

Abstract
Metformin has been a frontline therapy for type 2 diabetes (T2D) for many years. Its effectiveness in T2D treatment is mostly attributed to its suppression of hepatic gluconeogenesis; however, the mechanistic aspects of metformin action remain elusive. In addition to its glucose-lowering effect, metformin possesses other pleiotropic health-promoting effects that include reduced cancer risk and tumorigenesis. Metformin inhibits the electron transport chain (ETC) and ATP synthesis; however, recent data reveal that metformin regulates AMP-activated protein kinase (AMPK) and the mechanistic target of rapamycin complex 1 (mTORC1) by multiple, mutually nonexclusive mechanisms that do not necessarily depend on the inhibition of ETC and the cellular ATP level. In this review, we discuss recent advances in elucidating the molecular mechanisms that are relevant for metformin use in cancer treatment.
Keywords: metformin, AMPK, mTORC1, electron transport chain
Metformin in T2D Treatment
The occurrence of type 2 diabetes (T2D) has increased dramatically worldwide and is one of the factors contributing to the development of cardiovascular diseases, neuropathy, retinopathy, and nephropathy [1,2]. Metformin (N,N-dimethylbiguanide) is inexpensive, safe, and the most widely prescribed drug for T2D treatment [1]. Metformin, along with its derivatives phenformin and buformin belong to the biguanide (see Glossary) class of antidiabetic drugs. They were originally derived from galegine, a natural product from the plant Galega officinalis (French lilac), used for centuries in Europe as a herbal medicine. Phenformin and buformin were withdrawn from human therapy due to concerns of lactic acidosis. The incidence of lactic acidosis with metformin at therapeutic doses is rare [1,2]. However, metformin treatment is associated with gastrointestinal side effects in about 20–30% of patients, resulting in discontinuation of metformin treatment in about 5% of patients [3,4]. Metformin function in T2D treatment consists mostly of decreasing glucose production by gluconeogenesis in the liver and, to a lesser extent, increased insulin-mediated glucose uptake in the skeletal muscle [5].
The major molecular targets of metformin include complex I of the mitochondrial electron transport chain (ETC), adenosine monophosphate (AMP)-activated protein kinase (AMPK), and mechanistic target of rapamycin complex 1 (mTORC1; Figure 1, Key Figure). In addition, metformin inhibits the mitochondrial glycerol 3-phosphate dehydrogenase (G3PDH) [6]. G3PDH, an enzyme of the glycerolphosphate shuttle, is a major contributor of electrons to the ETC in the mitochondria. This enzyme is also required for gluconeogenesis from glycerol. However, the significance of metformin inhibition of G3PDH as a mechanism responsible for the therapeutic effect of metformin needs to be determined [7].
Figure 1.
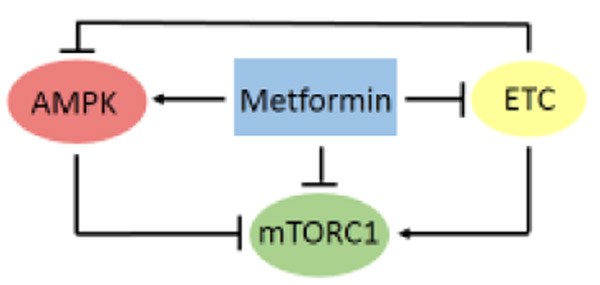
The Major Molecular Targets of Metformin. These are the ETC, AMPK, and mTORC1. ETC produces ATP, leading to AMPK downregulation. Metformin inhibits the ETC, resulting in reduced ATP synthesis. The elevated AMP/ATP ratio activates AMPK, which phosphorylates and inhibits mTORC1. The metformin-mediated inhibition of ATP synthesis also results in inhibition of mTORC1. Metformin also activates AMPK and inhibits mTORC1 by a mechanism that is independent of the ETC. Abbreviations: AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; ETC, electron transport chain; mTORC1, mechanistic target of rapamycin complex 1.
In humans, metformin is administered orally, is not metabolized, and is eliminated through renal excretion. The maximum recommended dose for treatment of T2D is 2.5 g per day (35 mg/kg body weight). The plasma concentration of metformin in patients taking 0.5 g of metformin daily is approximately 5 μM. A single dose of 1.5 g of metformin results in a maximal plasma concentration of 18 μM [8]. Based on experiments in animal models and positron emission tomography (PET) in humans, it is estimated that the metformin concentration in the human liver is about 50–100 μM [9,10]. Following a single oral dose, metformin is partially absorbed by the small intestine and distributed to many tissues; however, the luminal concentration in the gastrointestinal (GI) tract remains high. The plasma concentration peaks at about 3 hours and the mean plasma half-life is approximately 20 hours [8]. Since the portal vein brings blood from the GI tract to the liver, the metformin concentration in the liver and portal vein blood is significantly higher than in the general circulation or other organs [4,5,9].
Due to its hydrophilicity, metformin cannot simply diffuse through cell membranes and is transported inside the cell via uptake transporters. Metformin is a substrate for OCT1 (organic cation transporter 1), an uptake transporter primarily expressed in the hepatocytes [11]. Once inside the cells, metformin accumulates in the mitochondrial matrix, presumably because of its positive charge and the polarization of the mitochondrial inner membrane [12]. Tissues other than liver are more responsive to phenformin, a derivative of metformin, as phenformin is more hydrophobic than metformin, can passively diffuse through cell membranes, and its cellular uptake does not depend on OCT1. Phenformin is frequently considered to be a more potent version of metformin; however, it was banned from T2D treatment due to increased lactic acidosis [1,4,5].
