

Abstract
Initiation of expression of fibroblast growth factor receptor 1 (FGFR1) concurrent with loss of FGFR2 expression is a well-documented event in the progression of prostate cancer (PCa). Although it is known that some FGFR isoforms confer advantages in cell proliferation and survival, the mechanism by which the subversion of different FGFR isoforms contributes to PCa progression is incompletely understood. Here, we report that fibroblast growth factor (FGF) promotes NF-κB signaling in PCa cells and that this increase is associated with FGFR1 expression. Disruption of FGFR1 kinase activity abrogated both FGF activity and NF-κB signaling in PCa cells. Of note, the three common signaling pathways downstream of FGFR1 kinase, extracellular signal–regulated kinase 1/2 (ERK1/2), phosphoinositide 3-kinase (PI3K/AKT), and phosphoinositide phospholipase Cγ (PLCγ), were not required for FGF-mediated NF-κB signaling. Instead, transforming growth factor β–activating kinase 1 (TAK1), a central regulator of the NF-κB pathway, was required for FGFR1 to stimulate NF-κB signaling. Moreover, we found that FGFR1 promotes NF-κB signaling in PCa cells by reducing TAK1 degradation and thereby supporting sustained NF-κB activation. Consistently, Fgfr1 ablation in the transgenic adenocarcinoma of the mouse prostate (TRAMP) model reduced inflammation in the tumor microenvironment. In contrast, activation of the FGFR1 kinase in the juxtaposition of chemical-induced dimerization (CID) and kinase 1 (JOCK1) mouse model increased inflammation. As inflammation plays an important role in PCa initiation and progression, these findings suggest that ectopically expressed FGFR1 promotes PCa progression, at least in part, by increasing inflammation in the tumor microenvironment.
Keywords: fibroblast growth factor (FGF), prostate cancer, inflammation, NF-kappa B (NF-KB), tyrosine-protein kinase (tyrosine kinase), tumor promoter, tumor cell biology, receptor tyrosine kinase, mouse genetics, ectopic FGF signaling, tumor microenvironment
Introduction
Cancer-related inflammation is one of the hallmark changes found during cancer progression (1, 2). The tumor microenvironment is home to multiple types of inflammatory immune cells, including CD4+ T cells, macrophages, and neutrophils, which facilitate cancer progression and recurrence (3–6). Prostate cancer (PCa), 4 the second most frequently diagnosed cancer in men worldwide (7), is no exception. Emerging evidence demonstrates that chronic inflammation promotes the development of PCa to an advanced metastatic disease (8, 9) that is castration-resistant (10, 11).
Tumor cells produce multiple cytokines and chemokines to attract infiltrating lymphocytes to the tumor microenvironment. Among these are the upstream regulators of nuclear factor (NF)-κB, which was first discovered as a transcription factor that promotes expression of the immunoglobulin κ chain by binding with its enhancer during the maturation of B lymph cells (12). NF-κB plays important roles in the cytokine/chemokine-mediated immune response (8, 9, 13, 14). The NF-κB transcription factor family consists of five proteins, p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52, which associate with each other to form distinct homo- and heterodimeric complexes (15). In its resting state, NF-κB dimers are inactivated by interacting with the inhibitor of κB (IκB), preventing NF-κB nuclear translocation (12). Many cytokines and chemokines, including tumor necrosis factor α (TNFα) and interleukin-1, activate NF-κB. Upon activation by TNFα, the TNFα receptor recruits TNF receptor–associated factor 2/5 (TRAF2/5) and transforming growth factor β-activated kinase 1 (TAK1) (16). Activated TAK1 then phosphorylates and activates the inhibitor of κB kinase α/β (IKKα/β), leading to the phosphorylation and degradation of IκBα. The released NF-κB is then translocated into the nucleus and initiates target gene transcription (17).
The NF-κB pathway is constitutively activated in a significant portion of human PCa. This dysregulation correlates with the castration resistance of PCa (13). Consistent with clinical observations, the NF-κB pathway is also highly activated in androgen-independent prostate cancer (PC)-3 and Duke University (DU)145 cells, but not in androgen-dependent lymph node carcinoma of the prostate (LNCaP) cells (14). Moreover, NF-κB signaling promotes cytokine expression and increases tumor-infiltrating macrophages that facilitate PCa survival, metastasis, and angiogenesis (18, 19). However, the mechanism by which the NF-κB pathway is dysregulated in PCa cells is not understood.
The fibroblast growth factor (FGF) family consists of 18 tyrosine kinase receptor–binding ligands regulating a broad spectrum of cellular processes. FGFs bind and activate the FGF receptor (FGFR), transmembrane tyrosine kinases encoded by four highly homologous genes, denoted Fgfr1, Fgfr2, Fgfr3, and Fgfr4 (20). Ectopic expression and activation of the FGF signaling axis is associated with many diseases, including developmental disorders and cancer (21). Loss of resident FGFR2 and acquisition of ectopic FGFR1 is a common event in PCa progression (22). However, how this conversion of FGFR isoform expression contributes to PCa progression is not fully understood. In addition, the cross-talk between the FGF and NF-κB signaling pathways remains controversial. In some reports, FGF signaling positively regulates the NF-κB pathway (23–29), whereas in others, FGF signaling negatively regulates the NF-κB pathway (30–32). In addition, the mechanism by which FGFR regulates the NF-κB pathway is unclear, although downstream signaling pathways of FGFR kinases, such as the extracellular signal–regulated protein kinases (ERKs), phosphatidylinositol 3-kinase (PI3K)/AKT, or phospholipase C (PLC)-γ pathways, are known to promote activation of the NF-κB pathway.
In this report, we demonstrated that FGF augmented NF-κB signaling in FGFR1-expressing, but not in FGFR2-expressing, PCa cells; that FGFR1 augmented NF-κB signaling via tyrosine phosphorylation, thereby stabilizing TAK1 and sustaining NF-κB signaling; and that inflammation in the PCa microenvironment was associated with ectopically expressed FGFR1 kinase. Inflammation has long been known to promote cancer initiation and progression, and our results suggest that FGFR1 promotes inflammation in a receptor isoform–specific manner, which, at least in part, accounts for its activity to promote PCa progression.
Results
FGF1 promotes NF-κB signaling in DU145 cells that highly express FGFR1, but not in LNCaP cells that do not express FGFR1
Although the acquisition of ectopic FGFR1 and elevated inflammation are characteristic events in PCa progression, the cross-talk between FGF and NF-κB signaling remains controversial. To determine the presence or absence of cross-talk between ectopic FGFR1 signaling and the NF-κB pathway in PCa cells, DU145 cells that highly expressed FGFR1 and LNCaP cells that highly expressed FGFR2 were treated with FGF1 (Fig. 1A). Western blotting analyses revealed that, although FGF1 induced strong phosphorylation of fibroblast growth factor receptor substrate 2α (FRS2α) and ERK1/2 in both DU145 and LNCaP cells, a time-dependent phosphorylation of IKKα and IKKβ was observed only in DU145 cells (Fig. 1B). In the NF-κB pathway, phosphorylation activates IKKα/β, which in turn phosphorylates IκBα, leading to degradation of IκBα and, therefore, activation of NF-κB. Treatment of DU145 cells with FGF1 consistently increased the phosphorylation and reduced the abundance of IκBα. Interestingly, FGF1 also induced phosphorylation of p65 in DU145 cells (Fig. 1B), further indicating activation of the NF-κB pathway. In contrast, FGF1 failed to induce phosphorylation of IKKα, IKKβ, IκBα, or p65, as well as to reduce IκBα abundance in LNCaP cells (Fig. 1C). Together with the finding that DU145, but not LNCaP, cells highly expressed FGFR1, these results suggest that the cross-talk between the FGF and NF-κB pathways is FGFR1-dependent.
