

Abstract
Wound healing is essential for skin repair after injury, and consists of hemostasis, inflammation, re-epithelialization and remodeling phases. Successful re-epithelialization, which relies on proliferation and migration of epidermal keratinocytes, requires reduction in tissue inflammation. Therefore, understanding the molecular mechanism underlying the transition from inflammation to re-epithelialization will help to better understand the principles of wound healing. Currently, the in vivo functions of specific microRNAs in wound healing arenot fully understood. We observed that miR-31 expression is strongly induced in wound edge keratinocytes, and is directly regulated by the activity of NF-κB and STAT3 signaling pathways during inflammation phase. We utilized miR-31 loss-of-function mouse models to demonstrate that miR-31 promotes keratinocyte proliferation and migration. Mechanistically, miR-31 activates the RAS/MAPK signaling by directly targeting Rasa1, Spred1, Spred2 and Spry4, which are negative regulators of the RAS/MAPK pathway. Knockdown of these miR-31 targets at least partially rescues the delayed scratch wound re-epithelialization phenotype observed in vitro in miR-31 knockdown keratinocytes. Taken together, these findings identify miR-31 as an important cell-autonomous mediator during the transition from inflammation to re-epithelialization phases of wound healing, suggesting a therapeutic potential for miR-31 in skin injury repair.
Keywords: microRNA, keratinocyte proliferation and migration, wound healing, miR-31, RAS/MAPK
INTRODUCTION
Wound healing is the physiological response of skin to injury, which restores its integrity and some of its functions. Wound healing consists of hemostasis, inflammation, re-epithelialization and remodeling phases (Reinke and Sorg, 2012). Acute wounds are able to heal rapidly by properly executing all of the phases. However, repair process becomes defective in chronicwounds, which fail to heal and pose an increasingly urgent clinical challenge worldwide (Mustoe et al., 2006). Elucidating the molecular mechanism underlying normal wound healing can help understand the principle of skin repair and provide new insight into the pathogenesis of chronic wounds. Re-epithelialization is the crucial phase during wound healing (Pastar et al., 2014), when keratinocytes migrate from the edge towards the wound center, and form new epidermis over the provisional dermal scar. Many molecular factors governing wound re-epithelialization have been identified, including integrins (Grinnell, 1992, Koivisto et al., 2014), matrix metalloproteinases (MMPs) (Krampert et al., 2004), growth factors (Le et al., 2012), cytokines (Werner and Grose, 2003) and extracellular matrix (ECM) (Zhou et al., 2016). However, several aspects of the mechanism for re-epithelialization, including the role that microRNAs (miRNAs) play, remain incompletely understood.
MiRNAs are ~22nt-long non-coding RNAs that negatively regulate gene expression by either degrading mRNA of their target genes or repressing their translation. It has been reported that microRNAs are extensively involves in a variety of normal and pathological biological processes (Fahs et al., 2015). Recently, miRNAs have been shown to modulate wound re-epithelialization by regulating pathway activities tied with keratinocyte proliferation and migration. Many microRNAs dynamically change during the wound healing process (Banerjee and Sen, 2015, Horsburgh et al., 2017). Among them, miR-31 is of great interest, because it has been implicated in promoting epidermal hyperplasia in psoriasis (Xu et al., 2013, Yan et al., 2015), epithelial stem cell regulation in the mammary gland (Lv et al., 2017) and intestine (Tian et al., 2017), and because it functions as an oncogene in lung and colorectal cancers (Edmonds, 2016, Sun et al., 2013). Furthermore, in vitro evidence suggests that miR-31 likely promotes skin wound healing in humans by directly regulating epithelial membrane protein-1 (EMP-1) (Li et al., 2015). However, the in vivo functions of miR-31 during the key phases of wound inflammation and re-epithelialization have not been comprehensively accessed.
Here we found that miR-31 is induced in wound edge keratinocytes in response to inflammatory signaling through the activity of NF-κB and STAT3 pathways. We utilized miR-31 loss-of-function mouse models to demonstrate that miR-31 is the key regulator in promoting migration and proliferation of keratinocytes by targeting RAS/MAPK signaling pathway. Our data identifies miR-31 as an important cell-intrinsic regulator of transition from inflammation to re-epithelialization phases in wound keratinocytes in vivo.
Results
MiR-31 expression pattern during skin wound healing.
We compared miR-31 expression in whole tissue between heart, skin, spleen, lung, liver, kidney and duodenum in mice and found that it is expressed in the skin at a moderate level, compatible to that in the lung and liver (Figure S1a). We then examined miR-31 expression levels in skin at different time points after wounding. MiR-31 expression increased in the wound edge tissue during the inflammatory phase (PWD 1–3) and reached its highest level during the proliferative phase as compared to its level two hours after wounding (Figure 1a). In situ hybridization revealed that miR-31 signal primarily localizes to the keratinocytes at the wound edge (Figure 1b). Normal epidermis away from the wound edge was largely negative. Positive in situ signal was also seen in some cells in the dermis.
Figure 1. MiR-31 is induced by the NF-κB and STAT3 signaling.
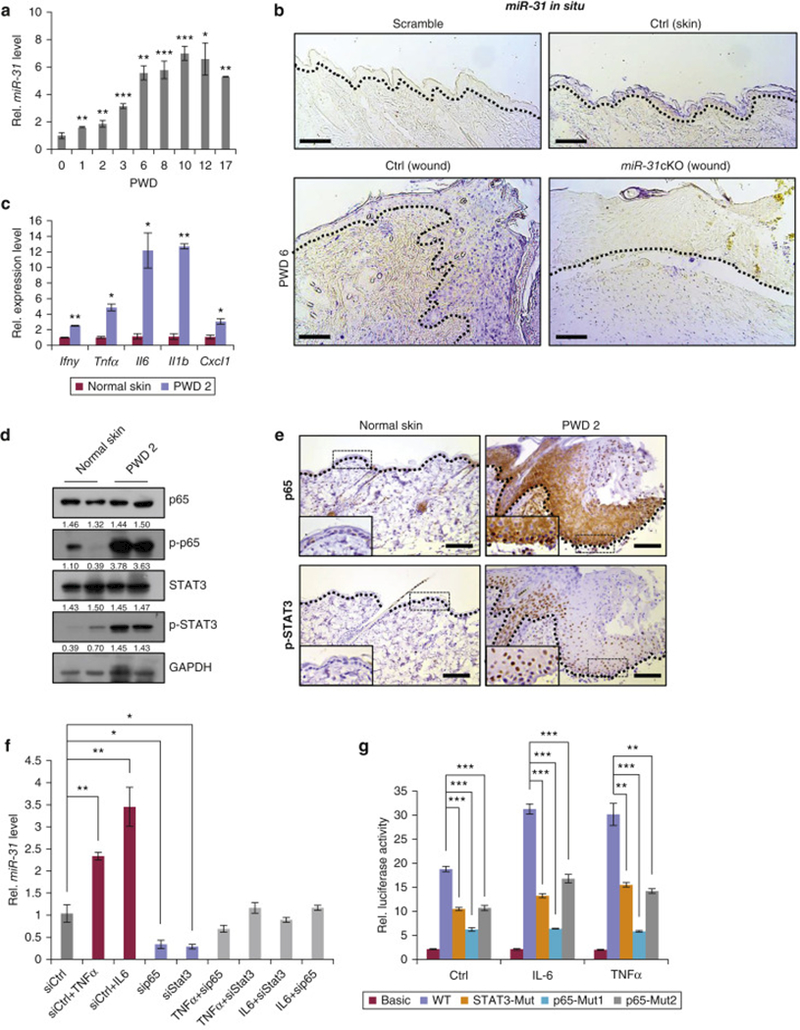
(a) qRT-PCR for miR-31 in the wound edge tissue at indicated time points. (b) In situ hybridization for miR-31 in skin wounds. Scale bar: 50 μm. (c-e) qRT-PCR analysis for Ifnγ, Tnfa, Il6, Il1b and Cxcl1 (c); Western blot for p65, p-p65, STAT3 and p-STAT3 (d); immunostaining for p65 and p-STAT3 (e) in skin wounds at PWD 2. (f) qRT-PCR for miR-31 in keratinocytes upon treatments of siCtrl, siCtrl+IL6, siCtrl+TNFα, sip65, siStat3, TNFα+sip65, TNFα+siStat3, IL6+siStat3 and IL6+sip65 for 24 hours. (g) Luciferase reporter assay for miR-31 promoter in HaCaT cells at indicated conditions. *p<0.05; **p <0.01; ***p<0.001. n ≥ 3 biological replicates for a,and c-h.
MiR-31 is directly regulated by the NF-κB and STAT3 signaling pathways.
Next, we examined how miR-31 is regulated after wounding. Our previous work has shown that miR-31 promoter contains p65 and STAT3 binding sites (Tian et al., 2017). Therefore, we tested if miR-31 is induced upon activation of NF-κB and STAT3 signaling pathways. Multiple inflammatory cytokines,including Ifnγ, Tnfα, Cxcl1, Il6 and Il1b were induced two days after wounding (Figure 1c) and NF-κB and STAT3 signaling pathways as judged by p-p65 and p-STAT3 levels respectively, were activated in the wound edge tissue (Figure 1d and 1e), where miR-31 also becomes highly induced. In agreement with miR-31 upregulation at PWD 6, we found that NF-κB and STAT3 signaling pathway are still activated at this time point (Figure S1b and S1c). It has also been reported that TNFα can activate NF-κB (Zhu et al., 2012), while IL-6 can activate STAT3 pathway (Venkatesan et al., 2017, Wang et al., 2003). Thus, we tested whether TNFα or IL-6 can trigger miR-31 expression in vitro. Indeed, both TNFα and IL-6 were able to induce miR-31 expression in mouse primary keratinocytes (Figure 1f), concomitant with upregulation of p-p65 and p-STAT3 (Figure S1d and S1e). In contrast, siRNA-mediated knockdown of p65 and Stat3 significantly repressed miR-31 expression, accompanied by the repression of NF-κB and STAT3 signaling (Figures 1f, S1f and S1g). We also found that both TNFα and IL-6 induced miR-31 up-regulation in HaCaT cells, while siRNA-mediated knockdown of p65 and Stat3 repressed miR-31 expression (Figure S1h and S1i). Moreover, luciferase reporter assay revealed that mutation of the p65 and STAT3 binding sites in miR-31 promoter suppressed luciferase activity. Consistently, upon TNFα (10 ng/mL) or IL-6 (20 ng/mL) stimulation, the above mutations also blocked luciferase activity induced by these cytokines (Figure 1g). Together, these data suggest that miR-31 expression is induced in keratinocytes after wounding by NF-κB and STAT3 signaling pathways.
Conditional deletion of miR-31 delays skin wound healing.
To investigate the function of miR-31 in wound healing in vivo, we generated K14-Cre;miR-31fl/fl (cKO) mice, in which miR-31 is specifically deleted in skin epithelia. The specificity and efficiency of miR-31 deletion in cKO epidermis was confirmed by qRT-PCR analysis, while no change in miR-31 levels was detected in cKO duodenum and liver (Figure 2a). MiR-31 cKO mice were fertile without apparent gross phenotypes. Skin histology in day P49 miR-31 cKO mice was comparable to that in littermate controls. There were no significant differences in epidermal thickness between cKO and control mice (Figure S2a). We also tested whether loss of miR-31 affects differentiation, apoptosis and cell proliferation. Immunofluorescence for K14, K10 and Loricrin showed that epidermal differentiation is not altered in P49 cKO epidermis (Figure S2b and S2c). Cleaved Caspase3 staining revealed no apoptotic cells at P49 both in cKO and control epidermis (Figure S2d). Immunofluorescence for PCNA and Ki67 showed that miR-31 deletion does not significantly affect proliferation dynamics in the basal layer of the epidermis (Figure S2e, S2f and S2g). Collectively, these findings suggest that miR-31 is not essential for key physiological functions of epidermis under normal conditions.
Figure 2. MiR-31 conditional deletion delays skin wound healing.
