

Abstract
Pyoverdines (PVDs) are important chromophore-containing siderophores of fluorescent pseudomonad bacteria such as the opportunistic human pathogen Pseudomonas aeruginosa in which they play an essential role in host infection. PVD biosynthesis encompasses a complex pathway comprising cytosolic nonribosomal peptide synthetases that produce a polypeptide precursor that periplasmic enzymes convert to the final product. The structures of most enzymes involved in PVD chromophore maturation have been elucidated, but the structure of the essential tyrosinase PvdP, a monooxygenase required for the penultimate step in PVD biosynthesis, is not known. Here, we closed this gap by determining the crystal structure of PvdP in an apo and tyrosine-complexed state at 2.1 and 2.7 Å, respectively. These structures revealed that PvdP is a homodimer, with each chain consisting of a C-terminal tyrosinase domain and an N-terminal eight-stranded β-barrel reminiscent of streptavidin that appears to have a structural role only. We observed that ligand binding leads to the displacement of a “placeholder” tyrosine that blocks the active site in the apo structure. This exposes a large, deep binding site that seems suitable for accommodating ferribactin, a substrate of PvdP in PVD biosynthesis. The binding site consists almost exclusively of residues from the tyrosinase domain. Of note, we also found that this domain is more closely related to tyrosinases from arthropods rather than to tyrosinases from other bacteria. In conclusion, our work unravels the structural basis of PvdP's activity in PVD biosynthesis, observations that may inform structure-guided development of PvdP-specific inhibitors to manage P. aeruginosa infections.
Keywords: siderophore, Pseudomonas aeruginosa (P. aeruginosa), infectious disease, copper, protein structure, tyrosinase, biosynthesis, enzyme, streptavidin
Introduction
The mobilization of iron from environmental sources is difficult because iron is usually deposited in insoluble Fe3+ compounds or otherwise tightly bound, e.g. to proteins. To overcome this growth-limiting factor, bacteria produce chelating agents (siderophores) that are capable of binding ferric iron tightly and transport it into their cells. A particularly well studied group of siderophores are the pyoverdines (PVDs), 2 which are pigments and important virulence factors of fluorescent pseudomonads. Almost 70 strain-specific PVDs have been described to date (1). PVDs consist of three parts: a short peptide backbone of 6–12 amino acids is bound to a fluorescent dihydroxyquinoline chromophore, which is connected to an additional acyl side chain of variable length (2). Ferric iron is trapped with high affinity in a stable 1:1 octahedral complex between two hydroxamate groups (occasionally β-hydroxyamino acids) of the peptide backbone and the catecholate groups of the chromophore (3). Three strain-specific pyoverdines (PVDI–III) are known (4) from Pseudomonas aeruginosa with PVDI from strain PAO1 being the best investigated. At least 12 enzymes are involved in PVD biosynthesis of this strain (see Fig. 1A). The initial steps are catalyzed by cytoplasmic nonribosomal peptide synthetases. PvdL, PvdI, PvdJ, and PvdD not only synthesize the PVD peptide backbone but also moieties that will eventually become the fluorescent chromophore (5). Because the composition of the peptide is strain-specific, accessory proteins like PvdA, PvdF, and PvdH provide noncanonical amino acid building blocks (6–8). It is believed that PvdE, which is an “export” ABC transporter in the inner membrane, then transports the nonfluorescent precursor to the periplasm for further maturation by the five enzymes PvdM, PvdN, PvdO, PvdP, and PvdQ (9–12). The myristoyl membrane anchor of the pyoverdin precursor is removed by the hydrolase PvdQ (13), and the fluorescent chromophore of PVD is furnished by PvdP (14) and PvdO (15) before the pyridoxal phosphate–containing PvdN modifies the acyl side chain at the 3-amino group of the chromophore (16–18). In addition to PvdQ and PvdN, the structures of PvdM (Protein Data Bank (PDB) entry code 3B40) and PvdO (19) have been determined. PvdM possesses structural similarity to metal-dependent amidohydrolases, but its exact function in PVD biosynthesis is currently unknown.
Figure 1.

A, overview of PVD biosynthesis from fatty acids (FA) and proteinogenic and nonproteinogenic amino acids. l-Asp-SA, l-Asp-semialdehyde; l-Dab, l-2,4-diaminobutyrate; l-Orn, l-ornithine; l-OH-Orn, l-N5-hydroxy-Orn; l-fOH-Orn, l-N5-formyl-N5-hydroxyornithine. The precursor of PVD assembles in the cytosol, undergoes maturation in the periplasm, and binds ferric ion outside of the cell. B, current understanding of chromophore formation in PVD biosynthesis from ferribactin (left). TE, thioesterase domain. PvdP and the tyrosyl moiety of PVD are highlighted in red.
The biosynthesis of the fluorescent dihydroxyquinoline moiety involves an oxidative cascade (20) in which the tyrosinase PvdP acts as a cresolase (monooxidase) to form a catechol from the d-Tyr moiety of the PVD precursor ferribactin first, followed by catecholase (oxidase) activity to create an o-quinone that undergoes intramolecular cyclization before PvdO performs a final oxidation to the fluorophore (Fig. 1B; Refs. 14 and 15). Although the identity between PvdP and other tyrosinases is low, a tyrosinase-typical type-3 dicopper center involving six essential, highly conserved histidines has been revealed by homology modeling. Based on these findings, it has been proposed that PvdP is the first member of a new tyrosinase family (14). However, no homology can be detected for a significant part of the N terminus of PvdP, suggesting that the N terminus contains a second domain of unknown function. Because PvdP is essential for PVD biosynthesis (11) and was found to be up-regulated in an acute burned mouse P. aeruginosa infection model (21), it may be a suitable target for anti-infectives, but the lack of high-resolution structures hampers the development of such inhibitors. We therefore conducted crystallization and X-ray diffraction experiments, revealing that the homodimeric PvdP indeed is a two-domain protein with an unprecedented architecture, consisting of an N-terminal streptavidin-like β-barrel and a C-terminal tyrosinase. The active site arranges into a new class of tyrosinases from a hitherto uncharacterized branch of type-3 copper proteins. Comparison of the apo structure with a ligand complex shows that the active site is blocked by a “placeholder” residue (Tyr531) in the apo form, which is typical for tyrosinases. Ligand binding displaces this placeholder and exposes an extensive binding site to host the large ferribactin substrate. The binding site consists mainly of residues from the tyrosinase domain and is lined by a small proportion of the N terminus of the second monomer, suggesting that the streptavidin-like domain primarily possesses a structural rather than a functional role.
Results
Structure determination
PvdP was produced without the N-terminal periplasmic localization signal by heterologous overexpression in Escherichia coli and purified via Ni2+-affinity chromatography followed by a size exclusion step. Because the removal of the N-terminal His6 tag by tobacco etch virus protease (TEV) was not successful, all subsequent experiments were performed with His6-TEV-PvdP(26–544) (where TEV indicates the TEV cleavage site). The protein was active as demonstrated by dopaquinone formation from l- or D-tyrosine in the presence of Cu(II)SO4, and the following enzyme kinetic parameters were determined: Km = 1.077 ± 0.103 mm, kcat = 228 ± 8 s−1 for l-Tyr and Km = 1.074 ± 0.209 mm, kcat = 197 ± 13 s−1 for d-Tyr (Fig. S1), indicative of nearly identical turnover of these surrogate substrates. Of note, although Km values are similar to previously reported numbers, kcat values were approximately 200-fold higher, which may be because the N-terminal periplasmatic localization signal was not omitted in the previous report (14).
Crystallization provided only thin plates that were very sensitive to handling and usually gave only strongly anisotropic diffraction patterns if any. Initial phases were derived from single anomalous diffraction data merged from three seleno-l-methionine–containing His6-PvdP crystals. Because soaking destroyed the apo crystals, cocrystallization was used to obtain protein–ligand complex structures. Ellipsoidal resolution cutoffs were applied to compensate for the anisotropic diffraction, allowing to refine the apo structure at 2.09 Å and the complex with l-tyrosine at 2.7 Å. Although the electron density was unambiguous in general, residues at the N and C termini, a loop between strands β6 and β7, and in parts of the flexible C-terminal region beyond amino acid 485 were not visible in both structures. Data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| PvdPapo | PvdPTyr | PvdPSeMet | |
|---|---|---|---|
| Data collection statistics | |||
| Beamline | PETRAIII P11 | BESSYII 14.2 | SLS X06DA/PETRAIII P11 |
| No. of crystals | 1 | 1 | 3 |
| Wavelength (Å) | 0.9794 | 1.282 | 0.9779/0.9794 |
| Space group | P21 | P21 | P21 |
| Unit cell dimensions | |||
| a, b, c (Å) | 97.35, 107.79, 107.94 | 77.33, 109.14, 82.51 | 96.19, 108.41, 108.41 |
| α, β, γ (°) | 90, 99.97, 90 | 90, 95.55, 90 | 90, 99.70, 90 |
| Resolution range (Å) (highest shell) | 48.07–2.09 (2.26–2.09) | 48.09–2.70 (3.03–2.70) | 48.34–3.50 (3.70–3.50) |
| Ellipsoidala resolution (Å) (direction)b | 2.84 (0.988 a* − 0.152 c*) | 3.73 (0.718 a* − 0.696 c*) | n.a.c |
| 2.19 (b*) | 2.7 (b*) | n.a. | |
| 2.09 (−0.032 a* + 0.999 c*) | 2.93 (0.64 a* + 0.768 c*) | n.a. | |
| Total no. of reflections (ellipsoidal)d | 578,302 (27,712) | 229,383 (8,710) | 2,663,558 (404,486) |
| No. of unique reflections (ellipsoidala)d | 85,942 (4,285) | 23,084 (1,154) | 54,434 (8,374) |
| Average multiplicityd | 6.7 (6.5) | 9.9 (7.5) | 48.9 (48.3) |
| Completenessd (%) | 66.3 (16.0) | 61.5 (10.5) | 99.9 (100.0) |
| Completeness (ellipsoidala)d,e (%) | 93.2 (76.7) | 92.1 (56.5) | n.a. |
| I/σ(I) (ellipsoidala)d | 10.7 (1.6) | 8.6 (1.6) | 18.1 (10.6) (spherical) |
| Rmeasd,f | 0.13 (1.28) | 0.32 (1.49) | 0.27 (0.54) |
| Rpimd,g | 0.051 (0.50) | 0.10 (0.54) | NDh |
| CC½i | 0.99 (0.54) | 0.99 (0.51) | 0.99 (0.99) |
| Refinement statistics | |||
| Resolution (Å) | 45.61–2.09 | 48.09–2.70 | |
| No. of reflections used | 85,901 | 22,962 | |
| Rworkj (%) | 21.41 | 20.46 | |
| Rfreek (%) | 24.10 | 26.18 | |
| No. of residues | |||
| Protein | 1,889 | 916 | |
| Water | 513 | 24 | |
| Zn2+ | 4 | ||
| l-Tyrosine | 2 | ||
| Mean B-factorl (Å2) | 48 | 34 | |
| All protein residues | 48 | 34 | |
| Ligands | 36 | ||
| Water molecules | 38 | 20 | |
| r.m.s.d. | |||
| Bond length (Å)m | 0.002 | 0.002 | |
| Bond angle (°)m | 0.495 | 0.460 | |
| Ramachandran plot (%) | |||
| Favored regionsm | 96.82 | 96.6 | |
| Allowed regionsm | 100 | 100 | |
| Outliersm | 0 | 0 | |
| MolProbity scorem | 1.00 | 0.95 | |
| PDB code | 6EYS | 6EYV | |
a Statistics refer to data truncated by STARANISO to remove weak reflections affected by anisotropy (46).
b The resolution limits for three directions in reciprocal space (a*, b*, c*) are indicated here. To accomplish this, STARANISO computed an ellipsoid postfitted by least squares to the cutoff surface, removing points where the fit was poor. Note that the cutoff surface is unlikely to be perfectly ellipsoidal, so this is only an estimate.
c n.a., not applicable.
d Values in parentheses are for the highest-resolution shell.
e The anisotropic completeness was obtained by least squares fitting an ellipsoid to the reciprocal lattice points at the cutoff surface defined by a local mean I/σ(I) threshold of 1.5, rejecting outliers in the fit due to spurious deviations (including any cusp) and calculating the fraction of observed data lying inside the ellipsoid so defined. Note that the cutoff surface is unlikely to be perfectly ellipsoidal, so this is only an estimate.
f Rmeas = Σhkl{N(hkl)/[N(hkl) − 1]}1/2 × Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣi Ii(hkl).
g Rpim = Σhkl{1/[N(hkl) − 1]}1/2 × Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
h Not determined.
i CC1/2 = Σ(x − 〈x〉)(y − 〈x〉)/[Σ(x − 〈x〉)2Σ(y − 〈y〉)2]1/2.
j Rwork = (Σhkl‖Fobs| − k|Fcalc‖)/(Σhkl|Fobs|).
k Rfree is the same as Rwork with 5% of reflections chosen at random and omitted from refinement.
l B-factors calculated with Moleman2 (55).
m Statistics calculated with the MolProbity web server (56) (http://molprobity.biochem.duke.edu/) (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.).
Overall architecture
PvdP crystallized in space group P21 with four or two monomers in the asymmetric unit of the apo or complex structure, respectively. These monomers possess an unprecedented two-domain architecture consisting of an N-terminal β-barrel domain (BBD) and a C-terminal tyrosinase domain (TYD) connected by a short linker (residues 189–192; Fig. 2, A and B). The interface between both domains consists of a continuous sequence on the TYD side (residues 292–339; helices α9, α10, and N terminus of α11), which assumes an L-shaped structure that matches the β-barrel on its nonsolvent-exposed face. Both domains share a 713-Å2 interface that is filled with water molecules and has an average gap width of 3.7–4.2 Å. Only a few interactions aside of van der Waals forces stabilize the interaction between both domains, namely five H-bonds: the backbone amide of Leu297 interacts with the carbonyl oxygen atom of Asp86; the side chain of Arg301 establishes three H-bonds with the backbone of a loop containing Leu104, Ala106, and Glu108; and His333 bridges to Asp56.
Figure 2.

A, overall structure of the PvdP dimer in the apo form. The N-terminal β-barrel domain is shown in yellow, the C-terminal tyrosinase domain is in blue. The typical four-helix bundle of type-3 copper proteins is shown in light blue, the C-terminal section that gets displaced in the l-tyrosine complex is in red. Histidines of the CuA and CuB sites and the placeholder residue Tyr531 are shown as sticks. Orange spheres indicate the expected positions of two Cu2+ ions deducted from the coordinates of Zn2+ in the l-tyrosine complex. B, topology diagram of the PvdP monomer. Helix α19 contains the placeholder residue Tyr531 and becomes disordered in the complex with l-tyrosine. The topology diagram was drawn with TopDraw (57), and all molecular representations were prepared with PyMOL (51).
The monomers contained in the asymmetric unit associate to homodimers in which the BBD of one monomer tightly interacts with the TYD of the other (Fig. 2A). The overall surface area of the dimer is 37,345 Å2. Analysis with PISA (22) indicates a dissociation energy, ΔGdiss, of 34.4 kcal/mol. Both molecules share an interface area of 3225 Å2 of which the N termini of the BBD (residues 36–47) contribute 1163 Å2. Removal of the N termini reduces ΔGdiss to 17.2 kcal/mol. All monomers are virtually identical and superimpose with a maximum Cα r.m.s.d. of 0.176 Å between the four chains of the apo structure and of 0.385 Å between the apo and the ligand-bound structure.
N-terminal domain (BBD)
The N-terminal domain (residues 36–188) comprises an eight-stranded antiparallel β-barrel that is closed off with helix α1 at one end and by the solvent-exposed loop L1 at the other. With the exception of one histidine (His142), the inside of the barrel is filled by the side chains of hydrophobic residues. Of note, Trp128 reaches especially deep into the barrel and blocks the passage (Fig. 3A). