered CSDs noninvasively through intact skull using optogenetics in channelrhodopsin-2 transgenic mice (Thy1-ChR2-YFP homozygous, line 18), and reproduced the EB leakage in the ipsilateral, but not the contralateral, hemisphere (Figure 1C, right). The small yet consistent increase in parenchymal EB in the contralateral hemisphere in all these experiments likely reflects diffusion of EB from the ipsilateral hemisphere over time, and possibly contamination with ipsilateral EB during tissue preparation. Lastly, because EB has significant shortcomings to assess BBB leakage (Saunders et al., 2015), we confirmed these results by measuring the extravasation of 70 kDa FITC-dextran administered intravascularly 1 hour before sacrifice. We found significant 70 kDa FITC-dextran leakage at 6 hours post-CSD, and complete closure of the BBB within 48 hours (Figure 1D). Altogether, these complementary methods converge to a transient opening of the BBB that starts within 6 hours after CSD and resolves within 48 hours.
Figure 1. BBB transiently opens after CSD.
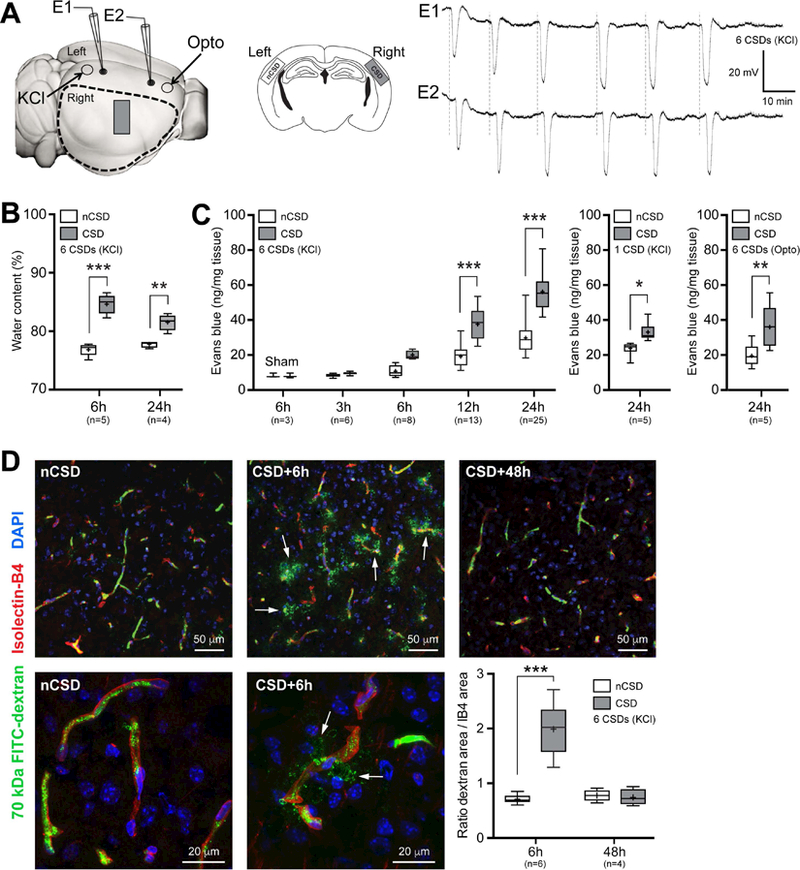
A) Left panel shows the experimental setup to induce CSDs by either topical KCl (300 mM) application onto occipital cortex via a burr hole, or optogenetic (Opto) stimulation of frontal cortex through intact skull. KCl-induced CSDs were recorded by two glass micropipettes placed serially along the CSD propagation path. Optogenetic CSDs were detected non-invasively by optical intrinsic signal imaging through intact skull. Dashed outline shows cortical tissue harvested for water content and Evans blue extravasation and ROCK mRNA measurements, and the rectangle shows the approximate position at which tissue sections were obtained for fluorescent confocal and electron microscopy studies, also shown in the middle panel on coronal sections in CSD and control (non-CSD, nCSD) cortices. All assessments were carried out in tissue remote from CSD induction and electrophysiological recording sites to avoid direct non-specific effects of trauma and high concentration KCl. Right panel shows representative intracortical microelectrode recordings of 6 CSDs induced approximately every 10 minutes by topical KCl application.
B) Parenchymal water content at 6 and 24 hours after 6 CSDs compared with nCSD side. **p<0.01, ***p<0.001, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparisons test.
C) Left panel shows cortical Evans blue extravasation in CSD and nCSD hemispheres 3 to 24 hours after 6 CSDs. Tissue Evans blue levels in sham controls at 6 hours are also shown. ***p<0.001, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparisons test. Middle panel shows the effect of a single CSD at 24 hours. *p<0.05, paired t-test. Right panel shows cortical Evans blue extravasation in CSD and nCSD hemispheres 24 hours after 6 optogenetic CSDs induced and recorded non-invasively. **p<0.01, paired t-test.
D) Histological analysis of 70 kDa FITC-Dextran (green) leakage from isolectin-B4 (IB4)-stained blood vessels (red) at 6 and 48 hours after 6 CSDs induced by KCl. Top and bottom histological panels display low and high magnifications, respectively, of different fields. Arrows point to extravascular 70 kDa FITC-Dextran. Lower right panel shows the ratio of total pixel area of 70 kDa FITC-Dextran to IB4 pixel area indicating vasculature. ***p<0.001, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparisons.
Whisker-box plots show full (whiskers) and interquartile (box) ranges, as well as the median (horizontal line) and mean (+).
We next examined the subcellular mechanisms underlying BBB leakage after CSD. Unlike peripheral endothelial cells, brain endothelial cells are characterized by highly specialized tight junctions (TJs) preventing water-soluble molecules from passing between adjacent cells, as well as by low rates of transcytosis from the vessel lumen to the brain parenchyma (Andreone et al., 2015; Chow and Gu, 2015). Therefore, after showing CSD-induced BBB permeability to large molecules, we examined the ultrastructural indices of BBB integrity following CSD in the same region of the cerebral cortex (i.e. remote from the KCl application site). Transmission electron microscopy (TEM) showed TJs as electron-dense linear structures where adjacent membranes are tightly opposed (Figure 2A). We did not find any change in TJ integrity, as both the space between endothelial cells and endothelial thickness at the TJ remained constant throughout our temporal analysis following CSDs (Figure 2A). Tight junctional length and angle did not appear significantly altered by CSD either (Figure 2B). We further examined functional integrity of TJs following intravascular horseradish peroxidase (HRP) injection (Andreone et al., 2017; Ben-Zvi et al., 2014; Reese and Karnovsky, 1967). Once exposed to diaminobenzidine and H2O2, an electron-dense reaction product precipitates and fills the vessel lumen. We found that TJ barrier function was preserved at all time points as HRP penetrated the intercellular spaces between neighboring endothelial cells only for short distances and stopped abruptly at TJ (a.k.a. kissing points; Figure 2C).
Figure 2. CSD does not affect tight junction integrity.