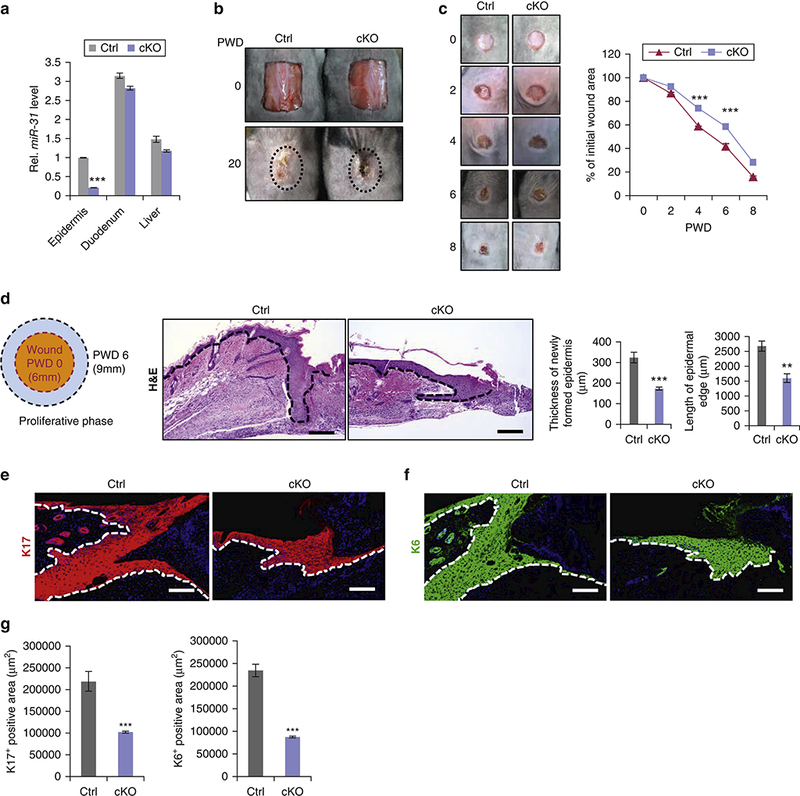
(a) qRT-PCR for miR-31 in epidermis, duodenum and liver of miR-31 cKO and control. (b) Representative images of 1×1.5 cm2 skin wounds on miR-31 cKO and control mice on PWD 0 and 20. (c) Representative images and quantification of wound sizes in cKO and control mice on PWD 0, 2, 4, 6 and 8 after 6mm injury. (d) Schematics showing wound tissue sample collection. Histology and quantification of newly formed epidermis in miR-31 cKO and control wounds on PWD 6. Scale bar: 50 μm. (e,f,g) Immunostaining for keratins K17 (e) and K6 (f) and their quantification (g) in miR-31 cKO and control wounds on PWD 6. *p<0.05; **p <0.01; n ≥ 3 biological replicates for a-g.
Considering prominent upregulation of miR-31 after wounding, we studied its role during skin wound healing. When animals were challenged with large, 1×1.5 cm2 full-thickness wounds, healing was delayed in miR-31 cKO mice as compared to control on PWD 20, evidenced by the retention of scab in mutants (n = 5 biological replicates for both control and cKO mice) (Figure 2b). We then compared healing dynamics of smaller, 6mm round wounds over time between cKO and control mice (n = 6 biological replicates for both control and cKO mice). Area measurements show that wound closure process is moderately, yet significantly delayed in cKO as compared to control mice (Figure 2c). On histology at PWD 6, epidermal edge was shorter and thinner in cKO mice (n = 6 biological replicates for both control and cKO mice) (Figure 2d), suggesting impaired keratinocyte proliferation and/or migration. Inagreement, analysis of tail skin wounds, which have minimal contraction (Falanga et al., 2004), also revealed delayed wound closure in cKO mice (n = 3 biological replicates for both control and cKO mice) (Figure S3a and S3b). Keratins K6 and K17 are normally induced in response to injury and are highly expressed in migrating keratinocytes near the wound edge (McGowan and Coulombe, 1998, Wevers et al., 1990). Consistently, we observed fewer K6- and K17- positive cells in cKO epidermis around the wound (Figure 2e, 2f and 2g). To understand the cellular basis of delayed wound healing in cKO mice, we examined proliferation by the wound edge keratinocytes on PWD 6. Following 90 min BrdU pulse, much fewer BrdU-positive keratinocytes were found in the cKO mice. Consistently, the numbers of Ki67-positive proliferating keratinocytes were also decreased in cKO mice (Figure 3a and S3c). Furthermore, WST-8 assays showed that miR-31 promotes proliferation of HaCaT keratinocytes in vitro (Figure 3b). These findings demonstrate that loss of miR-31 represses proliferation of keratinocytes around the wound edge.
Figure 3. MiR-31 promotes proliferation and migration of keratinocytes.
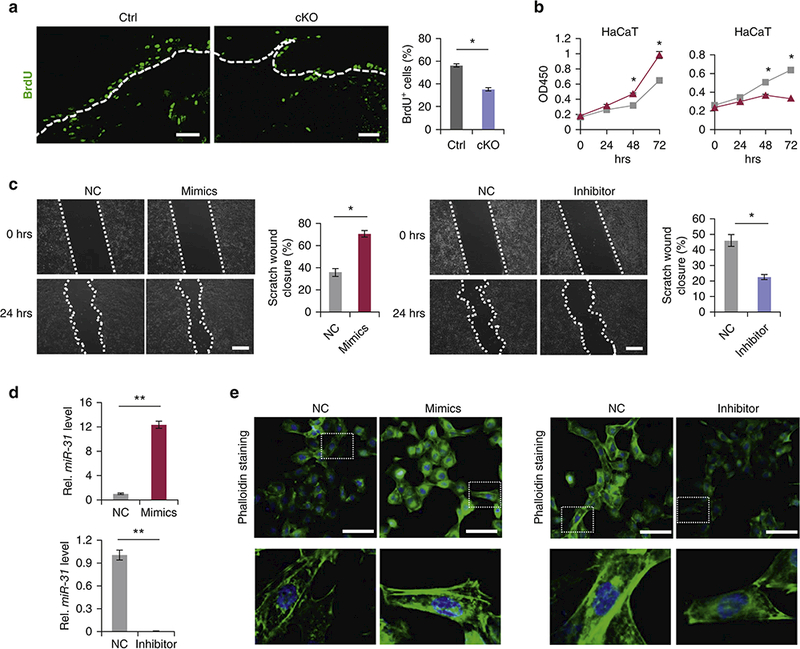
(a) Immunostaining for BrdU in the wound edge keratinocytes in miR-31 cKO and control wounds on PWD 6 following 90 min BrdU pulse. Scale bar: 25 μm. (b) WST-8 assay showing human HaCaT keratinocyte growth changes in response to miR-31 mimics (blue) and inhibitor (red). (c) In vitro scratch assay showing changes in migration potential of HaCaT cells in response to miR-31 mimics and inhibitor. (d) qRT-PCR for miR-31 to test the transfection efficiency for miR-31 mimics and inhibitor. **p < 0.01. (e) Phalloidin staining (green) in HaCaT keratinocytes transfected with miR-31 mimics (left) and inhibitor (right). Scale bar: 25 μm. n =3 biological replicates for a-e.
Next, we examined whether altered levels of miR-31 influences keratinocyte migrations. The in vitro scratch assay revealed that miR-31 inhibitor indeed significantly repress migration of HaCaT keratinocytes, whereas miR-31 mimics significantly promoted it (Figure 3c). High transfection efficiency for miR-31 mimics and inhibitor was confirmed by qRT-PCR analysis (Figure 3d). F-actin polymerizes to form a contractile cable at the cell’s leading edge, and drives epithelial sheet movement in a purse string manner (Martin and Parkhurst, 2004, Shimizu et al., 2005). In agreement with its role in promoting cell migration, phalloidin staining showed that miR-31 mimics upregulate F-actin fiber formation and, conversely, miR-31 inhibitor decreases it (Figure 3e). Taken together, these findings indicate that miR-31 accelerates the process of wound healing by promoting both the proliferation and migration of keratinocytes.
MiR-31 activates the RAS/MAPK (ERK1/2) pathway.
To gain a mechanistic insight into the molecular events underlying the delayed wound healing in cKO mice, we examined the effect of miR-31 alterations on the status of RAS/MAPK signaling pathway, previously shown to be essential for promoting cell proliferation and migration (Falanga et al., 2004, Lin et al., 2016). We examined phosphorylation status of ERK at the wound edge at PWD 6, and found p-ERK to be prominently downregulated in miR-31 cKO mice (Figure 4a). Cyclin D1 and c-Myc are the two downstream components of the RAS/MAPK (ERK1/2) pathway (Lavoie et al., 1996, Li et al., 2014, Luo et al., 2012). We observed that the expression levels for both Cyclin D1 and c-Myc were decreased at the edges of PWD 6 cKO wounds (Figure 4b and 4c). Slug, another downstream component of the RAS/MAPK pathway,which triggers epithelial-mesenchymal transition of keratinocytes (Aomatsu et al., 2012, Hwang et al., 2017, Kusewitt et al., 2009, Savagner, 2001), was also downregulated in miR-31 cKO wounds at PWD 6 (Figure 4d). Downregulation of Cyclin D1 (Ccdn1), c-Myc and Slug at the RNA level was further confirmed by qRT-PCR (Figure 4e). Western blot assay showed decreased phosphorylation of ERK, p-MEK, Cyclin D1, c-Myc and Slug in cKO mice (Figure 4f), supporting the notion that miR-31 upregulates RAS/MAPK pathway activity. To learn if miR-31 can activate RAS/MAPK pathway in vitro, we analyzed the effects of miR-31 alterations on RAS/MAPK signaling status in HaCaT cells. MiR-31 mimics increased the number of cells with nuclear p-ERK expression, while miR-31 inhibitor had an opposite effect (Figure 4g). Consistently, Western blot assay also showed that miR-31 mimics increased p-ERK protein level both with and without EGF addition, while miR-31 inhibitor decreased it (Figure 4h). Furthermore,protein levels of p-MEK, c-Myc, Cyclin D1 and Slug also increased after miR-31 mimics treatment, and reduced in response to miR-31 inhibitor (Figure 4i). U0126 is extensively used as the inhibitor of RAS/MAPK signaling (Marampon et al., 2011, Tong et al., 2003). To further test whether the effect of miR-31 on keratinocyte proliferation and migration is through the regulation of RAS/MAPK signaling pathway, we treated HaCaT cells in vitro under conditions of scrambled RNA (negative control, NC), miR-31mimics and miR-31 mimics with U0126. We found that U0126 repressed cell migration induced by miR-31 mimics (Figure S4a), concomitant with the repression of RAS/MAPK pathway (Figure S4b). In addition, WST-8 assay showed that U0126 at least partially attenuates increased cell proliferation caused by miR-31 mimics (Figure S4c). Collectively, our results demonstrate that miR-31 promotes wound healing by activating the RAS/MAPK signaling pathway.
Figure 4. MiR-31 activates RAS/MAPK signaling pathway.
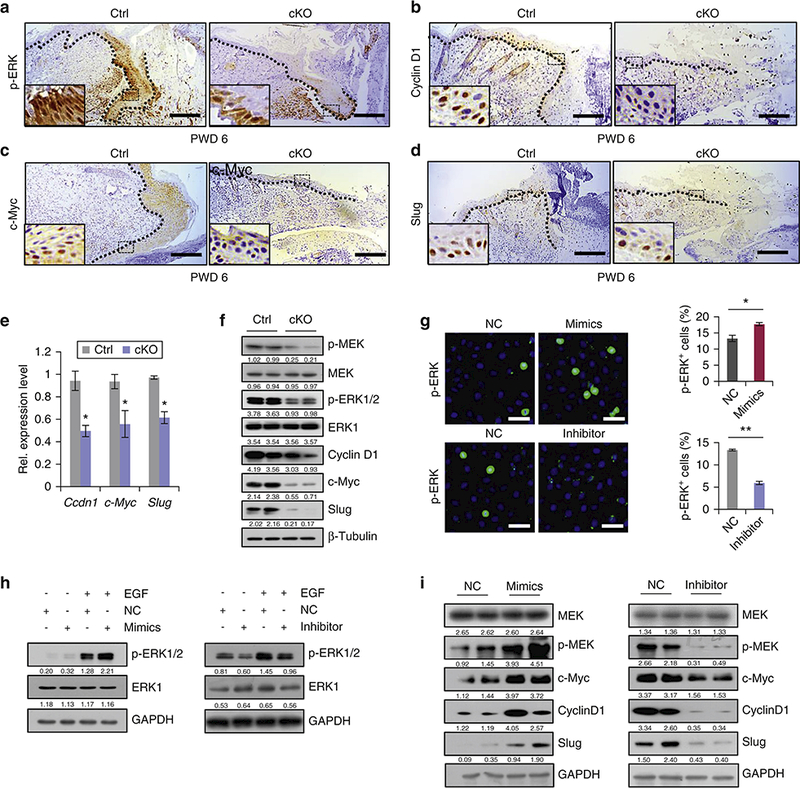
(a-d) Immunostaining for p-ERK, Cyclin D1, c-Myc and Slug in cKO wound edges on PWD 6. Scale bar: 50 μm. (e,f) qRT-PCR for Ccdn1, c-Myc and Slug, and Western blot for p-MEK, MEK, p-ERK1/2, ERK1, Cyclin D1, c-Myc and Slug in cKO wounds on PWD 6. (g) Immunostaining for p-ERK in HaCaT cells and quantification of p-ERK+ cells treated with miR-31 mimics or inhibitor. Scale bar: 25 μm. (h) Western blot for phosphorylated and total ERK in HaCaT cells transfected with miR-31 mimics or inhibitor. (i) Western blot for MEK, p-MEK, c-Myc, Cyclin D1 and Slug in HaCaT cells upon miR-31 mimics or inhibitor treatment. n =3 biological replicates for a-i. *p < 0.05; **p < 0.01.
MiR-31 directly targets negative regulators of RAS/MAPK signaling pathway.
MicroRNAs exert their functions by negatively regulating target genes. To identify miR-31 targets that may be responsible for its observed functionaleffects, we analyzed miR-31-binding sites in 3’UTRs of transcripts encoding negative regulators of RAS/MAPK pathway. Putative miR-31-binding sites were found in several genes including Rasa1, Spred1 and Spry4 (Figure S5a). We also aligned and analyzed the 3’UTR sequences of other family members of negative regulators of RAS/MAPK signaling, and found that 3’UTR of Spred2 also has a potential miR-31-binding region. To validate whether the above genes are actual targets of miR-31, first we examined their expression levels in wounds of miR-31 cKO mice. We found that Rasa1, Spred1, Spred2 and Spry4 are upregulated in cKO wounds both at the protein and RNA levels (Figure 5a and 5b). Further, we analyzed their expression levels in the HaCaT cells transfected with either miR-31 mimics or inhibitor. RASA1, SPRED1, SPRED2 and SPRY4 were downregulated in miR-31 mimics treated cells both at the protein and RNA levels (Figure 5c and 5d). Conversely, they were increased in response to miR-31 inhibitor (Figure 5c and 5e). To further test whether they are direct targets of miR-31, we constructed luciferase reporters for the Rasa1, Spred1, Spred2 and Spry4 3’UTRs, as well as reporter constructs in which the predicted miR-31 binding sites were mutated (Figure S5b). The luciferase reporter assay showed that miR-31 mimics significantly repressed luciferase activity of constructs containing Rasa1, Spred1, Spred2 and Spry4 3’UTR elements, while miR-31 inhibitor significantly up-regulated it.. Mutations in 3’UTRs abolished miR-31 binding and the repressive effect of miR-31 mimics, or the potentiating effect of miR-31 inhibitor (Figure 5f and 5g). Together, these results identify Rasa1, Spred1, Spred2 and Spry4 as the direct molecular targets of miR-31. In addition, we also found that , Emp-1, a previously reported target gene of miR-31 (Li et al., 2015) was significantly up-regulated in miR-31 cKO wounds (Figure S6). This suggests that miR-31 likely promotes wound healing via several pathways.
Figure 5. Identification of miR-31 direct targets.
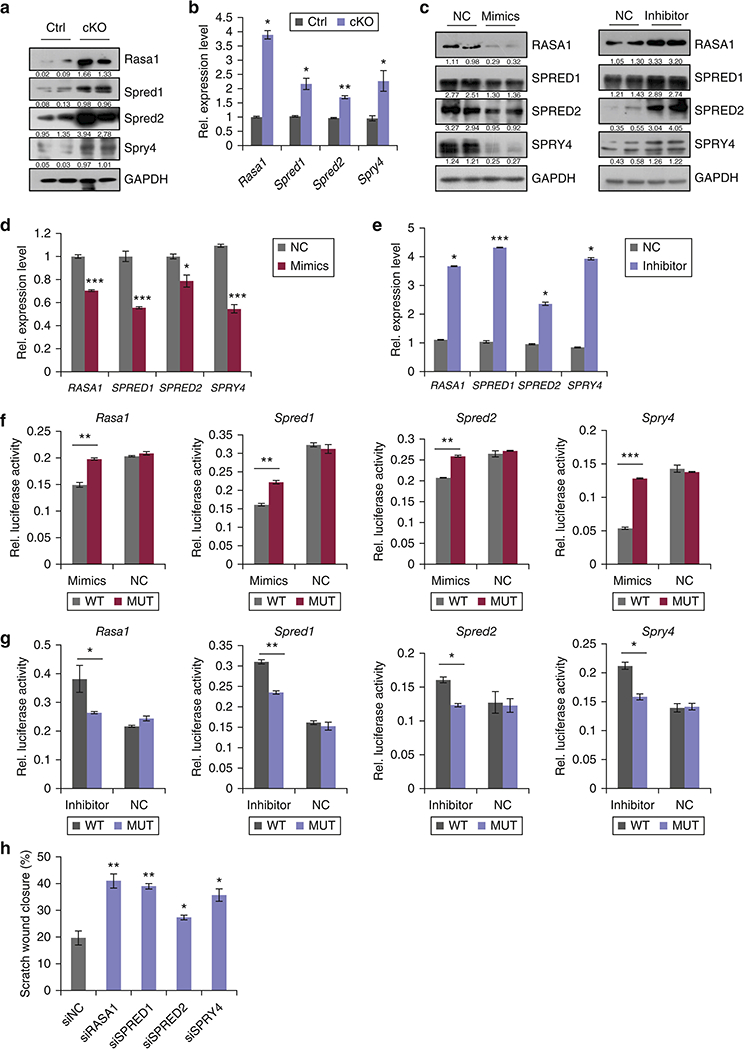
(a,b) Western blot (a) and qRT-PCR (b) for Rasa1, Spred1, Spred2 and Spry4 in miR-31 cKO and control wounds on PWD 6. (c-e) Western blot for RASA1, SPRED1, SPRED2 and SPRY4 (c), and qRT-PCR for Rasa1, Spred1, Spred2 and SPRY4 (d,e) in HaCaT cells transfected with miR-31 mimics or inhibitor. (f,g) Luciferase reporter activity of WT and mutant 3’UTR constructs of Rasa1, Spred1, Spred2 and Spry4 in HaCaT cells transfected with miR-31 mimics (f) and miR-31 inhibitor (g). (h) Statistical analysis on wound closure in in vitro scratch assay shown in supplementary Figure S6c. n =3 biological replicates for a-h. *p< 0.05;**p<0.01;***p<0.001.
Knockdown of Rasa1, Spred1, Spred2 and SPRY4 phenocopies the effect of miR-31 overexpression.
Next, we asked whether RASA1, SPRED1, SPRED2 and SPRY4 functionally mediate the observed biological function of miR-31. First, we generated siRNAs of RASA1, SPRED1, SPRED2 and SPRY4. ThesesiRNAs efficiently repressed their gene expression both at the protein and RNA levels (Figure S7a and S7b), and upregulated p-ERK level (Figure S7b), suggesting activation of RAS/MAPK pathway. In vitro scratch and WST-8 assays showed that inhibition of Rasa1, Spred1, Spred2 and SPRY4 with siRNAs significantly promotes cell migration and proliferation (Figure 5h, S7c and S7d).
We also tested whether silencing of these targets is able to rescue the miR-31 loss phenotypes. Indeed, siRNA-mediated silencing of any one of these targets was sufficient to rescue reduced cell migration and proliferation caused by miR-31 inhibitor (Figure 6a-6c). Thus, our results reveal that Rasa1, Spred1, Spred2 and SPRY4 are functional targets of miR-31, mediating its wound healing promoting effect. In summary, our findings demonstrate that inflammation-induced miR-31 drives keratinocyte proliferation and migration via RAS/MAPK signaling pathway, acting to promote wound re-epithelialization.
Figure 6. Repression of Rasa1, Spred1, Spred2 and SPRY4 can rescue the effects of miR-31 knockdown in keratinocytes.
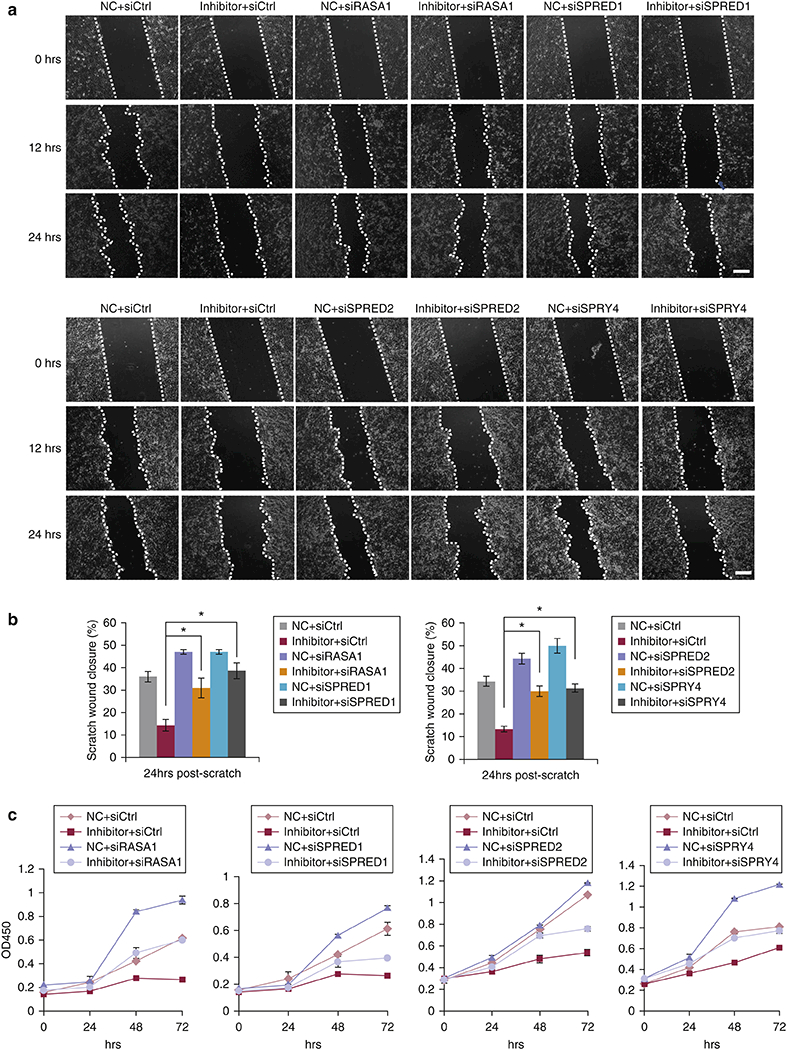
(a) In vitro scratch assays assessing the migration rate of HaCaT cells transfected with miR-31 inhibitor, inhibitor control and/or siRNAs for Rasa1, Spred1, Spred2 or SPRY4 for 24 hours. Representative images are shown at indicated time points. (b) Statistical analysis of scratch assay wound closure from panel (a). (c) WST-8 assays on HaCaT cells transfected with miR-31 inhibitor, inhibitor control and/or siRNA for Rasa1, Spred1, Spred2 or SPRY4. n = 3 biological replicates for a-c. ** p < 0.01 and * p < 0.05.
DISCUSSION
In this study, we utilized a miR-31 loss-of-function mouse model to demonstrate that inflammatory signals-responsive miR-31 promotes keratinocyte proliferation and migration during wound healing by directly regulating RAS/MAPK signaling pathway. To our knowledge, this study is the first to demonstrate the in vivo, physiological role of miR-31 in regulating wound healing using loss-of-function mouse model. Previously, Li et al. showed that miR-31 becomes upregulated in wound edge keratinocytes in human volunteers (Li et al., 2015). Using in vitro assays, Li et al. also showed that miR-31 promotes primary human keratinocyte proliferation and migration, and that miR-31 directly targets epithelial membrane protein 1 (EMP-1). Our mouse findings corroborate the expression pattern and the effect of miR-31 on wound edge keratinocytes, provide crucial in vivo functional data from the in miR-31 cKO animal model, and also identify several functionally important miR-31 targets. Together with Li et al’s findings, they demonstrate that miR-31 is able to promote wound healing via several pathways, significantly strengthening the evidence for the importance of miR-31 in wound healing.
Wound healing comprises four distinct but overlapping phases: hemostasis, inflammation, proliferation and remodeling (Gurtner et al., 2008). After skin wounding, epidermal keratinocytes produce inflammatory cytokines to activate immune response during the inflammatory phase (Strbo et al., 2014, Wilson et al., 2001). Our results also suggest that miR-31 starts to up-regulated at the leading migrating keratinocytes of wound edge in the inflammatory phase. Our molecular data shows that miR-31 is upregulated by both NF-κB and STAT3 pathways. Our findings of a tight association between miR-31 induction and NF-κB and STAT3 pathway activation during wound healing are important, as both of these pathways are activated by the inflammatory signals (Venkatesan et al., 2017, Yang et al., 2017). They suggest that miR-31 is an important mediator of inflammatory signals in promoting epithelial regeneration during wound healing. In our recent work on the gut, we showed that miR-31 was upregulated by STAT3 signaling activity upon radiation-induced intestinal injury and that it promoted intestinal epithelium regeneration (Tian et al., 2017). In the mammary gland, we showed that miR-31 mediates progesterone signaling through the NF-κB signaling pathway (Lv et al., 2017). Thus, miR-31 emerged as the stress-responsive microRNA downstream of NF-κB and STAT3 inflammatory signals. It plays essential role in epithelial remodeling in the context of diverse tissue injuries. In the skin, it has been reported that miR-31 can be induced by the NF-κB signaling activity and that it promotes epidermal hyperplasia in psoriasis (Yan et al., 2015). In contrast, miR-31 could modulate inflammatory signaling by targeting STK40 in psoriasis (Xu et al., 2013). Thus, complex feedback regulatory network between inflammatory signals and miR-31 likely operates in psoriasis.