Figure 1.
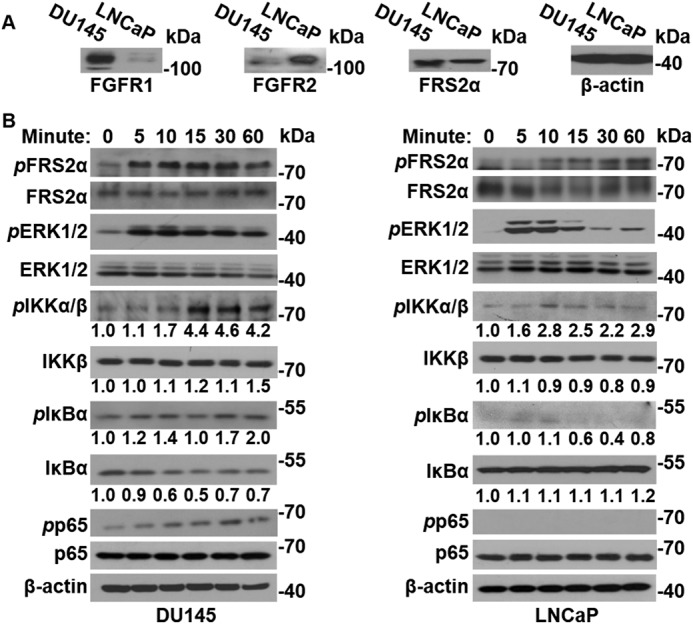
FGF augments NF-κB signaling in PCa cells that overexpress FGFR1. A, Western blot analyses of FGFR1 and FRS2α in DU145 and LNCaP cells. B, DU145 and LNCaP cells were treated with 50 ng/ml FGF1 for the indicated times. The cells were then lysed, and the cell lysates were analyzed by Western blotting for activation of FGF and NF-κB pathways. β-Actin was used as an internal loading control. pFRS2α, phosphorylated FRS2α; pERK1/2, phosphorylated ERK1/2; pIKKα/β, phosphorylated IKKα/β; pIκBα, phosphorylated IκBα; pp65, phosphorylated p65.
Disruption of the FGFR1 pathway compromises NF-κB signaling in PCa cells
To determine whether FGFR kinase activity was required for FGF to promote NF-κB signaling, DU145 cells were incubated with the FGFR kinase inhibitor, AZD4547, before FGF treatment. Western blotting analyses revealed that AZD4547 blunted the activity of TNFα, evidenced by reduced phosphorylation of IKKα/β and p65, and increased IκBα abundance (Fig. 2A). Furthermore, real-time RT-PCR analyses revealed that AZD4547 suppressed the activity of TNFα and FGF, reducing expression of TNFα and the chemokine CXC motif ligand 1 (CXCL-1), both of which are downstream targets of the NF-κB pathway (Fig. 2B). To determine whether FGFR1 was required for FGF to promote NF-κB signaling, the CRISPR/Cas9 gene editing method was used to ablate Fgfr1 in DU145 cells (designated DU145ΔR1). Unlike control DU145 cells, where TNFα induced nuclear translocation of p65 within 15 min, DU145ΔR1 cells failed to respond to TNFα with respect to p65 nuclear translocation (Fig. 3A). Immunostaining further demonstrated that TNFα induced p65 translocation in control DU145 cells, but not DU145ΔR1 cells (Fig. 3B). Forced expression of FGFR1 restored TNFα-induced p65 translocation in DU145ΔR1 cells. Similar to treatment with AZD4547, ablation of Fgfr1 compromised the TNFα-induced activation of NF-κB and abrogated the ability of FGF1 to augment TNFα signaling in DU145 cells (Fig. 3C). Together, these results indicate that FGFR kinase activity is required for cross-talk between the FGF and NF-κB signaling pathways.
Figure 2.
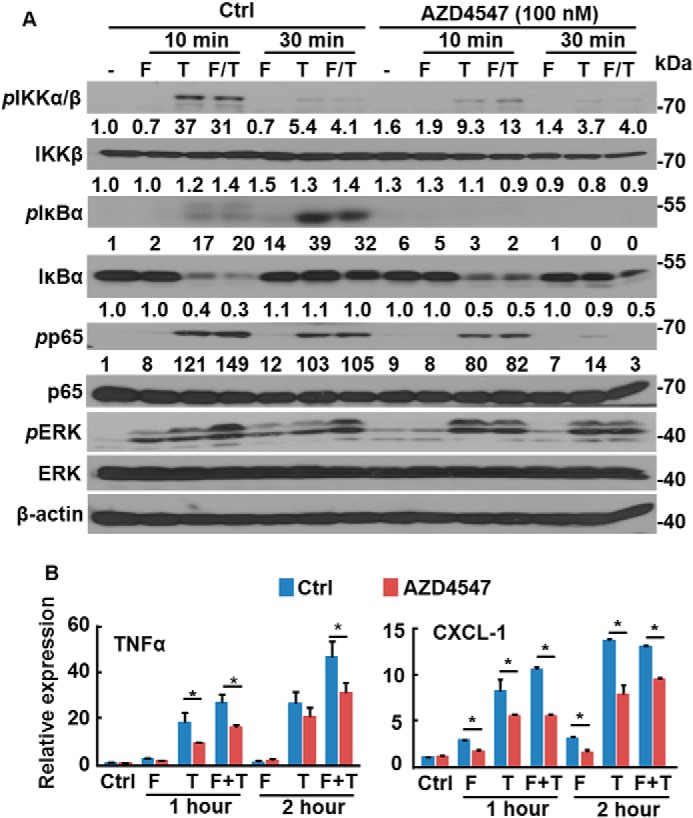
Inhibition of FGFR attenuates NF-κB signaling in PCa cells. DU145 cells were treated with the FGFR inhibitor, AZD4547, for 10 min and then 20 ng/ml FGF1 (F) and/or 10 ng/ml TNFα (T) for the indicated times. The cells were harvested for Western blotting (A) or real-time RT-PCR (B) analyses of the activation of FGF and NF-κB signaling pathways. Data are normalized with a β-actin–loading control and expressed as mean ± S.D. (error bars); *, p ≤ 0.05. Ctrl, solvent control; F, FGF1; T, TNFα; F/T, FGF1 and TNFα.
Figure 3.

FGFR1 is required for FGF to augment NF-κB signaling in PCa cells. A, WT, Fgfr1null (ΔR1), and caFGFR1-transfected Fgfr1null (ΔR1/R1OE) DU145 cells were treated with TNFα (10 ng/ml) for the indicated times. The cells were lysed, and the lysates were separated into cytosol and nuclear fractions. The abundance of p65 in the two fractions was assessed by Western blotting. PARP1/2 and β-tubulin were used for verification of the nuclear and cytosol fractions, respectively. B, control and Fgfr1null DU145 cells were treated with TNFα (10 ng/ml) for 30 min, and the presence of p65 was detected by immunostaining with an anti-p65 antibody. TO-PRO-3 was used for nuclear staining. Red arrows, nucleus. C, Fgfr1null and WT DU145 cells were treated with FGF1 (20 ng/ml) and/or TNFα (10 ng/ml) for the indicated times. RNA was extracted for RT-PCR analyses to determine the expression of TNFα and CXCL-1. *, p ≤ 0.05; data represent means ± S.D. (error bars) from three mice.
TAK1 is required for FGFR1 to augment NF-κB signaling
The ERK1/2, PI3K/AKT, and PLCγ pathways are the three common pathways downstream of FGFR. To determine whether these three pathways were required for FGF to augment NF-κB signaling, mouse embryonic fibroblasts (MEFs) were incubated with the ERK, AKT, or PLCγ inhibitors SL327, LY294002, or U73122, respectively, 1 h before FGF and TNFα treatment. Western blotting analyses revealed that suppression of either ERK, AKT, or PLCγ did not affect the ability of FGF1 to augment NF-κB signaling (Fig. 4A), despite the fact that the inhibitors suppressed phosphorylation of ERK and AKT or Ca2+ influx (data not shown), respectively. These results, indicating that inhibition of PI3K with LY294002 did not affect the phosphorylation of IKKα/β in MEFs, differ from previous results indicating that FGF1 activates NF-κB via the AKT pathway in breast cancer cells (28). This is likely another example of cell context–dependent signaling of the FGF pathway. However, the underlying mechanism remains to be determined.
Figure 4.

TAK1 is required for FGF to augment NF-κB signaling. A, MEFs were treated with 10 μm SL327 (ERKi), LY294002 (AKTi), U73122 (PLCγi), or (5Z)-7-oxozeaenol (TAKi) for 1 h and then with 20 ng/ml FGF1 (F) and/or 10 ng/ml TNFα (T) for 30 min. The cells were lysed, and the lysates were analyzed by Western blotting to determine the activation of FGF and NF-κB pathways. B, Tak1null (ΔTAK1) and TAK1-transfected Tak1null (ΔTAK1/TAK1OE) cells were treated with FGF1 and/or TNFα for 30 min before lysis for Western blotting analyses to determine the activation of FGF and NF-κB pathways.
TAK1 is a member of the mitogen-activated kinase kinase kinase family. TAK1 can be activated by multiple cytokines or by coexpression with the TAK1-binding protein (TAB1) (34). Upon activation, TAK1 phosphorylates IKKα/β, thereby activating the canonic NF-κB pathway. Whether TAK1 is required for the FGF pathway to cross-talk with NF-κB is not fully clear, although one report found that FGFR interacts with TAK1 in multiple myeloma and bladder cancer cells (23). To determine whether the FGF pathway enhanced NF-κB signaling via activation of TAK1, MEFs were treated with the TAK1 inhibitor, (5Z)-7-oxozeaenol. Inhibition of TAK1 abrogated the FGF1-induced phosphorylation of IKKα/β (Fig. 4A). These data suggest that TAK1 kinase activity is required for FGF to promote NF-κB activity in MEFs.
Furthermore, to determine whether TAK1 was required for FGF to augment NF-κB signaling, MEFs bearing null Tak1 alleles (MEFΔTAK1) were treated with FGF1 and TNFα. Western blot analysis revealed that both FGF1 and TNFα failed to induce phosphorylation of IKKα/β in MEFΔTAK1 cells (Fig. 4B). Forced expression of TAK1 in MEFΔTAK1 cells by stable transfection restored TNFα-induced IKKα/β phosphorylation as well as the ability of FGF1 to enhance TNFα-induced phosphorylation of IKKα/β (Fig. 4B).
Consistently, inhibiting PI3K/AKT, ERK1/2, or PLCγ, downstream pathways of FGFR1, only partially blocked TNFα-induced p65 nuclear translocalization in DU145 cells. In contrast, treating with FGFR1 and TAK1 inhibitor abolished p65 nuclear translocalization (Fig. 5). These results further indicate that TAK1, but not PI3K/AKT, ERK1/2, and PLCγ, is required for FGFR1 to augment NF-κB signaling.
Figure 5.

Suppression of pathways downstream of FGFR partially suppresses p65 nuclear localization. A, DU145 cells were pretreated with inhibitor for AKT (LY294002, 10 μm), ERK1/2 inhibitor (SL327, 10 μm), PLCγ (U73122, 2 μm), FGFR1 (AZD4547, 100 nm), or TAK1 ((5Z)-7-oxozeaend, 40 nm) for 1 h, followed by FGF1 (25 ng/ml) and TNFα (10 ng/ml) for 30 min. The cells were then fixed and immunostained with anti-p65 antibody. Representative images were presented. B, the nuclear and cytosol abundance of p65 were measured from a minimum of three fields from the same experiment. The average ratios of nuclear/cytosol p65 are presented as mean and S.D. (error bars). Scale bar, 50 μm. DAPI, 4′,6-diamidino-2-phenylindole.
FGFR1 forms a complex with TAK1 and leads to TAK1 tyrosine phosphorylation
To determine the mechanism by which FGFR1 cross-talked with the NF-κB pathway, FLAG-tagged FGFR1 was co-expressed with MYC- or HA-tagged components of the TNFα receptor complex in human embryonic kidney (HEK)293 cells. Immunoprecipitation with anti-MYC or anti-HA antibodies was employed to pull down FGFR1 that bound to the MYC- or HA-tagged proteins. The resulting complexes were then subjected to Western blotting analyses (Fig. 6A); FGFR1 was pulled down by TAK1 and the TAK1–TAB1 complex, but not by other components of the TNFα receptor complex. These results indicate the presence of an interaction between FGFR1 and TAK1.
Figure 6.

FGFR1 forms a complex with TAK1 and leads to TAK1 tyrosine phosphorylation. A, FLAG-tagged FGFR1 (FGFR1) was co-expressed in HEK293 cells with the MYC- or HA-tagged components of the TNFα receptor complex, as indicated. The cell lysates were used for immunoprecipitation with anti-MYC or anti-HA antibodies. The pulled-down complexes and input cell lysates were analyzed by Western blotting with the anti-FLAG, anti-MYC, or anti-HA antibodies. B, FLAG-FGFR1 and FLAG-caFGFR1 were co-expressed with MYC-tagged TAK1 or TRAF2 constructs in HEK293 cells. The cell lysates were subjected to immunoprecipitation with anti-MYC antibodies and Western blotting analyses. C, schematic of TAK1 structure domains. The four tyrosine phosphorylation sites are denoted by green dots. D and E, FLAG-caFGFR1 was co-expressed in HEK293 cells with the indicated MYC-tagged TAK1 mutants. The cell lysates were subjected to immunoprecipitation with anti-MYC antibodies and Western blotting with the indicated antibodies. −, vector-only negative control; 1, MYC-TRAF2; 2, HA-RIP1; 3, MYC-TAK1; 4, TAB1; 5, MYC-TAK1 + TAB1; 6, HA-IKKβ; 7, MYC-p65; IB, immunoblotting; IP, immunoprecipitation; 2Fa, Y124F and Y125F substitutions; 2Fb, Y143F and Y206F substitutions; 4F, all four tyrosine substitutions; BD, binding domain; CD, C-terminal binding domain; KD, kinase domain; 4G10, anti-phosphotyrosine antibody.
