

Abstract
Genetic testing is used for screening, diagnosis and prognosis of diseases consistent with a genetic etiology, and to guide drug therapy to improve drug efficacy and to avoid adverse effects (pharmacogenomics). This in practice aims to inform on DNA-related genetic test availability, interpretation, and recommended clinical actions based on results using evidence from clinical guidelines, when available. We discuss challenges that limit the widespread use of genetic information in the clinical care setting, including a small number of actionable genetic variants with strong evidence of clinical validity and utility, and the need for improving the health literacy of health care providers and public including for direct-to-consumer tests. Ethical, legal and social issues and incidental findings also need to be addressed. Because our understanding of genetic factors associated with disease and drug response is rapidly increasing, and new genetic tests are being developed that could be adopted by clinicians in the short term, we also provide extensive resources for information and education on genetic testing.
Keywords: genetic testing, pharmacogenetics, kidney disease, APOL1, drug selection, drug dosing, risk allele, pharmacokinetics, ancestry, mutation, polymorphism, pharmacogenomics, gene variant, review
Overview of genetic testing
The US National Institutes of Health (NIH) defines genetic testing as an analysis of human chromosomes, genes, or proteins in order to detect heritable disease for clinical purposes.2 This definition does not include tests used for research purposes. Genetic testing has been traditionally used for pre-natal screening, screening for carriers of a genetic disorder for reproductive purpose, and diagnosis of rare Mendelian disorders suspected based on clinical evidence or family history. Recent advances in high-throughput genomics have made large-scale genotyping and sequencing affordable. This has led to an increased number of genetic tests being developed, including tests for clinical use and commercially available direct-to-consumer genetic tests.3 In the emerging field of pharmacogenomics, genetic testing is proposed to guide drug therapy to improve drug efficacy or to avoid adverse effects. However, there are still several challenges that limit the widespread use of genetic information in the clinical care setting.
This review aims to inform on new developments on the field, including available tests, their utility to patients, as well as regulatory and ethnic issues related to genetic testing. We will focus on molecular genetic tests, the purpose of which is to identify DNA variation, including both polymorphisms (which are usually not pathogenic) and mutations that are associated with genetic disorders. We will cover genetic testing for Mendelian disorders, complex traits such as APOL1 (apolipoprotein L1)-associated CKD, and pharmacogenomics.
We will not address genetic testing for prenatal and newborn screening, diagnosis of rare or atypical diseases in newborns including chromosomal abnormalities, diagnosis of somatic mutations (DNA changes that occur after conception, e.g., in cancer), or genetic tests for metabolic diseases. The American College of Medical Genetics and Genomics (ACMG) provides practice guidelines that cover these topics (Table 1).
Table 1.
Resources for information on genetic testing
| Source | Information provided | Website or reference |
|---|---|---|
|
| ||
| ACMG: The American College of Medical Genetics and Genomics guidelines | Practice guidelines based on disease topics and policy statements regarding genetic testing. | http://www.acmg.net/ACMG/Publications/Practice_Guidelines/ACMG/Publications/Practice_Guidelines.aspx?hkey=b5e361a3-65b1-40ae-bb3e-4254fce9453a |
|
| ||
| ASHG: The American Society of Human Genetics | Genetic testing educational resources | https://www.ashg.org/education/genetic_testing.shtml |
|
| ||
| Centers for Disease Control and Prevention | ACCE model for evaluating genetic tests | https://www.cdc.gov/genomics/gtesting/ACCE/ |
|
| ||
| Center for Disease Control and Prevention | EGAPP recommendations | https://www.cdc.gov/genomics/gtesting/egapp/recommend/index.htm |
|
| ||
| Center for Disease Control and Prevention | Direct-to-consumer information | https://blogs.cdc.gov/genomics/2017/04/18/direct-to-consumer-2/ |
|
| ||
| CPIC: Clinical Pharmacogenetics Implementation Consortium | Pharmacogenetics guidelines | https://www.pharmgkb.org/view/dosing-guidelines.do?source=CPIC |
|
| ||
| NCBI ClinVar | Online genetic variant registry | https://www.ncbi.nlm.nih.gov/clinvar/ |
|
| ||
| FDA | Pharmacogenomic biomarkers in drug labeling | https://www.fda.gov/downloads/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/UCM545881.pdf |
|
| ||
| GTR: Genetic Testing Resource | Registry of available clinical tests and YouTube tutorials | https://www.ncbi.nlm.nih.gov/gtr/ |
| https://www.youtube.com/playlist?list=PL1C4A2AFF811F6F0B | ||
|
| ||
| IGNITE: Implementing GeNomics In practice Network | Consortium investigating barriers to implementation of genetic testing in clinical care | www.ignite-genomics.org |
|
| ||
| MedGen | Information on genetic disease, clinical characteristics, variants and genetic testing, professional guidelines | https://www.ncbi.nlm.nih.gov/medgen/ |
|
| ||
| NIH/NHGRI: The National Institutes of Health National Human Genome Research Institute | Genetic testing coverage and reimbursement, regulation, human subjects and privacy, informed consent, and legislation | https://www.genome.gov/27527652/genomic-medicine-and-health-care/ |
|
| ||
| NIH/NHGRI: The National Institutes of Health National Human Genome Research Institute | Handbook and toolkit for introductory training of physicians in genomic medicine | https://www.genome.gov/27569865/2017-news-feature-genomics-handbook-provides-customized-education-for-physicians/2017-news-feature-genomics-handbook-provides-customized-education-for-physicians/ |
|
| ||
| OMIM: Online Mendelian Inheritance in Man | Database of genes related to Mendelian disorders | http://omim.org/ |
|
| ||
| PharmGKB: The Pharmacogenomics Knowledgebase | Comprehensive resource that curates knowledge of genetic variation impacting drug response | https://www.pharmgkb.org/ |
|
| ||
| PGRN: The Pharmacogenomics Research Network | Tools to find potentially actionable variants for pharmacogenomics | http://www.pgrn.org |
| http://www.pgrn.org/tools.html | ||
Because our understanding of genetic factors associated with disease and drug response is expected to rapidly increase in the next few years, and new genetic tests are being developed that could be adopted by clinicians in the short term, we also provide extensive resources for information and education on genetic testing (Table 1).
Type of genetic tests
Several types of genetic tests are available including single-variant tests (e.g., diagnosis of the HBB p.Glu7Val mutation [a substitution of valine for glutamine at amino acid 7 of beta globin] that causes sickle cell disease), gene-based tests (single or multiple genes, e.g., genetic testing for autosomal dominant polycystic kidney disease [ADPKD] mutations in the PKD1 and PKD2 genes), and genetic panels (e.g., for genetic variants associated with drug metabolism). Genetic tests cover single-nucleotide variants, haplotypes (e.g., HLA region), deletion/insertion variants, copy number variants, and mutations in mitochondrial DNA. Whole-exome sequencing and whole-genome sequencing use next-generation sequencing (NGS) methods of high-throughput DNA analysis to identify Mendelian disorders when clinical features and family history are consistent with a genetic etiology.
