

Abstract
Kidney donors face a small but definite risk of end-stage renal disease 15-30 years postdonation. The development of proteinuria, hypertension with gradual decrease in kidney function in the donor after surgical resection of 1 kidney has been attributed to hyperfiltration. Genetic variations, physiological adaptations, and co-morbidities exacerbate the hyperfiltration-induced loss of kidney function in the years following donation. A focus on glomerular hemodynamics and capillary pressure has led to the development of drugs that target the renin-angiotensin-aldosterone system (RAAS), but these agents yield mixed results in transplant recipients and donors. Recent work on glomerular biomechanical forces highlights the differential effects of tensile stress and fluid flow shear stress (FFSS) from hyperfiltration. Capillary wall stretch due to glomerular capillary pressure increases tensile stress on podocyte foot processes that cover the capillary. In parallel, increased flow of the ultrafiltrate due to single nephron glomerular filtration rate elevates FFSS on the podocyte cell body. While tensile stress invokes the RAAS, FFSS predominantly activates the COX2-PGE2-EP2 axis. Distinguishing these 2 mechanisms is critical, as current therapeutic approaches focus on the RAAS system. A better understanding of the biomechanical forces can lead to novel therapeutic agents to target FFSS through the COX2-PGE2-EP2 axis in hyperfiltration-mediated injury. We present an overview of several aspects of the risk to transplant donors and discuss the relevance of FFSS in podocyte injury, loss of glomerular barrier function leading to albuminuria and gradual loss of renal function, and potential therapeutic strategies to mitigate hyperfiltration-mediated injury to the remaining kidney.
Introduction
Living donor kidney transplantation compared to deceased donor transplantation has better short- and long-term outcomes in the recipient. Improved outcome is corollary to better kidney quality, shorter cold ischemia time, planned surgery, and shorter time on dialysis. In addition, limited availability of deceased donor organs and the ever-increasing need for kidney allografts for patients on dialysis have led transplant centers to encourage living donation. Since outcomes for recipients of kidneys from living donors are highly encouraging, living donor kidney transplantation is the treatment of choice. However, living donation carries a small but distinct risk to the kidney donor. Several factors may contribute to the risk of kidney disease in donors.
Some kidney donors will develop proteinuria, hypertension, decreased eGFR and End Stage Renal Disease (ESRD) from injury to the remaining kidney [1-7]. This injury is mainly attributed to hyperfiltration. Glomerular hyperfiltration in the remaining functional nephrons not only results from but also contributes to ongoing and repetitive glomerular injury. Hyperfiltration is recognized as a phenomenon of adaptive changes under both physiological and pathological conditions in the kidney. It involves changes in renal hemodynamics, glomerular hypertrophy, afferent arteriolar vasodilation, and/or efferent arteriolar constriction [8,9]. However, the exact mechanism by which hyperfiltration-induced injury is mediated remains unclear. Hyperfiltration is considered to be one of the underlying key mechanisms of renal damage in obesity, diabetes, and hypertension. These factors further exacerbate the deleterious effects of adaptive hyperfiltration on the structure-function integrity of the glomerulus. Development of 1 or more of these co-morbidities in kidney donors further aggravates the hyperfiltration-mediated injury. Affecting not only transplant donors, hyperfiltration also contributes to negative long-term outcomes in transplant recipients who are subjected to further injury through immunological and nonimmunological mechanisms.
Despite decades of familiarity with the concept of hyperfiltration, the mechanical and rheological aspects of hyperfiltration and their effects on glomerular structures such as podocytes remain unclear. The cellular and biological mechanism of hyperfiltration-mediated glomerular injury needs to be further investigated. In the following sections, we highlight the significance of hyperfiltration in living kidney donors and transplant recipients. We summarize hyperfiltration-induced morphological, hemodynamic and molecular changes observed in unilaterally nephrectomized rodent models. We also outline the results of recent in vitro and in vivo experiments to highlight the significance of hyperfiltration-associated biomechanical forces for a better understanding of the mechanism of hyperfiltration-mediated injury in donors.
Glomerular hemodynamic changes in a single kidney
Glomerular filtration is a complex interplay of renal blood flow, ultrafiltration pressure (PUF), and ultrafiltration coefficient (Kf), which is a product of area and hydraulic permeability (Lp) that determines the Single Nephron Glomerular Filtration Rate (SNGFR). SNGFR is derived from the following equation where x represents the normalized length of the glomerular capillary with the afferent-end designated by 0 and the efferent-end by 1; P and π are the hydraulic and osmotic pressures, respectively, at distance x along the capillary length; σ is the reflection coefficient which has a range from 0 to 1 and; Kf.
Based on this equation, increased Kf (dependent on Lp and area) and/or ∆P (determined by PGC and ∆π) would be the key determinants of increased SNGFR in hyperfiltration. These basic characteristics of glomerular hemodynamics are altered in a single kidney. Studies involving animal models and human subjects have relied on these parameters to determine changes in glomerular hemodynamics associated with kidney disease. Unilateral nephrectomy in rodents is an established approach to study the effects of hyperfiltration. Animal models provide the convenience of direct measurement of the components of the above equation and validation of the calculated SNGFR in humans. Celsi et al [10] showed that unilateral nephrectomy in 5-day old Sprague-Dawley rats resulted in an increased SNGFR and PUF in the early period (observed in 20-day old rats). An increase in glomerular area and a decrease in Lp were additional changes observed at a later stage (60-day old rats). Further, Celsi et al [11] showed that high ultrafiltration pressure was not an absolute requirement for increasing SNGFR in unilaterally nephrectomized rats. The reported increase in SNGFR following unilateral nephrectomy ranges from 30%-36% in mice and 57%-86% in rats [10-14].
In the ALTOLD (Assessing Long Term Outcome in Living Kidney Donors) study, GFR, measured by plasma iohexol clearance, increased at a rate of 1.47 ± 5.02 ml/min/year in donors between 6 and 36 months compared to a decrease of 0.36 ± 7.55 ml/min/year (p=0.005) in controls on follow up [15]. The measured GFR continued to increase in kidney donors up to 36 months after donation, suggesting an ongoing increase in SNGFR following kidney donation.
In line with animal studies, Lenihan et al [16] found the mean single kidney GFR increased from 47 (predonation) to 64 (early postdonation) to 66 (late postdonation) ml/min/1.73 m2 with a parallel increase in single kidney renal plasma flow of 235, 314, and 335 ml/min/1.73m2, renal cortical volume of 103, 131, and 139 cm3, and calculated Kf with a fixed glomerular transcapillary hydraulic pressure gradient (∆P) at 40 mmHg of 4.9, 7.1 and 7.0 ml/(min.mmHg) in 21 transplant donors. The study highlights the increase of SNGFR in transplant donors by ~40% with no or minimal increase in ∆P [16]. Thus, adaptive hyperfiltration in a single kidney results in a significant increase in SNGFR in humans (calculated) and in animal models (directly measured).
Long-term outcome for kidney donors
Living kidney donation is now a safe procedure following stringent donor screening. However, the long-term outcome for all kidney donors may not be as benign as previously reported [4,5]. Of historical note, Ronald Lee Herrick, the first living donor, in 1954, spent the last decade of his life on dialysis. As early as 1984, Hakim et al [17] identified proteinuria and hypertension without a significant increase in serum creatinine after long-term (≥10 years) follow up of 52 transplant donors. Timsit et al [18] showed ~30% decrease in eGFR following unilateral nephrectomy in 124 renal cancer patients and in 124 transplant donors. At 4 years, 16.2% and 5.3% of post-kidney resection renal cancer patients and transplant donors had an eGFR of <45 ml/min/1.73m2. Ibrahim et al [19] reported eGFR <60, <45, and <30 ml/min/1.73m2 in 35.6%, 10.9%, and 2.6%, respectively, as well as proteinuria in 6.1% of Caucasian donors within 10 years of kidney donation.
Differences between early and recent studies underscore the shift in thinking about the remaining kidney in the transplant donor. Earlier studies were based on small cohorts, use of general population as control, short duration follow-up, and serum creatinine as the main indicator of renal function. In contrast, recent reports are based on population risk models developed using large data sets. Mjøen et al [4] compared 1,901 transplant donors to 32,621 control healthy, eligible nondonors, and reported an adjusted hazard ratio of 11.38 for ESRD. The crude incidence of ESRD in these Norwegian donors was 302/million person-years compared to ~100/million person-years. Similarly, Muzaale et al [5] compared 96,217 transplant donors to 20,024 healthy nondonors. The estimated lifetime risk of ESRD was 90 per 10 000 donors and 14 per 10 000 healthy nondonors compared to 326 per 10 000 in the general US population. The Chronic Kidney Disease Prognosis Consortium reported that the risk of kidney disease at 15 years after donation is 3.5-5.3 times greater than that in the control population [7]. Li et al [20] reported a pooled rate of 1.1% for ESRD, 5.7% for proteinuria, and 20.9% for microalbuminuria at >10 years following donation. A meta-analysis comprising 52 studies with follow-up for 24 years reported a significant increase in relative risk for ESRD of 8.83 (95%CI 1.02-20.93) [1]. These findings have led to a closer examination of the statistical methods, selection of control cohorts, hereditary risk of ESRD between related donors, and other aspects of these studies. Overall, consensus appears to be emerging that at least some kidney donors may develop proteinuria, decreased eGFR and ESRD in the long-term, and that novel diagnostic and preventative approaches need to be developed.
Influence of age, gender and ethnicity on the remaining kidney in donors
Younger transplant donors (<35 years) of all ethnicities are at a higher risk of developing ESRD as they survive into old age [21,22]. In a population model, Kiberd [23] found the cumulative risk of ESRD in Caucasian male donors to increase by +1.01, +0.89, +0.67, and +0.50 at ages 20, 40, 50, and 60 years, respectively. A similar significant increase was seen in African American males, and in women of both ethnicities. Muzaale et al [5] reported a greater risk of ESRD among African American donors (50.8 per 10 000) compared to Hispanics (25.9 per 10 000) and Caucasians (22.7 per 10 000) as compared with nondonors. The calculated risk was 0.24% for African American men, 0.15% for African American women, 0.06% for Caucasian men, and 0.04% for Caucasian women [7]. Thus, young African American men likely face the highest risk for ESRD in the years following kidney donation [7,18,23]. Longer follow up of Caucasian male donors from Minnesota showed a cumulative incidence of ESRD per 10,000 donors at 15, 30, and 40 years is 13.5, 68.7, and 78.7, respectively [19]. Massie et al [24] reported median (interquartile range) predicted risk of ESRD of 1 (1-2), 6 (4-11), 16 (10-29), and 34 (20-59) cases per 10 000 donors at 5, 10, 15, and 20 years post-kidney donation, respectively. Matas et al [22] reported the cumulative incidence of ESRD (95%CI) of 3.0 (0.0-8.9), 14.4 (2.9-28.5), 50.9 (22.8-83.3), 109.1 (61.0-159.3), and 381.0 (246.8-534.4) cases per 10 000 donors at 10, 15, 20, 30, and 40 years post-kidney donation, respectively. The mean interval to ESRD was 27.1 ± 9.8 years [22]. These studies suggest that it takes ~15-30 years for hyperfiltration-mediated kidney injury to become manifest in transplant donors.