Inflammatory phase is followed by the proliferative phase, when keratinocytes proliferate and migrate to execute wound re-epithelialization (Takeo et al., 2015). Here we demonstrate that miR-31 promotes keratinocyte proliferation and migration by activating RAS/MAPK signaling pathway. Recently, this pathway has been shown to play a crucial role during skin wound healing (Li et al., 2017). In addition, RAS/MAPK activation by miR-31 has been found in the lung, colorectal and pancreatic cancer cells (Edmonds, 2016, Kent et al., 2016, Sun et al., 2013). It appears that miR-31 mediates epithelial regeneration and tumorigenesis in different tissues via common molecular mechanism.
In summary, our findings demonstrate that miR-31 is induced by inflammatory signals and that it promotes keratinocyte proliferation and migration during skin wound healing. We identified miR-31 as an important mediator of transition from inflammation to re-epithelialization phase (Figure S8), highlighting essential role of miR-31 in wound healing in vivo.
MATERIALS AND METHODS
Additional details are available in the Supplementary Materials.
Mouse tissue collection.
Prior to wounding, 7- to 8-week-old mice were shaved. Biopsy punches were used to inflict 6mm and 2mm full-thickness wounds in the dorsal and tail skin respectively. 1×1.5 cm2 full-thickness large wounds were created on dorsal skin for observing the retention of scab on PWD 20. Mice were sacrificed and tissue biopsies at the wound site and intact skin were collected for analysis 6 days after wounding. All mice were kept under specific pathogen-free (SPF) conditions in accordance with the guidelines of the Institutional Animal Care and Use Committee of China Agricultural University (approval number: SKLAB-2011–04-03).
miR-31 in situ hybridizations.
miR-31 in situ hybridization was performed using previously described protocol (Jorgensen et al., 2010, Yuan et al., 2015). Double DIG-labeled miR-31 and scrambled LNA probes (Exiqon) were hybridized at 61°C. In situ signal was detected by staining with Anti-Digoxigenin-AP antibody (Roche) and developed using BM purple substrate (Roche).
Luciferase assay for miR-31 promoter activity.
For promoter reporter assay, pGL3-basic vector (Promega) was used to clone the promoter of miR-31 as previously described (Tian et al., 2017). HaCaT cells were seeded in a 24-well plate at the density of 5×104 cells per well one day before transfection and then each well was transfected with a mixture of 500 ng pGL3 luciferase vector and 50 ng pRL-TK renilla vector using Lipofectamine 2000 Transfection Reagent (Invitrogen). 24 hours post transfection, cells weretreated with inflammatory cytokines for another 24 hours before being lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). The ratio of firefly luciferase to renilla luciferase was calculated for each well.
Statistical analysis.
The data were analyzed in triplicate or greater and the means obtained were used for student’s t-tests. Asterisks denote statistical significance (*p < 0.05; ** p < 0.01; *** p<0.001). All datas are reported as mean ± SD. Means and standard errors were from at least three independent biological replicates experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank Xueyun Bi, Jiuzhi Xu, Yu Zheng, Xiaole Sheng, Pengbo Lou, Chunlei Shao and Min Deng for the kindly helps in sharing the reagents and methods. ZY is supported by the National Natural Science Foundation of China (No. 81572614, 31271584); Beijing Nature Foundation Grant (5162018); the Major Project for Cultivation Technology (2016ZX08008001, 2014ZX08008001); Basic Research Program (2015QC0104, 2015TC041, 2016SY001, 2016QC086); SKLB Open Grant (2018SKLB6–10). MVP is supported by Pew Charitable Trust grant and NIH grants R01-AR067273 and R01-AR069653.
Abbreviations used:
- PWD
post-wounding
- WT
wild type
- NC
negative control
- cKO
conditional knockout
Footnotes
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
REFERENCE
- Aomatsu K, Arao T, Abe K, Kodama A, Sugioka K, Matsumoto K, et al. Slug is upregulated during wound healing and regulates cellular phenotypes in corneal epithelial cells. Investigative ophthalmology & visual science 2012;53(2):751–6. [DOI] [PubMed] [Google Scholar]
- Banerjee J, Sen CK. MicroRNA and Wound Healing. Adv Exp Med Biol 2015;888:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds MD, Boyd Kelli L, Tamara Moyo, Ramkrishna Mitra, Robert Dusaynski, Arrate MP, et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J Clin Invest. 2016;126(1):349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahs F, Bi X, Yu FS, Zhou L, Mi QS. New insights into microRNAs in skin wound healing. IUBMB life 2015;67(12):889–96. [DOI] [PubMed] [Google Scholar]
- Falanga V, Schrayer D, Cha J, Butmarc J, Carson P, Roberts AB, et al. Full-thickness wounding of the mouse tail as a model for delayed wound healing: accelerated wound closure in Smad3 knock-out mice. Wound repair and regeneration : official publication of the Wound Healing Society and the European Tissue Repair Society 2004;12(3):320–6. [DOI] [PubMed] [Google Scholar]
- Grinnell F Wound repair, keratinocyte activation and integrin modulation. J Cell Sci 1992;101 ( Pt 1):1–5. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 2008;453(7193):314–21. [DOI] [PubMed] [Google Scholar]
- Horsburgh S, Fullard N, Roger M, Degnan A, Todryk S, Przyborski S, et al. MicroRNAs in the skin: role in development, homoeostasis and regeneration. Clinical science 2017;131(15):1923–40. [DOI] [PubMed] [Google Scholar]
- Hwang JH, Park SJ, Ko WG, Kang SM, Lee DB, Bang J, et al. Cordycepin induces human lung cancer cell apoptosis by inhibiting nitric oxide mediated ERK/Slug signaling pathway. Am J Cancer Res 2017;7(3):417–32. [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Baker A, Moller S, Nielsen BS. Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods 2010;52(4):375–81. [DOI] [PubMed] [Google Scholar]
- Kent OA, Mendell JT, Rottapel R. Transcriptional Regulation of miR-31 by Oncogenic KRAS Mediates Metastatic Phenotypes by Repressing RASA1. Mol Cancer Res 2016;14(3):267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivisto L, Heino J, Hakkinen L, Larjava H. Integrins in Wound Healing. Advances in wound care 2014;3(12):762–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampert M, Bloch W, Sasaki T, Bugnon P, Rulicke T, Wolf E, et al. Activities of the matrix metalloproteinase stromelysin-2 (MMP-10) in matrix degradation and keratinocyte organization in wounded skin. Molecular biology of the cell 2004;15(12):5242–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusewitt DF, Choi C, Newkirk KM, Leroy P, Li Y, Chavez MG, et al. Slug/Snai2 is a downstream mediator of epidermal growth factor receptor-stimulated re-epithelialization. The Journal of investigative dermatology 2009;129(2):491–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. The Journal of biological chemistry 1996;271(34):20608–16. [DOI] [PubMed] [Google Scholar]
- Le M, Naridze R, Morrison J, Biggs LC, Rhea L, Schutte BC, et al. Transforming growth factor Beta 3 is required for excisional wound repair in vivo. PloS one 2012;7(10): e48040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Li X, Wang A, Meisgen F, Pivarcsi A, Sonkoly E, et al. MicroRNA-31 Promotes Skin Wound Healing by Enhancing Keratinocyte Proliferation and Migration. The Journal of investigative dermatology 2015;135(6):1676–85. [DOI] [PubMed] [Google Scholar]
- Li T, Song T, Ni L, Yang G, Song X, Wu L, et al. The p-ERK-p-c-Jun-cyclinD1 pathway is involved in proliferation of smooth muscle cells after exposure to cigarette smoke extract. Biochemical and biophysical research communications 2014;453(3):316–20. [DOI] [PubMed] [Google Scholar]
- Li X, Li D, Wikstrom JD, Pivarcsi A, Sonkoly E, Stahle M, et al. MicroRNA-132 promotes fibroblast migration via regulating RAS p21 protein activator 1 in skin wound healing. Scientific reports 2017;7(1):7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zhou S, Zhao T, Ju T, Zhang L. TRPM7 channel regulates ox-LDL-induced proliferation and migration of vascular smooth muscle cells via MEK-ERK pathways. FEBS letters 2016;590(4):520–32. [DOI] [PubMed] [Google Scholar]
- Luo H, Rankin GO, Juliano N, Jiang BH, Chen YC. Kaempferol inhibits VEGF expression and in vitro angiogenesis through a novel ERK-NFkappaB-cMyc-p21 pathway. Food chemistry 2012;130(2):321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv C, Li F, Li X, Tian Y, Zhang Y, Sheng X, et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressingWnt signaling antagonists. Nature communications 2017;8(1):1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marampon F, Gravina GL, Di Rocco A, Bonfili P, Di Staso M, Fardella C, et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Molecular cancer therapeutics 2011;10(1):159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development 2004;131(13):3021–34. [DOI] [PubMed] [Google Scholar]
- McGowan K, Coulombe PA. The wound repair-associated keratins 6, 16, and 17. Insights into the role of intermediate filaments in specifying keratinocyte cytoarchitecture. Subcell Biochem 1998;31:173–204. [PubMed] [Google Scholar]
- Mustoe TA, O’Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plastic and reconstructive surgery 2006;117(7 Suppl):35S–41S. [DOI] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG. Epithelialization in Wound Healing: A Comprehensive Review. Advances in wound care 2014;3(7):445–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke JM, Sorg H. Wound repair and regeneration. European surgical research 2012;49(1):35–43. [DOI] [PubMed] [Google Scholar]
- Savagner P Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. BioEssays 2001;23(10):912–23. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol 2005;168(6):941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strbo N, Yin N, Stojadinovic O. Innate and Adaptive Immune Responses in Wound Epithelialization. Advances in wound care 2014;3(7):492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Yu F, Ma Y, Zhao R, Chen X, Zhu J, et al. MicroRNA-31 activates the RAS pathway and functions as an oncogenic MicroRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). The Journal of biological chemistry 2013;288(13):9508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeo M, Lee W, Ito M. Wound healing and skin regeneration. Cold Spring Harbor perspectives in medicine 2015;5(1):a023267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Ma X, Lv C, Sheng X, Li X, et al. Stress responsive miR-31 is a major modulator of mouse intestinal stem cells during regeneration and tumorigenesis. eLife 2017;6:e29538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Fan HY, Chen DY, Song XF, Schatten H, Sun QY, et al. Effects of MEK inhibitor U0126 on meiotic progression in mouse oocytes:microtuble organization, asymmetric division and metaphase II arrest. Cell research 2003;13(5):375–83. [DOI] [PubMed] [Google Scholar]
- Venkatesan T, Choi YW, Lee J, Kim YK. Pinus densiflora needle supercritical fluid extract suppresses the expression of pro-inflammatory mediators iNOS, IL-6 and IL-1beta, and activation of inflammatory STAT1 and STAT3 signaling proteins in bacterial lipopolysaccharide-challenged murine macrophages. DARU Journal of Pharmaceutical Sciences 2017;25(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV, et al. IL-6 Induces NF- B Activation in the Intestinal Epithelia. The Journal of Immunology 2003;171(6):3194–201. [DOI] [PubMed] [Google Scholar]
- Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003;83(3):835–70. [DOI] [PubMed] [Google Scholar]
- Wevers A, Arnold HG, Bonnekoh B, Schulze HJ, Mahrle G. Increased expression of proliferation keratins K6 and K16 in unaffected skin in exacerbated psoriasis. Z Hautkr 1990;65(5):471–2, 5. [PubMed] [Google Scholar]
- Wilson SE, Mohan RR, Mohan RR, Ambrosio R Jr., Hong J, Lee J, et al. The corneal wound healing response: cytokine-mediated interaction of the epithelium, stroma, and inflammatory cells. Prog Retin Eye Res 2001;20(5):625–37. [DOI] [PubMed] [Google Scholar]
- Xu N, Meisgen F, Butler LM, Han G, Wang XJ, Soderberg-Naucler C, et al. MicroRNA-31 is overexpressed in psoriasis and modulates inflammatory cytokine and chemokine production in keratinocytes via targeting serine/threonine kinase 40. Journal of immunology 2013;190(2):678–88. [DOI] [PubMed] [Google Scholar]
- Yan S, Xu Z, Lou F, Zhang L, Ke F. NF-kappaB-induced microRNA-31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nature communications 2015;6:7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Gao X, Cao Y, Guo Q, Li S, Zhu Z, et al. Anti-Inflammatory Effects of Boldine and Reticuline Isolated from Litsea cubeba through JAK2/STAT3 and NF-kappaB Signaling Pathways. Planta Med 2017; 10.1055/s-0043-113447. [DOI] [PubMed] [Google Scholar]
- Yuan S, Li F, Meng Q, Zhao Y, Chen L, Zhang H, et al. Post-transcriptional Regulation of Keratinocyte Progenitor Cell Expansion, Differentiation and Hair Follicle Regression by miR-22. PLoS Genet 2015;11(5): e1005253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Wang N, Xue Y, Ding T, Liu X, Mo X, et al. Electrospun tilapia collagen nanofibers accelerating wound healing via inducing keratinocytes proliferation and differentiation. Colloids and surfaces B, Biointerfaces 2016;143:415–22. [DOI] [PubMed] [Google Scholar]
- Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-alpha. Nature medicine 2012;18(7):1077–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.