To determine the structure domains of TAK1 required for interaction with FGFR1, either MYC-tagged full-length TAK1, kinase domain, or C-terminal sequence downstream of the kinase domain was co-expressed with FLAG-tagged FGFR1 or a constitutively active mutant of FGFR1 (caFGFR1) in HEK293 cells. Co-immunoprecipitation analysis demonstrated that only the full-length TAK1 and the kinase domain pulled down FGFR1 (Fig. 6B), suggesting that TAK1 interacted with FGFR1 via its kinase domain. In addition, the relative amount of caFGFR1 pulled down was greater than that of WT FGFR1, suggesting that kinase activity enhanced the interaction between FGFR and TAK1.
Further in silico screening identified four potential tyrosine phosphorylation sites in the kinase domain of TAK1 (Fig. 6C). Site-directed mutagenesis was utilized (Tyr → Phe, singly and in combination) to determine the phosphorylation status of these tyrosine residues and whether the phosphorylation was FGFR1-dependent. Western blotting analyses revealed that TAK1 was tyrosine-phosphorylated only when co-expressed with caFGFR1 in 293 cells (Fig. 6D), indicating that the phosphorylation was FGFR1 kinase–dependent. Mutations of each individual tyrosine residue alone, as well as two tyrosine residues together (2Fa and 2Fb, as indicated) only partially reduced phosphorylation. However, no tyrosine phosphorylation was detected with the 4F mutant (all four tyrosine residues substituted with phenylalanine) (Fig. 6E). The results indicate that all four tyrosine residues were phosphorylated.
Elimination of the four tyrosine phosphorylation sites of TAK1 accelerates its lysosome-mediated degradation
TAK1 is activated through binding to TAB1. However, no significant difference was observed in TAB1 binding between WT TAK1 and the TAK1 4F mutant (data not shown). Lys-48 ubiquitination–mediated lysosome degradation is involved in inactivation of TAK1 activity (35, 36), and Lys-63 ubiquitination on Lys-158 is involved in TAK1 activation (36–38). Therefore, we investigated whether the ubiquitination of TAK1 was regulated by tyrosine phosphorylation. Interestingly, we found that the ubiquitination of TAK1 was increased in DU145ΔR1 cells when they were treated with lysosome inhibitor MG132 (Fig. 7A). Consistently, the 4F mutant also exhibited increased ubiquitination in DU145 cells in the presence of MG132. To determine whether tyrosine phosphorylation suppresses ubiquitin-mediated degradation of TAK1, both TAK1 WT and tyrosine phosphorylation site mutants were co-expressed with caFGFR1 in HEK293 cells. The cells were treated with cycloheximide to halt protein synthesis followed by the addition of the proteasome inhibitor, MG132, for the indicated times. Western blot analysis was then used to assess the abundance of WT and mutant TAK1 at different time points with or without treating with MG132 (Fig. 7, B and C). Quantitative analyses of TAK1 intensity revealed that the 4F mutant exhibited a faster down-regulation than other TAK1 constructs. Treating the cells with lysosome inhibitor, MG132, diminished the differences in down-regulation, indicating that the 4F mutant was down-regulated faster than WT and other mutants by proteasome-mediated degradation. These results suggest that tyrosine phosphorylation suppresses degradation of TAK1 and therefore sustains NF-κB signaling.
Figure 7.

Tyrosine phosphorylation of TAK1 prevents its ubiquitination and degradation. A, control (Ctrl) and Fgfr1null (ΔFGFR1) DU145 cells were stably transfected with MYC-tagged TAK1 (WT) or TAK1 4F mutant (4F). The cells were treated overnight with MG132 to block lysosomal degradation where indicated. The cells were lysed and subjected to immunoprecipitation with anti-MYC antibodies. The pulled-down complexes and input cell lysates were analyzed by Western blotting with anti-ubiquitin and anti-MYC antibodies. B and C, the indicated MYC-tagged TAK1 mutants were transiently expressed in HEK293 cells. The cells were treated with MG132 for 24 h followed by cycloheximide to halt protein synthesis. The cells were lysed at the indicated time points, and the lysates were subjected to Western blotting analyses with anti-MYC antibodies (B). The intensity of MYC bands was analyzed with ImageJ, normalized with that of β-actin, and plotted (C). IB, immunoblot; IP, immunoprecipitation; Ub, ubiquitin; CHX, cycloheximide.
Ablation of Fgfr1 in mouse PCa reduces inflammation in the tumor microenvironment
To investigate how ectopic FGFR1 contributed to inflammation in the tumor microenvironment, sections of PCa derived from TRAMP mice with or without ablation of Fgfr1 alleles were immunostained with antibodies against cyclooxygenase (COX)-2 (expressed only in inflammatory tissues) or F4/80 (a macrophage-specific marker) (Fig. 8A). The results clearly demonstrated that ablation of Fgfr1 reduced the abundance of COX-2 and F4/80 in PCa. In addition, similar experiments were carried out with the juxtaposition of CID and kinase 1 (JOCK1) PCa mouse model, where PCa was induced through chemical-induced dimerization of the FGFR1 kinase domain–FK506-binding protein fusion protein (39). We found that activation of the fusion protein with chemical-induced dimerization also increased the staining of COX-2 and F4/80 in the prostate tissues (Fig. 8B). Together, these results demonstrate that activation of FGFR1 in prostate cells increases expression of COX-2 and infiltration of macrophages, both of which are indicative of inflammation, and therefore support the in vitro finding that FGFR1 promotes inflammation.
Figure 8.

Ablation of FGFR1 compromises and activation of FGFR1 promotes inflammation in the PCa microenvironment. Tissue sections from TRAMP and Fgfr1null TRAMP (TRAMPΔR1) (A) or JOCK1 with or without chemical-induced dimerization (+CID and −CID, respectively) (B) were immunostained with anti-COX-2 or anti-F4/80 antibodies, as indicated. For the JOCK1 model, five pairs of mice with or without FGFR1 kinase with CID were used. For the TRAMP model, three tumors with Fgfr1 deletion and four tumors with intact Fgfr1 alleles were used in this study. The representative confocal microscope images were presented. TO-PRO-3 was used for nuclear counterstaining.