Thus, adaptive hyperfiltration does not appear to be completely benign for all transplant donors. A 50% reduction in total renal mass following kidney donation can result in ESRD and a need for renal replacement therapy in the long-term. Quantitative approaches are being increasingly employed to determine the risk of ESRD following kidney donation. These prediction models are based on the data from donor follow up for 4-16 years but lack the information on family history of ESRD that limits their value for predicting ESRD in younger donors with and without a family history of ESRD. Such models will hopefully be used to complement, but not replace, the comprehensive evaluation of a potential donor [25-27]. These risk prediction models based on the work of Grams et al [7], Massie et al [24] and Ibrahim et al [19] can be found at http://www.transplantmodels.com/esrdrisk/, http://www.transplantmodels.com/donesrd/ and http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2015091018/-/DCSupplemental, respectively, and are available for clinical use.
The prevalence of ESRD or chronic kidney disease (CKD) Stage 5 in the general adult population is only 0.1% compared to 0.2% for CKD Stage 4, 4.3% for CKD Stage 3, 3.0% for CKD Stage 2, and 3.3% for CKD Stage 1 [28]. CKD Stage 5/ESRD represents only 0.2% of all adults with CKD [28]. Therefore, ESRD is only the tip of the iceberg that is CKD. While recent studies clearly identify the risk of ESRD, donors with variable degrees of CKD ranging from CKD Stage 2 to 4 still need to be adequately studied.
Amplification of hyperfiltration-mediated injury by co-morbid conditions in donors
Genetic background, hypertension, diabetes, aging, obesity, low nephron endowment at birth, smoking, and proteinuria have been implicated in the progression of CKD. The following paragraphs summarize the significance of these co-morbid conditions in hyperfiltration-mediated injury.
Apolipoprotein 1 (APOL1) and kidney donation
Several studies have observed that African American donors are at the highest risk for developing CKD [21,23,24,29,30]. Apolipoprotein 1 (APOL1) gene variants (G1 and G2) have been associated with poor outcomes in African-American [31]. APOL1 is a part of the innate immune response to Trypanosoma brucei infection [31,32]. The G1 and G2 variants make host APOL1 resistant to neutralization by serum resistance-associated protein of Trypanosoma brucei [32]. African-American with 2 copies of the APOL1 gene variants in contrast to those with 0 or only 1 copy were found to be at a greater risk of ESRD and, to have a more rapid decline in eGFR as reported by the African American Study of Kidney Disease and Hypertension (AASK), the Chronic Renal Insufficiency Cohort (CRIC) study, Coronary Artery Risk Development in Young Adults (CARDIA), Atherosclerosis Risk in Communities (ARIC), and the Chronic Kidney Disease in Children (CKiD) [33-36]. The ARIC study found large variability and substantial overlap in eGFR trajectory among Caucasians, African Americans with low-risk APOL1 status, and African Americans with high-risk APOL1 status, making it difficult to predict kidney disease [36].
Since the high-risk APOL1 genotype occurs in ~13% of the African American population [37,38] targeting potential kidney donors in this at-risk population has been proposed [39]. Doshi et al [40] examined the effect of the high-risk APOL1 genotype in African American living kidney donors, and found lower eGFR at baseline and follow up, and faster decline in eGFR compared to African American living donors with low-risk APOL1 genotype. In the same study they also compared donors matched to nondonors by APOL1 genotype, and found no difference in the rate of eGFR decline or an interaction by APOL1 status. Although the high-risk APOL1 genotype in living kidney donors was associated with a greater decline in eGFR, its trajectory was similar between donors and nondonors. Thus, the influence of the APOL1 genotype on the outcome for kidney donors is still under investigation. The question of whether African-Americans carrying high-risk APOL1 gene variants should be allowed to donate or not remains unresolved [41-43]. In addition to the high-risk genetic background, African-Americans have a relatively high prevalence of hypertension and diabetes, which would likely amplify the effect of hyperfiltration [29,30].
Hypertension and kidney donation
Matas et al [22] reported that for the majority of living donors ESRD occurred later in life with new onset diabetes and hypertension that change the slope of eGFR. This connection suggests an amplification of hyperfiltration-mediated injury by these co-morbidities. In a meta-analysis the weighted mean was found to be higher for systolic and diastolic blood pressure, 6 mm Hg [95%CI, 2 to 11 mm Hg] and 4 mm Hg [95%CI, 1 to 7 mm Hg], respectively [44]. A study linked to pharmacy claims reported that 17.8% of the donors had filled at least 1 antihypertensive medication within 5 years following donation [45]. In this study the use of antihypertensive medication was lower after kidney donation compared to the unscreened general population. Prevalence of hypertension following kidney donation is higher in African-American compared to Caucasian [45-48]. In a head-to-head comparison, African-American donors were found to have a higher incidence of hypertension compared to healthy non-donor African-American (40.8% vs. 17.9%, RR 2.4, 95%CI 1.7-3.4) [48]. In comparison with healthy controls, living donors have higher rates of hypertension (16.3% vs. 11.9%, HR 1.4, 95%CI 1.2-1.7) [49]. Thus, kidney donation may accelerate the risk of progression of hypertension in the donor, and more so in African-Americans.
The loss of normal nocturnal fall in BP during the sleep state (or nondipping) on ambulatory blood pressure monitoring (ABPM) has been shown to be a risk factor for cardiovascular events, but only a small number of studies reporting ABPM in living donors are available. Although the blood pressure on ABPM was not elevated after donation, 39.4% of living kidney donors were found to be nondippers [50]. In a short follow up for 6-12 months, 2 studies found no significant difference in adverse outcomes among donors who were nondippers at baseline [51,52]. Another study found similar systolic and diastolic BP measurements, and nondipping status between donors and controls at 10 years following donation [53]. The EARNEST study is an ongoing prospective, multicenter, longitudinal, parallel group study of 440 living kidney donors and 440 healthy controls involving measurement of arterial stiffness, office blood pressure, ABPM, and a series of biomarkers for cardiovascular and bone mineral disease at baseline and at 12 months [54]. Results of this study may provide further information on blood pressure changes and cardiovascular outcomes in donors.
Diabetes and kidney donation
Diabetes is the leading cause of ESRD. In diabetic kidney disease, a subject may progress through the classical 5 stages: (1) Early functional phase with reversible glomerular hyperfiltration, (2) Normal GFR and normal urine albumin excretion (< 30 mg/day), (3) Incipient nephropathy, defined by normal GFR and microalbuminuria (30–300 mg/day), (4) Macroalbuminuria (>300 mg/day), and (5) ESRD [55]. Approximately 5-10 years following the onset of diabetes, 20 - 40% patients develop microalbuminuria. Of these patients 80-90% progress to more advanced stages. After 10-20 years, 20-40% of patients develop macroalbuminuria, and about half of them progress to ESRD within 5 years [55]. The United Kingdom Prospective Diabetes Study (UKPDS64) reported that following the diagnosis of diabetes, microalbuminuria occurred at 2.0% per year, progression from microalbuminuria to macroalbuminuria at 2.8% per year, and progression from macroalbuminuria to elevated plasma creatinine or ESRD at 2.3% per year [56]. At 10 years following the diagnosis of diabetes, prevalence rates were at 24.9% for microalbuminuria, 5.3% for macroalbuminuria, and 0.8% for elevated plasma creatinine or ESRD [56]. Thus, the timescale for development of diabetic nephropathy is very similar to those of kidney disease in kidney donors.
Hyperfiltration is a maladaptive response in diabetes which predisposes the glomerulus to irreversible damage of nephrons and development of progressive renal disease [57]. The initial phase of diabetic kidney disease is characterized by glomerular hyperfiltration with an absolute increase in whole-kidney GFR [58]. GFR increases by 10-67% in Type 1 diabetes and 6-73% in Type 2 diabetes [58-60]. Increased SNGFR is believed to maintain whole-kidney filtration within the normal range in diabetic subjects to compensate for reduced nephron number (renal mass) and/or as a response to metabolic and (neuro)hormonal stimuli from diabetes itself [58]. This elevated SNGFR then contributes to the decline in renal function [58]. A de novo development of diabetes and hypertension following kidney donation increases the risk of proteinuria by ~4-fold, and for ESRD by ~2-fold [7].
Aging and kidney donation
In Caucasians, but not in African Americans, the risk for lower eGFR increases with age at donation [6]. Denic et al [61] showed a decrease in nonsclerotic (functional) nephrons and cortical volume with age in normal healthy adults. Older donors (>55 years) compared to younger donors (<45 years) have lower GFR, decreased renal cortical volume, and lower whole kidney Kf attributed to a decrease in the number of functional nephrons prior to donation [62]. Thus, an age-associated decrease in the number of functional nephrons would likely exacerbate hyperfiltration in the remaining kidney in the donor as well as in the recipient.
Obesity and kidney donation
Several studies have found that obesity increases the risk of adverse outcomes in kidney donors [6,7,24,29,30]. The estimated risk of ESRD at 20 years postdonation is reportedly 93.9 per 10 000 in obese (BMI >30 kg/m2) compared to 39.7 per 10 000 in nonobese donors [63]. Ibrahim et al [19] showed that for every 1 unit increase in BMI there is an associated 3%-10% greater risk for decreased eGFR and increased proteinuria. In donors with BMI >30 kg/m2 for every 5 kg/m2, Grams et al [7] found that the risk of ESRD increased by 16% (HR 1.16, 95%CI 1.04-1.29), while Massie et al [24] reported an increase of 61% (HR 1.61, 95%CI 1.29-2.00). Chagnac et al [64] showed that obese (BMI >38 kg/m2) nondiabetic individuals have increased GFR, renal plasma flow, and urinary albumin excretion rate. Dextran-sieving profiles suggested that increased SNGFR (ie, hyperfiltration) in obese individuals was due to increased ΔP from dilatation of the afferent arteriole.
Low nephron number and kidney donation
The significance of low nephron endowment in the progression of CKD, hypertension, and cardiovascular outcomes for both adults and children has been highlighted over the past decade [65-79]. Thus, low nephron endowment, loss of nephrons with age, hypertension, diabetes, and obesity would further amplify the hyperfiltration-associated glomerular changes in living donors. While one cannot control aging, sex, ethnicity, and genetic background, careful monitoring to control postdonation co-morbidities (eg, excessive weight, hypertension and diabetes), and guidance to avoid smoking and nephrotoxic drugs are within the physician’s reach.