Whole-exome sequencing with confirmation of relevant genetic variants by Sanger sequencing may be the most cost-effective approach to genetic clinical diagnosis when multiple loci are possible explanations for a particular syndrome. However, this approach may identify mutations for which the clinical significance has not been established (variants of unknown significance, or VUS) for which return of results to patients is uncertain.4 Genome-wide genotype arrays and whole-genome sequencing are also available as direct-to-consumer products for disease risk prediction in individuals without a suspected genetic condition. The clinical utility of these tests in asymptomatic healthy individuals is unclear, and there are potential harms related to reporting incidental findings and/or genetic variants for which the clinical consequences are unknown, e.g., VUS.5
Genetic test validity and utility
In choosing a genetic test, one needs to assess the following: (1) how well the test performs to detect the genetic variation or mutation of interest (technical performance), (2) how well the variant or mutation tested accurately and reliably predicts the clinical disease (clinical validity), and (3) what is the evidence that the genetic test improves clinical outcomes or has added value for patient management decisions (clinical utility) (Figure 1). These criteria for the evaluation of a genetic test are based on the Center for Disease Control and Prevention ACCE model, which also includes ethical, legal, and social implications of genetic testing (Table 1).
Figure 1.
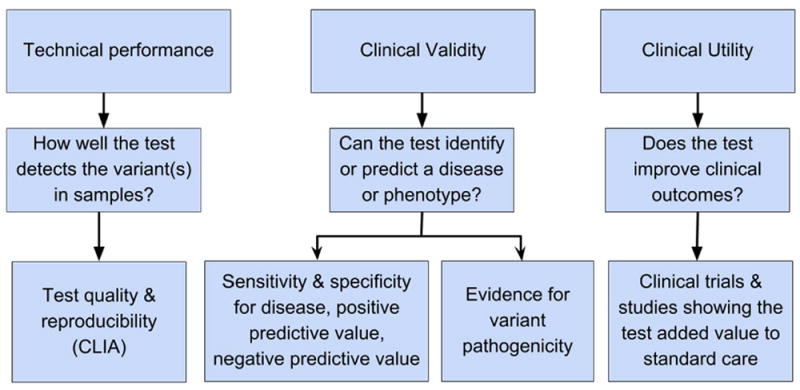
Evaluation of a clinical test. The three aspects that need to be evaluated are technical performance, clinical validity and utility. Each aspect relates to a distinct function of the test (second row) and yields data on particular aspects of test performance and utility (third row).
In the United States, the technical performance or safety and effectiveness of a test is regulated by the Food and Drug Administration (FDA) under the Federal Food, Drug, and Cosmetic Act as medical devices for tests sold as kits. However, for tests marketed as a laboratory-developed test and performed by a single laboratory, the FDA has practiced “enforcement discretion”. The Centers for Medicare and Medicaid Services (CMS) is responsible for regulating the clinical laboratories performing genetic testing in the U.S., ensuring their compliance with Clinical Laboratory Improvement Amendments (CLIA). Some tests are also regulated by states. The sequencing and interpretation pipeline is accredited by the College of American Pathologists (CAP).
The clinical validity of a test depends on the evidence of the variant’s association with disease, and includes functional information for the variant tested, i.e., loss of function mutations that disrupt the function of protein-coding genes are more likely to influence phenotypes or lead to a clinical disease. However, the function of many variants is unclear. The ClinVar archive at the National Center for Biotechnology Information (NCBI) lists known human genetic variants and provides information about which variants have been associated with human disease, and which ones have no phenotype identified to date for interpretation of the clinical significance of variants found in patient samples including pathogenicity. The evidence for clinical validity of variants listed in ClinVar varies (Table 1).
The clinical utility is the evidence that the test improves clinical outcomes and therefore helps with patient management decisions. For example, most tested variants have little evidence from randomized clinical trials to establish their clinical utility.
Online genetic variant registries
The MEDLINE resources of the National Library of Medicine include another highly useful repository of genetic information that pertain to gene, diseases and diagnoses, in addition to ClinVar. The Online Mendelian Inheritance in Man (OMIM) includes human genes and genetic phenotypes and despite its name, is not restricted to genes with Mendelian inheritance. Currently, over 15,000 disorders are listed.
Genetic test indications
Mendelian disorders
Several Mendelian and mitochondrial disorders have kidney phenotypes that manifest at childhood or adulthood and may initially go undiagnosed. These disorders result from mutations that have high penetrance (proportion of individuals presenting with the disease or phenotype among carriers of the mutation and the mode of inheritance) and varying expressivity (different clinical/pathologic manifestations from mild to severe disease). Their diagnosis may have implications for therapy and screening of family members for counseling or for kidney donation.
Mendelian disorders result from complex mutations in genes, which provide challenges for genetic testing. The choice to pursue genetic testing should be guided by the benefit of the knowledge obtained. For example, highly penetrant mutations in over 50 different genes in nuclear or mitochondrial DNA have been identified for focal segmental glomerulosclerosis (FSGS).6 Children with FSGS due to genetic mutations are less likely to respond to glucocorticoids and less likely to have disease recurrence after kidney transplantation. Whole-exome sequencing is a more comprehensive approach for diagnosis of these mutations unless there is a known mutation segregating in families. There is a need for more studies on the clinical validity and utility of genetic testing for patients and families, particularly when the test is used for screening of genetic causes of FSGS.
For some known genetic disorders, there may be an alternative method of diagnosis for the disease. For example, ADPKD is the most commonly diagnosed inherited kidney disease, accounting for 5-10% of all patients on renal replacement therapy (recently reviewed by Chebib and Torres7). Most cases of ADPKD are due to mutations in the PKD1 (80-85%) and the PKD2 (15-20%) genes. Recently, mutations in the GANAB gene were identified in whole-exome sequencing in affected family members 8 and mutations in an additional seven genes were identified for hereditary polycystic kidney and liver disease.9 Over 2,300 and 270 mutations have been reported for the PKD1 and PKD2 genes, respectively.9a These can be tested using Sanger sequencing, NGS, and commercially available testing services.10 However, in clinical practice, ADPKD diagnosis is based on imaging of the kidneys and age-related ultrasound diagnostic criteria, or use of alternative kidney imaging such as magnetic resonance imaging or computed tomography rather than genetic testing.11 This is because genetic testing may not provide a definitive diagnosis if detected mutations have unknown pathogenicity, the frequency of mutation detection often is equivalent to the frequency of disease detection by imaging, and there are issues related to cost and insurance coverage of the test. A recent KDIGO (Kidney Disease: Improving Global Outcomes) conference recommended genetic testing in special situations such as for diagnosis of ADPKD when renal imaging is inconclusive, in cases of early and severe clinical presentation and those with a negative family history (potential de novo mutations), and for screening in the setting of reproductive counseling.11 Genetic testing in these cases usually includes consultation with genetic medical experts and genetic counselors.
APOL1-associated CKD
APOL1 G1 and G2 alleles are African-ancestry specific variants recently identified as risk factors for CKD. 12,13 G1 encodes two highly correlated nonsynonymous (amino acid changing) variants, whereas G2 encodes a 6-nucleotide deletion. Small indels (insertions or deletions of bases in the genome) such as G2 can be captured using sequencing, although different platforms have different reliability for indels. Individuals carrying two APOL1 risk genotypes (high-risk; about 13% of African Americans12,13 and 2% of U.S. Hispanics/Latinos of Caribbean background14) have an increased risk for end-stage kidney disease (odds ratio of ~ 7) and for FSGS (odds ratio of 10 to 29 including HIV nephropathy).12,13 Among CKD participants of the African American Study of Kidney Disease and Hypertension (AASK), APOL1 high-risk carriers had a 1.9-fold risk of a composite outcome of ESRD or a doubling of the serum creatinine level.15 However, among African Americans in the general population, APOL1 high-risk status was associated with 1.5-fold risk for CKD.16
Using published data on APOL1 and CKD in African Americans, we estimated the sensitivity and specificity, and the likelihood ratios (LR) that a test result would be expected in a patient with disease compared to a patient without disease using different scenarios related to the purpose of the test (Figure 2). When genetic testing is performed for diagnosis, a positive APOL1 test result has moderate to low LR (true positive/false positive) for FSGS and ESRD, respectively, and would only increase the probability of disease by ~30%. However, a negative test can be useful to rule out APOL1-related FSGS disease and therefore for counselling. When APOL1 is tested in African Americans at-risk of CKD or progression to later stages of CKD, a positive test has a low true positive to false positive ratio, and is not helpful for screening or prognosis of CKD. A negative test in these settings is also not helpful, as it cannot discriminate a true negative from a false negative result (LR close to 1). These are due to differences in the magnitude of the genotype-disease association (odds ratio) and prevalence of the disease in these different settings.