The precise molecular mechanism of metformin action in T2D treatment remains somewhat controversial [5,7,13–16]. The controversy concerns the role of AMPK; there are two actively debated mechanisms of metformin action in T2D treatment: AMPK-dependent and - independent. According to the AMPK-dependent mechanism, metformin at therapeutically achievable concentrations activates AMPK, resulting in phosphorylation of CRTC2 [cAMP-response element-binding protein (CREB)-regulated transcriptional coactivator 2] and CBP (CREB-binding protein), disassembly of CREB coactivator complex, and inhibition of transcription of gluconeogenic genes [15,17,18]. In agreement with this AMPK-dependent mechanism, knockout of liver kinase B1 (LKB1), the upstream kinase that phosphorylates and activates AMPK, abolishes metformin inhibition of hepatic glucose production [18]. Consistent with these findings, low concentrations of metformin, which do not affect the AMP/adenosine triphosphate (ATP) ratio, inhibit glucose production in hepatocytes through an AMPK-dependent mechanism [14]. One argument against this AMPK-dependent mechanism of metformin action is that it does not explain how metformin activates AMPK without inhibiting ETC and altering concentrations of ATP and AMP. The AMPK-dependent mechanism of metformin action in T2D treatment has been challenged over the past decade, and accumulating evidence has indicated that metformin inhibits hepatic gluconeogenesis by an AMPK-independent mechanism. The key study has demonstrated that metformin inhibits glucose production in AMPK- and LKB1-null hepatocytes [19]. Moreover, activation of AMPK by small molecule activators does not lower blood glucose level or glucose production [6,19–22]. Nevertheless, several metformin effects, such as inhibition of lipogenesis and activation of lipid catabolism are attributed to AMPK activation [23–25].
Metformin as an Anticancer Therapeutic
Untreated T2D is associated with an increased cancer risk, attributed mostly to the growth-promoting effect of chronically elevated plasma glucose and insulin levels [1–3]. The risk due to preexisting diabetes is most significant for cancers of the liver, pancreas, endometrium, colon, breast, and bladder [26]. The interest in metformin for cancer prevention and treatment is based on clinical studies that show that the use of metformin is associated with significantly lower cancer incidence in diabetic patients [27–32]. Several clinical trials using metformin as a treatment in non-diabetic cancer patients have produced encouraging results [33–36]. Analysis of clinical trials registered on http://ClinicalTrials.gov in June 2018 has revealed 75 studies that use metformin in cancer treatment. Out of these 75 studies, 13 were completed, and the results of seven studies of metformin in cancer treatment of non-diabetic patients were published or deposited in http://ClinicalTrials.gov. These seven studies were conducted in various disease settings, and included different groups of patients, treatment regiments, and means of assessment; they are summarized in Table 1. Several of these studies found a decrease in the expression of Ki-67, a marker of cell proliferation, after metformin treatment [37,39,40]. By contrast, another study found no effect of metformin on Ki-67 expression [38], and one study found elevated Ki-67 expression after metformin treatment (NCT01433913). Another study found a significantly decreased prostate specific antigen (PSA) after treatment of prostate cancer patients with metformin (NCT01620593). Some of these results are promising; however, large-scale, randomized, double-blind, and placebo-controlled studies are needed to conclusively address efficacy of metformin in different cancers.
Table 1.
Clinical Trials Using Metformin for Treatment of Cancer
| Title | NCT number |
Organ site | Primary Outcome | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| Clinical and biologic effects of metformin in early stage breast cancer |
NCT00897884 | Breast | 2.97% decrease in Ki-67 | [37] | ||||
| Pre-surgical trial of the combination of metformin and atorvastatin in newly diagnosed operable breast cancer |
NCT01980823 | Breast | No reduction in Ki-67 | [38] | ||||
| Preoperative window study of metformin for the treatment of endometrial cancer |
NCT01911247 | Uterus | 11.75% decrease in Ki-67 | [39] | ||||
| A study of pre-operative metformin in prostate cancer |
NCT00881725 | Prostate | 29.5% decrease in Ki-67 | [40] | ||||
| Metformin hydrochloride in preventing esophageal cancer in patients with Barrett esophagus |
NCT01447927 | Esophagus | No significant change in markers of protein synthesis |
[41] | ||||
| Castration compared to castration plus metformin as first line treatment for patients with advanced prostate cancer |
NCT01620593 | Prostate | Significant decrease in PSA (58.2 ng/ml in castration/placebo; 8.4 ng/ml in castration/metformin) |
http://ClinicalTrials.gov | ||||
| Metformin hydrochloride in treating patients with prostate cancer undergoing surgery |
NCT01433913 | Prostate | Elevated Ki-67 in the metformin group; no significant difference in apoptosis between the placebo and metformin groups |
http://ClinicalTrials.gov | ||||
The promising results prompted studies aimed at elucidating the mechanisms of metformin function in the context of cancer prevention and treatment. Over the past several years, these studies have revealed a significantly more complex picture of metformin action and demonstrated that metformin at the cellular level regulates metabolism in a comprehensive way: it inhibits the ETC and ATP production, stimulates catabolism by activating AMPK, and inhibits anabolism by inhibiting mTORC1 (Figure 1) [5]. Due to the central position and interconnections of ETC, AMPK, and mTORC1 in regulation of metabolism at cellular as well as organismal level, the assignment of target(s) that are relevant for T2D and cancer is almost impossible.
Mechanisms of metformin’s antitumor effect can be classified as AMPK- and mTORC1-independent, AMPK-, and mTORC1-dependent. The AMPK- and mTORC1-independent mechanism has been attributed to the decreased glucose and insulin blood levels and decreased production of biosynthetic precursors generated by the tricarboxylic acid (TCA) cycle. In addition, by inhibiting the ETC, metformin reduces production of reactive oxygen species (ROS), oxidative stress, and DNA damage [5]. Metformin also downregulates the expression of transcription factors specificity protein (Sp)1, Sp3, and Sp4 and pro-oncogenic, Sp-regulated genes. Sp1, Sp3, and Sp4 are highly expressed in several cancers, including pancreatic cancer, and are targets for several anticancer drugs [42]. The AMPK-dependent mechanism of metformin is mediated through direct inhibitory phosphorylation of mTORC1 subunits, inhibition of lipid synthesis and NFκB pathway, and increased protein acetylation. The mTORC1-dependent mechanisms are due to metformin-mediated and AMPK-independent inhibition of mTORC1. By targeting AMPK and mTORC1, the two master metabolic regulators with key functions in human physiology and pathophysiology, metformin has the potential to be used in combination therapies for cancer, metabolic diseases, neurodegeneration, and aging-related diseases [43–48]. In addition to effects at the cellular level, metformin has systemic effects that contribute to its potential as an anticancer therapeutic. These systemic effects include inhibition of tumor development by reducing insulin/insulin-like growth factor (IGF)-1 signaling, reducing proinflammatory cytokine levels, reducing expression of cell adhesion molecules, suppressing Warburg effect, and releasing of lactate by tumors [2,5].
Metformin Inhibition of Complex I of the ETC
The only direct target of metformin defined at the molecular level is complex I of the mitochondrial ETC. Molecular modeling of the metformin analog targeted to mitochondria revealed that this metformin analog likely inhibits the flow of electrons from the catalytic site of complex I to the ubiquinone binding site; the same is likely true for metformin [49]. Regardless, the immediate effect of metformin’s action is decrease in oxidation of reduced nicotinamide adenine dinucleotide (NADH) and ATP production. This effect has been demonstrated in a more convincing way in vitro than in vivo [12,19,50]. The main argument against this mechanism is that the inhibition of complex I of the ETC in vitro requires relatively high metformin concentrations, which may not be achievable in the serum of human patients [13]. However, this apparent controversy can be explained. The concentration of metformin in the mitochondrial matrix of certain tissues, such as liver, is significantly higher than the concentrations found in the serum due to the positive charge of metformin and the polarization of the mitochondrial inner membrane [12,51,52]. When the concentration of metformin in the serum is relatively low, the inhibition of the ETC in the liver cells is delayed due to the extra time required to achieve the effective concentration of metformin in mitochondria [12,52]. The importance of accumulation of metformin in mitochondria for inhibition of complex I of the ETC is underscored by the markedly increased effectiveness of mitochondrially targeted metformin. Targeting of metformin to mitochondria is accomplished by the tagging of metformin to lipophilic cationic groups, which increase hydrophobicity and the positive charge of metformin [53]. In addition to increased ETC inhibition, the mitochondrially targeted metformin is very effective in killing pancreatic cancer cells [49,54]. It is also known that metformin at high concentrations, while inhibiting the ETC, also activates AMPK and inhibits mTORC1 through both AMPK-dependent and -independent mechanisms (see below sections ‘Metformin Inhibition of mTORC1 by AMPK-Dependent Mechanism’ and ‘Metformin Inhibition of mTORC1 by AMPK-Independent Mechanism’). Moreover, high concentrations of metformin (250 μM and higher) lead to an increase of AMP/ATP ratio, which directly suppresses gluconeogenesis, even in the absence of AMPK [19].
Inhibition of ETC results in an inability to oxidize NADH to nicotinamide adenine dinucleotide (NAD+) and downregulation of the TC A cycle due to NAD+ depletion. In agreement with the inhibition of the ETC and TCA cycles, metformin decreases glucose oxidation and increases dependency on reductive glutamine metabolism [55–57]. Increasing glutamine metabolism rescues the proliferative defect induced by metformin, and vice versa; inhibition of glutamine metabolism synergizes with metformin [56,58]. Additionally, inhibition of ETC results in activation of glycolysis as an alternative pathway for ATP production and increased production of lactate to allow recycling of NADH into NAD+. Metformin decreases cellular levels of all TCA cycle intermediates by inhibiting the oxidation of NADH to NAD+ and dramatically inhibits synthesis of ribonucleotide and deoxyribonucleotide triphosphates [59]. Metformin mediated inhibition of ETC results in an elevated NADH/NAD+ ratio due to the inability to use oxygen as the terminal acceptor of electrons. However, providing an alternative electron acceptor, such as pyruvate, allows cells that do not have functional ETC to proliferate in culture [60,61].
In normal cells with functional ETC, the malate-aspartate shuttle delivers cytosolic NADH into mitochondria as a source of electrons for ETC. The shuttle involves export of aspartate from mitochondria to the cytosol, transamination of aspartate with α-ketoglutarate to yield oxaloacetate, and reduction of oxaloacetate to malate using the cytosolic NADH (Figure 2A). Malate is subsequently transported into mitochondria and oxidized into oxaloacetate, regenerating the NADH in the mitochondrial matrix. Oxaloacetate is then transaminated with α-ketoglutarate to yield aspartate by the mitochondrial aspartate aminotransferase (mAST). Aspartate is then exported from mitochondria to the cytosol. When the ETC is inhibited with metformin, mitochondrial NAD+ is depleted while NADH accumulates, and the malate-aspartate shuttle appears to run in the opposite direction, exporting reducing equivalents in the form of malate from the mitochondrial matrix to the cytosol (Figure 2B). Malate is converted in the cytosol into oxaloacetate, which is then transaminated into aspartate by the cytosolic aspartate aminotransferase (cAST). Thus, when ETC is inhibited, cells depend on cAST for synthesis of aspartate, as supplementation of aspartate allows cells with ETC defect to proliferate [62,63]. Since aspartate is required for protein synthesis and is a precursor for purine and pyrimidine synthesis [64], it would be expected that aspartate and nucleotides would be limiting to cells treated with metformin, phenformin, or other ETC inhibitors. This hypothesis was proven only partially correct under in vivo conditions. A comparison of metabolomics analyses of tumor tissues removed from ovarian cancer patients treated with therapeutic doses of metformin and not treated with metformin revealed depletion of TCA cycle intermediates and accumulation of NADH; however, aspartate was not depleted [65]. This can perhaps be explained by nutrient limitation experienced by cells in the tumor environment [65]. As the metformin concentration in the tumor tissues was in the low micromolar range, these results also indicate that the micromolar concentration of metformin in tissues of patients treated with metformin is sufficient to affect the ETC and support the notion that complex I of the ETC is the target of metformin in non-hepatic tissues also [65].
Figure 2.
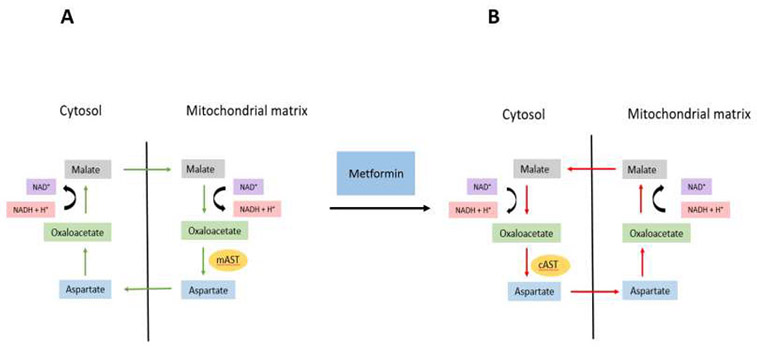
Inhibition of the ETC Reverses the Direction of the Malate-Aspartate Shuttle. (A) In the absence of metformin, the malate-aspartate shuttle removes NADH from cytosol and regenerates it in mitochondria to be used as a substrate for the ETC. Since NADH cannot be transported across the mitochondrial inner membrane, the reducing equivalents are transported from the cytosol to the mitochondria in the form of malate (indicated by green arrows). Under these conditions, aspartate is produced in mitochondria by the mitochondrial aspartate aminotransferase mAST. (B) When the ETC is inhibited by metformin or phenformin, mitochondrial NADH accumulates and malate is transported in the opposite direction from mitochondria to cytosol (indicated by red arrows). Under these conditions, cytosolic aspartate aminotransferase cAST is required for synthesis of aspartate and cell proliferation. Abbreviations: cAST, cytosolic aspartate aminotransferase; ETC, electron transport chain; mAST, mitochondrial aspartate aminotransferase; NADH, nicotinamide adenine dinucleotide.
Metformin Activation of AMPK
AMPK is an energy sensor and master regulator of metabolism, functioning as a fuel gauge and monitoring systemic and cellular energy status [66–68]. Activation of AMPK occurs when the intracellular AMP/ATP ratio increases. In general, AMPK inhibits anabolism to minimize energy consumption and activates catabolism to increase ATP production.
AMPK targets a number of metabolic enzymes and transporters, such as glucose transporter (GLUT)1 and GLUT4, glycogen synthase (GS), acetyl-CoA carboxylase (ACC), and hydroxymethylglutaryl-CoA reductase (HMGCR). AMPK also regulates metabolism at the transcriptional level by phosphorylating sterol regulatory element-binding protein 1 (SREBP1), carbohydrate-responsive element-binding protein (ChREBP), transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), and transcription factor forkhead box O3 (FOXO3) [66,67].
The antitumor effects of metformin-activated AMPK are at least partially independent of the role of AMPK in regulation of gluconeogenesis. AMPK activity is important for proper control of cell proliferation; loss of LKB1 occurs frequently in cancer and germline mutation in LKB1 is responsible for Peutz-Jeghers syndrome, a cancer-predisposing condition [69]. In addition, AMPK phosphorylates p53, but the role of metformin in p53 activation is not clear [70].
Activation of AMPK inhibits fatty acid synthesis and tumor growth, since rapid growth of cancer cells requires an increased rate of fatty acid synthesis to accommodate assembly of cellular membranes [71–74]. One of the corresponding mechanisms is inhibitory phosphorylation of ACC by AMPK. The nucleocytosolic acetyl-CoA, produced from citrate by citrate lyase, is a critical precursor for de novo synthesis of fatty acids. ACC catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, the first and rate-limiting reaction in the de novo synthesis of fatty acids. AMPK also phosphorylates and inhibits HMG-CoA reductase, which catalyzes the rate-limiting step in cholesterol synthesis [75].
In addition to inhibition of lipid synthesis, AMPK activation with metformin increases acetylation of histone and non-histone proteins. Histone acetylation requires acetyl-CoA in the nucleocytosolic compartment as a substrate for histone acetyltransferases. As detailed above, activation of AMPK with metformin decreases conversion of acetyl-CoA to malonyl-CoA, resulting in an increased pool of nucleocytosolic acetyl-CoA, increased acetylation of histone and non-histone proteins, and altered gene expression [76,77]. Surprisingly, this is not the only epigenetic effect of metformin; metformin also affects DNA and histone methylation [78,79]. Finally, regulation of mTORC1 by AMPK (discussed below) represents perhaps the most important antineoplastic effect of metformin-activated AMPK, since mTORC1 inhibition downregulates protein synthesis and cell proliferation.
Metformin Inhibition of mTORC1 by AMPK-Dependent Mechanism
mTOR is a conserved serine/threonine protein kinase from the phosphatidylinositol-3-kinase (PI3K) family, which was discovered as a target of rapamycin [80–83]. mTOR is found in all eukaryotes and forms the catalytic subunit of mTORC1. mTORC1 is regulated by nutrients and growth factors, and functions as a master regulator of cell growth and metabolism by phosphorylating hosts of targets. [44,84]. Since mTORC1 functions downstream of many oncogenic pathways that are frequently mutated in cancer, many tumors display elevated activity of mTORC1 [44].
The AMPK-dependent mechanisms of mTORC1 inhibition are mediated by phosphorylation of tuberous sclerosis complex (TSC) and Raptor subunit of mTORC1 [85,86] (Figure 3). TSC functions as a GTPase activating protein (GAP) for the small GTPase Ras homolog enriched in brain (Rheb), which directly binds and activates mTORC1 [87–89]. Thus, by downregulating Rheb, TSC inhibits mTORC1 and downregulation of TSC, which therefore leads to activation of mTORC1. In addition to integrating signals from several growth factor pathways, TSC is also regulated by AMPK. When AMPK is activated by nutrient loss or by metformin [90], AMPK directly phosphorylates TSC2 on serine residues that are distinct from those regulated by growth factor pathways, resulting in TSC activation and mTORC1 inhibition. In addition to TSC2, AMPK also phosphorylates mTORC1 subunit Raptor, leading again to mTORC1 inhibition [86].
Figure 3.
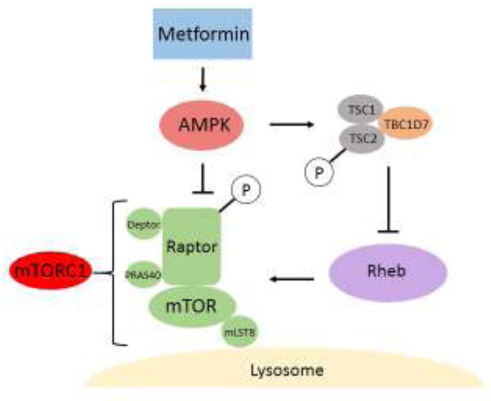
Metformin Inhibits mTORC1 in an AMPK-Dependent Manner. Metformin-activated AMPK downregulates mTORC1 signaling by at least two mechanisms. First, AMPK inhibits mTORC1 by phosphorylating the raptor subunit of mTORC1. Second, AMPK activates the tuberous sclerosis complex (TSC), composed of TSC1, TSC2, and TBC1D7, by phosphorylating the TSC2 subunit. This, in turn, inhibits Rheb. Abbreviations: AMPK, AMP-activated protein kinase; mTORC1, mechanistic target of rapamycin complex 1; Rheb, Ras homolog enriched in the brain.
Reciprocal Regulation of AMPK and mTORC1 by Metformin in the Lysosomal Pathway
Amino acids activate mTORC1 through the Ras-related guanosine triphosphatase (GTPase) complex (Rag), which is recruited to the lysosomal membrane by interacting with the Ragulator complex [91–94] (Figure 4). When stimulated by amino acids, the Ragulator complex acts as a guanine nucleotide exchange factor (GEF) for Rags and converts them to their active nucleotide-bound state. In this active state, RagA or RagB have bound guanosine triphosphate (GTP) and RagC or RagD have bound guanosine diphosphate (GDP), and the Rag heterodimer recruits mTORC1 to the lysosomal membrane by binding through Raptor [95,96]. The activation of Ragulator by lysosomal amino acids requires lysosomal v-ATPase, which interacts with Ragulator and stimulates the GEF activity of Ragulator and activation of Rags. For full activity, however, mTORC1 requires Rheb, also located at the lysosomal membrane (Figure 4). Thus, mTORC1 is active only when the Rags are activated by amino acids and Rheb is activated by growth factors [44].
Figure 4.
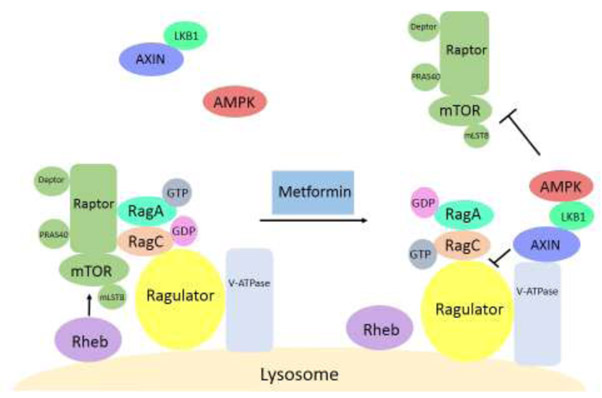
Reciprocal Regulation of AMPK and mTORC1 by Metformin in the Lysosomal Pathway. mTORC1 resides at the lysosomal membrane through interacting with the Rag heterodimer (RagA and RagC) and is activated by amino acids that are sensed by the Rags, Ragulator, and v-ATPase. mTORC1 is also activated by growth factors through the GTPase Rheb. Metformin induces recruitment of AMPK to the lysosomal membrane and promotes formation of a complex consisting of Ragulator, v-ATPase, AXIN, LKB1, and AMPK. AXIN inhibits the GEF activity of Ragulator towards Rags, leading to mTORC1 dissociation from the Ragulator and lysosome. The overall effect of metformin is activation of AMPK and inhibition of mTORC1 at the lysosome. Abbreviations: AMPK, AMP-activated protein kinase; GEF, guanine nucleotide exchange factor; GTPase, guanosine triphosphatase; mTORC1, mechanistic target of rapamycin complex 1; Rag, Ras-related GTPase complex.
Metformin at therapeutically achievable concentrations promotes the formation of a complex containing v-ATPase, Ragulator, scaffold protein AXIN, LKB1, and AMPK on the lysosome surface, resulting in AMPK activation. At the same time, the GEF activity of Ragulator towards Rags is inhibited by AXIN, resulting in inhibition of mTORC1 and its dissociation from Ragulator and lysosome [97–99]. Ragulator is thus a key component of a molecular switch between inversely regulated AMPK and mTORC1 pathways and between catabolism and anabolism. The ability to operate the switch between AMPK and mTORC1 at the Ragulator endows metformin with the ability to regulate both pathways at the same time. (Figure 4).
Metformin Inhibition of mTORC1 by AMPK-Independent Mechanism
Metformin also inhibits mTORC1 independently of AMPK in a Rag GTPase-dependent manner [100]. It appears that the activation of RagC requires that RagC enters the nucleus, where it binds GDP. RagC, with bound GDP, then exits the nucleus and together with RagA or RagB bound to GTP activates mTORC1. Administration of metformin results in functional alteration of the nuclear pore complex (NPC), which leads to nuclear exclusion of RagC (Figure 5). Consistent with this mechanism, a majority of the GAP activity for RagC resides in the nucleus [101] and folliculin, the GAP for RagC, is localized to the nucleus [102]. The NPC thus emerged as another target of metformin that responds to ETC inhibition and reduced ATP level.
Figure 5.
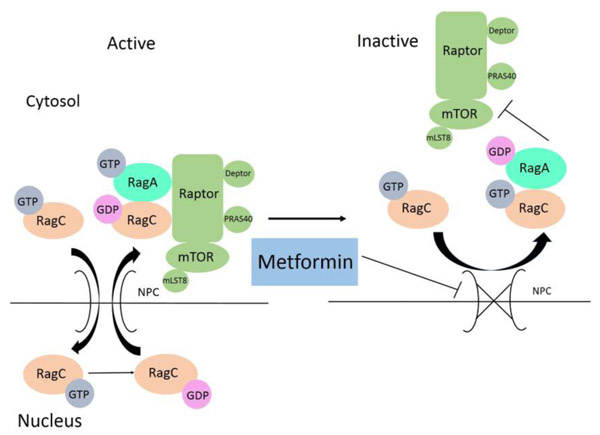
Metformin Inhibits mTORC1 by Preventing RagC Activation. Rag heterodimer activates mTORC1 when RagA (or RagB) binds GTP and RagC (or RagD) binds GDP. In order to be activated, RagC needs to enter the nucleus, where it acquires GDP and becomes competent for mTORC1 activation. When the cellular level of ATP is reduced due to metformin inhibition of the ETC, function of the nuclear pore complex is altered, resulting in nuclear exclusion of RagC, failure of RagC to bind GDP, and, consequently, mTORC1 inhibition. Abbreviations: ATP, adenosine triphosphate; ETC, electron transport chain; GDP, guanosine diphosphate; GTP, guanosine triphosphate; mTORC1, mechanistic target of rapamycin complex 1; Rag, Ras-related GTPase complex.
Concluding Remarks
By targeting AMPK and mTORC1 simultaneously, metformin shifts metabolism from anabolism towards catabolism; this may underlie its pleiotropic health-promoting effects. The long-term effects of metformin treatment resemble the effects of dietary restriction, possibly leading to reduced cancer incidence and extended lifespan (see Outstanding Questions). Given the critical functions of AMPK and mTORC1 in human physiology and pathophysiology and the outstanding safety profile of metformin, the potential therapeutic uses of metformin as a single therapy or in combination with other drugs that target mTORC1, AMPK, and other pathways that regulate metabolism and growth are very significant. Possible partner drugs for metformin may include mTOR inhibitors. Rapalogs, the first generation of mTOR inhibitors, are used by recipients of organ transplants and cancer patients and include rapamycin (known in the clinic as Sirolimus or Rapamune) and rapamycin derivatives temsirolimus and everolimus. Temsirolimus, introduced in 2007 by Pfizer, and everolimus, introduced in 2009 by Novartis, are approved for treatment of renal cell carcinoma. Since rapalogs are allosteric inhibitors, they inhibit phosphorylation of only a subset of mTORC1 substrates and have only limited efficacy in cancer treatment [103]. To overcome these limitations, second and third generations of mTOR inhibitors have been developed and are in clinical trials. The second generation of mTOR inhibitors are ATP-competitive inhibitors that suppress mTORC1 catalytic activity and appear to be more effective than rapalogs in preclinical cancer models. The third generation of mTOR inhibitors is represented by recently described 'RapaLink', in which rapamycin is linked to the ATP-competitive inhibitor [104].
Outstanding Questions.
Does the predominant mechanism of AMPK activation by metformin depend on ETC inhibition and elevated AMP/ATP ratio, or does metformin activate AMPK independently of the intracellular AMP/ATP ratio?
What is the predominant mechanism of mTORC1 inhibition by metformin? Does the mechanism depend on (i) AMPK, (ii) nuclear exclusion of RagC, or (iii) dissociation from the lysosomal membrane?
Is the mechanism of AMPK activation and mTORC1 inhibition by metformin specific for different cell types and tissues? Is it affected by different physiological conditions?
What is the metformin concentration in biopsy samples from cancer patients? Can analyses of these biopsies, including metabolomic, transcriptomic, and proteomic approaches reveal the predominant molecular target of metformin?
Can metformin be used in combination with mTORC1 inhibitors as cancer therapy?
Does metformin use in non-diabetic patients lower cancer incidence? If it does, what is the responsible mechanism?
Future studies should explore the utility of combination therapies involving metformin in treatment of cancer, metabolic diseases, neurodegeneration, and age-related diseases. Development of future therapeutic strategies will also depend on the progress in elucidating whether metformin activates AMPK and inhibits mTORC1 through the cellular ATP level or independently of the cellular energy status and whether both mechanisms coexist in different cell types. These efforts will be facilitated by the development of novel, more efficient, and specific analogues of metformin, including mitochondrially-targeted versions.
Highlights.
Metformin use is associated with lower cancer incidence in diabetic patients and is considered for cancer therapy of non-diabetic patients.
Metformin inhibits ETC and elevates AMP/ATP ratio, resulting in AMPK activation and mTORC1 inhibition.
In addition, at metformin concentrations that do not affect the ETC and AMP/ATP ratio, administration of metformin results in AMPK activation and mTORC1 inhibition by mechanisms not involving ETC.
Metformin-induced inhibition of ATP synthesis affects the nuclear pore complex, which leads to nuclear exclusion of RagC. Without being able to pass through the nucleus, RagC cannot be potentiated for mTORC1 activation.
Acknowledgments
The work in the Vancura lab is supported by NIH GM120710 grant.
Glossary
- Adenosine monophosphate (AMP)-activated protein kinase (AMPK)
a heterotrimeric serine/threonine protein kinase activated by a low cellular energy charge (high AMP/ATP ratio) that activates catabolism and inactivates anabolism.
- Anabolism
biosynthetic pathways that require energy to build more complex molecules from simpler precursors.
- Biguanide
an organic molecule. Several derivatives of biguanide, including metformin, are used as oral antihyperglycemic drugs for the treatment of type 2 diabetes.
- Catabolism
metabolic pathways that break down more complex molecules to produce energy and small molecules for biosynthetic reactions.
- Electron transport chain (ETC)
a series of proteins in the mitochondrial inner membrane, involved in transducing electrons from NADH to molecular oxygen.
- Gluconeogenesis
a metabolic pathway for glucose synthesis from non-carbohydrate carbon sources.
- Guanosine triphosphatases (GTPases)
enzymes that bind and hydrolyze guanosine triphosphate (GTP); they play important roles in many different signaling pathways.
- Insulin-like growth factor (IGF-1)
a hormone similar to insulin with anabolic effects.
- Lactic acidosis
increased concentration of lactate in the body, resulting in acidification of tissues; biguanides may cause lactic acidosis by promoting reduction of pyruvate into lactate.
- Lipogenesis
synthesis of fatty acids from acetyl-CoA and triacylglycerols from fatty acids.
- Mechanistic target of rapamycin complex 1 (mTORC1)
a serine/threonine protein kinase in the PI3K-related kinase family; mTORC1 is regulated by nutrients and growth factors and is a master regulator of cell growth and metabolism; mTORC1 is composed of catalytic subunit mTOR, Raptor, mammalian lethal with SEC13 protein 8 (mLST8), PRAS40, and DEP domain-containing mTOR-interacting protein (DEPTOR).
- Nicotinamide adenine dinucleotide (NAD+)
oxidized form of NADH; carrier of electrons in redox reactions.