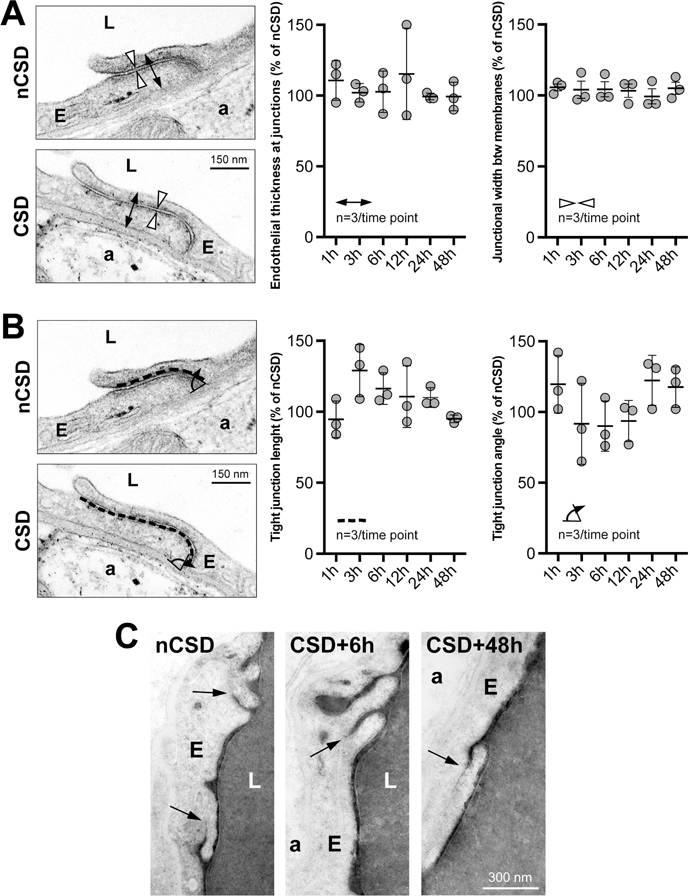
Endothelial tight junctions remain sealed throughout the period of CSD-induced BBB breakdown, as assessed by their normal ultrastructure (A and B) and by their ability to retain intra-arterially injected horseradish peroxidase (C; dark precipitate revealed with DAB). Double arrows show the measurement of endothelial thickness at junctions, arrowheads show the measurement of junctional width, dashed line shows the measurement of endothelial length at junctions, curved arrow shows the measurement of tight junctional angle, arrows indicate kissing points where endothelial membranes are tightly opposed with no evidence of HRP leakage. p>0.05, one-way ANOVA. Data are mean ± S.E.M. N=3 animals with 20 replicates in each animal.
While TJs remained unaffected by CSD, we found a remarkable increase in endothelial transcytosis (Figure 3). Within 6 hours after CSD, the density of endothelial vesicles increased by more than 50% compared with the contralateral hemisphere, peaked around 6–12 hours post-CSD, and gradually normalized over the next 24 hours (Figure 3A). This increase was evident in both cytoplasmic and plasma membrane-connected vesicles (Figure 3B; see also luminal and abluminal plasma membrane-connected vesicles). Importantly, neither endothelial cross sectional area nor capillary diameter was altered at any time point after CSD, as relevant denominators for vesicle density (Figure 3C). Presence of vesicles filled with HRP, HRP exocytosis on the abluminal side, and accumulation within the basement membrane all confirmed the directional transport from vessel lumen to brain parenchyma via transcytosis (Figure 4).
Figure 3. CSD transiently activates endothelial transcytosis.
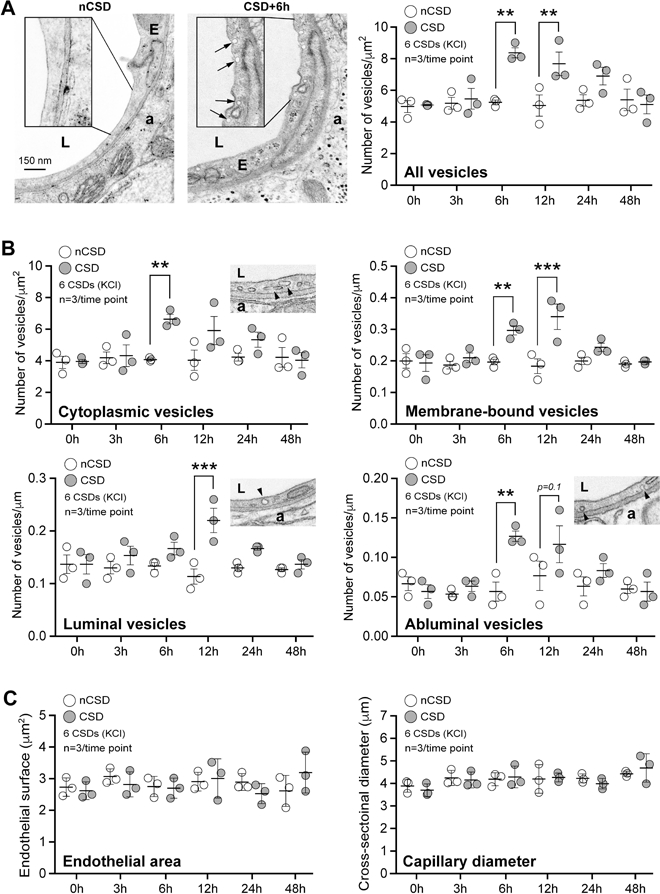
A) Transmission electron microscopic analysis revealed a significant increase in endothelial vesicle number 6 and 12 hours after KCl-induced CSDs. Left panel shows individual vesicles (arrows) on CSD hemisphere. Right panel shows the time course of total endothelial vesicle density. *p<0.05, **p<0.01, ***p<0.001, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparison test.
B) Cytoplasmic, membrane-bound (all), luminal and abluminal endothelial pinocytic vesicle density shown separately following KCl-induced repetitive CSD. *p<0.05, **p<0.01, ***p<0.001, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparison test.
C) Endothelial cell area (i.e. cytoplasm) and capillary diameter (i.e. lumen) following KCl-induced repetitive CSD. p>0.05, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparisons test.
Figure 4. Directional transport of transcytosis from vessel lumen to brain parenchyma.