Figure 2. Clinical validity of APOL1 testing for disease diagnosis and prognosis in African Americans.
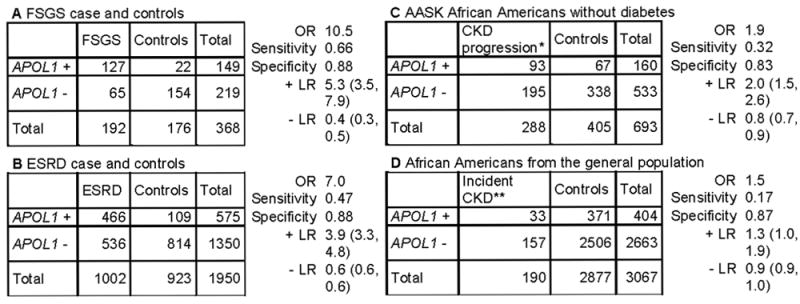
We estimated the sensitivity, specificity, positive likelihood ratio (+LR, true positive/false positive) and negative likelihood ratio (-LR, true negative/false negative) for APOL1 testing (and 95% confidence intervals) using published data. The settings designated as A and B are related to FSGS and ESRD diagnosis, respectively. C and D are genetic testing used for prognosis. The +LR is moderate to low when a genetic testing is performed for diagnosis of APOL1-related FSGS (A) and ESRD (B), respectively, and so a positive test may be useful only for diagnosis of APOL1-related FSGS. The -LR is close to zero in setting A, so a negative test could help to rule out APOL1-related FSGS. When testing African Americans with non-diabetic CKD (C) or screening African Americans without CKD in the general population (D), the +LR is low, supporting the low yield for an APOL1 genetic testing used for prognosis or screening. In addition, a negative test is not helpful to rule out future disease or counseling in settings C and D since the -LR is close to 1. Data obtained from table 1 in Genovese et al12 (A & B), table 2 in Parsa et al15 (C) and table 2 in Foster et al 16 (D). APOL1 + is high-risk genotypes and APOL1 – is low risk genotypes. We assumed the disease is causally related to APOL1 high-risk genotypes, although other genetic variants could contribute to CKD. *incident ESRD or doubling of serum creatinine **An eGFR < 60 ml/min/1.73 m2 at follow-up
In contrast to Mendelian disorders described above, the disease penetrance of APOL1-related CKD is low, with only about 20% of individuals carrying high-risk genotypes developing CKD,17 and there is a large variability in clinical disease manifestations and progression among those developing CKD. With regard to kidney transplantation, recipients of donor kidneys obtained from individuals carrying high-risk APOL1 genotypes may have shorter allograft survival.18 A critical question that is an area of active investigation relates to the frequency of APOL1 high-risk alleles in CKD in living donors and the etiologic role that these variants might play.
Importantly, the clinical and environmental factors leading to development of APOL1-related CKD and effective therapies for prevention or treatment of individuals at risk are not yet established. Screening of individuals for APOL1 alleles for prognosis will require assessments of the risk/benefit of genetic testing in the clinical setting and engagement of patients and African American communities in these medical decisions, given potential harms.19 There is currently insufficient evidence to recommend genetic testing of APOL1 for diagnosis and prognosis of CKD. To answer some of the questions related to organ donation, NIH recently established the Long-term Kidney Transplantation Outcomes Research Network (APOLLO), which will assess the role of APOL1 genetic variants as susceptibility factors in kidney transplant recipients of organs from African American donors, and the clinical outcome of kidney donors who carry APOL1 variants.
Pharmacogenetics
Overview
Genes influence the response to pharmacologic agents and genetic tests may help to guide treatment strategies such as avoiding side-effects and other harmful complications. Genetic testing may provide information on drug effectiveness (e.g., non-responders) and effects on drug metabolism (faster versus slower metabolizers) that could allow individualized drug dose. In addition, pharmacogenetic testing can be used to identify individuals who are at-risk of severe idiosyncratic adverse events, therefore helping in medical decisions for choice of therapy including using alternative strategies for treatment. Although there has been great interest in pharmacogenetics, there is a large gap in the knowledge on actionable variants (those which results can change treatment), and the use of genetic testing in clinical care is still limited to few variants and drugs.
The Clinical Pharmacogenetics Implementation Consortium (CPIC) is an international consortium that develops peer-reviewed guidelines for pharmacogenetic testing based on evidence from randomized controlled trials and other clinical studies.20 CPIC recommendations focus on gene-drug pairs for CLIA-approved genetic tests that show evidence for the need to change drug dose or consider an alternative drug but does not recommend whether a test should be ordered. So far, CPIC has published 36 pharmacogenetic drug guidelines that includes variants in 15 genes.
Table 2 summarizes the CPIC guidelines (updated at https://cpicpgx.org/guidelines/) for genetic testing for selected drug-gene pairs that have strong levels of evidence based on clinical validity and utility. Below we discuss a few examples of pharmacogenetic testing based on clinical indication.
Table 2.
Selected drug-gene pairs with a guideline recommendation.
| Drug (Clinical use) | Genetic testing indication | Positive genetic test resultsˆ | Negative genetic test resultsˆ | Evidence for clinical utility | Treatment guidance |
|---|---|---|---|---|---|
| Abacavir (1st- line treatment of HIV infection) | Screening to avoid immune-mediated hypersensitivity reactions (occurs in ~6% of pts) | 1-2 copies of HLA-B*57:01 | 0 copies of HLA-B*57:01 | PREDICT-1: NPV=100%, PPV=47.9%, evidence for cost-effectiveness; recommended by FDA 26,30 | PharmGKB GL72; drug contraindicate d for pts w/HLA-B*57:01 |
| Allopurinol (treatment of gout) | Screening to avoid severe cutaneous adverse reactions | 1-2 copies of HLA-B*58:01 | 0 copies of HLA-B*58:01 | Significantly increased risk (ORs of 80-580) for pts w/ HLA-B*58:01 variant48-53 | PharmGKB GL73; drug contraindicate d for pts w/ HLA-B*58:01 |
| Atazanavir (antiretroviral protease inhibitor) | Screening for drug-related jaundice | Intermed metabolizer: presence of 1 UGT1A1 decreased-function allele (*6, *28, *37) w/ UGT1A1*1 (normal function) or UGT1A1*36 (increased function), 1 copy of rs887829 T allele; poor metabolizer: 2 decreased-function alleles (UGT1A1*6, *28, *37) or 2 copies of rs887829 T alleles | UGT1A1*1/*1; *1/*36; *36/*36; rs887829 C/C | AIDS Clinical Trials Group protocol A5257: high likelihood of developing jaundice resulting in atazanavir discontinuation w/ UGT1A1 poor metabolizers 54,55 | PharmGKB GL74 |
| Azathioprine (immunosuppr essant used in solid organ transplant & immunological disorders) | Dose adjustment for variants related to low or deficient drug metabolism | TPMT intermed activity (3%-14% of pts): 1 functional allele (*1) + 1 nonfunctional allele (*2, *3A, *3B, *3C, *4); low/deficient activity (1 in 178-3,736 pts): 2 nonfunctional alleles (*2, *3A, *3B, *3C, *4) | TPMT*1/*1 | Substantial evidence that dose adjustments based on TPMT reduce adverse effects w/out compromising desired therapeutic effects 56 | PharmGKB GL75 |
| Carbamazepin e (anticonvulsan t, also used in trigeminal neuralgia) | Screening for Steven-Johnson syndrome & toxic epidermal necrolysis | 1-2 copies of HLA-B*15:02 | 0 copies of HLA-B*15:02 | Significant association of HLA-B*15:02 genotype in pts w/ Asian ancestry w/ carbamazepine-induced Steven-Johnson syndrome & toxic epidermal necrolysis vs carbamazepine-tolerant pts & healthy controls 34 | PharmGKB GL76; use alt drug for pts testing positive if naïve |
| Clopidogrel (antiplatelet drug) | Screening before percutaneous coronary angioplasty for effectiveness (non-response) | CYP2C19 ultra-rapid metabolizer (~5%-30% of pts): 2 increased-activity alleles (*17) or 1 functional allele (*1) + 1 increased-activity allele (*17); intermed metabolizer (~18%-45% of pts): 1 functional allele (*1) + 1 LOF allele (*2-*8), or 1 LOF allele (*2-*8) + 1 increased-activity allele (*17); poor metabolizer (~2-15% of pts; *4-*8 rarely seen): 2 LOF alleles (*2-*8) | CYP2C19*1/*1 | Substantial literature implicating LOF CYP2C19 alleles in adverse clopidogrel responses, FDA black box warning on drug label 57; TRITON-TIMI 38: vs noncarriers, pts w/ reduced-function CYP2C19 allele have HR of 4.79 (1.40-16.37) for death from CV causes, 1.38 (0.94-2.02) for nonfatal MI, 3.93 (0.66-23.51) for nonfatal stroke, & 3.09 (1.19-8.00) for stent thrombosis 58 | PharmGKB GL77; CYP2C19 ultra-rapid metabolizer: use standard dose; intermed & poor metabolizer: use alt drug |
| Codeine (analgesic) | Screening for efficacy (poor metabolizer, insufficient pain relief) & toxicity (ultra-rapid metabolizer, due to increased metabolism to morphine) | CYP2D6 ultra-rapid metabolizer (~1-2% of pts): 2+ copies of functional alleles (*1/*1xN, *1/*2xN); intermed metabolizer (~2-11% of pts): 1 reduced & 1 nonfunctional allele (*4/*10, *5/*41); poor metabolizer (~5-10% of pts): nonfunctional alleles (*4/*4, *4/*5, *5/*5, *4/*6) | CYP2D6 *1/*1, *1/*2, *2/*2, *1/*41, *1/*4, *2/*5, *10/*10 | Substantial evidence for decreased analgesia in poor metabolizers & severe or life-threatening toxicity following normal doses of codeine in ultrarapid metabolizers 59 | PharmGKB GL78 |
| Ivacaftor (cystic fibrosis treatment) | Screening for drug efficacy (non-response) | 2 copies of CFTR F508del (rs113993960 or rs199826652 genotype del/del) | 0-1 copies of CFTR F508del; 1-2 copies of the following CFTR variantsˆˆ: E56K, P67L, R74W, D110E, D110H, R117C, E193K, L206W, R347H, R352Q, A455E, D579G, S945L, S977F, F1052V, K1060T, A1067T, G1069R, R1070Q, R1070W, F1074L, D1152H, D1270N, G551D | CFTR F508del 2 copies: no significant reduction in sweat chloride concentrations, drug provides no clinical & lung function improvement 23; G551D/G551D or G551D/F508del: improvement of lung function, weight, risk of pulmonary exacerbation, & reduction in sweat chloride concentrations w/ drug use; moderate evidence for improvement w/ 1+ copy of variants inˆˆ except G551D | PharmGKB GL79; 2 copies of CFTR F508del: ivacaftor not recommended ; G551D/G551 D or G551D/F508d el: ivacaftor recommended |
| Phenytoin (anticonvulsan t) | Screening to avoid cutaneous adverse reactions of Stevens-Johnson syndrome & toxic epidermal necrolysis | 1-2 copies of HLA-B*15:02; CYP2C9 intermed metabolizer (~8% of pts): *1/*2, *1/*3; CYP2C9poor metabolizer (~1% of pts): *2/*2, *3/*3, *2/*3 | 0 copies of HLA-B*15:02; CYP2C9*1/*1 | Significantly increased risk of Steven-Johnson syndrome & toxic epidermal necrolysis w/ HLA-B*15:02 variant in Asian groups (OR, 4.26 [1.93-9.39])60,61; phenytoin maintenance doses reduced by 23%-38% in heterozygous pts & by 31-52% in homozygous pts for decreased-function CYP2C9 alleles62-64 | PharmGKB GL80; HLA-B*15:02: drug contraindicated for phenytoin-naïve pts |
| Simvastatin (lipid-lowering drug) | Screening to avoid simvastatin-induced myopathy (myalgias occur in 1-5% of exposed pts) | Intermed function in plasma clearance of simvastatin: 1 copy of decreased-function allele of SLCO1B1 (*5, *15, *17) or rs4149056 T/C; low function: 2 copies of SLCO1B1*5, *15, *17 or rs4149056 C/C | Normal function: SLCO1B1*1a/*1a, *1a/*1b, *1b/*1b or rs4149056 T/T | RCT (SEARCH) & clinical practice-based cohorts (HPS): ORs for myopathy of 4.5 & 2.6, respectively, per copy of the minor C rs4149056 allele w/ simvastatin (less evidence for other statins); STRENGTH: highest overall effect size of drug discontinuation for any adverse effect, myalgia, muscle cramping, or elevated serum CK levels >3-fold of ULN for simvastatin (OR 2.8 [1.3-6.0]) compared to atorvastatin (OR 1.6 [0.7-3.7]) or pravastatin (OR 1.06 [0.22-4.8])65-68 | PharmGKB GL81 |
| Tacrolimus (immunosuppr essant) | Dose adjustments for variants related to enzyme expression | CYP3A5 extensive metabolizer: *1/*1; intermed metabolizer: *1/*3, *1/*6, *1/*7 | CYP3A5 poor metabolizer: *3/*3, *6/*6, *7/*7, *3/*6, *3/*7, *6/*7 | Strong evidence based on more than 50 studies for use of tacrolimus in kidney, heart, lung, HSCT, & liver Tx pts where the donor & recipient genotypes are identical; 69 ongoing clinical trials: ClinicalTrials.gov | PharmGKB GL82 |
| Warfarin (oral anticoagulant) | Dose adjustment & efficacy based on metabolism (CYP2C9) & sensitivity (VKORC1) | Non-African ancestry: CYP2C9*2/*3, *3/*3 (poor metabolizer) or both increased sensitivity (VKORC1 1639 A/A or A/G) & CYP2C9 poor metabolizer; African ancestry: CYP2C9*5 or *6 or *8 or *11, rs12777823 A/G or A/A, CYP4F2*3 | CYP2C9*1 + 0 copies of other alleles listed as positive test; 0 copies of CYP2C9 rs12777823 A allele (only in African ancestry); 0 copies of VKORC1 1639 A allele; 0 copies of CYP4F2*3ˆˆˆ | EU-PACT & COAG trials for genotype-guided dose adjustments; GIFT trial showed the effectiveness and safety of genotyped-guided dosing for VTE & major bleeding (27% reduction) 21,70,71 | PharmGKB GL83 |
Footnote: Based on information from the Clinical Pharmacogenetics Implementation Consortium guidelines (CPIC) and PharmGKB. The most updated version of these guidelines can be found at https://cpicpgx.org/guidelines/.