Discussion
Inflammation is a characteristic change in the tumor microenvironment, which promotes cancer initiation and progression. In addition, acquisition of ectopic FGFR1 expression is a common event during PCa progression. In this report, we demonstrated that ectopic FGFR1 promoted inflammation by preventing the proteasome-mediated degradation of TAK1 and therefore sustaining NF-κB signaling. Furthermore, we found that ablation of Fgfr1 in mouse PCa reduced inflammation and infiltration of macrophages into the tumor microenvironment. Therefore, these results reveal a novel mechanism by which ectopic FGF signaling contributes to PCa progression.
AKT, ERK, and PLCγ pathways are well characterized downstream signaling cascades in FGF signaling transduction. However, these are also common downstream pathways for multiple receptor tyrosine kinases, including epithelial growth factor, platelet-derived growth factor, and vascular endothelial growth factors. The mechanism by which FGFR1 elicits receptor-specific signals is poorly understood. Our data clearly demonstrate that cross-talk between FGF and NF-κB signaling is independent of ERK, PI3K/AKT, and PLCγ pathways and that FGFR1 directly interacts with TAK1. Interestingly, FGFR2, although highly homologous to FGFR1, did not interact with TAK1. 5 Together, these results reveal a novel pathway whereby FGFR1 elicits receptor isoform-specific signals. Inflammation and NF-κB signaling are complex processes and can be regulated by multiple cell signaling pathways at multiple levels. In this report, we demonstrated that FGFR1 promoted NF-κB by sustaining the stability of TAK1. However, we did not exclude the possibility that FGFR1 may also be promoting NF-κB signaling via other mechanisms.
Interestingly, our data reveal that FGF alone only weakly activates NF-κB signaling. However, it augments TNFα activity, suggesting that FGF signaling provides another layer of controlling NF-κB activation. DU145, MEF, and HEK293 cells may have distinct background TNFα and FGF signaling (autocrine); therefore, FGF alone exhibits a different effect on NF-κB activation in these three cell lines.
TNFα receptor adaptor RIP, TRAF2, and TRAF6 proteins are required for TNF-induced Lys-63–linked polyubiquitination of TAK1 (40–43). These polyubiquitin chains serve as a scaffold to recruit the TAK1 and IKK complexes and facilitate phosphorylation of IKKβ by TAK1, leading to activation of NF-κB. On the other hand, Lys-63–linked polyubiquitin chains can be disassembled by deubiquitination enzymes, including CYLD and A20, which terminates IKK activation (44). In addition, CYLD and A20 also induce Lys-48–linked polyubiquitination of TAK1, which mediates TAK1 degradation. Whether RIP, TRAF2, and TRAF6 are involved in the complex of FGFR1 and TAK1 and whether FGFR1 regulates assembly and disassembly of Lys-63– or Lys-48–linked polyubiquitin chains is unknown, and these remain as interesting questions. Furthermore, the mechanism by which FGFR1 induces tyrosine phosphorylation of TAK1 and thereby controls its ubiquitination and subsequent activation or proteasome-mediated degradation remains to be elucidated.
Chronic inflammation in the tumor microenvironment is a key contributor to tumor initiation and progression. Acquisition of ectopic FGFR1 concomitant with the loss of FGFR2 is a common event in PCa progression (22). Our data demonstrated that FGFR1, but not FGFR2, promoted NF-κB signaling and, therefore, inflammation. This is consistent with the fact that normal prostate tissue has no detectable inflammation nor expressed resident FGFR2, whereas PCa has a highly inflammatory tumor microenvironment and ectopically expresses FGR1 (22, 45). Our results further demonstrate that FGFR1 elicits isoform-specific signals to promote inflammation; the FGFR isoform-specific interaction between FGFR1 and TAK1 accounts for the FGFR isoform-specific cross-talk. However, the existence of additional mechanisms underlying the receptor isoform-specific interaction between FGF and NF-κB signaling pathways remains to be elucidated.
In addition to promotion of cell proliferation and survival, we also demonstrated that FGFR1 promoted tumor angiogenesis and cross-talk with stroma cells (46, 47). Furthermore, we found that ectopically expressed FGFR1 promoted tumor microenvironment remodeling, a promoter for PCa initiation and progression (47). In line with this, data in Fig. 8 clearly demonstrated that ablation of Fgfr1 reduced inflammation in the TRAMP tumor. In contrast, activation of the FGFR1 transgene promoted inflammation in JOCK1 PCa. Overexpression of FGFR1 and ectopic FGF signaling has been found in >80% of PCa (21). Further efforts are needed to determine whether overexpression of FGFR1 is associated with elevated inflammation in human PCa. Multiple FGFR kinase inhibitors, including TKI258, have been used in clinical trials for PCa and other cancers (21). Whether these inhibitors suppress inflammation in the tumor microenvironment of PCa and whether the effects are associated with FGFR1 expression remain to be determined.
In conclusion, FGFR1 augments NF-κB signaling by preventing the proteasome-mediated degradation of TAK1. Disruption of FGFR1 compromised NF-κB signaling and reduced inflammation in the tumor microenvironment. As the acquisition of ectopic FGFR1 is a key event in PCa progression, inhibition of ectopic FGFR1 signaling may retard PCa progression through suppressing chronic inflammation, therefore providing a potential new strategy for controlling PCa progression.
Materials and methods
Animals
Mice were housed under the Program of Animal Resources of the Institute of Biosciences and Technology in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Texas A&M University institutional animal care and use committee. Mice were bred and genotyped as described previously (48–50).
Cell culture
DU145, LNCaP, HEK293, and MEF cells were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 incubators. Tak1null MEFs were a gift from Dr. J. Yang at Baylor College of Medicine. For the cell signaling assay, ERK inhibitor SL327 (EMD Millipore, Billerica, MA), PI3K inhibitor LY294002 (EMD Millipore), PLCγ inhibitor U73122 (Selleckchem, Houston, TX), or FGFR inhibitor AZD4547 (Selleckchem) was added to the medium at the indicated concentrations. For cell transfections, cells cultured overnight (1 × 105 cells/well in 6-well plates) were transfected with 5 μg of pcDNAZeo plasmid (Invitrogen) bearing the indicated cDNA constructs and 5 μl of Lipofectamine (Invitrogen). The cells were then incubated at 37 °C for 24 h before analysis.
Western blot analysis
Cells were lysed in radioimmune precipitation assay (RIPA) buffer (50 mm Tris-HCl buffer, pH 7.4, 1% Nonidet P-40, 150 mm NaCl, 0.25% sodium deoxycholate, 1 mm EGTA, and 1 mm phenylmethylsulfonyl fluoride), and the extracted proteins were harvested by centrifugation (4,000 × g). Samples containing 30 μg of protein were separated by SDS-PAGE and blotted onto polyvinylidene difluoride membranes for Western blotting analyses with the indicated antibodies. The dilutions and sources of the antibodies are as follows. Anti-FRS2α (1:1,000), anti-phosphorylated FRS2α (1:1,000), anti-ERK1/2 (1:1,000), anti-phosphorylated ERK1/2 (1:2,000), anti-AKT (1:2,000), anti-phosphorylated AKT (1:1,000), anti-IKKβ (1:1,000), anti-p65 (1:1,000), anti-poly(ADP-ribose) polymerase 1/2 (PARP1/2) (1:2,000), anti-TAK1 (1:1,000), anti-ubiquitin (1:1,000), anti-HA (1:1,000), anti-MYC (1:1,000), anti-β-tubulin (1:1,000), and anti-β-actin (1:2,000) were all obtained from Santa Cruz Biotechnology, Inc. Anti-FGFR1 (1:1,000) was obtained from Abcam (Cambridge, MA). Anti-phosphorylated IKKα/β (1:1,000), anti-IκBα (1:1,500), anti-phosphorylated IκBα (1:1,500), anti-phosphorylated p65 (1:1,000), and anti-TAB1 (1:1,000) were obtained from Cell Signaling Technology (Danvers, MA). The anti-phosphotyrosine antibody 4G10 (1:2,500) was obtained from Merck Millipore (Danvers, MA). Anti-FLAG antibody (1:2,500) was obtained from Sigma-Aldrich.
The membranes were washed with TBS with Tween 20 (TBST) buffer to remove nonspecific antibodies and then incubated with horseradish peroxidase–conjugated rabbit antibody at room temperature for 1 h. The specifically bound antibodies were then visualized using ECL-Plus chemiluminescent reagents from Thermo Fisher Scientific. The films were scanned with a densitometer for quantitation.
Immunoprecipitation
Cells were lysed with RIPA buffer as described earlier. The cell lysates were incubated with anti-HA (1:1,000), anti-MYC (1:1,000), anti-TAB1 (1:1,000), or anti-FLAG (1:2,500) antibodies and protein A beads overnight at 4 °C as described previously (51). The immunocomplexes were washed with RIPA buffer four times, boiled with 30 μl of SDS sample buffer for 5 min, and then subjected to Western blot analysis with the indicated antibodies as described previously.
RNA expression
Total RNA was isolated from cells using TRIzol RNA isolation reagents from Life Technologies. The first-strand cDNAs were reverse transcribed from the RNA template using SuperScript III reverse transcriptase (Invitrogen) and random primers according to the manufacturer's protocols. Real-time PCR analyses were carried out using the Fast SYBR Green Master Mix (Life Technologies) as instructed by the manufacturer. The relative abundance of mRNA was calculated using the comparative threshold cycle method and normalized to β-actin as the internal control. The mean ± S.D. among at least three individual experiments are shown.
Site-directed mutagenesis
The QuikChange Lightning multisite-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) was used for generating TAK1 mutant cDNAs. Primer sequences used to generate the mutants were as follows: Y124F, GTG CTG AAC CAT TGC CAT TTT ATA CTG CTG CCC ACG CAA; Y125F, GTG CTG AAC CAT TGC CAT ATT TTA CTG CTG CCC ACG CAA; Y124F/Y125F, GTG CTG AAC CAT TGC CAT TTT TTA CTG CTG CCC ACG CAA; Y143F, GTT CCC AAG GAG TGG CTT TTC TTC ACA GCA TGC AAC CCA; Y206F, TTT TTG AAG GTA GTA ATT TCA GTG AAA AAT GTG ACG TCT.
Gene ablation
The lentivirus-based CRISPR-Cas9 system was used to ablate Fgfr1 alleles in DU145 cells. The sequence of single guide RNA (sgRNA) was AACTTGTTCCGATGGTTATC. Two days after infection with the lentivirus, the virus-containing cells were selected by growing in medium containing 2 μg/ml puromycin.
Protein stability assay
Stable transfectants of HEK293 cells expressing MYC-tagged TAK1 or TAK1 mutants were treated with cycloheximide for the indicated times. The abundance of MYC-tagged proteins was examined via Western blot analysis. The relative levels of specific bands were quantitated by ImageJ.
Immunostaining
PCa sections were rehydrated and stained with hematoxylin and immunostained as described previously (33). For p65 immunostaining, DU145 cells were fixed with 4% paraformaldehyde for 2 h at room temperature. The antigens were retrieved by boiling in citrate buffer (10 mm, pH 8.0) for 20 min. The rabbit anti-COX-2 antibody (1:1,000) was obtained from Cell Signaling Technology. Mouse anti-p65 (1:1,000) and rat anti-F4/80 (1:1,000) were obtained from Abcam. The ExtrAvidin peroxidase system (Sigma-Aldrich) and fluorescence-conjugated secondary antibodies (Invitrogen) were used to visualize specifically bound antibodies. For immunofluorescence staining, the nuclei were counterstained with TO-PRO-3 before observation under a confocal microscope (Zeiss LSM 510).
Statistical analysis
Statistical analysis was performed using a two-tailed t test, with significance set to p < 0.05. Error bars indicate S.D.
Author contributions
C. W., Y. K., and F. W. conceptualization; C. W., Y. K., S. L., S. P., Z. L., H. Z., Z. F., C. Z., and F. W. data curation; C. W., Y. K., S. L., and F. W. formal analysis; C. W., Y. K., S. L., S. P., Z. L., H. Z., C. Z., J. L., and F. W. investigation; C. W., Y. K., S. L., Z. L., H. Z., Z. F., C. Z., J. L., and F. W. methodology; C. W. and F. W. writing-original draft; C. W., Y. K., and F. W. project administration; F. W. funding acquisition.
Acknowledgment
We thank Lei An for excellent technical support.
This work was supported by National Institutes of Health Grants CA96824, DE023106, TAMU1400302, and CPRIT 110555 (to F. W.); a gift from Agilent Technologies; National Natural Science Foundation of China Grants 81101712, 31371470, and 81270761; and Natural Science Foundation of Zhejiang Province of China Grant LY16H140004 (to C. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
C. Wang, Y. Ke, S. Liu, S. Pan, Z. Liu, H. Zhang, Z. Fan, C. Zhou, J. Liu, and F. Wang, unpublished data.
- PCa
- prostate cancer
- NF
- nuclear factor
- IκB
- inhibitor of κB
- TNFα
- tumor necrosis factor α
- TRAF
- TNF receptor–associated factor
- TAK1
- transforming growth factor β–activating kinase 1
- IKK
- inhibitor of κB kinase
- LNCaP
- lymph node carcinoma of the prostate
- FGF
- fibroblast growth factor
- FGFR
- FGF receptor
- ERK
- extracellular signal–regulated kinase
- PI3K
- phosphoinositide 3-kinase
- PLC
- phospholipase C
- FRS2α
- fibroblast growth factor receptor substrate 2α
- MEF
- mouse embryo fibroblast
- HA
- hemagglutinin
- HEK
- human embryonic kidney
- caFGFR1
- constitutively active mutant of FGFR1
- COX
- cyclooxygenase
- RIPA
- radioimmune precipitation assay
- PARP
- poly(ADP-ribose) polymerase
- CID
- chemical-induced dimerization
- JOCK1
- juxtaposition of CID and kinase 1.