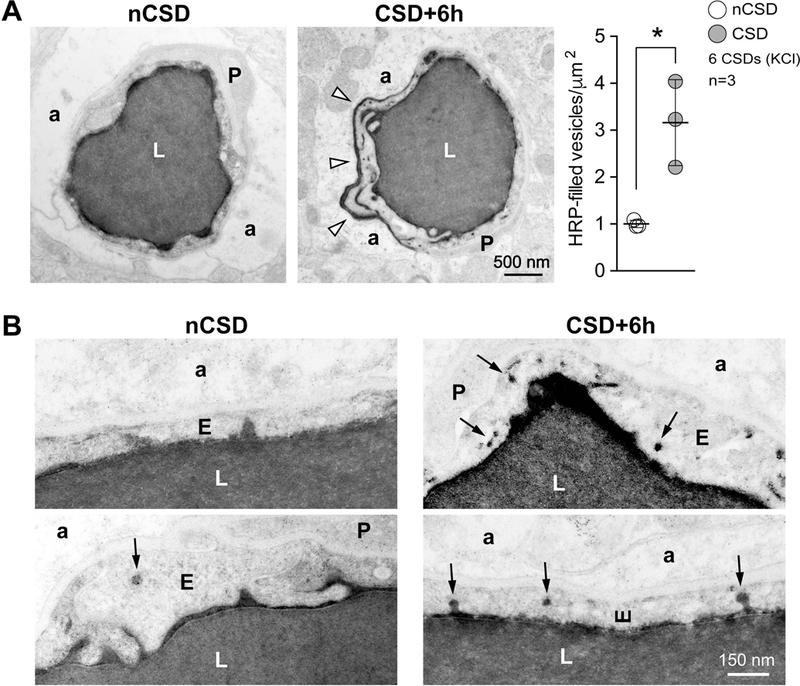
A) Left panel shows transmission electron micrographs from control (nCSD) versus CSD hemispheres illustrating leakage of intraluminal horseradish peroxidase (HRP) into the extravascular space (arrow heads). Right panel shows the density of HRP-filled vesicles within the endothelial cytoplasm 6 hours after repetitive KCl-induced CSDs. *p<0.05, paired t-test.
B) High-magnification transmission electron micrographs illustrating increased HRP transcytosis. Arrows indicate single HRP-filled vesicles either budding from the luminal membrane or traveling through the endothelial cytoplasm.
All data represent mean ± S.E.M. N=3 animals with 20 replicates in each animal. a, astrocytic endfoot; E, endothelium; L, lumen; nCSD, non-CSD hemisphere.
The smaller size (~50 nm) and lack of electron-dense coating of the vesicles were consistent with caveolae rather than clathrin-coated vesicles. To distinguish between clathrin- and caveolin-dependent mechanisms of CSD-induced transcytosis, we assessed 70 kDa FITC-dextran extravasation in cav-1-/- mice subjected to CSDs, and found that CSD-induced vascular leakage was completely absent in cav-1-/- mice compared with age-matched WT controls of the same genetic background (Figure 5). Because pericytes regulate the BBB in part by downregulating transcytosis (Armulik et al., 2010; Bell et al., 2010; Villasenor et al., 2017), we also examined pericyte coverage and integrity, as well as their relationship to CSD-induced leakage. We found no change in the microvascular coverage by pericytes 6 hours after CSD, at a time when transcytosis was already markedly increased (Figure 6A). Indeed, 70 kDa FITC-dextran often extravasated around capillary surfaces covered by pericytes (Figure 6B). Ultrastructurally, pericytes appeared intact with normal attachment to the vessel wall and no sign of cellular distress (Figure 6C,D). Basement membrane thickness also remained unaffected by CSDs (Figure 6D). Together, these data establish that CSD-induced BBB disruption is caveolae-dependent and does not involve TJ breakdown.
Figure 5. Caveolin-1 is required for CSD-induced BBB disruption.
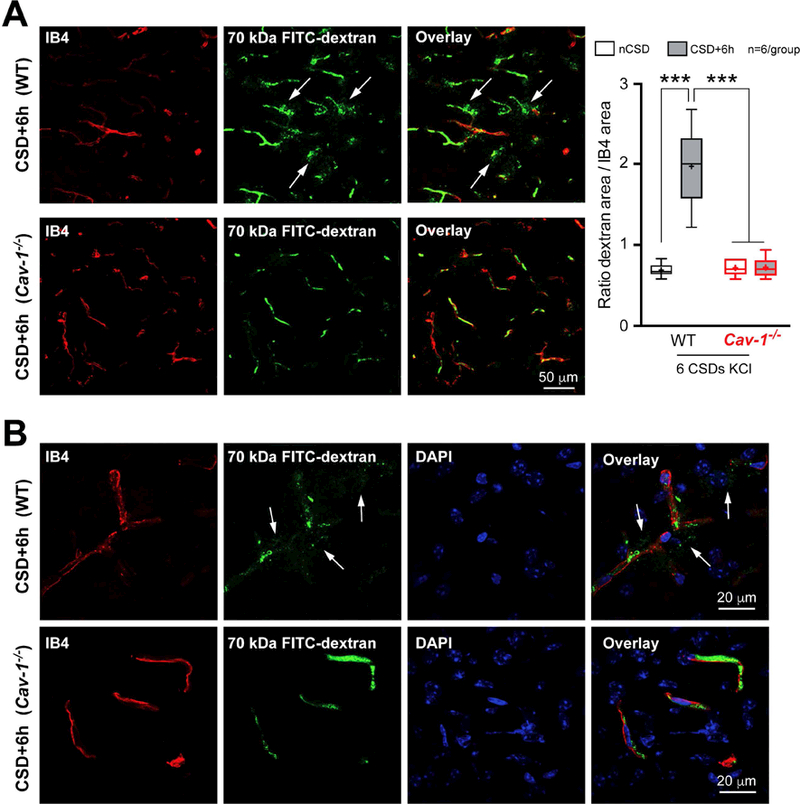
A) Left panel shows low magnification images of cortical 70 kDa FITC-Dextran (green) leakage around blood vessels (red) in WT (upper panels) and absence of it in Cav-1-/- (lower panels) mice 6 hours after KCl-induced CSDs. Right, Quantification of 70 kDa FITC-Dextran extravasation in the cerebral cortex. Whisker-box plots show full (whiskers) and interquartile (box) ranges, as well as the median (horizontal line) and mean (+). ***p<0.001 by one-way ANOVA followed by Turkey’s post-hoc test.
B) Higher magnification images to better illustrate the absence of leakage in Cav-1-/- mice (lower panels) compared to WT mice (upper panels) 6 hours after KCl-induced CSDs.
DAPI, 4,6-Diamidino-2-phenylindole; IB4, isolectin-B4; nCSD, non-CSD hemisphere.
Figure 6. CSD does not affect pericyte integrity.
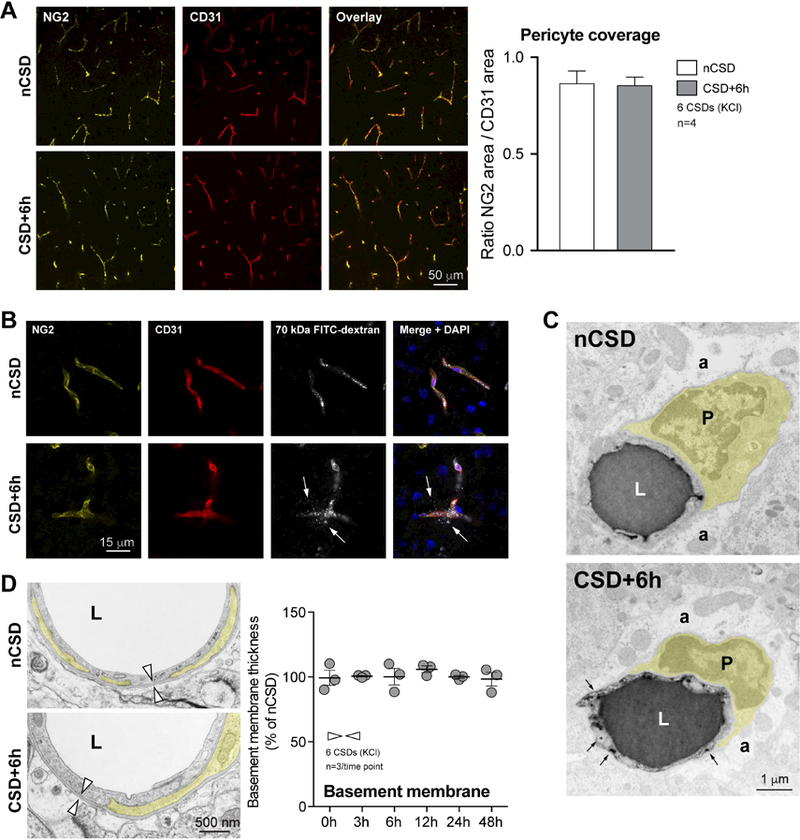
A) Left panel shows low magnification images of double immunofluorescence for pericyte marker NG2 and endothelial marker CD31 in the contra-lateral (nCSD, upper panels) versus CSD (lower panels) hemispheres. Right panel shows quantification of endothelial coverage by pericytes, expressed as surface area ratio. p>0.05, paired t-test.
B) Fluorescent microscopic images of pericytes (NG2, yellow) and endothelial cells (CD31, red) in relation to 70 kDa FITC-Dextran fluorescence (grey). Nuclei are stained with DAPI (blue). White arrows indicate FITC-Dextran leakage at places where pericyte coverage of the endothelium appears intact.
C) Transmission electron micrographs illustrating normal pericyte attachment in both the affected (CSD+6h) and non-affected (nCSD) hemispheres. Pericytes are pseudo-colored in yellow. Arrows indicate HRP-filled endothelial vesicles.
D) Left panel shows electron micrographs of normal pericyte attachment to the vessel wall and the basement membrane thickness (arrowheads) in both CSD and nCSD hemispheres. Right panel shows stable basement membrane thickness after CSD. p>0.05, one-way ANOVA. N=3 animals with 20 replicates in each animal.
Data represent mean ± S.E.M. a, astrocytic endfoot; CD31, cluster of differentiation-31; DAPI, 4,6-Diamidino-2-phenylindole; E, endothelium; L, lumen; nCSD, non-CSD hemisphere; NG2, neural/glial antigen 2; P, pericyte.
To further understand the molecular mechanisms regulating caveolae-mediated transcytosis and BBB leakage after CSD, we next examined ROCK as a critical regulator of actin cytoskeleton and endothelial permeability. Pre-treatment with isoform-nonselective ROCK inhibitor fasudil (10 mg/kg) abolished the increase in pinocytotic vesicle density after CSD compared to vehicle-treated animals (Figure 7A), and prevented HRP transcytosis (Figure 7B), without altering the endothelial area or capillary diameter (Figure 7C). As a result, CSD-induced increase in water content, EB leakage and 70 kDa FITC-dextran extravasation were all significantly reduced or abolished by fasudil pre-treatment (Figure 8). Importantly, electrophysiological properties of CSDs (i.e. amplitude and duration) were normal in both cav-1-/- mice and mice treated with ROCK inhibitors (Table 1).
Figure 7. Rho-kinase inhibition blocks CSD-induced transcytosis.
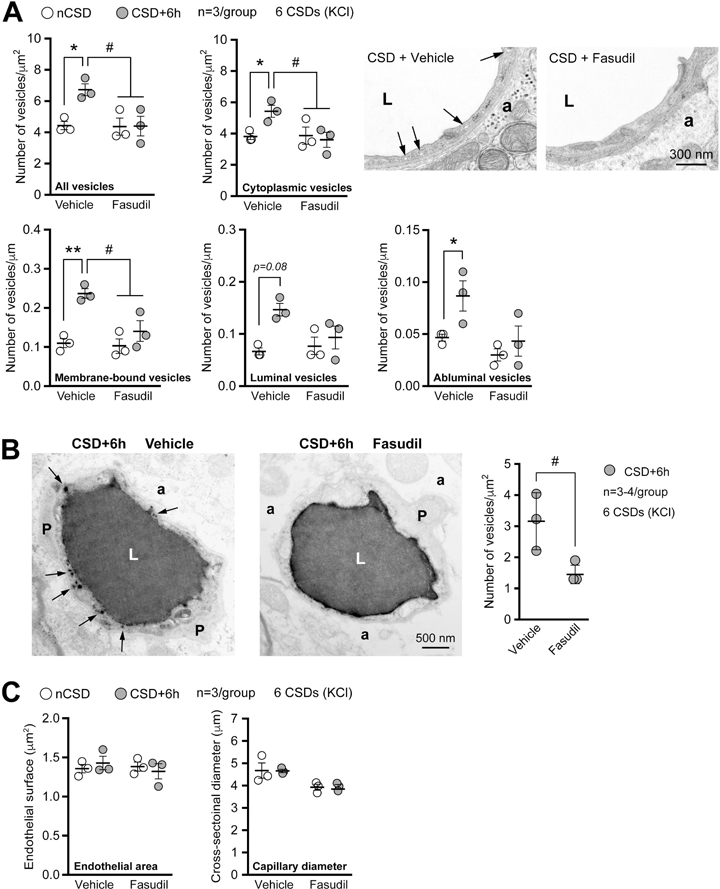
A) Left panel shows the total, cytoplasmic, membrane-bound (all), luminal and abluminal endothelial vesicles 6h after KCl-induced repetitive CSD. Mice were pre-treated with either vehicle or non-selective rho-kinase (ROCK) inhibitor Fasudil (10 mg/Kg, i.p.). Right panel shows representative transmission electron micrographic examples. Arrows indicate endothelial vesicles. *,#p<0.05, **p<0.01, two-way ANOVA (hemisphere x treatment as factors) and Sidak’s post-hoc multiple comparisons test.
B) Left panel shows transmission electron micrographs of HRP-filled blood vessels demonstrating the effect of ROCK inhibition on endothelial transcytosis. Arrows indicate HRP-filled vesicles, conspicuously absent in fasudil-treated mice. Right panel shows the effect of ROCK inhibition on HRP transcytosis. #p<0.05, t-test.
C) Endothelial cell area (i.e. cytoplasm) and capillary diameter (i.e. lumen) 6 hours after KCl-induced CSDs in mice treated with vehicle or fasudil. p>0.05, two-way ANOVA (treatment x hemisphere as factors) followed by Sidak’s post-hoc multiple comparisons test.
Data represent mean ± S.E.M. N=3 animals with 10 replicates per hemisphere in each animal.
a, astrocytic endfoot; L, lumen; nCSD, non-CSD hemisphere; P, pericyte.
Figure 8. Rho-kinase inhibition prevents CSD-induced BBB disruption.
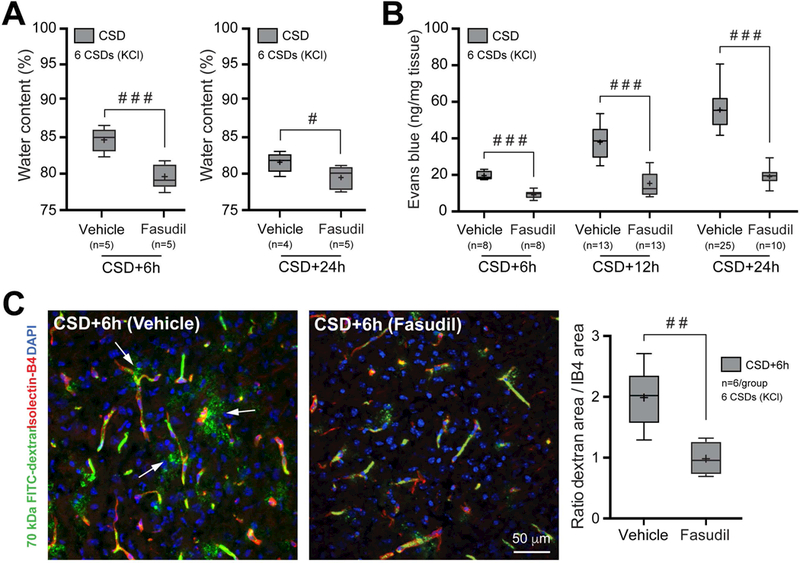
A) Brain water content measured 6 hours after repetitive CSD in mice treated with either vehicle or ROCK inhibitor Fasudil (10mg/Kg, i.p.). #p<0.01, ###p<0.001, unpaired t-test.
B) Evans blue leakage measured 6 hours after repetitive CSD in mice treated with either vehicle or Fasudil. ###p<0.001, two-way ANOVA (time x hemisphere as factors) followed by Sidak’s post-hoc multiple comparison test.
C) Left panel shows extravasation of 70 KDa FITC-dextran 6 hours after repetitive CSD in animals treated with either vehicle or Fasudil. Right panel shows 70 kDa FITC-dextran extravasation as area ratio. ##p<0.01, unpaired t-test.
Whisker-box plots show full (whiskers) and interquartile (box) ranges, as well as the median (horizontal line) and mean (+). Control groups are replotted from Figure 1 for comparison. DAPI, 4,6-Diamidino-2-phenylindole; IB4, isolectin-B4.
Table 1.
Arterial blood pressure and electrophysiological measures of CSD
| Mean arterial pressure (mmHg) |
CSD amplitude (mV) | CSD duration (sec) | |
|---|---|---|---|
| Control | 87±4 | 25±1 | 34±4 |
| Fasudil | 85±4 | 25±2 | 33±7 |
| KD025 | 82±4 | 24±2 | 31±2 |
| Caveolin-1 knockout | NA | 25±2 | 30±4 |
Suppression of BBB opening by fasudil might reflect a permissive role for ROCK in CSD-induced vascular permeability. To address this question, we performed real time qPCR and found increased mRNA expression of both ROCK1 and ROCK2 isoforms in ipsilateral (CSD) cortical homogenates compared to the contralateral (non-CSD) hemisphere 6 hours after CSD (Figure 9A), strongly suggesting that ROCK is upregulated by CSD, rather than being just a permissive factor, as a critical step in BBB opening. To determine the ROCK isoform responsible for BBB opening, we next tested the ROCK2-selective inhibitor KD025 (200 mg/kg twice daily for 48 hours before CSD) (Lee et al., 2014) and found it to be as effective as fasudil in preventing the BBB leakage (Figure 9B). This is consistent with data suggesting that ROCK2 is the predominant endothelial isoform regulating barrier function (Beckers et al., 2015; De Silva et al., 2016).
Figure 9. CSD upregulates rho-kinase expression, and selective rho-kinase 2 inhibition prevents CSD-induced Evans blue leakage.
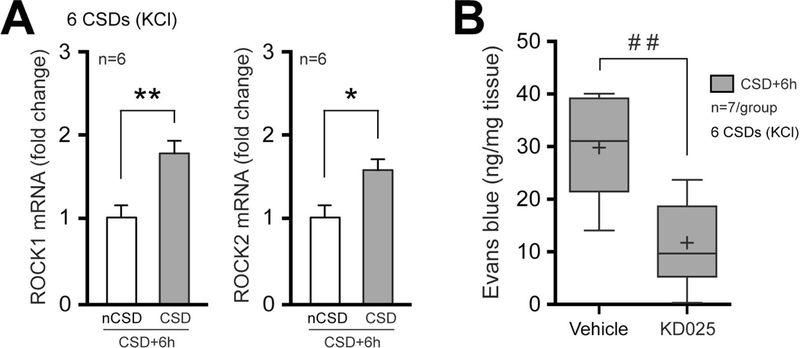
A) Expression (mRNA) of ROCK isoforms 1 & 2 6 hours after CSD, as measured by RT-qPCR in cerebral cortex extracts. Data represent mean ± S.E.M. *p<0.05, **p<0.01, paired t-test.
B) Evans blue leakage measured 6 hours after CSD in mice treated with either vehicle or ROCK2-selective inhibitor KD025. Whisker-box plots show full (whiskers) and interquartile (box) ranges, as well as the median (horizontal line) and mean (+). ##p<0.01, unpaired t-test.
Last but not the least, CSDs are known to cause a transient oxygen supply-demand mismatch raising the possibility that tissue hypoxia may be the trigger for BBB opening (Piilgaard and Lauritzen, 2009; Takano et al., 2007; Yuzawa et al., 2012). To test this, we performed additional experiments by employing normobaric hyperoxia (100% oxygen inhalation) during the period of CSD induction (1 hour), which is known to abrogate the relative tissue hypoxia observed during CSD (Takano et al., 2007). Hyperoxic animals showed nearly identical increase in EB leakage 24 hours after CSDs (%192 ± 9 of contralateral; n=6) compared with the normoxic group (%196 ± 9 of contralateral), arguing against hypoxia as a relevant trigger for BBB opening during CSD.
DISCUSSION
Our data demonstrate that CSD disrupts the BBB exclusively via endothelial transcytosis, without affecting endothelial TJs, basement membrane or pericytes. We also show that CSD-induced transcytosis is caveolin-1- dependent, and that both transcytosis and leakage after CSD require ROCK2 activity as a gatekeeper, likely through phosphorylation of caveolin-1 (Li et al., 2014). Altogether, our data establish the endothelial mechanism of BBB disruption by CSD.