Alt, alternative; NPV, negative predictive value; PPV. Positive predictive value; COAG, Clarification of Optimal Anticoagulation Through Genetics; CV, cardiovascular; HPS, Heart Protection Study; HSCT, hematopoietic stem cell transplant; intermed, intermediate; MI, myocardial infarction; SEARCH, Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine; STRENGTH, Statin Response Examined by Genetic Haplotype Markers; EU-PACT, European Pharmacogenetics of Anticoagulant Therapy; PREDICT-1, Prospective Randomized Evaluation of DNA Screening in a Clinical Trial; TRITON-TIMI, Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis in Myocardial Infarction; LOF, loss of function; RCT, randomized controlled trial; Tx, transplant; ULN, upper limit of normal.
Laboratory reporting of genetic variants varies; a positive result would be one guiding treatment modifications, while a negative result would one with no modification in drug regimen needed.
SNP numbers are as follows for the following amino acid changes: E56K (rs397508256), P67L (rs368505753), R74W (rs115545701), D110E (rs397508537), D110H (rs113993958), R117C (rs77834169), E193K (rs397508759), L206W (rs121908752), R347H (rs77932196), R352Q (rs121908753), A455E (rs74551128), D579G (rs397508288), S945L (rs397508442), S977F (rs141033578), F1052V (rs150212784), K1060T (rs397508513), A1067T (rs121909020), G1069R (rs200321110), R1070Q (rs78769542), R1070W (rs202179988), F1074L (rs186045772), D1152H (rs75541969), D1270N (rs11971167), G551D (rs75527207)
additional variants need to be genotyped in those of African ancestry
Genetic testing to guide drug dose
Genetic variants can alter drug-metabolism (pharmacokinetics) and therefore guide drug dose adjustments to avoid under-treatment or side effects from high drug exposure. For example, the oral anticoagulant warfarin, widely used for the prevention and treatment of thromboembolic disorders, has large inter-individual variability in dosing to achieve therapeutic range related to both dietary and genetic factors. Warfarin has a narrow therapeutic range, and both low and high international normalized ratio (INR) can be harmful. Testing for genetic variants in genes related to warfarin metabolism (CYP2C9), the target enzyme for warfarin action (VKORC1, described in the section on genetic testing for drug effectiveness) and in pathways related to vitamin K recycling (CYP4F2) are currently recommended by CPIC to guide the dosing of warfarin.21 Variants in these three genes account for up to 18%, 30%, and 11%, respectively, of the variability in warfarin dose in individuals of European ancestry. Genetic testing is available for CYP2C9*2 and *3 (more common in European ancestry; CYP2C9*2 is absent in Asians), CYP2C9*5, *6, *8, and *11 (more common in African ancestry), and CYP4F2*3.
Genetic testing to guide drug choice
Genetic testing can help treatment decisions by identifying sensitivity or resistance to drugs used for the condition being treated (drug effectiveness). For example, ivacaftor is an FDA-approved drug for cystic fibrosis that regulates the activity of the cystic fibrosis transmembrane conductance regulator (CFTR) channel and has been shown to improve lung function in clinical trials.22 Approximately 85-90% of individuals of European ancestry with cystic fibrosis carry at least one copy of the F508del variant, which is a functional variant that leads to an abnormal CFTR protein. Individuals carrying two copies of the CFTR F508del mutation showed no improvement in clinical symptoms or in lung function after ivacaftor treatment (non-responders), and an alternative treatment is recommended for these patients. Additional variants associated with lack of response to the drug and variants associated with drug efficacy (33 CFTR variants) are shown in Table 2. Current guidelines recommend genetic testing for CFTR variants before initiation of ivacaftor treatment based on clinical efficacy studies.23,24
Another example of genetic variants affecting drug response and currently recommended for genetic testing are variants in the VKORC1 gene, which encodes the enzyme vitamin K epoxide reductase, the target for the oral anticoagulant warfarin. A common variant in VKORC1 (reference single-nucleotide polymorphism [rs] identifier 9923231, 1639G>A [substitution of guanine by adenine at nucleotide 1639]) is associated with increased sensitivity to warfarin. Patients who carry this variant require lower doses of warfarin to achieve the target INR. Rare protein-changing variants in VKORC1 have shown to confer warfarin resistance but are usually not tested for. Guidelines for dose drug adjustments based on the common VKORC1 variant and other genetic variants related to warfarin pharmacokinetics have been recently updated (Table 1).21 It is expected that additional genetic testing will be developed to guide the choice of drugs based on drug efficacy in clinical care.
Genetic testing to avoid idiosyncratic drug events
Adverse events may themselves result in morbidity and, in severe cases, mortality, and can contribute to drug treatment non-adherence. Some adverse events with underlying genetic susceptibility (e.g., Stevens-Johnson syndrome, toxic epidermal necrolysis, and drug-induced liver disease) may be prevented by genetic testing.25 For example, genetic testing is already integral to the use of some antiretroviral agents in HIV clinical care management in the U.S.26 Abacavir is generally well-tolerated; however it may cause an immunologically mediated hypersensitivity reaction driven by activation of HLA-B*57:01.27,28 Abacavir hypersensitivity reactions related to HLA-B*57:01 occur in 3 to 5% of patients during the first 6 weeks of treatment.29 Testing for the HLA-B*57:01 allele is now recommended by the FDA before initiating antiviral treatment that includes abacavir based on results of PREDICT-1 (Prospective Randomized Evaluation of DNA Screening in a Clinical Trial).30 The trial randomized HIV-infected patients to a prospective-screening group, which excluded HLA-B*57:01–positive patients from abacavir treatment, or a control group, which used a standard-of-care approach to abacavir use without prospective screening. Immunologically confirmed hypersensitivity reactions occurred in 0% in the prospective-screening group vs. 2.7% in the control group (P<0.001), with a NPV of 100% and a positive predictive value (PPV) of 47.9%. Additional studies have shown the cost-effectiveness of testing for HLA-B*57:01 before starting abacavir treatment.31-33
Genetic testing for HLA-B*15:02 is recommended before using the anticonvulsant drug carbamazepine to avoid life-threatening Stevens-Johnson syndrome and toxic epidermal necrolysis. Individuals with one or two copies of the HLA-B*1502 allele, which is common in Oceanic, East Asian, and South/Central Asian populations (1 to 10%) but not in Europeans, are at risk of cutaneous reactions, and avoidance of the drug is recommended for patients not previously exposed to the drug.34
Information on commercially available genetic testing
The Genetic Testing Registry (GTR) is a NIH-developed voluntary registry of genetic tests and genetic testing services, both academic and commercial. GTR includes information on the test’s purpose, methodology, validity, usefulness, and laboratory contacts and credentials (Table 1). The information on quality and utility for clinical use of tests available in this database varies. The most recent data from GTR from December 2017 shows 54,290 tests for 16,406 genes and 10,974 conditions performed by 509 labs. As an example, 16 clinical tests for APOL1 are currently listed at GTR, offered by 12 CLIA-certified laboratories. Seven are a single gene test for APOL1, at a cost of $200 to $1,000, and include whole-gene sequencing, sequencing of APOL1 exon 6 only, or direct genotyping of G1/G2 variants. Information on positive and negative results are not provided, and the reporting of VUS that will likely will be identified in sequencing data varies.
Direct-to-consumer testing
Healthcare attitudes continue to shift towards empowering patients, who have become more proactive in managing their care. As a result, direct-to-consumer genetic testing has become a popular option. The FDA has expressed concern over direct-to-consumer tests being used by the public to self-diagnose without the intervention of healthcare providers, potentially leading to self-treatment or cessation of current medication.35 In recent years, the FDA has sent warning letters to direct-to-consumer genetic test companies, including 23andMe and DNA4Life, stating that their genotyping tests needed approval as a medical device prior to marketing.36-38 Both of these companies genotype pharmacogenomic variants, and in the United Kingdom, 23andMe still provides results for some pharmacogenomics tests. Direct-to-consumer pharmacogenomics testing is unique in that results become actionable once a drug is prescribed, ranging from the point of testing or potentially many years later, leading to advocacy for preemptive genotyping pharmacogenomics panels. Arguably, direct-to-consumer genotyping will act as a powerful conduit for integrating pharmacogenomics into practice as evidence for its clinical utility is being collected.39 With the expansion of such testing, educational initiatives on direct-to-consumer genotyping are essential to improving the health literacy of healthcare providers and the public.40
Challenges and limitations of genetic testing
Some of the challenges related to the adoption of genetic testing for diagnosis and treatment of patients are the lack of demonstrated clinical validity and utility of some genetic variants (such as most of the genetic variants identified in genome-wide association studies) and the existence of alternative approaches that have lower costs (e.g., dosing blood levels of the drug instead of adjusting based on genetic testing). For a limited number of variants with strong evidence for preemptive screening, mostly in pharmacogenomics, efforts should focus on increased access of information for health care providers through education on test availability, interpretation, and recommended clinical actions based on results, including existing guidelines from CPIC and/or scientific societies. For example, pharmacogenomic tests could be reported as positive (see Table 2 for list of variants) with recommendations on clinical actions. Concerns related to patient safety due to incorrect test ordering, misinterpretation and lack of follow-up of findings have also been raised41. Incorporating genetic testing into electronic medical records will facilitate genetic-driven clinical medical decisions.
Most diseases or traits including CKD are polygenic, i.e., influenced by multiple genes. Therefore, genetic panel testing including several variants may be more efficient for use in screening and diagnosis of genetic disease and in pharmacogenomics. Because the prevalence of variants varies in ancestral populations and some variants may be rare in some ethnic groups, genetic testing will likely be more cost-effective if targeting patients from a racial/ethnic group with a high prevalence of the variant. For example, the HLA-B*57:01 allele related to abacavir toxicity is common in European ancestry individuals (allele frequency of 8%) but rare in individuals of African ancestry (0.3%).42 Conversely, genetic panels including variants from multiple ancestral populations will be more suitable for widespread use in clinical care in countries with a large ancestry admixture such as the U.S.
Ethical and legal aspects
The implementation of genetic testing in clinical care will require establishing a facile process for ordering genetic testing in hospital and clinics, access to expertise to address ethical, legal community, patient, and family concerns,19 in addition to information on cost and coverage by insurance. To address concerns related to discrimination of individuals for insurance purpose or by society, anti-discrimination laws have been passed in the U.S. These include the Genetic Information Nondiscrimination Act (GINA), which prevents insurers and employers from using genetic information in decisions about health-care coverage or employment, and the Patient Protection and Affordable Care Act, which blocks insurers from denying coverage or raising premiuems due to pre-existing conditions. Some of the challenges on implementation of genetic testing are being studied in the National Institutes of Health-funded Implementing GeNomics In practice (IGNITE) Network. 43
Discussion with patients and families
Discussions with patients and families should be tailored to the specific purpose for ordering the genetic test (diagnosis, prediction, screening) and guided by information on risk and benefits in these settings. Topics include discussion on whether the genetic results will alter the clinical management of the patient or provide information important for the care of the patient or family members who may carry the genetic mutation, alternative options to genetic testing, and costs. Engagement of a genetic counselor both before ordering the test and when test results are available is recommended, as these counselors have familiarity with testing technical procedures and interpretation of results, and extensive experience in providing counseling to families and in addressing patients’ concerns. Genetic testing for APOL1 is currently controversial outside research settings, with little evidence available on benefits or harms that would support specific recommendations. African American patients should be informed on the limited evidence for clinical utility of the genetic testing of APOL1 variants for CKD, and the current lack of specific treatment for CKD patients who are tested positive. Discussion should also include the lack of data on the utility of APOL1 genetic testing for screening CKD in healthy African Americans including kidney donors.
Case study.
A 67-year-old white male with CKD stage G3, coronary heart disease, hypertension and hyperlipidemia was started on allopurinol 300 mg/d for gout.1 Additional medications were metoprolol, simvastatin, enalapril and aspirin. After one week of allopurinol treatment, nausea and a skin rash developed. He was admitted to the hospital three days later with a generalized maculopapular exanthema, fever to 39°C, and a perip heral leukocyte count of 24,000 cells/μL with 20% eosinophils. His right great toe had red swelling and was painful. Serum creatinine was 1.8 mg/dl (160 μmol/l) and urinalysis was negative for nitrites and positive for leukocytes. Broad spectrum antibiotics were started. Blood and urine cultures all gave negative results. He received a diagnosis of allopurinol-induced hypersensitivity and was started on prednisolone 1 mg/kg/d. His symptoms improved sufficiently to be discharged from the hospital after two weeks. However, his skin lesions had not healed and he was admitted six weeks later with sepsis and multi-organ failure.
Case Review.
Allopurinol-induced hypersensitivity is an uncommon and devastating adverse effect of allopurinol, which has a 25% mortality rate.44 Allopurinol reactions can manifest as Stevens-Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms (DRESS), and severe cutaneous adverse reactions (SCAR). This patient has clinical and laboratory findings compatible with allopurinol hypersensitivity syndrome. Two case series have described hypersensitivity following exposure to allopurinol in 78 individuals45 and 101 individuals.37 Common features are erythematous skin rash, eosinophilia, hepatitis, and reduced kidney function. The treatment of allopurinol hypersensitivity syndrome is mostly supportive and includes future avoidance of the drug. Oxypurinol, the active metabolite of allopurinol, is involved in allopurinol-associated hypersensitivity by inducing T-cell response and hypersensitivity. An HLA-B*58:01 variant has been associated with hypersensitivity to allopurinol and has an allele frequency of 6-7% in Asian populations (particularly those in East Asia) and 1% in European descent populations.44 The odds ratio for hypersensitivity associated to allopurinol with the HLA-B*58:01 allele is estimated to be 80 to 580:1. Other risk factors include recent initiation of therapy and impaired renal function, which elevates levels of allopurinol and oxypurinol. Oxypurinol preferentially binds to the peptide binding groove of HLA-B*58:01,44 and likely forms a drug-peptide-HLA complex that is highly immunogenic, initiating the hypersensitivity reaction. The 2012 American College of Rheumatology Guidelines for treatment of gout recommends preemptive genetic testing for patients of Korean descent with CKD stage 3 or worse (allele frequency of 5%), and persons of Han-Chinese or Thai descent irrespective of kidney function.46 The 2015 updated CPIC guidelines states that allopurinol is contraindicated in persons of any ethnicity with a positive genetic test for HLA-B*58:01.47 In light of the morbidity and mortality of allopurinol-associated hypersensitivity, testing for HLA-B*58:01 variants is indicated prior to allopurinol therapy initiation. In the case study, genetic testing was not performed, and so a role for HLA-B*58:01 cannot be known for certain.