Relevance of hyperfiltration-mediated injury in kidney transplant recipients
Recent decades have witnessed significant improvements in the short-term outcomes for recipients of kidney transplantation, but progress toward improving long-term outcomes remains a challenge. Unfortunately, almost half of the allografts are lost within 10 years posttransplantation [80]. Interstitial fibrosis with tubular atrophy (previously known as chronic allograft nephropathy) and death while the graft is functioning are the 2 major causes of graft loss in recipients that have remained unchanged [81,82].
The transplanted kidney in the recipient is also exposed to hyperfiltration-mediated injury. Additionally, the transplanted kidney in the recipient, is also exposed to a host of adverse conditions, including immunological and nonimmunological injury such as acute and chronic rejection, recurrence of the disease, acute and chronic infections, ischemia-reperfusion injury, drug toxicity, renal artery stenosis, urinary tract obstruction, hypertension, diabetes, donor APOL1 genotype, etc. Immunological and nonimmunological injuries further accelerate the progression of CKD in transplant recipients compared to donors. We believe that the development of therapies to mitigate hyperfiltration-mediated injury will benefit the transplant recipients as well as donors.
African-American transplant recipients with high-risk APOL1 genotype compared to low-risk APOL1 genotype showed no difference in allograft survival [83]. On the other hand, Reeves-Daniel et al [84] reported shorter allograft survival in recipients from deceased donor kidneys with high-risk APOL1 genotype. This observation has been validated by large cohort studies that reported shorter renal allograft survival and higher follow-up serum creatinine concentrations in recipients of kidneys from donors with high-risk APOL1 genotype compared to low-risk APOL1 genotype [85,86].
Old versus newer paradigms for hyperfiltration-mediated injury
Early work by Brenner and colleagues used the 5/6 nephrectomy rat model to demonstrate a strong link between increased ∆P or PGC and kidney injury, and to highlight the importance of hyperfiltration-mediated injury [8,9]. Subsequent seminal work from Brenner and colleagues led to characterization of the renin-angiotensin-aldosterone system (RAAS). With these ground-breaking findings, several RAAS blockers were developed to mitigate hyperfiltration-mediated kidney injury in humans [8,9]. As mentioned earlier, most research on adaptive hyperfiltration to date has focused on glomerular pressure and renal hemodynamics. Such singular focus on glomerular capillary pressure has also uncovered the unexplained role of several factors that contribute to hyperfiltration-mediated glomerular injury leading to CKD.
With the benefit of hindsight gained from previous work by others on hemodynamic parameters, our group and others in the field are reexamining hyperfiltration. This new paradigm explains the effect of hyperfiltration in terms of glomerular biomechanical forces, namely tensile stress and fluid flow shear stress (FFSS) on the podocyte [87-92]. We propose that renal injury in transplant donors primarily stems from increased biomechanical forces. A better understanding of these biomechanical forces corresponding to persistent hyperfiltration will likely lead to identification of novel therapeutic targets and drug development that would complement currently used RAAS blockers in mitigating hyperfiltration-mediated injury.
Biomechanical forces associated with glomerular ultrafiltration
Glomerular filtration barrier, the site of plasma filtration, is constituted by glomerular capillary endothelial cells, basement membrane (GBM), and podocytes (visceral epithelial cells). Mesangial cells also contribute to the barrier function by stabilizing the glomerular structure. Structural and functional integrity of all components of the barrier are essential but podocytes are distinct in several ways. Podocytes are large cell bodies within Bowman’s space covering the capillary with foot processes that form slit junctions by interdigitating with foot processes from other podocytes. Slit junctions are critical for a size-based filtration of plasma components. However, in contrast to mesangial and endothelial cells, podocytes are terminally differentiated cells with low mitotic potential. Changes in the capillary pressure and the flow of the glomerular ultrafiltrate subject podocytes to pressure and flow-associated biomechanical forces that may result in their detachment [90-93]. These rheological factors and the inability to replace the lost cells render podocytes the most vulnerable component of the glomerular filtration barrier vis- à-vis mesangial and endothelial cells.
The passage of blood through the capillary and the flow of the ultrafiltrate along the podocyte surface can be explored using principles of fluid mechanics. Blood flow through capillary loops generates force radially at 90° to the direction of flow, causing the capillary wall to stretch and dilate which, in turn, radiates the force to podocyte foot processes attached to the GBM on the outer aspect of the capillary (Figure 1). Glomerular capillary pressure within the vascular compartment is the major determinant of tensile stress exerted over the basolateral aspect of podocytes [90,94]. The stretch on podocyte foot processes attached to the GBM results in tensile stress on podocytes. Kriz and Lemley [87] have suggested that the burden for counteracting transmural pressure gradients (or tensile stress) falls largely on the GBM, which can create significant elastic counterforce to balance the effects of increased transmural pressure. In vitro methods to study tensile stress are based on techniques to induce elongation or compression of cells. The most common method involves growing podocytes on a flexible membrane of specially constructed cell culture dishes. Negative pressure (vacuum) is applied to create a cyclical or sustained stretch to adherent cells on the flexible membrane. The stretch can be applied in 1 or 2 dimensions, resulting in uniaxial or biaxial stress. Other variables in these experiments include the (a) frequency (cycles/second, Hz), (b) magnitude (percent change), (c) pattern of application (square-wave or sine-wave), and (d) the duration of stretch. Additional methods to study axial stress include application of hydrostatic force, hypo-osmotic stretch, or magnetic pull [95-97].
Figure 1.
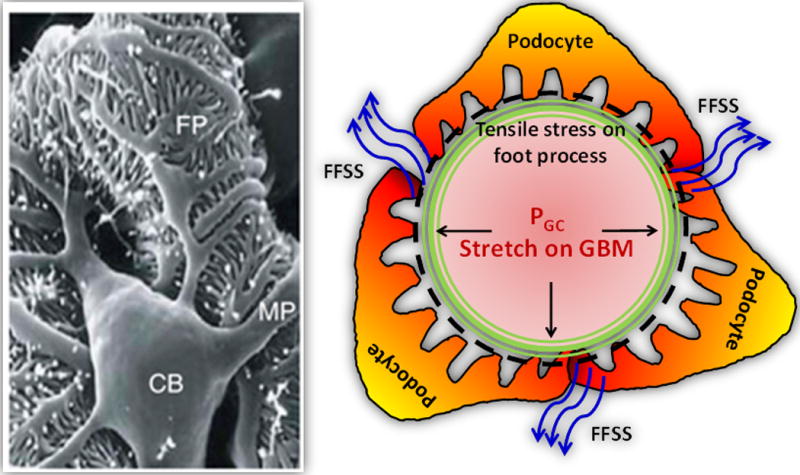
The large podocyte cell body (CB) localizes in the glomerular urinary space anchored to the capillary surface through major processes (MP) that branch into foot processes (FP) to cover the glomerular basement membrane (GBM) of the capillary wall (shown by black dashed circle). Foot processes experience the stretch caused by glomerular capillary pressure (PGC, straight line arrows), resulting in tensile stress on the podocyte. The ultrafiltrate flowing over the cell body and major processes of the podocyte exerts fluid flow shear stress (FFSS) on podocytes (shown by blue curved arrows). Early hyperfiltration in the remaining kidney of donors results in increased FFSS on podocytes from increased single nephron glomerular filtration rate. These changes are later exacerbated by increased tensile stress on podocytes from a rise in glomerular capillary pressure (PGC).
Ultrafiltrate flow generates another biomechanical force, namely FFSS associated with hyperfiltration that has direct consequences for the structural integrity of podocytes [87,94,98,99]. We and others have shown that an increase in FFSS alters podocyte structure that changes glomerular filtration barrier function leading to an increase in urinary protein excretion [87,98]. FFSS is a measure of the frictional force from a fluid acting on a body in the path of that fluid. In humans, the flow of 180 L/day of ultrafiltrate over the cell body and major processes exerts FFSS on podocytes (Figure 1) [90,99]. The major determinant of FFSS over the podocytes is SNGFR [90,92]. In vitro methods to study fluid flow shear stress (τ) use technique(s) for controlled flow of a fluid column over cells. A commonly used method involves fluid flow over the cells grown on glass slides. In these experiments the height (h), width (w), and viscosity (η) of the fluid column (eg, cell culture medium) are constant [98,100]. The rate of flow (q) of the fluid column determines the magnitude of FFSS on the cell given by the equation for parallel plate geometry, . Fluid column can be patterned as laminar, pulsatile, or oscillating. Another method to apply FFSS is by rotational flow of fluid on an orbital shaker [101]. FFSS at the bottom of the dish is calculated using the equation ), where α is the radius of orbit rotation, ρ is the density of the fluid medium, η is the viscosity of the fluid medium, and f is the frequency of rotations per second.
Hyperfiltration increases tensile stress as well as FFSS on podocytes within the glomerulus [98-104]. The engineering aspects of stresses and strains involved in generation of biomechanical forces on podocytes within the glomerulus during glomerular filtration have been reviewed in detail by Srivastava et al [91]. Our findings (vide infra) as well as those reported by Kriz and Lemley [88] suggest that FFSS is the main detrimental biomechanical force in glomerular injury and plays a major role in detaching podocytes from the glomerular basement membrane. In contrast to tensile stress, FFSS directly impacts podocyte structural integrity and stability, and contributes to podocyte loss. Therefore, study of the effect(s) of biomechanical forces on podocytes appears to be the next logical step to advance our understanding of hyperfiltration-mediated glomerular injury.
Activation of the Renin-Angiotensin-Aldosterone System (RAAS) by tensile stress and of the Cyclooxygenase 2 - Prostaglandin E2 - EP2 Receptor (COX2-PGE2-EP2) axis by fluid flow shear stress
Studies addressing the role of glomerular biomechanical forces suggest that tensile stress activates the RAAS whereas FFSS engages the COX2-PGE2-EP2 axis [98,104]. Capillary pressure-related changes and the activation of RAAS have been investigated for over half a century [105-112]. Briefly, podocytes express all components of the RAAS as well as receptors for angiotensin II [104]. Tensile stress results in increased secretion of angiotensin II, influx of Ca2+, up-regulation of the angiotensin-II receptor (AT1R), COX2, EP4, osteopontin, and activation of p38 MAPK, ERK1/2 and JNK [94,104,113,114]. Characterization of the RAAS led to development of several RAAS blockers that include angiotensin-converting enzyme inhibitors (ACEI), angiotensin receptor blockers (ARB), mineralocorticoid receptor antagonists, and direct renin inhibitors. ACEI and ARB, alone or in combination, have been shown to delay the progression of CKD/ESRD in diabetic nephropathy [115-117]. Subsequently RAAS blockers were shown to be effective in delaying the progression of CKD/ESRD in nondiabetic proteinuric kidney diseases [118-121]. These beneficial effects of RAAS blockers have been attributed to reduced hyperfiltration-mediated kidney injury. However, RAAS blockers interfere with the autoregulation of RBF and GFR, especially in patients with bilateral renal artery stenosis or those with stenosis in a solitary kidney requiring angiotensin II to maintain renal perfusion [122,123]. Thus, physicians hesitate using RAAS blockers in patients with solitary functioning kidney [124]. Compelling data on the efficacy of RAAS blockers in nontransplant populations led to studies of transplant recipients. These studies do not support the use of RAAS blockers for improving clinical outcomes in transplant recipients and suggest that these drugs may, at times, be detrimental to graft function and survival [124-128]. In view of the pharmacological effects of RAAS blockers on glomerular hemodynamics, the role of these drugs in controlling proteinuria and hypertension in transplant donors and recipients continues to be controversial [129-132]. A recent meta-analysis could neither support nor refute the use of RAAS blockers in transplant recipients [133]. Accumulating evidence shows beneficial effects of RAAS blockers in many proteinuric CKD patients, but not in subjects with sickle cell disease [134,135], autosomal dominant polycystic kidney disease [136,137], or congenital anomalies of the kidney and urinary tract [138].