The identity of the factor(s) linking the parenchymal (i.e., neuroglial) depolarization during CSD to the endothelium to stimulate transcytosis is unknown. CSD is associated with major shifts in extracellular ion concentrations (e.g. decreased pH, increased extracellular K+) and massive increases in numerous neurotransmitters (e.g. glutamate) that can overwhelm the usual reuptake and clearance mechanisms (Ayata and Lauritzen, 2015). As a result, elevated extracellular levels of ions and neurotransmitters are likely to traverse the perivascular diffusional barriers (e.g. astrocyte end feet) and reach the microvascular endothelial cells. Astrocytic end feet may in fact facilitate perivascular K+ rise by siphoning (Leis et al., 2005; Newman, 1986). Moreover, the secondary depolarization of perivascular nerves during CSD might release their vasoactive transmitters (e.g. substance P) (Ayata and Lauritzen, 2015). Prior studies suggest that some of the CSD-induced ultrastructural changes can be pharmacologically modulated (e.g. paracetamol, serotonergic transmission) (Maneesri et al., 2004; Saengjaroentham et al., 2015; Yisarakun et al., 2014). Given the multitude of potential signaling molecules simultaneously released during CSD in vivo, identifying the signal that triggers the endothelial phenotype change may require in vitro models.
An alternative explanation might be CSD-induced tissue hypoxia. However, CSD causes only a brief (<30 seconds) oxygen supply-demand mismatch within otherwise normal brain parenchyma, (Yuzawa et al., 2012), and normobaric hyperoxia, which effectively prevents the oxygen supply-demand mismatch during CSD (Takano et al., 2007), did not block the BBB opening in our study, arguing against this possibility. In addition, non-invasive optogenetic induction of CSD also effectively disrupted the BBB, suggesting that tissue injury is not required.
Our findings have implications for migraine. CSD is the electrophysiological event underlying aura (Ayata, 2010). Clinically, severe migraine auras, where multiple, long lasting or reverberating CSDs may occur (e.g. hemiplegic migraine) (Eikermann-Haerter et al., 2009; Eikermann-Haerter et al., 2011; Ferrari et al., 2015), have been reported to show neuroimaging signs of BBB opening (Cha et al., 2007; Dreier et al., 2005; Kors et al., 2001). Increased endothelial pinocytosis during classical migraine attacks has been suggested more than 40 years ago, in part based on CSF/plasma protein ratios (Harper et al., 1977; Kangasniemi et al., 1974) However, clinical studies have thus far failed to detect BBB opening after migraine attacks, likely due to technical limitations and study design (Amin et al., 2017; Hougaard et al., 2017; Schankin et al., 2016). Because a single CSD was also capable of triggering BBB leakage in our study, newer MRI techniques to image “free water”, such as diffusion based-models (i.e. neurite orientation dispersion and density imaging, NODDI) or T2 mapping, may be sensitive enough to detect BBB opening as a footprint even in classical migraine with aura, especially when properly timed. Since BBB opening was delayed by several hours after CSD, plasma protein and water leakage are unlikely to be relevant for the headache phase. However, chronic episodic leakage of plasma constituents during frequent migraine aura attacks may be a potential mechanism for progressive neurovascular conditions linked to migraine with aura (e.g. periventricular white matter disease) (Mawet et al., 2015). Lastly, opening of the BBB after CSD (i.e. aura) might provide an entry route for novel migraine therapeutics that do not normally cross the BBB, such as antibodies targeting the CGRP system (Tso and Goadsby, 2017).
An abundance of experimental and clinical data indicates that CSD plays a critical role in the pathophysiology of ischemic, hemorrhagic and traumatic brain injury as well (Lauritzen et al., 2011). Recurrent CSDs in injured brain promote secondary tissue loss and neurological deficits, making CSD a clinically relevant therapeutic target. Our data suggest that CSD alone (i.e. without attendant tissue injury) recapitulates the morphological features and mechanisms of BBB opening after injury (Cipolla et al., 2004; Dietrich et al., 1987; Haley and Lawrence, 2017; Ito et al., 1980; Knowland et al., 2014; Krueger et al., 2013; Lossinsky and Shivers, 2004; Nahirney et al., 2016). The overlap is substantial. For example, both caveolin-1 expression and phosphorylation are increased after an ischemic insult, and caveolin-1 knockout mice do not develop increased transcytosis or early BBB leakage after ischemia (Knowland et al., 2014; Nag et al., 2007). Cerebral ischemia upregulates the RhoA/ROCK pathway as well (Brabeck et al., 2003; Erdo et al., 2004; Trapp et al., 2001), and RhoA/ROCK inhibitors attenuate edema in experimental models of brain injury (Fu et al., 2014; Fujii et al., 2012; Gibson et al., 2014; Huang et al., 2012; Niego et al., 2017). Also consistent with our findings after CSD, cerebral ischemia did not alter basement membrane thickness, pericyte coverage, capillary diameter or endothelial cell area (Haley and Lawrence, 2017). Indeed, opening of the paracellular pathway occurs only during late stages of severe ischemic injury, and is not triggered by CSD alone. As such, CSD is an example where transcellular and paracellular BBB opening dissociates (Kang et al., 2013). Importantly, in our experiments where only a small number of CSDs were induced over one hour, BBB opening was transient, with spontaneous recovery within 48 hours. In the injured brain, recurrent CSDs continue to develop for many hours and even days after the initial insult (Hartings et al., 2003; Kudo et al., 2016), which might explain the longer-lasting BBB opening after brain injury. Since CSDs originating in injured brain often propagate into the normal tissue, they may explain the BBB opening in surrounding non-injured brain as well (Lapilover et al., 2012; Stoll et al., 2009). Altogether, these data implicate CSDs in brain injury as a contributor to early BBB opening to large molecules and vasogenic edema that often becomes a clinically significant mechanism of secondary injury and sometimes herniation (Ayata and Ropper, 2002). Therefore, therapeutic targeting of CSDs may diminish edema in injured brain.
Lastly, we propose that the increase in endothelial transcytosis is only one sign of a dramatic endothelial change after CSD, i.e. the tip of the iceberg. The transformation emerges after a delay of a few hours, presumably regulated at the transcriptional, translational and/or post-translational levels. Therefore, CSD may not be as innocuous as it is often believed to be, and the cerebrovascular endothelial phenotype shift may have implications for the increased cerebrovascular event risk in migraineurs with aura.
ACKNOWLEDGEMENTS
Fellowship Award from the International Headache Society, Scholarship Award from the Frontiers in Headache Research, the American Headache Society to H.S. The Mahoney postdoctoral fellowship to B.L., Japanese Heart Foundation and Bayer Yakuhin Research Grant Abroad to F.O. The National Institute of Neurological Disorders and Stroke at the National Institutes of Health (R25NS065743 to D.Y.C.), Fidelity Biosciences Research Initiative to C.G., The National Institute of Health (DP1 NS092473 Pioneer Award to C.G.), The research of C.G. was also supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute. The National Institute of Neurological Disorders and Stroke at the National Institutes of Health (P01NS055104 and R01NS102969 to C.A.), Fondation Leducq, Heitman Foundation and Ellison Foundation to C.A.
Footnotes
POTENTIAL CONFLICTS OF INTEREST
Each author certifies that he or she has no funding or commercial association that might pose a conflict of interest in connection with submitted manuscript.
REFERENCES
- Amin FM, Hougaard A, Cramer SP, Christensen CE, Wolfram F, Larsson HBW, and Ashina M (2017). Intact blood-brain barrier during spontaneous attacks of migraine without aura: a 3T DCE-MRI study. Eur J Neurol 24, 1116–1124. [DOI] [PubMed] [Google Scholar]
- Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, Deik AA, Ginty DD, Clish CB, and Gu C (2017). Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 94, 581–594 e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Lacoste B, and Gu C (2015). Neuronal and vascular interactions. Annu Rev Neurosci 38, 25–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561. [DOI] [PubMed] [Google Scholar]
- Ayata C (2010). Cortical spreading depression triggers migraine attack: pro. Headache 50, 725–730. [DOI] [PubMed] [Google Scholar]
- Ayata C (2013). Pearls and pitfalls in experimental models of spreading depression. Cephalalgia 33, 604–613. [DOI] [PubMed] [Google Scholar]
- Ayata C, and Lauritzen M (2015). Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol Rev 95, 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, and Ropper AH (2002). Ischaemic brain oedema. J Clin Neurosci 9, 113–124. [DOI] [PubMed] [Google Scholar]
- Beckers CM, Knezevic N, Valent ET, Tauseef M, Krishnan R, Rajendran K, Hardin CC, Aman J, van Bezu J, Sweetnam P, et al. (2015). ROCK2 primes the endothelium for vascular hyperpermeability responses by raising baseline junctional tension. Vascular pharmacology 70, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, and Zlokovic BV (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, and Gu C (2014). Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabeck C, Mittelbronn M, Bekure K, Meyermann R, Schluesener HJ, and Schwab JM (2003). Effect of focal cerebral infarctions on lesional RhoA and RhoB expression. Arch Neurol 60, 1245–1249. [DOI] [PubMed] [Google Scholar]
- Cha YH, Millett D, Kane M, Jen J, and Baloh R (2007). Adult-onset hemiplegic migraine with cortical enhancement and oedema. Cephalalgia 27, 1166–1170. [DOI] [PubMed] [Google Scholar]
- Chow BW, and Gu C (2015). The molecular constituents of the blood-brain barrier. Trends Neurosci 38, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DY, Sadeghian H, Qin T, Lule S, Lee H, Karakaya F, Goins S, Oka F, Yaseen MA, Houben T, et al. (2018). Determinants of Optogenetic Cortical Spreading Depolarizations. Cereb Cortex [DOI] [PMC free article] [PubMed]
- Cipolla MJ, Crete R, Vitullo L, and Rix RD (2004). Transcellular transport as a mechanism of blood-brain barrier disruption during stroke. Front Biosci 9, 777–785. [DOI] [PubMed] [Google Scholar]
- De Silva TM, Kinzenbaw DA, Modrick ML, Reinhardt LD, and Faraci FM (2016). Heterogeneous Impact of ROCK2 on Carotid and Cerebrovascular Function. Hypertension 68, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WD, Busto R, Watson BD, Scheinberg P, and Ginsberg MD (1987). Photochemically induced cerebral infarction. II. Edema and blood-brain barrier disruption. Acta neuropathologica 72, 326–334. [DOI] [PubMed] [Google Scholar]
- Dreier JP (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17, 439–447. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Jurkat-Rott K, Petzold GC, Tomkins O, Klingebiel R, Kopp UA, Lehmann-Horn F, Friedman A, and Dichgans M (2005). Opening of the blood-brain barrier preceding cortical edema in a severe attack of FHM type II. Neurology 64, 2145–2147. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Dilekoz E, Kudo C, Savitz SI, Waeber C, Baum MJ, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, and Ayata C (2009). Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest 119, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Lee JH, Yuzawa I, Liu CH, Zhou Z, Shin HK, Zheng Y, Qin T, Kurth T, Waeber C, et al. (2012). Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation 125, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR, Hoffmann U, Dilekoz E, Waeber C, Ferrari MD, et al. (2011). Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci 31, 5755–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdo F, Trapp T, Mies G, and Hossmann KA (2004). Immunohistochemical analysis of protein expression after middle cerebral artery occlusion in mice. Acta Neuropathol (Berl) 107, 127–136. [DOI] [PubMed] [Google Scholar]
- Ferrari MD, Klever RR, Terwindt GM, Ayata C, and van den Maagdenberg AM (2015). Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol 14, 65–80. [DOI] [PubMed] [Google Scholar]
- Fu Z, Chen Y, Qin F, Yang S, Deng X, Ding R, Feng L, Li W, and Zhu J (2014). Increased activity of Rho kinase contributes to hemoglobin-induced early disruption of the blood-brain barrier in vivo after the occurrence of intracerebral hemorrhage. Int J Clin Exp Pathol 7, 7844–7853. [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Duris K, Altay O, Soejima Y, Sherchan P, and Zhang JH (2012). Inhibition of Rho kinase by hydroxyfasudil attenuates brain edema after subarachnoid hemorrhage in rats. Neurochem Int 60, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CL, Srivastava K, Sprigg N, Bath PM, and Bayraktutan U (2014). Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J Neurochem 129, 816–826. [DOI] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, Wang X, Rosenberg GA, Lo EH, and Moskowitz MA (2004). Cortical spreading depression activates and upregulates MMP-9. J Clin Invest 113, 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley MJ, and Lawrence CB (2017). The blood-brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab 37, 456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AM, MacKenzie ET, McCulloch J, and Pickard JD (1977). Migraine and the blood-brain barrier. Lancet 1, 1034–1036. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Rolli ML, Lu XC, and Tortella FC (2003). Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J Neurosci 23, 11602–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben T, Loonen IC, Baca SM, Schenke M, Meijer JH, Ferrari MD, Terwindt GM, Voskuyl RA, Charles A, van den Maagdenberg AM, and Tolner EA (2016). Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J Cereb Blood Flow Metab [DOI] [PMC free article] [PubMed]
- Hougaard A, Amin FM, Christensen CE, Younis S, Wolfram F, Cramer SP, Larsson HBW, and Ashina M (2017). Increased brainstem perfusion, but no blood-brain barrier disruption, during attacks of migraine with aura. Brain 140, 1633–1642. [DOI] [PubMed] [Google Scholar]
- Huang B, Krafft PR, Ma Q, Rolland WB, Caner B, Lekic T, Manaenko A, Le M, Tang J, and Zhang JH (2012). Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis 46, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito U, Ohno K, Yamaguchi T, Takei H, Tomita H, and Inaba Y (1980). Effect of hypertension on blood-brain barrier. Change after restoration of blood flow in post-ischemic gerbil brains. An electronmicroscopic study. Stroke 11, 606–611. [DOI] [PubMed] [Google Scholar]
- Kang EJ, Major S, Jorks D, Reiffurth C, Offenhauser N, Friedman A, and Dreier JP (2013). Blood-brain barrier opening to large molecules does not imply blood-brain barrier opening to small ions. Neurobiol Dis 52, 204–218. [DOI] [PubMed] [Google Scholar]