Acknowledgments
Support: This work was supported in part by R01 MD012765, R56 DK104806 and R21 HL123677 (to NF), and by the NIDDK Intramural Research Program, NIH, Bethesda, MD (JBK).
Footnotes
Financial Disclosure: The authors declare that they have no relevant financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laurisch S, Jaedtke M, Demir R, et al. Allopurinol-induced hypersensitivity syndrome resulting in death. Med Klin (Munich) 2010;105(4):262–266. doi: 10.1007/s00063-010-1037-3. [DOI] [PubMed] [Google Scholar]
- 2.U.S. Food and Drug Administration. [October 28, 2016];Antiretroviral drugs used in the treatment of HIV infection. http://www.fda.gov/ForPatients/Illness/HIVAIDS/Treatment/ucm118915.htm.
- 3.Rockwell KL. Direct-to-Consumer Medical Testing in the Era of Value-Based Care. JAMA. 2017;317(24):2485–2486. doi: 10.1001/jama.2017.5929. [DOI] [PubMed] [Google Scholar]
- 4.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–255. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 5.Evans JP, Powell BC, Berg JS. Finding the Rare Pathogenic Variants in a Human Genome. JAMA. 2017;317(18):1904–1905. doi: 10.1001/jama.2017.0432. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2017;12(3):502–517. doi: 10.2215/CJN.05960616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chebib FT, Torres VE. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am J Kidney Dis. 2016;67(5):792–810. doi: 10.1053/j.ajkd.2015.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porath B, Gainullin VG, Cornec-Le Gall E, et al. Mutations in GANAB, Encoding the Glucosidase IIalpha Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet. 2016;98(6):1193–1207. doi: 10.1016/j.ajhg.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornec-Le Gall E, Torres VE, Harris PC. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol. 2018;29(1):13–23. doi: 10.1681/ASN.2017050483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Autosomal Dominant Polycystic Kidney Disease Mutation Database [Google Scholar]
- 10.Tan AY, Michaeel A, Liu G, et al. Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J Mol Diagn. 2014;16(2):216–228. doi: 10.1016/j.jmoldx.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman AB, Devuyst O, Eckardt KU, et al. Kidney Int; Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference; 2015. pp. 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. Journal of the American Society of Nephrology : JASN. 2011;22(11):2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer HJ, Stilp AM, Laurie CC, et al. African Ancestry-Specific Alleles and Kidney Disease Risk in Hispanics/Latinos. J Am Soc Nephrol. 2017;28(3):915–922. doi: 10.1681/ASN.2016030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsa A, Kao WH, Xie D, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med. 2013;369(23):2183–2196. doi: 10.1056/NEJMoa1310345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. Journal of the American Society of Nephrology : JASN. 2013;24(9):1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dummer PD, Limou S, Rosenberg AZ, et al. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Semin Nephrol. 2015;35(3):222–236. doi: 10.1016/j.semnephrol.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freedman BI, Pastan SO, Israni AK, et al. APOL1 Genotype and Kidney Transplantation Outcomes From Deceased African American Donors. Transplantation. 2016;100(1):194–202. doi: 10.1097/TP.0000000000000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young BA, Fullerton SM, Wilson JG, et al. Clinical Genetic Testing for APOL1: Are we There Yet? Semin Nephrol. 2017;37(6):552–557. doi: 10.1016/j.semnephrol.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209–217. doi: 10.2174/1389200215666140130124910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson JA, Caudle KE, Gong L, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Pharmacogenetics-Guided Warfarin Dosing: 2017 Update. Clin Pharmacol Ther. 2017;102(3):397–404. doi: 10.1002/cpt.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clancy JP, Johnson SG, Yee SW, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for ivacaftor therapy in the context of CFTR genotype. Clin Pharmacol Ther. 2014;95(6):592–597. doi: 10.1038/clpt.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.PharmGKB. Annotation of CPIC Guideline for ivacaftor and CFTR. [12/15/2017]; https://www.pharmgkb.org/guideline/PA166114461.
- 25.Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med. 2001;250(3):186–200. doi: 10.1046/j.1365-2796.2001.00879.x. [DOI] [PubMed] [Google Scholar]
- 26.Martin MA, Klein TE, Dong BJ, et al. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther. 2012;91(4):734–738. doi: 10.1038/clpt.2011.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Small CB, Margolis DA, Shaefer MS, Ross LL. HLA-B*57:01 allele prevalence in HIV-infected North American subjects and the impact of allele testing on the incidence of abacavir-associated hypersensitivity reaction in HLA-B*57:01-negative subjects. BMC Infect Dis. 2017;17(1):256. doi: 10.1186/s12879-017-2331-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes AR, Spreen WR, Mosteller M, et al. Pharmacogenetics of hypersensitivity to abacavir: from PGx hypothesis to confirmation to clinical utility. Pharmacogenomics J. 2008;8(6):365–374. doi: 10.1038/tpj.2008.3. [DOI] [PubMed] [Google Scholar]
- 29.Temesgen Z, Beri G. HIV and drug allergy. Immunol Allergy Clin North Am. 2004;24(3):521–531. viii. doi: 10.1016/j.iac.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358(6):568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 31.Schackman BR, Scott CA, Walensky RP, Losina E, Freedberg KA, Sax PE. The cost-effectiveness of HLA-B*5701 genetic screening to guide initial antiretroviral therapy for HIV. AIDS. 2008;22(15):2025–2033. doi: 10.1097/QAD.0b013e3283103ce6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes DA, Vilar FJ, Ward CC, Alfirevic A, Park BK, Pirmohamed M. Cost-effectiveness analysis of HLA B*5701 genotyping in preventing abacavir hypersensitivity. Pharmacogenetics. 2004;14(6):335–342. doi: 10.1097/00008571-200406000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Kauf TL, Farkouh RA, Earnshaw SR, Watson ME, Maroudas P, Chambers MG. Economic efficiency of genetic screening to inform the use of abacavir sulfate in the treatment of HIV. Pharmacoeconomics. 2010;28(11):1025–1039. doi: 10.2165/11535540-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 34.Leckband SG, Kelsoe JR, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for HLA-B genotype and carbamazepine dosing. Clin Pharmacol Ther. 2013;94(3):324–328. doi: 10.1038/clpt.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howard HC, Borry P. Direct-to-consumer pharmacogenomic testing. Pharmacogenomics. 2011;12(10):1367–1370. doi: 10.2217/pgs.11.100. [DOI] [PubMed] [Google Scholar]
- 36.Post FA, Tebas P, Clarke A, et al. Brief Report: Switching to Tenofovir Alafenamide, Coformulated With Elvitegravir, Cobicistat, and Emtricitabine, in HIV-Infected Adults With Renal Impairment: 96-Week Results From a Single-Arm, Multicenter, Open-Label Phase 3 Study. J Acquir Immune Defic Syndr. 2017;74(2):180–184. doi: 10.1097/QAI.0000000000001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arellano F, Sacristan JA. Allopurinol hypersensitivity syndrome: a review. Ann Pharmacother. 1993;27(3):337–343. doi: 10.1177/106002809302700317. [DOI] [PubMed] [Google Scholar]
- 38.Lepist EI, Zhang X, Hao J, et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014;86(2):350–357. doi: 10.1038/ki.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu M, Lewis CM, Traylor M. Pharmacogenetic testing through the direct-to-consumer genetic testing company 23andMe. BMC Med Genomics. 2017;10(1):47. doi: 10.1186/s12920-017-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Wouden CH, Carere DA, Maitland-van der Zee AH, et al. Consumer Perceptions of Interactions With Primary Care Providers After Direct-to-Consumer Personal Genomic Testing. Ann Intern Med. 2016;164(8):513–522. doi: 10.7326/M15-0995. [DOI] [PubMed] [Google Scholar]