Research on the role of FFSS is nascent but has yielded promising results that encourage a search for additional molecular targets to treat hyperfiltration-induced renal injury. COX1 and 2 activities on arachidonic acid generate several metabolites with prostaglandin E2 (PGE2) as a major prostaglandin. PGE2 is a ligand of 4 transmembrane G-protein coupled receptors, namely EP1-EP4. We have shown that podocytes express EP2 and EP4 [102]. PGE2-EP receptor complexes can activate several signaling pathways [139]. Our laboratory has led the efforts to explore the molecular mediators of changes in podocytes caused by increased FFSS. During the past decade we have found that podocytes exposed to FFSS in vitro and in vivo show upregulation of COX-2 but not COX-1, induction of PGE2 receptor EP2 but not EP4, elevated PGE2 levels, and derangement of the actin cytoskeleton. Additionally, glomeruli exposed to FFSS showed an increase in glomerular albumin permeability (Palb) [98,100,102,103]. Indomethacin blocked each of these effects of FFSS [98,100]. Thus, the activity of the COX2-PGE2-EP2 axis in podocytes and glomeruli was upregulated following an increase in FFSS in both cell and animal studies [98,100,102]. The relevance of the COX2-PGE2-EP2 axis is further supported by studies using animal models of hyperfiltration of 5/6 nephrectomy, high dietary protein, and diabetes which show increased renal COX-2 expression, renal PGE2 synthesis, and urinary PGE2 [140-147]. We recently reported that FFSS on podocytes recruits the Akt-GSK3β-β-catenin, ERK1/2, and p38MAPK signaling pathways, but not the cAMP-PKA pathway [139]. Activation of the COX2-PGE2-EP2 axis through membrane-localized EP2 receptor and its link to genome activation in the nucleus through transcriptional factors is a subject of ongoing studies.
Each of these and other mechanisms may be important in how podocytes respond to FFSS and tensile stress. The known significance of tensile stress and the RAAS is the basis for using RAAS blockers. Our work suggests that research on FFSS may lead to additional targets such as EP2 and/or downstream signaling molecules [139]. EP2 receptor activation has been found to be deleterious in chronic inflammatory neurodegenerative diseases and colon cancer [148,149]. Currently under development, EP2 antagonists to treat these diseases will also be relevant for kidney donors. Thus, studies on biomechanical forces within the glomerulus will likely identify novel non-RAAS pathways activated in response to hyperfiltration. We postulate that modulating EP2 receptor would be a valuable target to mitigate hyperfiltration-mediated injury, in addition to the known RAAS blockers in kidney donors.
Hyperfiltration continuum model in progression of chronic kidney injury in donors
In most CKD, a gradual loss of functional nephrons over time leads to persistent hyperfiltration in the remaining glomeruli. Adaptive changes initially appear in the form of increased SNGFR due to greater renal blood flow and Kf. Animal studies show that hyperfiltration is associated with increased SNGFR due to higher Kf and blood flow during the initial phase followed by a gradual increase in ∆P or PGC in the later period [9-14,112]. Similarly, a gradual loss of functional nephrons in CKD results in a proportional intensification of ongoing hyperfiltration. In both animal and human studies as discussed earlier, an early increase in SNGFR is followed by an increase in PGC as the kidney suffers progressive loss of function from mild to moderate and severe. Over time, the loss of functional nephrons results in glomerular hypertension from increased PGC. We conceptualize a rapid decline in GFR in the later stages of CKD from acceleration in hyperfiltration-mediated injury with continuing and cumulative loss of functional nephrons. This association between increased PGC, ie, tensile stress and proteinuria, has been recognized since Brenner’s findings and addressed in animal and clinical studies with RAAS blockers [8,9]. However, the role of increased SNGFR due to ultrafiltrate flow, ie, FFSS, was not a part of earlier investigations on hyperfiltration-mediated glomerular injury.
We have developed a model to outline the significance of FFSS and tensile stress. This model emphasizes that a gradual decline in GFR from 100% to 0% and persistent hyperfiltration, increased FFSS from SNGFR, and increased tensile stress from PGC all play important roles in causing progressive damage to podocytes and loss of glomerular function. As shown in Figure 2, hyperfiltration-mediated changes may be conceptualized as a continuum where both FFSS and tensile stress contribute sequentially and additively to podocyte injury. In Figure 2, the arrow (top) indicates the average reported GFR in the early period following kidney donation. The study by Lenihan et al [16] highlighted that SNGFR in transplant donors increased by ~40% with none-to-minimal increase in ∆P. Timsit et al [18] found ~30% decrease in eGFR following unilateral nephrectomy in transplant donors and renal cancer. Thus, an increase in SNGFR corresponds to higher FFSS over podocytes and precedes the rise in PGC (and tensile stress) in this continuum (see lower half of Figure 2). We have observed a 1.5- to 2-fold increase in FFSS following unilateral nephrectomy in animals and, based on available human studies, hypothesize an increase in FFSS in a kidney donor [103].
Figure 2.
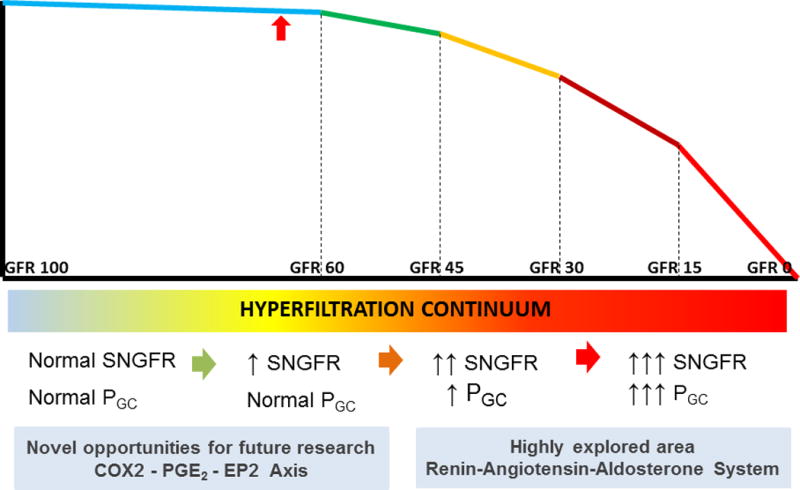
Hyperfiltration-mediated hemodynamic changes may be conceptualized as a continuum. In most kidney diseases, a gradual loss of functional nephrons over time leads to hyperfiltration in the remaining glomeruli. Adaptive changes initially appear in the form of increased single nephron glomerular filtration rate (SNGFR) from changes in renal blood flow and ultrafiltration coefficient (Kf). Over time, progressive loss of functional nephrons results in glomerular hypertension from increased glomerular capillary pressure (PGC). We theorize that in the later stages of CKD with continuing loss of functional nephrons there will be a more rapid decline in GFR from acceleration in hyperfiltration-mediated injury. We propose that the cyclooxygenase 2 enzyme - prostaglandin E2 - prostanoid receptor EP2 (COX2-PGE2-EP2) axis and components of the renin-angiotensin-aldosterone system (RAAS) are relevant in the early and late stages of the hyperfiltration continuum, respectively (see text for details). The red arrow marks the average reported GFR in the early period following kidney donation. Early molecular, biochemical, and signaling changes in the podocyte prior to an elevation of PGC are not known. Slow progression of the disease provides an opportunity for innovative intervention to protect the remaining kidney of donors.
However, early molecular, biochemical, and signaling changes in podocytes prior to an increase in PGC are not known. Figure 2 also highlights the need for research to investigate the early response in podocytes when SNGFR rises without an increase in PGC. In this regard, the COX2-PGE2-EP2 axis is a candidate pathway for further studies. We propose that the COX2-PGE2-EP2 axis and components of the RAAS are relevant in the early and late stages of the hyperfiltration continuum, respectively. Outlining the slow progression of CKD enables us to model hyperfiltration-associated changes with the inclusion of potential contributing factors that amplify hyperfiltration, such as obesity, hypertension, diabetes, etc. and their temporal entry into the hyperfiltration continuum. Conceptualizing hyperfiltration as a continuum rather than a distinct event also allows inclusion of overlapping adaptive and maladaptive changes that, in turn, generate cellular responses within the glomerulus (Figure 2). The gradual nature of glomerular changes becomes apparent in the proposed model of hyperfiltration as a continuum and provides an opportunity for innovative intervention for protecting the remaining kidney in transplant donors. Thus, the key to improving outcomes in kidney donors lies not only in understanding the mechanistic basis, but also in identifying new targets for therapeutic interventions in hyperfiltration-mediated glomerular injury. This model highlights the opportunity to explore new targets by including changes in glomerular biomechanical forces occurring over the continuum.
Summary
In the foregoing text we sought to connect multiple topics related to epidemiology, clinical information, physiology, animal models, cell biology and molecular signaling in hyperfiltration-mediated injury resulting from loss of 1 kidney in living donors. Each of these sections could be explored in greater detail but we aimed at highlighting the diverse factors that influence the outcome following kidney donation. Further, accumulating data on living donors is just beginning to shed light on the long-term functional changes in the remaining kidney. Hyperfiltration-mediated injury ran as a common thread linking subsections of this article to advance the recent work on biomechanical forces. We thus bring attention to the significance of biomechanical forces in hyperfiltration-mediated injury in donors in this overview. The specific role of biomechanical forces, ie, tensile stress and FFSS, needs to be further validated in transplant donors. The progression of CKD/ESRD following kidney donation is also influenced by ethnicity (APOL1 gene variants), age, hypertension, obesity, smoking, etc. Whether the known risk factors such as obesity, hypertension, diabetes, etc. would amplify or simply add to the effect of biomechanical forces, needs to be further explored. The new paradigm based on biomechanical forces to understand the mechanism of hyperfiltration-mediated injury identifies COX2-PGE2-EP2 axis as a potential target for intervention to complement the currently used RAAS blockers in mitigating injury. It would be too early to conclude but EP2 receptor antagonists currently being investigated in clinical studies raise the possibility of intervention for kidney donors at greater risk for progression of CKD/ESRD.
Living kidney donation is an indispensable option for patients with ESRD. However, donors with the remaining kidney are at a small but distinct risk for developing ESRD themselves. Identifying the subset of donors at greater risk of CKD (a) before and (b) after transplantation will help minimize the risk to donors. In this regard, low nephron endowment at birth sets the foundation for nephron deficiency (or inadequacy) through kidney donation [75-79]. The impact of nephron mass reduction following donation would be amplified in those with unidentified low nephron number prior to kidney donation. Technological advances have made it possible to estimate nephron number in animals using MRI [150]. Progress in imaging techniques will provide sophisticated information on the status of a donor kidney prior to transplantation to exclude candidates with low nephron endowment. Due to technical limitations, this method is not yet ready for human use. In addition, the usefulness of prescreening African American donors for high risk APOL1 gene variants predonation and of withholding donation is under discussion [41,43]. Biomarkers that are associated with progression of CKD may be useful in identifying donors at increased risk. Urinary epidermal growth factor has recently been identified as a biomarker to identify patients at greater risk of progression while in early stages of CKD [151]. In a small group of children with solitary functioning kidney, we have found an increase in urinary PGE2 and a decrease in PGI2 [90]. The usefulness of these novel imaging techniques and biomarkers needs to be validated in large transplant donor cohorts.
As the number of living donors is consistently increasing by ~5,500 each year, there is growing recognition that kidney donation exposes the donor to a small but definite risk for CKD. The foregoing discussion in this overview has outlined the risk(s) to the remaining kidney of living transplant donors and transplant recipients. Inability to successfully treat such patients underscores the limitations of current treatment regimens. Thus, in addition to mandating a stringent follow up, it would be prudent to invest resources to advance our understanding of the mechanistic basis of injury from adaptive hyperfiltration, and to develop tools to identify donors at risk before and after donation. Further research to understand the mechanism(s) of injury, to identify novel targets, and to develop the next generation of therapeutics to preserve kidney function in transplant donors (Figure 3) will encourage and support kidney donation. A treatment strategy to mitigate FFSS-mediated injury will build on existing knowledge supported by clear scientific rationale for optimizing long-term kidney function in donors.
Figure 3.
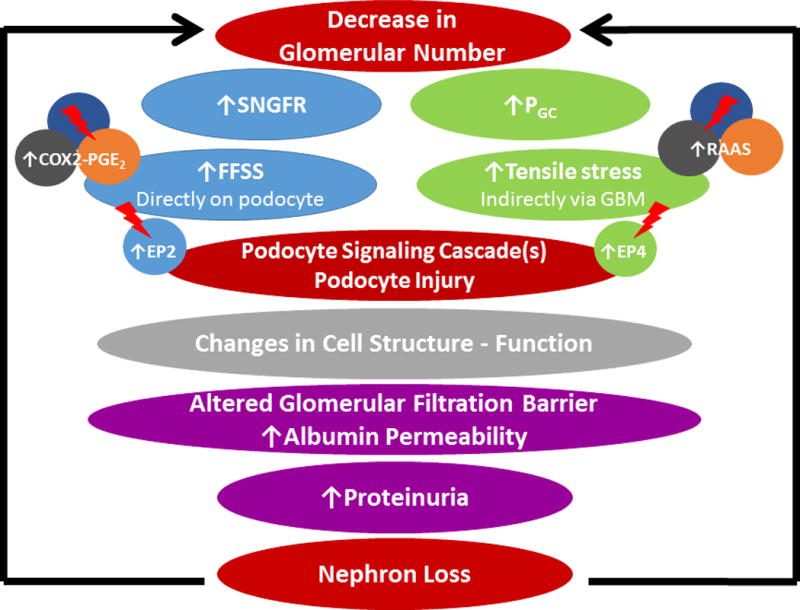
Outline of a conceptual model of hyperfiltration-mediated injury in the remaining kidney in transplant donors. The 2 biomechanical forces, namely fluid flow shear stress (FFSS) caused by single nephron glomerular filtration rate (SNGFR) and tensile stress caused by glomerular capillary pressure (PGC), mediate different pathways that can be potentially targeted by drug therapy (shown by lightning arrow). These forces have been shown to cause damage to the glomerular filtration barrier, leading to proteinuria and nephron loss. This begets additional nephron loss through further increasing the biomechanical forces, thus setting up a spiraling cascade.
Acknowledgments
We gratefully acknowledge the contribution of many investigators whose work could not be cited here. Space limits do not permit inclusion of references to all the work that helped our progress with the ideas discussed in this article.
Funding
This work was supported by NIDDK R01DK107490 (Srivastava, Sharma), the Department of Veterans Affairs, the Veterans Health Administration, Office of Research and Development, VA BX001037 (Savin, Sharma), DK 1RO1 DK064969 (McCarthy, Sharma), the Sam and Helen Kaplan Research Fund in Pediatric Nephrology (Alon, Srivastava), and the Midwest Biomedical Research Foundation (Savin, Sharma).
Abbreviations
- CKD
Chronic Kidney Disease
- COX2
Cyclooxygenase 2 Enzyme
- eGFR
Estimated Glomerular Filtration Rate
- EP2
Prostanoid Receptor EP2
- EP4
Prostanoid Receptor EP4
- ESRD
End-Stage Renal Disease
- FFSS
Fluid Flow Shear Stress
- Kf
Glomerular ultrafiltration coefficient
- Lp
Hydraulic permeability
- Palb
Glomerular albumin permeability
- PGC
Glomerular capillary pressure
- PGE2
Prostaglandin E2
- PUF
Glomerular ultrafiltration pressure
- RAAS
Renin-Angiotensin-Aldosterone System
- SNGFR
Single Nephron Glomerular Filtration Rate
- Δπ
Difference in osmotic pressure
Footnotes
Contribution
Drs. Srivastava, Hariharan, Alon, McCarthy, R. Sharma, El-Meanawy, Savin, and M. Sharma have worked closely together for many years. This overview is based on our earlier work and ongoing discussions within the group. All authors have been part of the conceptualization, design, review, revision and approval of the final manuscript as submitted. Drs. Srivastava and M. Sharma drafted the initial and completed the final versions of the manuscript for the group.
Disclosure
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States Government. The authors declare no conflicts of interest.
References
- 1.O’Keeffe LM, Ramond A, Oliver-Williams C, et al. Mid- and long-term health risks in living kidney donors: a systematic review and meta-analysis. Ann Intern Med. 2018;168:276–284. doi: 10.7326/M17-1235. [DOI] [PubMed] [Google Scholar]
- 2.Boudville N, Prasad GV, Knoll G, et al. Donor nephrectomy outcomes research (DONOR) network. Meta-analysis: risk for hypertension in living kidney donors. Ann Intern Med. 2006;145:185–196. doi: 10.7326/0003-4819-145-3-200608010-00006. [DOI] [PubMed] [Google Scholar]
- 3.Garg AX, Muirhead N, Knoll G, et al. Donor nephrectomy outcomes research (DONOR) network. Proteinuria and reduced kidney function in living kidney donors: A systematic review, meta-analysis, and meta-regression. Kidney Int. 2006;70:1801–1810. doi: 10.1038/sj.ki.5001819. [DOI] [PubMed] [Google Scholar]
- 4.Mjøen G, Hallan S, Hartmann A, et al. Long-term risks for kidney donors. Kidney Int. 2014;86:162–167. doi: 10.1038/ki.2013.460. [DOI] [PubMed] [Google Scholar]
- 5.Muzaale AD, Massiei AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311:579–586. doi: 10.1001/jama.2013.285141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathillas O, Attman PO, Aurell M, Brynger H. Glomerular filtration rate, hypertension and proteinuria after renal ablation: a long-term follow-up study in kidney donors. Scand J Urol Nephrol Suppl. 1988;108:49–55. [PubMed] [Google Scholar]
- 7.Grams ME, Sang Y, Levey AS, et al. Kidney-failure risk projection for the living kidney-donor candidate. N Engl J Med. 2016;374:411–421. doi: 10.1056/NEJMoa1510491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenner BM, Lawler EV, Mackenzie HS. The hyperfiltration theory: a paradigm shift in nephrology. Kidney Int. 1996;49:1774–1777. doi: 10.1038/ki.1996.265. [DOI] [PubMed] [Google Scholar]
- 9.Brenner BM. Nephron adaptation to renal injury or ablation. Am J Physiol. 1985;249:F324–337. doi: 10.1152/ajprenal.1985.249.3.F324. [DOI] [PubMed] [Google Scholar]
- 10.Celsi G, Larsson L, Seri I, Savin V, Aperia A. Glomerular adaptation in uninephrectomized young rats. Pediatr Nephrol. 1989;3:280–285. doi: 10.1007/BF00858530. [DOI] [PubMed] [Google Scholar]
- 11.Celsi G, Savin J, Henter JI, Sohtell M. The contribution of ultrafiltration pressure for glomerular hyperfiltration in young nephrectomized rats. Acta Physiol Scan. 1991;141:483–487. doi: 10.1111/j.1748-1716.1991.tb09109.x. [DOI] [PubMed] [Google Scholar]
- 12.Levine DZ, Iacovitti M, Robertson SJ, Mokhtar GA. Modulation of single-nephron GFR in the db/db mouse model of type 2 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol. 2006;290:R975–981. doi: 10.1152/ajpregu.00693.2005. [DOI] [PubMed] [Google Scholar]
- 13.Bank N, Alterman L, Aynedjian HS. Selective deep nephron hyperfiltration in uninephrectomized spontaneously hypertensive rats. Kidney Int. 1983;24:185–191. doi: 10.1038/ki.1983.143. [DOI] [PubMed] [Google Scholar]
- 14.Hayslett JP, Kashgarian M, Epstein FH. Mechanism of change in the excretion of sodium per nephron when renal mass is reduced. J Clin Invest. 1969;48:1002–1006. doi: 10.1172/JCI106056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasiske BL, Anderson-Haag T, Israni AK, et al. A prospective controlled study of living kidney donors: three-year follow-up. Am J Kidney Dis. 2015;66:114–124. doi: 10.1053/j.ajkd.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenihan CR, Busque S, Derby G, Blouch K, Myers BD, Tan JC. Longitudinal study of living kidney donor glomerular dynamics after nephrectomy. J Clin Invest. 2015;125:1311–1318. doi: 10.1172/JCI78885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hakim RM, Goldszer RC, Brenner BM. Hypertension and proteinuria: long-term sequelae of uninephrectomy in humans. Kidney Int. 1984;25:930–936. doi: 10.1038/ki.1984.112. [DOI] [PubMed] [Google Scholar]
- 18.Timsit MO, Nguyen KN, Rouach Y, et al. Kidney function following nephrectomy: similitude and discrepancies between kidney cancer and living donation. Urol Oncol. 2012;30:482–486. doi: 10.1016/j.urolonc.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Ibrahim HN, Foley RN, Reule SA, et al. Renal function profile in white kidney donors: The first 4 decades. J Am Soc Nephrol. 2016;27:2885–2893. doi: 10.1681/ASN.2015091018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li SS, Huang YM, Wang M, et al. A meta-analysis of renal outcomes in living kidney donors. Medicine (Baltimore) 2016;95:e3847. doi: 10.1097/MD.0000000000003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibney EM, Parikh CR, Garg AX. Age, gender, race, and associations with kidney failure following living kidney donation. Transplant Proc. 2008;40:1337–1340. doi: 10.1016/j.transproceed.2008.03.104. [DOI] [PubMed] [Google Scholar]
- 22.Matas AJ, Berglund DM, Vock DM, Ibrahim HN. Causes and timing of end-stage renal disease after living kidney donation. Am J Transplant. 2018;18:1140–1150. doi: 10.1111/ajt.14671. [DOI] [PubMed] [Google Scholar]
- 23.Kiberd BA. Estimating the long term impact of kidney donation on life expectancy and end stage renal disease. Transplant Res. 2013;2:2. doi: 10.1186/2047-1440-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massie AB, Muzaale AD, Luo X, et al. Quantifying postdonation risk of ESRD in living kidney donors. J Am Soc Nephrol. 2017 Sep;28:2749–2755. doi: 10.1681/ASN.2016101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maggiore U, Budde K, Heemann U, et al. Long-term risks of kidney living donation: review and position paper by the ERA-EDTA DESCARTES working group. Nephrol Dial Transplant. 2017;32:216–223. doi: 10.1093/ndt/gfw429. [DOI] [PubMed] [Google Scholar]
- 26.Gaillard F, Baron S, Timsit MO, et al. What is the significance of end-stage renal disease risk estimation in living kidney donors? Transpl Int. 2017;30:799–806. doi: 10.1111/tri.12931. [DOI] [PubMed] [Google Scholar]
- 27.Grams ME, Garg AX, Lentine KL. Kidney-failure risk projection for the living kidney-donor candidate. N Engl J Med. 2016;374:2094–2095. doi: 10.1056/NEJMc1603007. [DOI] [PubMed] [Google Scholar]
- 28.National Kidney Foundation. K/DOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification and Stratification. Am J Kidney Dis. 2002;39(suppl 1):S1–S266. [PubMed] [Google Scholar]
- 29.Lentine KL, Schnitzler MA, Xiao H, et al. Racial variation in medical outcomes among living kidney donors. N Engl J Med. 2010;363:724–732. doi: 10.1056/NEJMoa1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lentine KL, Patel A. Risks and outcomes of living donation. Adv Chronic Kidney Dis. 2012;19:220–228. doi: 10.1053/j.ackd.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. ProcNatl Acad Sci U S A. 2014;111:E2130–E2139. doi: 10.1073/pnas.1400699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsa A, Kao WH, Xie D, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med. 2013;369:2183–2196. doi: 10.1056/NEJMoa1310345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng DK, Robertson CC, Woroniecki RP, et al. APOL1-associated glomerular disease among African-American children: a collaboration of the Chronic Kidney Disease in Children (CKiD) and Nephrotic Syndrome Study Network (NEPTUNE) cohorts. Nephrol Dial Transplant. 2017;32:983–990. doi: 10.1093/ndt/gfw061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Locke JE, Sawinski D, Reed RD, et al. Apolipoprotein L1 and Chronic Kidney Disease Risk in Young Potential Living Kidney Donors. Ann Surg. 2018;267:1161–1168. doi: 10.1097/SLA.0000000000002174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grams ME, Rebholz CM, Chen Y, et al. Race, APOL1 risk, and eGFR decline in the general population. J Am Soc Nephrol. 2016;27:2842–2850. doi: 10.1681/ASN.2015070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013;24:1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol. 2011;22:2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen DM, Mittalhenkle A, Scott DL, Young CJ, Norman DJ. African American living-kidney donors should be screened for APOL1 risk alleles. Transplantation. 2011;92:722–725. doi: 10.1097/TP.0b013e31822eec39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doshi MD, Ortigosa-Goggins M, Garg AX, et al. APOL1 genotype and renal function of black living donors. J Am Soc Nephrol. 2018;29:1309–1316. doi: 10.1681/ASN.2017060658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riella LV, Sheridan AM. Testing for high-risk APOL1 alleles in potential living kidney donors. Am J Kidney Dis. 2015;66:396–401. doi: 10.1053/j.ajkd.2015.04.046. [DOI] [PubMed] [Google Scholar]
- 42.Young BA, Fullerton SM, Wilson JG, et al. Clinical genetic testing for APOL1: Are we there yet? Semin Nephrol. 2017;37:552–557. doi: 10.1016/j.semnephrol.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newell KA, Formica RN, Gill JS, et al. Integrating APOL1 gene variants into renal transplantation: considerations arising from the American Society of Transplantation Expert Conference. Am J Transplant. 2017;17:901–911. doi: 10.1111/ajt.14173. [DOI] [PubMed] [Google Scholar]
- 44.Boudville N, Prasad GVR, Knoll GA, et al. Meta-analysis risk for hypertension in living kidney donors. Ann Intern Med. 2006;145:185–196. doi: 10.7326/0003-4819-145-3-200608010-00006. [DOI] [PubMed] [Google Scholar]
- 45.Lentine KL, Schnitzler MA, Garg AX, et al. Understanding antihypertensive medication use after living kidney donation through linked national registry and pharmacy claims data. Am J Nephrol. 2014;40:74–183. doi: 10.1159/000365157. [DOI] [PubMed] [Google Scholar]
- 46.Lentine KL, Schnitzler MA, Xiao H, et al. Racial variation in medical outcomes among living kidney donors. N Engl J Med. 2010;363:724–732. doi: 10.1056/NEJMoa1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lentine KL, Schnitzler MA, Xiao H, et al. Consistency of racial variation in medical outcomes among publicly and privately insured living kidney donors. Transplantation. 2014;97:316–324. doi: 10.1097/01.TP.0000436731.23554.5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doshi MD, Goggins MO, Li L, Garg AX. Medical outcomes in African American live kidney donors: a matched cohort study. Am J Transplant. 2013;13:111–118. doi: 10.1111/j.1600-6143.2012.04303.x. [DOI] [PubMed] [Google Scholar]
- 49.Garg AX, Prasad GV, Thiessen-Philbrook HR, et al. Donor Nephrectomy Outcomes Research (DONOR) Network: Cardiovascular disease and hypertension risk in living kidney donors: an analysis of health administrative data in Ontario, Canada. Transplantation. 2008;86:399–406. doi: 10.1097/TP.0b013e31817ba9e3. [DOI] [PubMed] [Google Scholar]
- 50.Yazawa M, Kido R, Shibagaki Y, et al. Kidney function, albuminuria and cardiovascular risk factors in post-operative living kidney donors: a single-center, cross-sectional study. Clin Exp Nephrol. 2011;15:514–521. doi: 10.1007/s10157-011-0441-1. [DOI] [PubMed] [Google Scholar]
- 51.Ramesh Prasad GV, Lipszyc D, Sarker S, Huang M, Nash MM, Rapi L. Twenty four-hour ambulatory blood pressure profiles 12 months post living kidney donation. Transpl Int. 2010;23:771–776. doi: 10.1111/j.1432-2277.2009.01040.x. [DOI] [PubMed] [Google Scholar]
- 52.DeLoach SS, Meyers KE, Townsend RR. Living donor kidney donation: another form of white coat effect. Am J Nephrol. 2012;35:75–79. doi: 10.1159/000335070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yalin SF, Trabulus S, Seyahi N, Cengiz M, Cicik ME, Altiparmak MR. Ambulatory Blood Pressure Monitoring in living kidney donors: what changes in ten years? Clin Transplant. 2018;32:e13224. doi: 10.1111/ctr.13224. [DOI] [PubMed] [Google Scholar]
- 54.Moody WE, Tomlinson LA, Ferro CJ, et al. Effect of a reduction in glomerular filtration rate after nephrectomy on arterial stiffness and central hemodynamics: rationale and design of the EARNEST study. Am Heart J. 2014;167:141–149. doi: 10.1016/j.ahj.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mora-Fernández C, Domínguez-Pimentel V, de Fuentes MM, Górriz JL, Martínez-Castelao A, Navarro-González JF. Diabetic kidney disease: from physiology to therapeutics. J Physiol. 2014;592:3997–4012. doi: 10.1113/jphysiol.2014.272328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR, UKPDS GROUP Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64) Kidney Int. 2003;63:225–232. doi: 10.1046/j.1523-1755.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- 57.Premaratne E, Verma S, Ekinci EI, Theverkalam G, Jerums G, MacIsaac RJ. The impact of hyperfiltration on the diabetic kidney. Diabetes Metab. 2015;41:5–17. doi: 10.1016/j.diabet.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Tonneijck L, Muskiet MH, Smits MM, et al. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J Am Soc Nephrol. 2017;28:1023–1039. doi: 10.1681/ASN.2016060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christiansen JS, Gammelgaard J, Frandsen M, Parving HH. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetologia. 1981;20:451–456. doi: 10.1007/BF00253406. [DOI] [PubMed] [Google Scholar]
- 60.Nelson RG, Bennett PH, Beck GJ, et al. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med. 1996;335:1636–1642. doi: 10.1056/NEJM199611283352203. [DOI] [PubMed] [Google Scholar]
- 61.Denic A, Lieske JC, Chakkera HA, et al. The substantial loss of nephrons in healthy human kidneys with aging. J Am Soc Nephrol. 2017;28:313–320. doi: 10.1681/ASN.2016020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan JC, Busque S, Workeneh B, et al. Effects of aging on glomerular function and number in living kidney donors. Kidney Int. 2010;78:686–692. doi: 10.1038/ki.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Locke JE, Reed RD, Massie A, et al. Obesity increases the risk of end-stage renal disease among living kidney donors. Kidney Int. 2017;91:699–703. doi: 10.1016/j.kint.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chagnac A, Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol Renal Physiol. 2000;278:F817–822. doi: 10.1152/ajprenal.2000.278.5.F817. [DOI] [PubMed] [Google Scholar]
- 65.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 66.Wang JT, Evans JC, Meigs JB, et al. Low-grade albuminuria and the risks of hypertension and blood pressure progression. Circulation. 2005;111:1370–1376. doi: 10.1161/01.CIR.0000158434.69180.2D. [DOI] [PubMed] [Google Scholar]