References
- 1. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2. Avtandilov G. G., Rainova L. V., Preobrazhenskaia T. M., Chervonnnaia L. V., and Ershikova Iu E. (1989) [Administrative and professional requirements for the clinical pathologist at a general hospital]. Arkh. Patol. 51, 81–84 [PubMed] [Google Scholar]
- 3. Strasner A., and Karin M. (2015) Immune infiltration and prostate cancer. Front. Oncol. 5, 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gannon P. O., Poisson A. O., Delvoye N., Lapointe R., Mes-Masson A. M., and Saad F. (2009) Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 348, 9–17 10.1016/j.jim.2009.06.004 [DOI] [PubMed] [Google Scholar]
- 5. Ebelt K., Babaryka G., Figel A. M., Pohla H., Buchner A., Stief C. G., Eisenmenger W., Kirchner T., Schendel D. J., and Noessner E. (2008) Dominance of CD4+ lymphocytic infiltrates with disturbed effector cell characteristics in the tumor microenvironment of prostate carcinoma. Prostate 68, 1–10 10.1002/pros.20661 [DOI] [PubMed] [Google Scholar]
- 6. Wu C. T., Hsieh C. C., Lin C. C., Chen W. C., Hong J. H., and Chen M. F. (2012) Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J. Mol. Med. 90, 1343–1355 10.1007/s00109-012-0916-x [DOI] [PubMed] [Google Scholar]
- 7. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., and Forman D. (2011) Global cancer statistics. CA Cancer J. Clin. 61, 69–90 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 8. Huang S., Pettaway C. A., Uehara H., Bucana C. D., and Fidler I. J. (2001) Blockade of NF-κB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 20, 4188–4197 10.1038/sj.onc.1204535 [DOI] [PubMed] [Google Scholar]
- 9. Jiang J., Slivova V., Jedinak A., and Sliva D. (2012) Gossypol inhibits growth, invasiveness, and angiogenesis in human prostate cancer cells by modulating NF-κB/AP-1 dependent- and independent-signaling. Clin. Exp. Metastasis 29, 165–178 10.1007/s10585-011-9439-z [DOI] [PubMed] [Google Scholar]
- 10. Jin R., Yamashita H., Yu X., Wang J., Franco O. E., Wang Y., Hayward S. W., and Matusik R. J. (2015) Inhibition of NF-κB signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene 34, 3700–3710 10.1038/onc.2014.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nadiminty N., Tummala R., Liu C., Yang J., Lou W., Evans C. P., and Gao A. C. (2013) NF-κB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol. Cancer Ther. 12, 1629–1637 10.1158/1535-7163.MCT-13-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gilmore T. D. (2006) Introduction to NF-κB: players, pathways, perspectives. Oncogene 25, 6680–6684 10.1038/sj.onc.1209954 [DOI] [PubMed] [Google Scholar]
- 13. Fradet V., Lessard L., Bégin L. R., Karakiewicz P., Masson A. M., and Saad F. (2004) Nuclear factor-κB nuclear localization is predictive of biochemical recurrence in patients with positive margin prostate cancer. Clin. Cancer Res. 10, 8460–8464 10.1158/1078-0432.CCR-04-0764 [DOI] [PubMed] [Google Scholar]
- 14. Gasparian A. V., Yao Y. J., Kowalczyk D., Lyakh L. A., Karseladze A., Slaga T. J., and Budunova I. V. (2002) The role of IKK in constitutive activation of NF-κB transcription factor in prostate carcinoma cells. J. Cell Sci. 115, 141–151 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh S., May M. J., and Kopp E. B. (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–260 10.1146/annurev.immunol.16.1.225 [DOI] [PubMed] [Google Scholar]
- 16. Kishimoto K., Matsumoto K., and Ninomiya-Tsuji J. (2000) TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 275, 7359–7364 10.1074/jbc.275.10.7359 [DOI] [PubMed] [Google Scholar]
- 17. Pahl H. L. (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18, 6853–6866 10.1038/sj.onc.1203239 [DOI] [PubMed] [Google Scholar]
- 18. Wang J., Cai Y., Shao L. J., Siddiqui J., Palanisamy N., Li R., Ren C., Ayala G., and Ittmann M. (2011) Activation of NF-κB by TMPRSS2/ERG fusion isoforms through Toll-like receptor-4. Cancer Res. 71, 1325–1333 10.1158/0008-5472.CAN-10-2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueno T., Toi M., Saji H., Muta M., Bando H., Kuroi K., Koike M., Inadera H., and Matsushima K. (2000) Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 6, 3282–3289 [PubMed] [Google Scholar]
- 20. McKeehan W. L., Wang F., and Kan M. (1998) The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog. Nucleic Acid Res. Mol. Biol. 59, 135–176 [DOI] [PubMed] [Google Scholar]
- 21. Corn P. G., Wang F., McKeehan W. L., and Navone N. (2013) Targeting fibroblast growth factor pathways in prostate cancer. Clin. Cancer Res. 19, 5856–5866 10.1158/1078-0432.CCR-13-1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang F., Luo Y., and McKeehan W. (2013) The FGF signaling axis in prostate tumorigenesis. in Molecular Oncology: Causes of Cancer and Targets for Treatment (Gellman E. P., Sawyers C. L., and Rauscher F. J. 3rd, eds) pp. 186–189, Cambridge University Press, London [Google Scholar]
- 23. Salazar L., Kashiwada T., Krejci P., Meyer A. N., Casale M., Hallowell M., Wilcox W. R., Donoghue D. J., and Thompson L. M. (2014) Fibroblast growth factor receptor 3 interacts with and activates TGFβ-activated kinase 1 tyrosine phosphorylation and NFκB signaling in multiple myeloma and bladder cancer. PLoS One 9, e86470 10.1371/journal.pone.0086470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Byrd V. M., Ballard D. W., Miller G. G., and Thomas J. W. (1999) Fibroblast growth factor-1 (FGF-1) enhances IL-2 production and nuclear translocation of NF-κB in FGF receptor-bearing Jurkat T cells. J. Immunol. 162, 5853–5859 [PubMed] [Google Scholar]
- 25. Tang C. H., Yang R. S., Chen Y. F., and Fu W. M. (2007) Basic fibroblast growth factor stimulates fibronectin expression through phospholipase Cγ, protein kinase Cα, c-Src, NF-κB, and p300 pathway in osteoblasts. J. Cell Physiol. 211, 45–55 10.1002/jcp.20896 [DOI] [PubMed] [Google Scholar]
- 26. Kanaya S., Nemoto E., Sakisaka Y., and Shimauchi H. (2013) Calcium-mediated increased expression of fibroblast growth factor-2 acts through NF-κB and PGE2/EP4 receptor signaling pathways in cementoblasts. Bone 56, 398–405 10.1016/j.bone.2013.06.031 [DOI] [PubMed] [Google Scholar]
- 27. Niu J., Chang Z., Peng B., Xia Q., Lu W., Huang P., Tsao M. S., and Chiao P. J. (2007) Keratinocyte growth factor/fibroblast growth factor-7-regulated cell migration and invasion through activation of NF-κB transcription factors. J. Biol. Chem. 282, 6001–6011 10.1074/jbc.M606878200 [DOI] [PubMed] [Google Scholar]