- Kangasniemi P, Riekkinen P, Penttinen R, Ivaska K, and Rinne UK (1974). Enzyme changes in the cerebrospinal fluid and serum and their correlation to the breakdown of bradykinin during different stages of headache attacks of migraine patients. Headache 14, 139–148. [DOI] [PubMed] [Google Scholar]
- Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, and Dalkara T (2013). Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339, 1092–1095. [DOI] [PubMed] [Google Scholar]
- Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, and Agalliu D (2014). Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, Love S, van den Maagdenberg AM, Haan J, Frants RR, and Ferrari MD (2001). Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol 49, 753–760. [DOI] [PubMed] [Google Scholar]
- Krueger M, Hartig W, Reichenbach A, Bechmann I, and Michalski D (2013). Blood-brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS ONE 8, e56419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo K, Zhao L, and Nowak TS Jr. (2016). Peri-infarct depolarizations during focal ischemia in the awake Spontaneously Hypertensive Rat. Minimizing anesthesia confounds in experimental stroke. Neuroscience 325, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste B, Comin CH, Ben-Zvi A, Kaeser PS, Xu X, Costa Lda F, and Gu C (2014). Sensory-related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron 83, 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapilover EG, Lippmann K, Salar S, Maslarova A, Dreier JP, Heinemann U, and Friedman A (2012). Peri-infarct blood-brain barrier dysfunction facilitates induction of spreading depolarization associated with epileptiform discharges. Neurobiol Dis 48, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, and Strong AJ (2011). Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 31, 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao A (1944). Spreading depression of activity in the cerebral cortex. J Neurophysiol 7, 359–390. [DOI] [PubMed] [Google Scholar]
- Lee JH, Zheng Y, von Bornstadt D, Wei Y, Balcioglu A, Daneshmand A, Yalcin N, Yu E, Herisson F, Atalay YB, et al. (2014). Selective ROCK2 Inhibition In Focal Cerebral Ischemia. Annals of clinical and translational neurology 1, 2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leis JA, Bekar LK, and Walz W (2005). Potassium homeostasis in the ischemic brain. Glia 50, 407–416. [DOI] [PubMed] [Google Scholar]
- Li Z, Liu YH, Xue YX, Liu LB, and Wang P (2014). Low-dose endothelial monocyte-activating polypeptide-ii increases permeability of blood-tumor barrier by caveolae-mediated transcellular pathway. J Mol Neurosci 52, 313–322. [DOI] [PubMed] [Google Scholar]
- Lossinsky AS, and Shivers RR (2004). Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histology and histopathology 19, 535–564. [DOI] [PubMed] [Google Scholar]
- Maneesri S, Patamanont J, Patumraj S, and Srikiatkhachorn A (2004). Cortical spreading depression, meningeal inflammation and trigeminal nociception. Neuroreport 15, 1623–1627. [DOI] [PubMed] [Google Scholar]
- Mawet J, Kurth T, and Ayata C (2015). Migraine and stroke: In search of shared mechanisms. Cephalalgia 35, 165–181. [DOI] [PubMed] [Google Scholar]
- Nag S, Venugopalan R, and Stewart DJ (2007). Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta neuropathologica 114, 459–469. [DOI] [PubMed] [Google Scholar]
- Nahirney PC, Reeson P, and Brown CE (2016). Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab 36, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (1986). Regional specialization of the membrane of retinal glial cells and its importance to K+ spatial buffering. Ann N Y Acad Sci 481, 273–286. [DOI] [PubMed] [Google Scholar]
- Niego B, Lee N, Larsson P, De Silva TM, Au AE, McCutcheon F, and Medcalf RL (2017). Selective inhibition of brain endothelial Rho-kinase-2 provides optimal protection of an in vitro blood-brain barrier from tissue-type plasminogen activator and plasmin. PLoS ONE 12, e0177332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piilgaard H, and Lauritzen M (2009). Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab [DOI] [PubMed]
- Reese TS, and Karnovsky MJ (1967). Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 34, 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saengjaroentham C, Supornsilpchai W, Ji-Au W, Srikiatkhachorn A, and Maneesri-le Grand S (2015). Serotonin depletion can enhance the cerebrovascular responses induced by cortical spreading depression via the nitric oxide pathway. Int J Neurosci 125, 130–139. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Mollgard K, and Habgood MD (2015). Markers for blood-brain barrier integrity: how appropriate is Evans blue in the twenty-first century and what are the alternatives? Front Neurosci 9, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schankin CJ, Maniyar FH, Seo Y, Kori S, Eller M, Chou DE, Blecha J, Murphy ST, Hawkins RA, Sprenger T, et al. (2016). Ictal lack of binding to brain parenchyma suggests integrity of the blood-brain barrier for 11C-dihydroergotamine during glyceryl trinitrate-induced migraine. Brain 139, 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, Huang PL, Boas DA, Liao JK, Moskowitz MA, and Ayata C (2007). Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab 27, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Kleinschnitz C, Meuth SG, Braeuninger S, Ip CW, Wessig C, Nolte I, and Bendszus M (2009). Transient widespread blood-brain barrier alterations after cerebral photothrombosis as revealed by gadofluorine M-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab 29, 331–341. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, and Nedergaard M (2007). Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10, 754–762. [DOI] [PubMed] [Google Scholar]
- Trapp T, Olah L, Holker I, Besselmann M, Tiesler C, Maeda K, and Hossmann KA (2001). GTPase RhoB: an early predictor of neuronal death after transient focal ischemia in mice. Mol Cell Neurosci 17, 883–894. [DOI] [PubMed] [Google Scholar]
- Tso AR, and Goadsby PJ (2017). Anti-CGRP Monoclonal Antibodies: the Next Era of Migraine Prevention? Curr Treat Options Neurol 19, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor R, Kuennecke B, Ozmen L, Ammann M, Kugler C, Gruninger F, Loetscher H, Freskgard PO, and Collin L (2017). Region-specific permeability of the blood-brain barrier upon pericyte loss. J Cereb Blood Flow Metab, 271678X17697340. [DOI] [PMC free article] [PubMed]
- von Bornstadt D, Houben T, Seidel JL, Zheng Y, Dilekoz E, Qin T, Sandow N, Kura S, Eikermann-Haerter K, Endres M, et al. (2015). Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 85, 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yisarakun W, Supornsilpchai W, Chantong C, Srikiatkhachorn A, and Maneesri-le Grand S (2014). Chronic paracetamol treatment increases alterations in cerebral vessels in cortical spreading depression model. Microvasc Res [DOI] [PubMed]
- Yuzawa I, Sakadzic S, Srinivasan VJ, Shin HK, Eikermann-Haerter K, Boas DA, and Ayata C (2012). Cortical spreading depression impairs oxygen delivery and metabolism in mice. J Cereb Blood Flow Metab 32, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]