- 67.Christinasen JS, Gammelgaard J, Frandsen M, Parving HH. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetlogia. 1981;20:451–456. doi: 10.1007/BF00253406. [DOI] [PubMed] [Google Scholar]
- 68.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure: less of one, more the other? Am J Hypertens. 1988;1:335–347. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 69.Ingelfinger JR. Is microanatomy destiny? N Engl J Med. 2003;348:99–100. doi: 10.1056/NEJMp020168. [DOI] [PubMed] [Google Scholar]
- 70.Neuringer JR, Brenner BM. Glomerular hypertension: cause and consequence of renal injury. J Hypertens. 1992;10:S91–S97. [PubMed] [Google Scholar]
- 71.Hallan S, Euser AM, Irgens LM, Finken MJ, Holmen J, Dekker FW. Effect of intrauterine growth restriction on kidney function at young adult age: the Nord Trøndelag Health (HUNT 2) Study. Am J Kidney Dis. 2008;51:10–20. doi: 10.1053/j.ajkd.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez MM, Gomez A, Abitbol C, Chandar J, Montané B, Zilleruelo G. Comparative renal histomorphometry: a case study of oligonephropathy of prematurity. Pediatr Nephrol. 2005;20:945–949. doi: 10.1007/s00467-004-1800-x. [DOI] [PubMed] [Google Scholar]
- 73.Abitbol CL, Bauer CR, Montané B, Chandar J, Duara S, Zilleruelo G. Long-term follow-up of extremely low birth weight infants with neonatal renal failure. Pediatr Nephrol. 2003;18:887–893. doi: 10.1007/s00467-003-1186-1. [DOI] [PubMed] [Google Scholar]
- 74.Abitbol CL, Ingelfinger JR. Nephron mass and cardiovascular and renal disease risks. Semin Nephrol. 2009;29:445–454. doi: 10.1016/j.semnephrol.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 75.Barker DJ, Bagby SP, Hanson MA. Mechanisms of disease: in utero programming in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:700–707. doi: 10.1038/ncpneph0344. [DOI] [PubMed] [Google Scholar]
- 76.Painter RC, Roseboom TJ, van Montfrans GA, et al. Microalbuminuria in adults after prenatal exposure to the Dutch famine. J Am Soc Nephrol. 2005;16:189–194. doi: 10.1681/ASN.2004060474. [DOI] [PubMed] [Google Scholar]
- 77.Hoy WE, Rees M, Kile E, Mathews JD, Wang Z. A new dimension to the Barker hypothesis: low birthweight and susceptibility to renal disease. Kidney Int. 1999;56:1072–1077. doi: 10.1046/j.1523-1755.1999.00633.x. [DOI] [PubMed] [Google Scholar]
- 78.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–941. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 79.Phipps K, Barker DJ, Hales CN, Fall CH, Osmond C, Clark PM. Fetal growth and impaired glucose tolerance in men and women. Diabetologia. 1993;36:225–228. doi: 10.1007/BF00399954. [DOI] [PubMed] [Google Scholar]
- 80.Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2013 Annual Data Report: kidney. Am J Transplant. 2015;15:1–34. doi: 10.1111/ajt.13195. [DOI] [PubMed] [Google Scholar]
- 81.Pascual M, Theruvath T, Kawai T, Tolkoff-Rubin N, Cosimi AB. Strategies to improve long-term outcomes after renal transplantation. N Engl J Med. 2002;346:580–590. doi: 10.1056/NEJMra011295. [DOI] [PubMed] [Google Scholar]
- 82.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011;11:450–462. doi: 10.1111/j.1600-6143.2010.03283.x. [DOI] [PubMed] [Google Scholar]
- 83.Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant. 2012;12:1924–1928. doi: 10.1111/j.1600-6143.2012.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reeves-Daniel AM, Depalma JA, Bleyer AJ, et al. The APOL1 Gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11:1025–1030. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freedman BI, Pastan SO, Israni AK, et al. APOL1 genotype and kidney transplantation outcomes from deceased African American donors. Transplantation. 2016;100:194–202. doi: 10.1097/TP.0000000000000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant. 2015;15:1615–1622. doi: 10.1111/ajt.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol. 2015;26:258–269. doi: 10.1681/ASN.2014030278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kriz W, Lemley KV. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017;91:1283–1286. doi: 10.1016/j.kint.2017.02.032. [DOI] [PubMed] [Google Scholar]
- 89.Endlich N, Endlich K. The challenge and response of podocytes to glomerular hypertension. Semin Nephrol. 2012;32:327–341. doi: 10.1016/j.semnephrol.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 90.Sharma M, Sharma R, McCarthy ET, Savin VJ, Srivastava T. Hyperfiltration-associated biomechanical forces in glomerular injury and response: Potential role for eicosanoids. Prostaglandins Other Lipid Mediat. 2017;132:59–68. doi: 10.1016/j.prostaglandins.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Srivastava T, Thiagarajan G, Alon US, et al. Role of biomechanical forces in hyperfiltration-mediated glomerular injury in congenital anomalies of the kidney and urinary tract. Nephrol Dial Transplant. 2017;32:759–765. doi: 10.1093/ndt/gfw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Endlich K, Kliewe F, Endlich N. Stressed podocytes-mechanical forces, sensors, signaling and response. Pflugers Arch. 2017;469:937–949. doi: 10.1007/s00424-017-2025-8. [DOI] [PubMed] [Google Scholar]
- 93.Pichler Sekulic S, Sekulic M. Rheological influence upon the glomerular podocyte and resultant mechanotransduction. Kidney Blood Press Res. 2015;40:176–187. doi: 10.1159/000368493. [DOI] [PubMed] [Google Scholar]
- 94.Endlich N, Kress KR, Reiser J, et al. Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol. 2001;12:413–422. doi: 10.1681/ASN.V123413. [DOI] [PubMed] [Google Scholar]
- 95.Coers W, Vos JT, Huitema S, Dijk F, Weening JJ. Biological alterations of rat podocytes cultured under basolateral hydrostatic pressure. Pathobiology. 1996;64:222–232. doi: 10.1159/000164052. [DOI] [PubMed] [Google Scholar]
- 96.Anderson M, Kim EY, Hagmann H, Benzing T, Dryer SE. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am J Physiol Cell Physiol. 2013;305:C276–C289. doi: 10.1152/ajpcell.00095.2013. [DOI] [PubMed] [Google Scholar]
- 97.Glogauer M, Ferrier J. A new method for application of force to cells via ferric oxide beads. Pflugers Arch. 1998;435:320–327. doi: 10.1007/s004240050518. [DOI] [PubMed] [Google Scholar]
- 98.Srivastava T, Alon US, Cudmore PA, et al. Cyclooxygenase-2, Prostaglandin E2 and Prostanoid receptor EP2 in fluid flow shear stress mediated injury in solitary kidney. Am J Physiol Renal Physiol. 2014;307:F1323–1333. doi: 10.1152/ajprenal.00335.2014. [DOI] [PubMed] [Google Scholar]
- 99.Friedrich C, Endlich N, Kriz W, Endlich K. Podocytes are sensitive to fluid shear stress in vitro. Am J Physiol Renal Physiol. 2006;291:F856–F865. doi: 10.1152/ajprenal.00196.2005. [DOI] [PubMed] [Google Scholar]
- 100.Srivastava T, McCarthy ET, Sharma R, et al. Prostaglandin E2 is crucial in the podocytes response to fluid flow shear stress. J Cell Commun Signal. 2010;4:79–90. doi: 10.1007/s12079-010-0088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huang C, Bruggeman LA, Hydo LM, Miller RT. Shear stress induces cell apoptosis via a c-src phospholipase D-mTOR pathway in cultured podocytes. Exp Cell Res. 2012;318:1075–1085. doi: 10.1016/j.yexcr.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Srivastava T, McCarthy ET, Sharma R, et al. Fluid flow shear stress upregulates prostanoid receptor EP2 but not EP4 in murine podocytes. Prostaglandins Other Lipid Mediat. 2013;104–105:49–57. doi: 10.1016/j.prostaglandins.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 103.Srivastava T, Celsi GE, Sharma M, et al. Fluid flow shear stress over podocytes is increased in the solitary kidney. Nephrol Dial Transplant. 2014;29:65–72. doi: 10.1093/ndt/gft387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Durvasula RV, Petermann AT, Kiromura K, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 105.Liu D, Wang LN, Li HX, Huang P, Qu LB, Chen FY. Pentoxifylline plus ACEIs/ARBs for proteinuria and kidney function in chronic kidney disease: a meta-analysis. J Int Med Res. 2017;45(2):383–398. doi: 10.1177/0300060516663094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen J, Huang YM, Song XN, et al. Protection against death and renal failure by renin-angiotensin system blockers in patients with diabetes and kidney disease. J Renin Angiotensin Aldosterone Syst. 2016;17 doi: 10.1177/1470320316656481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao HJ, Li Y, Liu SM, et al. Effect of calcium channels blockers and inhibitors of the renin-angiotensin system on renal outcomes and mortality in patients suffering from chronic kidney disease: systematic review and meta-analysis. Ren Fail. 2016;38:849–856. doi: 10.3109/0886022X.2016.1165065. [DOI] [PubMed] [Google Scholar]
- 108.Qin Y, Chen T, Chen Q, et al. The effect of angiotensin-converting enzyme inhibitor/angiotensin receptor blocker use on mortality in patients with chronic kidney disease: a meta-analysis of observational studies. Pharmacoepidemiol Drug Saf. 2016;25:503–511. doi: 10.1002/pds.3941. [DOI] [PubMed] [Google Scholar]
- 109.Anderson S, Brenner BM. Therapeutic benefit of converting-enzyme inhibition in progressive renal disease. Am J Hypertens. 1988;1(4 Pt 2):380S–383S. doi: 10.1093/ajh/1.4.380s. [DOI] [PubMed] [Google Scholar]
- 110.Anderson S, Rennke HG, Brenner BM. Antihypertensive therapy must control glomerular hypertension to limit glomerular injury. J Hypertens Suppl. 1986;4:S242–S244. [PubMed] [Google Scholar]
- 111.Anderson S, Rennke HG, Brenner BM. Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest. 1986;77:1993–2000. doi: 10.1172/JCI112528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anderson S, Meyer TW, Rennke HG, Brenner BM. Control of glomerular hypertension limits glomerular injury in rats with reduced renal mass. J Clin Invest. 1985;76:612–619. doi: 10.1172/JCI112013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martineau LC, McVeigh LI, Jasmin BJ, Kennedy CR. p38 MAP kinase mediates mechanically induced COX-2 and PG EP4 receptor expression in podocytes: implications for the actin cytoskeleton. Am J Physiol Renal Physiol. 2004;286:F693–701. doi: 10.1152/ajprenal.00331.2003. [DOI] [PubMed] [Google Scholar]