- 41.Korngiebel DM, Fullerton SM, Burke W. Patient safety in genomic medicine: an exploratory study. Genet Med. 2016;18(11):1136–1142. doi: 10.1038/gim.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orkin C, Sadiq ST, Rice L, Jackson F team UE. Prospective epidemiological study of the prevalence of human leukocyte antigen (HLA)-B*5701 in HIV-1-infected UK subjects. HIV Med. 2010;11(3):187–192. doi: 10.1111/j.1468-1293.2009.00762.x. [DOI] [PubMed] [Google Scholar]
- 43.Weitzel KW, Alexander M, Bernhardt BA, et al. The IGNITE network: a model for genomic medicine implementation and research. BMC Med Genomics. 2016;9:1. doi: 10.1186/s12920-015-0162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamp LK, Day RO, Yun J. Allopurinol hypersensitivity: investigating the cause and minimizing the risk. Nat Rev Rheumatol. 2016;12(4):235–242. doi: 10.1038/nrrheum.2015.132. [DOI] [PubMed] [Google Scholar]
- 45.Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity Description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76(1):47–56. doi: 10.1016/0002-9343(84)90743-5. [DOI] [PubMed] [Google Scholar]
- 46.Khanna D, Khanna PP, Fitzgerald JD, et al. American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken) 2012;64(10):1447–1461. doi: 10.1002/acr.21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saito Y, Stamp LK, Caudle KE, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for human leukocyte antigen B (HLA-B) genotype and allopurinol dosing: 2015 update. Clin Pharmacol Ther. 2016;99(1):36–37. doi: 10.1002/cpt.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A. 2005;102(11):4134–4139. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727–732. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 50.Lonjou C, Borot N, Sekula P, et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics. 2008;18(2):99–107. doi: 10.1097/FPC.0b013e3282f3ef9c. [DOI] [PubMed] [Google Scholar]
- 51.Kang HR, Jee YK, Kim YS, et al. Positive and negative associations of HLA class I alleles with allopurinol-induced SCARs in Koreans. Pharmacogenet Genomics. 2011;21(5):303–307. doi: 10.1097/FPC.0b013e32834282b8. [DOI] [PubMed] [Google Scholar]
- 52.Kaniwa N, Saito Y, Aihara M, et al. HLA-B locus in Japanese patients with anti-epileptics and allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics. 2008;9(11):1617–1622. doi: 10.2217/14622416.9.11.1617. [DOI] [PubMed] [Google Scholar]
- 53.Hershfield MS, Callaghan JT, Tassaneeyakul W, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for human leukocyte antigen-B genotype and allopurinol dosing. Clin Pharmacol Ther. 2013;93(2):153–158. doi: 10.1038/clpt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gammal RS, Court MH, Haidar CE, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for UGT1A1 and Atazanavir Prescribing. Clin Pharmacol Ther. 2016;99(4):363–369. doi: 10.1002/cpt.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vardhanabhuti S, Ribaudo HJ, Landovitz RJ, et al. Screening for UGT1A1 Genotype in Study A5257 Would Have Markedly Reduced Premature Discontinuation of Atazanavir for Hyperbilirubinemia. Open Forum Infect Dis. 2015;2(3):ofv085. doi: 10.1093/ofid/ofv085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89(3):387–391. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scott SA, Sangkuhl K, Stein CM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94(3):317–323. doi: 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360(4):354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- 59.Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther. 2014;95(4):376–382. doi: 10.1038/clpt.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caudle KE, Rettie AE, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clin Pharmacol Ther. 2014;96(5):542–548. doi: 10.1038/clpt.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheung YK, Cheng SH, Chan EJ, Lo SV, Ng MH, Kwan P. HLA-B alleles associated with severe cutaneous reactions to antiepileptic drugs in Han Chinese. Epilepsia. 2013;54(7):1307–1314. doi: 10.1111/epi.12217. [DOI] [PubMed] [Google Scholar]
- 62.Hung CC, Lin CJ, Chen CC, Chang CJ, Liou HH. Dosage recommendation of phenytoin for patients with epilepsy with different CYP2C9/CYP2C19 polymorphisms. Ther Drug Monit. 2004;26(5):534–540. doi: 10.1097/00007691-200410000-00012. [DOI] [PubMed] [Google Scholar]
- 63.van der Weide J, Steijns LS, van Weelden MJ, de Haan K. The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics. 2001;11(4):287–291. doi: 10.1097/00008571-200106000-00002. [DOI] [PubMed] [Google Scholar]
- 64.Hung CC, Huang HC, Gao YH, et al. Effects of polymorphisms in six candidate genes on phenytoin maintenance therapy in Han Chinese patients. Pharmacogenomics. 2012;13(12):1339–1349. doi: 10.2217/pgs.12.117. [DOI] [PubMed] [Google Scholar]
- 65.Ramsey LB, Johnson SG, Caudle KE, et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin Pharmacol Ther. 2014;96(4):423–428. doi: 10.1038/clpt.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Group SC, Link E, Parish S, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359(8):789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 67.Heart Protection Study Collaborative G. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- 68.Voora D, Shah SH, Spasojevic I, et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009;54(17):1609–1616. doi: 10.1016/j.jacc.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Birdwell KA, Decker B, Barbarino JM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin Pharmacol Ther. 2015;98(1):19–24. doi: 10.1002/cpt.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pirmohamed M, Burnside G, Eriksson N, et al. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 2013;369(24):2294–2303. doi: 10.1056/NEJMoa1311386. [DOI] [PubMed] [Google Scholar]
- 71.Do EJ, Lenzini P, Eby CS, et al. Genetics informatics trial (GIFT) of warfarin to prevent deep vein thrombosis (DVT): rationale and study design. Pharmacogenomics J. 2012;12(5):417–424. doi: 10.1038/tpj.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.PharmGKB. Annotation of CPIC Guideline for abacavir and HLA-B. https://www.pharmgkb.org/guideline/PA166104997.
- 73.PharmGKB. Annotation of CPIC Guideline for allopurinol and HLA-B. https://www.pharmgkb.org/guideline/PA166105003.
- 74.PharmGKB. Annotation of CPIC Guideline for atazanavir and UGT1A1. https://www.pharmgkb.org/guideline/PA166128738.
- 75.PharmGKB. Annotation of CPIC Guideline for azathioprine and TPMT. https://www.pharmgkb.org/guideline/PA166104933.
- 76.PharmGKB. Annotation of CPIC Guideline for carbamazepine and HLA-A,HLA-B. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166105008.
- 77.PharmGKB. Annotation of CPIC Guideline for clopidogrel and CYP2C19. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166104948.
- 78.PharmGKB. Annotation of CPIC Guideline for codeine and CYP2D6. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166104996.
- 79.PharmGKB. Annotation of CPIC Guideline for ivacaftor and CFTR. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166114461.
- 80.PharmGKB. Annotation of CPIC Guideline for phenytoin and CYP2C9,HLA-B. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166122806.
- 81.PharmGKB. Annotation of CPIC Guideline for simvastatin and SLCO1B1. [March 11, 2108]; http://www.editorialmanager.com/ajkd/default.aspx.
- 82.PharmGKB. Annotation of CPIC Guideline for tacrolimus and CYP3A5. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166124619.
- 83.PharmGKB. Annotation of CPIC Guideline for warfarin and CYP2C9,CYP4F2,VKORC1. [March 11, 2108]; https://www.pharmgkb.org/guideline/PA166104949.