- Prostate specific antigen (PSA)
a protein secreted by the epithelial cells of the prostate gland. The serum level of PSA is elevated in prostate cancer.
- Rag GTPase (Rag)
heterodimeric GTPases (RagA or RagB binds RagC or RagD) tethered to the lysosomal membrane by binding to the Ragulator complex. When Rags are activated by amino acids, they bind Raptor and recruit mTORC1 to the lysosomal surface.
- Ragulator
heteropentameric complex at the lysosomal surface that interacts with Rags and facilitates recruitment of mTORC1 to the lysosomal membrane.
- Raptor
one of the core components of mTORC1 that facilitates substrate recruitment to mTORC1 and is required for its correct subcellular localization.
- Ras homolog enriched in brain (Rheb)
small GTPase that directly binds and activates mTORC1. At least some portion of cellular Rheb localizes to the lysosomal surface.
- Reduced nicotinamide adenine dinucleotide (NADH)
carrier of reducing equivalents in metabolism and a substrate for ETC.
- Tricarboxylic acid (TCA) cycle
a key cyclic metabolic pathway that produces NADH molecules for ETC.
- Tuberous sclerosis complex (TSC)
heterotrimeric complex composed of TSC1, TSC2, and TBC1D7 that functions as GAP for Rheb. Several growth factor pathways regulate TSC by stimulating TSC2 phosphorylation, which in turn inhibits TSC by dissociating it from the lysosome.
- Warburg effect
metabolic adaptation of many types of cancer cells characterized by a high rate of glycolysis followed by lactic acid fermentation even in the presence of oxygen.
Footnotes
Disclaimer Statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Viollet B et al. (2012) Cellular and molecular mechanisms of metformin: an overview. Clin. Sci. (Lond) 122, 253–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pryor R and Cabreiro F (2015) Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem. J 471, 307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirpichnikov D et al. (2002) Metformin: an update. Ann. Intern. Med 137, 25–33 [DOI] [PubMed] [Google Scholar]
- 4.Rena G et al. (2017) The mechanism of action of metformin. Diabetologia 60, 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pernicova I and Korbonits M (2014) Metformin-mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol 10, 143–156 [DOI] [PubMed] [Google Scholar]
- 6.Madiraju AK et al. (2014) Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baur JA and Birnbaum MJ (2014) Control of gluconeogenesis by metformin: does redox trump energy charge? Cell Metab 20, 197–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker GT et al. (1981) Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br. J. Clin. Pharmac 12, 235–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilcock C and Bailey CJ (1994) Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 24, 49–57 [DOI] [PubMed] [Google Scholar]
- 10.Gormsen LC et al. (2016) In vivo imaging of human 11C-metformin in peripheral organs: dosimetry, biodistribution, and kinetic analyses. J. Nucl. Med 57, 1920–1926 [DOI] [PubMed] [Google Scholar]
- 11.Shu Y et al. (2007) Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest 117, 1422–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Owen MR et al. (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J 348, 607–614 [PMC free article] [PubMed] [Google Scholar]
- 13.He L and Wondisford FE (2015) Metformin action: concentrations matter. Cell Metab 21, 159–162 [DOI] [PubMed] [Google Scholar]
- 14.Cao J et al. (2014) Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem 289, 20435–20446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He L et al. (2009) Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 137, 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller RA et al. (2013) Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou G et al. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw RJ et al. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foretz M et al. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller RA et al. (2013) Biguanides suppress hepatic glucagon signaling by decreasing production of cyclic AMP. Nature 494, 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasenour CM et al. (2014) 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J. Biol. Chem 289, 5950–5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cokorinos EC et al. (2017) Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab 25, 1147–1159 [DOI] [PubMed] [Google Scholar]
- 23.Fullerton MD et al. (2013) Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature Medicine 19, 1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ford RJ et al. (2015) Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem. J 468, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boudaba N et al. (2018) AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28, 194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giovannucci E et al. (2010) Diabetes and cancer: a consensus report. CA Cancer J. Clin 60, 207–221. [DOI] [PubMed] [Google Scholar]
- 27.Evans JM et al. (2005) Metformin and reduced risk of cancer in diabetic patients. Br. Med. J 330, 1304–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emami RA et al. (2013) Metformin and cancer: from the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci 34, 126–135 [DOI] [PubMed] [Google Scholar]
- 29.Prasad S et al. (2016) Serendipity in cancer drug discovery: rational or coincidence? Trends Pharmacol. Sci 37, 435–450 [DOI] [PubMed] [Google Scholar]
- 30.Zingales V et al. (2017) Metformin: a bridge between diabetes and prostate cancer. Front. Oncol 7, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou XL et al. (2017) Association between metformin and the risk of gastric cancer in patients with type 2 diabetes mellitus: a meta-analysis of cohort studies. Oncotarget 8, 55622–55631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell JM et al. (2017) Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res. Rev 40, 31–44 [DOI] [PubMed] [Google Scholar]
- 33.Hosono K et al. (2010) Metformin suppresses colorectal aberrant crypt foci in a shortterm clinical trial. Cancer Prev. Res. (Phila.) 3, 1077–1083 [DOI] [PubMed] [Google Scholar]
- 34.Hadad S et al. (2011) Evidence for biological effects of metformin in operable breast cancer: a preoperative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat 128, 783–794 [DOI] [PubMed] [Google Scholar]
- 35.Camacho L et al. (2015) Metformin in breast cancer – an evolving mystery. Breast Cancer Res 17, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higurashi T et al. (2016) Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicenter double-blind, placebo-controlled, randomized phase 3 trial. Lancet Oncol 17, 475–483 [DOI] [PubMed] [Google Scholar]
- 37.Niraula S et al. (2012) Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat 135, 821–830 [DOI] [PubMed] [Google Scholar]
- 38.Kalinsky K et al. (2014) Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Investig. 32, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuler KM et al. (2015) Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med 4, 161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshua AM et al. (2014) A pilot (“window of opportunity”) neoadjuvant study of metformin in localized prostate cancer. Prostate Cancer Prostatic Dis 17, 252–258 [DOI] [PubMed] [Google Scholar]
- 41.Chak A et al. (2015) Metformin does not reduce markers of cell proliferation in esophageal tissues of patients with Barrett’s esophagus. Clin. Gastroenterol. Hepatol 13, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Safe S et al. (2018) Metformin-induced anticancer activities: recent insights. Biol. Chem 399, 321–335 [DOI] [PubMed] [Google Scholar]