- 28. Vandermoere F., El Yazidi-Belkoura I., Adriaenssens E., Lemoine J., and Hondermarck H. (2005) The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IκB kinase-β in breast cancer cells. Oncogene 24, 5482–5491 10.1038/sj.onc.1208713 [DOI] [PubMed] [Google Scholar]
- 29. Bushdid P. B., Chen C. L., Brantley D. M., Yull F., Raghow R., Kerr L. D., and Barnett J. V. (2001) NF-κB mediates FGF signal regulation of msx-1 expression. Dev. Biol. 237, 107–115 10.1006/dbio.2001.0356 [DOI] [PubMed] [Google Scholar]
- 30. Lee M. S., Choi S. E., Ha E. S., An S. Y., Kim T. H., Han S. J., Kim H. J., Kim D. J., Kang Y., and Lee K. W. (2012) Fibroblast growth factor-21 protects human skeletal muscle myotubes from palmitate-induced insulin resistance by inhibiting stress kinase and NF-κB. Metabolism 61, 1142–1151 10.1016/j.metabol.2012.01.012 [DOI] [PubMed] [Google Scholar]
- 31. Liu M. H. (2015) FGF-21 alleviates diabetes-associated vascular complications: inhibiting NF-κB/NLRP3 inflammasome-mediated inflammation? Int. J. Cardiol. 185, 320–321 10.1016/j.ijcard.2015.03.165 [DOI] [PubMed] [Google Scholar]
- 32. Flati V., Pastore L. I., Griffioen A. W., Satijn S., Toniato E., D'Alimonte I., Laglia E., Marchetti P., Gulino A., and Martinotti S. (2006) Endothelial cell anergy is mediated by bFGF through the sustained activation of p38-MAPK and NF-κB inhibition. Int. J. Immunopathol. Pharmacol. 19, 761–773 10.1177/039463200601900406 [DOI] [PubMed] [Google Scholar]
- 33. Xin L., Lukacs R. U., Lawson D. A., Cheng D., and Witte O. N. (2007) Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells 25, 2760–2769 10.1634/stemcells.2007-0355 [DOI] [PubMed] [Google Scholar]
- 34. Hirata Y., Takahashi M., Morishita T., Noguchi T., and Matsuzawa A. (2017) Post-translational modifications of the TAK1-TAB complex. Int. J. Mol. Sci. 18, 205 10.3390/ijms18010205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fan Y., Shi Y., Liu S., Mao R., An L., Zhao Y., Zhang H., Zhang F., Xu G., Qin J., and Yang J. (2012) Lys48-linked TAK1 polyubiquitination at lysine-72 downregulates TNFα-induced NF-κB activation via mediating TAK1 degradation. Cell. Signal. 24, 1381–1389 10.1016/j.cellsig.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ahmed N., Zeng M., Sinha I., Polin L., Wei W. Z., Rathinam C., Flavell R., Massoumi R., and Venuprasad K. (2011) The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat. Immunol. 12, 1176–1183 10.1038/ni.2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fan Y., Yu Y., Shi Y., Sun W., Xie M., Ge N., Mao R., Chang A., Xu G., Schneider M. D., Zhang H., Fu S., Qin J., and Yang J. (2010) Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor α- and interleukin-1β-induced IKK/NF-κB and JNK/AP-1 activation. J. Biol. Chem. 285, 5347–5360 10.1074/jbc.M109.076976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sorrentino A., Thakur N., Grimsby S., Marcusson A., von Bulow V., Schuster N., Zhang S., Heldin C. H., and Landström M. (2008) The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 10.1038/ncb1780 [DOI] [PubMed] [Google Scholar]
- 39. Acevedo V. D., Gangula R. D., Freeman K. W., Li R., Zhang Y., Wang F., Ayala G. E., Peterson L. E., Ittmann M., and Spencer D. M. (2007) Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 12, 559–571 10.1016/j.ccr.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 40. Skaug B., Jiang X., and Chen Z. J. (2009) The role of ubiquitin in NF-κB regulatory pathways. Annu. Rev. Biochem. 78, 769–796 10.1146/annurev.biochem.78.070907.102750 [DOI] [PubMed] [Google Scholar]
- 41. Landström M. (2010) The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 42, 585–589 10.1016/j.biocel.2009.12.023 [DOI] [PubMed] [Google Scholar]
- 42. Chen Z. J. (2012) Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 246, 95–106 10.1111/j.1600-065X.2012.01108.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui J., Chen Y., Wang H. Y., and Wang R. F. (2014) Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccin. Immunother. 10, 3270–3285 10.4161/21645515.2014.979640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coornaert B., Carpentier I., and Beyaert R. (2009) A20: central gatekeeper in inflammation and immunity. J. Biol. Chem. 284, 8217–8221 10.1074/jbc.R800032200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Demaria S., Pikarsky E., Karin M., Coussens L. M., Chen Y. C., El-Omar E. M., Trinchieri G., Dubinett S. M., Mao J. T., Szabo E., Krieg A., Weiner G. J., Fox B. A., Coukos G., Wang E., et al. (2010) Cancer and inflammation: promise for biologic therapy. J. Immunother. 33, 335–351 10.1097/CJI.0b013e3181d32e74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu J., You P., Chen G., Fu X., Zeng X., Wang C., Huang Y., An L., Wan X., Navone N., Wu C. L., McKeehan W. L., Zhang Z., Zhong W., and Wang F. (2016) Hyperactivated FRS2α-mediated signaling in prostate cancer cells promotes tumor angiogenesis and predicts poor clinical outcome of patients. Oncogene 35, 1750–1759 10.1038/onc.2015.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang Y., Jin C., Hamana T., Liu J., Wang C., An L., McKeehan W. L., and Wang F. (2015) Overexpression of FGF9 in prostate epithelial cells augments reactive stroma formation and promotes prostate cancer progression. Int. J. Biol. Sci. 11, 948–960 10.7150/ijbs.12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin Y., Liu G., Zhang Y., Hu Y. P., Yu K., Lin C., McKeehan K., Xuan J. W., Ornitz D. M., Shen M. M., Greenberg N., McKeehan W. L., and Wang F. (2007) Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development 134, 723–734 10.1242/dev.02765 [DOI] [PubMed] [Google Scholar]
- 49. Zhang Y., Zhang J., Lin Y., Lan Y., Lin C., Xuan J. W., Shen M. M., McKeehan W. L., Greenberg N. M., and Wang F. (2008) Role of epithelial cell fibroblast growth factor receptor substrate 2α in prostate development, regeneration and tumorigenesis. Development 135, 775–784 10.1242/dev.009910 [DOI] [PubMed] [Google Scholar]
- 50. Wang C., Chang J. Y., Yang C., Huang Y., Liu J., You P., McKeehan W. L., Wang F., and Li X. (2013) Type 1 fibroblast growth factor receptor in cranial neural crest cells-derived mesenchyme is required for palatogenesis. J. Biol. Chem. 288, 22174–22183 10.1074/jbc.M113.463620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang F., Lu W., McKeehan K., Mohamedali K., Gabriel J. L., Kan M., and McKeehan W. L. (1999) Common and specific determinants for fibroblast growth factors in the ectodomain of the receptor kinase complex. Biochemistry 38, 160–171 10.1021/bi981758m [DOI] [PubMed] [Google Scholar]