- 114.Faour WH, Thibodeau JF, Kennedy CR. Mechanical stretch and prostaglandin E2 modulate critical signaling pathways in mouse podocytes. Cell Signal. 2010;22:1222–1230. doi: 10.1016/j.cellsig.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 115.Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet. 2015;385:2047–2056. doi: 10.1016/S0140-6736(14)62459-4. [DOI] [PubMed] [Google Scholar]
- 116.Ren F, Tang L, Cai Y, et al. Meta-analysis: the efficacy and safety of combined treatment with ARB and ACEI on diabetic nephropathy. Ren Fail. 2015;37:548–561. doi: 10.3109/0886022X.2015.1012995. [DOI] [PubMed] [Google Scholar]
- 117.Catalá-López F, Macías Saint-Gerons D, González-Bermejo D, et al. Cardiovascular and Renal Outcomes of Renin-Angiotensin System Blockade in Adult Patients with Diabetes Mellitus: A Systematic Review with Network Meta-Analyses. PLoS Med. 2016;13:e1001971. doi: 10.1371/journal.pmed.1001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ruggenenti P, Perna A, Gherardi G, et al. Renoprotective properties of ACE inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet. 1999;354:359–364. doi: 10.1016/S0140-6736(98)10363-X. [DOI] [PubMed] [Google Scholar]
- 119.MacKinnon M, Shurraw S, Akbari A, Knoll GA, Jaffey J, Clark HD. Combination therapy with an angiotensin receptor blocker and an ACE inhibitor in proteinuric renal disease: a systematic review of the efficacy and safety data. Am J Kidney Dis. 2006;48:8–20. doi: 10.1053/j.ajkd.2006.04.077. [DOI] [PubMed] [Google Scholar]
- 120.Susantitaphong P, Sewaralthahab K, Balk EM, Eiam-ong S, Madias NE, Jaber BL. Efficacy and safety of combined vs. single renin-angiotensin-aldosterone system blockade in chronic kidney disease: a meta-analysis. Am J Hypertens. 2013;26:424–441. doi: 10.1093/ajh/hps038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Adarkwah CC, Gandjour A, Akkerman M, Evers S. To treat or not to treat? Cost-effectiveness of ace inhibitors in non-diabetic advanced renal disease – a Dutch perspective. Kidney Blood Press Res. 2013;37:168–180. doi: 10.1159/000350142. [DOI] [PubMed] [Google Scholar]
- 122.Mason NA. Angiotensin-converting enzyme inhibitors and renal function. DICP. 1990;24:496–505. doi: 10.1177/106002809002400511. [DOI] [PubMed] [Google Scholar]
- 123.Hricik DE, Browing PJ, Kopelman R, Goomo WE, Madias NE, Dzau VJ. Captopril induced functional renal insufficiency in patients with bilateral renal artery stenosis or renal artery stenosis in a solitary kidney. N Engl Med. 1983;308:373–376. doi: 10.1056/NEJM198302173080706. [DOI] [PubMed] [Google Scholar]
- 124.Hiremath S, Fergusson D, Doucette S, Mulay AV, Knoll GA. Renin angiotensin system blockade in kidney transplantation: a systematic review of the evidence. Am J Transplant. 2007;7:2350–2360. doi: 10.1111/j.1600-6143.2007.01928.x. [DOI] [PubMed] [Google Scholar]
- 125.Knoll GA, Fergusson D, Chassé M, et al. Ramipril versus placebo in kidney transplant patients with proteinuria: a multicentre, double-blind, randomised controlled trial. Lancet Diabetes Endocrinol. 2016;4:318–326. doi: 10.1016/S2213-8587(15)00368-X. [DOI] [PubMed] [Google Scholar]
- 126.Morales JM, Marcén R, Andrés A, et al. Renal transplantation in the modern immunosuppressive era in Spain: four-year results from a multicenter database focus on post-transplant cardiovascular disease. Kidney Int Suppl. 2008;111:S94–S99. doi: 10.1038/ki.2008.547. [DOI] [PubMed] [Google Scholar]
- 127.Dahlberg R, Muth B, Samaniego M, Hofmann RM, Pirsch J, Djamali A. One-year serum albumin is an independent predictor of outcomes in kidney transplant recipients. J Ren Nutr. 2010;20:392–397. doi: 10.1053/j.jrn.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Opelz G, Zeier M, Laux G, Morath C, Döhler B. No improvement of patient or graft survival in transplant recipients treated with angiotensin-converting enzyme inhibitors or angiotensin II type 1 receptor blockers: a collaborative transplant study report. J Am Soc Nephrol. 2006;17:3257–3262. doi: 10.1681/ASN.2006050543. [DOI] [PubMed] [Google Scholar]
- 129.Cross NB, Webster AC. Angiotensin-converting enzyme inhibitors-beneficial effects seen in many patient groups may not extend to kidney transplant recipients. Transplantation. 2016;100:472–473. doi: 10.1097/TP.0000000000001140. [DOI] [PubMed] [Google Scholar]
- 130.Heinze G, Mitterbauer C, Regele H, et al. Angiotensin-converting enzyme inhibitor or angiotensin II type 1 receptor antagonist therapy is associated with prolonged patient and graft survival after renal transplantation. J Am Soc Nephrol. 2006;17:889–899. doi: 10.1681/ASN.2005090955. [DOI] [PubMed] [Google Scholar]
- 131.Hernández D, Muriel A, Abraira V, et al. Renin-angiotensin system blockade and kidney transplantation: a longitudinal cohort study. Nephrol Dial Transplant. 2012;27:417–422. doi: 10.1093/ndt/gfr276. [DOI] [PubMed] [Google Scholar]
- 132.Rossi AP, Vella JP. Hypertension, living kidney donors, and transplantation: where are we today? Adv Chronic Kidney Dis. 2015;22:154–164. doi: 10.1053/j.ackd.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 133.Hiremath S, Fergusson DA, Fergusson N, Bennett A, Knoll GA. Renin-Angiotensin System Blockade and Long-term Clinical Outcomes in Kidney Transplant Recipients: A Meta-analysis of Randomized Controlled Trials. Am J Kidney Dis. 2017;69:78–86. doi: 10.1053/j.ajkd.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 134.Sasongko TH, Nagalla S, Ballas SK. Angiotensin-converting enzyme (ACE) inhibitors for proteinuria and microalbuminuria in people with sickle cell disease. Cochrane Database Syst Rev. 2015;6:CD009191. doi: 10.1002/14651858.CD009191.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Roy NB, Fortin PM, Bull KR, et al. Interventions for chronic kidney disease in people with sickle cell disease. Cochrane Database Syst Rev. 2017;7:CD012380. doi: 10.1002/14651858.CD012380.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371:2255–2266. doi: 10.1056/NEJMoa1402685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371:2267–2276. doi: 10.1056/NEJMoa1402686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ardissino G, Viganò S, Testa S, et al. ItalKid Project No clear evidence of ACEi efficacy on the progression of chronic kidney disease in children with hypodysplastic nephropathy-report from the ItalKid Project database. Nephrol Dial Transplant. 2007;22:2525–2530. doi: 10.1093/ndt/gfm237. [DOI] [PubMed] [Google Scholar]
- 139.Srivastava T, Dai H, Heruth DP, et al. Mechanotransduction Signaling in Podocytes from Fluid Flow Shear Stress. Am J Physiol Renal Physiol. 2018;314:F22–F34. doi: 10.1152/ajprenal.00325.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang JL, Cheng HF, Zhang MZ, McKanna JA, Harris RC. Selective increase of cyclooxygenase-2 expression in a model of renal ablation. Am J Physiol. 1998;275(4 Pt 2):F613–F622. doi: 10.1152/ajprenal.1998.275.4.F613. [DOI] [PubMed] [Google Scholar]
- 141.Nath KA, Chmielewski DH, Hostetter TH. Regulatory role of prostanoids in glomerular microcirculation of remnant nephrons. Am J Physiol. 1987;252(5 Pt 2):F829–F837. doi: 10.1152/ajprenal.1987.252.5.F829. [DOI] [PubMed] [Google Scholar]
- 142.Stahl RA, Kudelka S, Paravicini M, Schollmeyer P. Prostaglandin and thromboxane formation in glomeruli from rats with reduced renal mass. Nephron. 1986;42:252–257. doi: 10.1159/000183676. [DOI] [PubMed] [Google Scholar]
- 143.Griffin KA, Bidani AK, Picken M, Ellis VR, Churchill PC. Prostaglandins do not mediate impaired autoregulation or increased rennin secretion in remnant rat kidneys. Am J Physiol. 1992;263:F1057–F1062. doi: 10.1152/ajprenal.1992.263.6.F1057. [DOI] [PubMed] [Google Scholar]
- 144.Komers R, Lindsley JN, Oyama TT, Anderson S. Cyclo-oxygenase-2 inhibition attenuates the progression of nephropathy in uninephrectomized diabetic rats. Clin Exp Pharmacol Physiol. 2007;34:36–41. doi: 10.1111/j.1440-1681.2007.04534.x. [DOI] [PubMed] [Google Scholar]
- 145.Sanchez PL, Salgado LM, Ferreri NR, Escalante B. Effect of cyclooxygenase-2 inhibition on renal function after renal ablation. Hypertension. 1999;34(4 Pt 2):848–853. doi: 10.1161/01.hyp.34.4.848. [DOI] [PubMed] [Google Scholar]
- 146.Yao B, Xu J, Qi Z, Harris RC, Zhang MZ. Role of renal cortical cyclooxygenase-2 expression in hyperfiltration in rats with high-protein intake. Am J Physiol Renal Physiol. 2006;291:F368–374. doi: 10.1152/ajprenal.00500.2005. [DOI] [PubMed] [Google Scholar]
- 147.Horiba N, Kumano E, Watanabe T, Shinkura H, Sugimoto T, Inoue M. Subtotal nephrectomy stimulates cyclooxygenase 2 expression and prostacyclin synthesis in the rat remnant kidney. Nephron. 2002;91:134–141. doi: 10.1159/000057615. [DOI] [PubMed] [Google Scholar]
- 148.Ma X, Aoki T, Tsuruyama T, Narumiya S. Definition of Prostaglandin E2-EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth. Cancer Res. 2015;75:2822–2832. doi: 10.1158/0008-5472.CAN-15-0125. [DOI] [PubMed] [Google Scholar]
- 149.Ganesh T. Prostanoid receptor EP2 as a therapeutic target. J Med Chem. 2014;57:4454–4465. doi: 10.1021/jm401431x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Baldelomar EJ, Charlton JR, Beeman SC, et al. Phenotyping by magnetic resonance imaging nondestructively measures glomerular number and volume distribution in mice with and without nephron reduction. Kidney Int. 2016;89:498–505. doi: 10.1038/ki.2015.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ju W, Nair V, Smith S, et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med. 2015;7:316ra193. doi: 10.1126/scitranslmed.aac7071. [DOI] [PMC free article] [PubMed] [Google Scholar]