- 43.Lamming DW et al. (2013) Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Invest 123, 980–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxton RA and Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barzilai N et al. (2016) Metformin as a tool to target aging. Cell Metab 23, 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben-Sahra I and Manning BD (2017) mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol 45, 72–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Day EA et al. (2017) AMPK as a therapeutic target for treating metabolic diseases. Trends Endocrinol. Metab 28, 545–560 [DOI] [PubMed] [Google Scholar]
- 48.Blagosklonny MV (2017) From rapalogs to anti-aging formula. Oncotarget 8, 35492–35507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boukalova S et al. (2016) Mitochondrial targeting of metformin enhances its activity against pancreatic cancer. Mol. Cancer Ther 15, 2875–2886 [DOI] [PubMed] [Google Scholar]
- 50.El-Mir MY et al. (2000) Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem 275, 223–228 [DOI] [PubMed] [Google Scholar]
- 51.Foretz M et al. (2014) Metformin: from mechanisms to action to therapies. Cell Metab 20, 953–966 [DOI] [PubMed] [Google Scholar]
- 52.Bridges HR et al. (2014) Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J 462, 475–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy MP et al. (2007) Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol 47, 629–656 [DOI] [PubMed] [Google Scholar]
- 54.Cheng G et al. (2016) Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res 76, 3904–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wise DR and Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci 35, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fendt SM et al. (2013) Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res 73, 4429–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vander Heiden MG and DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JH et al. (2018) Effects of metformin on colorectal cancer stem cells depends on alterations in glutamine metabolism. Sci. Rep 8, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janzer A et al. (2014) Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 111, 10574–10579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris M (1980) Pyruvate blocks expression of sensitivity to antimycin A and chloramphenicol. Somatic Cell Genet 5, 833–845 [DOI] [PubMed] [Google Scholar]
- 61.King MP and Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503 [DOI] [PubMed] [Google Scholar]
- 62.Birsoy K et al. (2015) An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sullivan LB et al. (2015) Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lane AN and Fan TW (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43, 2466–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X et al. (2016) Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab 24, 728–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herzig S and Shaw R (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol 19, 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hardie DG (2018) Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 15, 20170774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kjobsted R et al. (2018) AMPK in skeletal muscle function and metabolism. FASEB J 32, 1741–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korsse SE et al. (2013) Targeting LKB1 signaling in cancer. Biochim. Biophys. Acta 1835, 194–210 [DOI] [PubMed] [Google Scholar]
- 70.Liang J et al. (2007) The energy-sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol 9, 218–224 [DOI] [PubMed] [Google Scholar]
- 71.Zadra G et al. (2010) New strategies in prostate cancer: targeting lipogenic pathways and the energy sensor AMPK. Clin. Cancer Res 16, 3322–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flavin R et al. (2010) Metabolic alterations and targeted therapies in prostate cancer. J. Pathol 223, 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li W et al. (2015) Targeting AMPK for cancer prevention and treatment. Oncotarget 6, 7365–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ikhlas S et al. (2017) Metformin: insights into its anticancer potential with special reference to AMPK dependent and independent pathways. Life Sci 185, 53–62 [DOI] [PubMed] [Google Scholar]
- 75.Carling D et al. (1987) A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 223, 217–222 [DOI] [PubMed] [Google Scholar]
- 76.Galdieri L et al. (2016) Activation of AMP-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J. Biol. Chem 291, 25154–25166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vancura A and Vancurova I (2017) Metformin induces protein acetylation in cancer cells. Oncotarget 8, 39939–39940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cuyas E et al. (2018) Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell e12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cuyas E et al. (2018) Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 37, 963–970 [DOI] [PubMed] [Google Scholar]
- 80.Brown EJ et al. (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758 [DOI] [PubMed] [Google Scholar]
- 81.Sabatini DM et al. (1994) RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78, 35–43 [DOI] [PubMed] [Google Scholar]
- 82.Sabers CJ et al. (1995) Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem 270, 815–822 [DOI] [PubMed] [Google Scholar]
- 83.Vezina C et al. (1975) Rapamycin (AY-22,989), a new antifungal antibiotic. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot 28, 721–726 [DOI] [PubMed] [Google Scholar]
- 84.Duvel K et al. (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Inoki K et al. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 86.Gwinn DM et al. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dibble CC et al. (2012) TBCD7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Long X et al. (2005) Rheb binds and regulates the mTOR kinase. Curr. Biol 15, 702–713 [DOI] [PubMed] [Google Scholar]
- 89.Sancak Y et al. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 90.Dowling RJ et al. (2007) Metformin inhibits mammalian target of rapamycin-dependent translation in breast cancer cells. Cancer Res 67, 10804–10812 [DOI] [PubMed] [Google Scholar]
- 91.Menon S et al. (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sancak Y et al. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bar-Peled L et al. (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bar-Peled L et al. (2013) A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim E et al. (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol 10, 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sancak Y et al. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang YL et al. (2013) AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab 18, 546–555 [DOI] [PubMed] [Google Scholar]
- 98.Zhang YL et al. (2014) The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab 20, 526–540 [DOI] [PubMed] [Google Scholar]
- 99.Zhang YL et al. (2016) Metformin activates AMPK through the lysosomal pathway. Cell Metab 24, 521–522 [DOI] [PubMed] [Google Scholar]
- 100.Kalender A et al. (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 11, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu L et al. (2016) An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell 167, 1705–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsun ZY et al. (2013) The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 52, 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tabarnero J et al. (2008) Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: A phase I tumor pharmacodynamics study in patients with advanced solid tumors. J. Clin Oncol 26, 1603–1610 [DOI] [PubMed] [Google Scholar]
- 104.Rodrik-Outmezguine VS et al. (2016) Overcoming mTOR resistance mutations with a new generation mTOR inhibitor. Nature 534, 272–276 [DOI] [PMC free article] [PubMed] [Google Scholar]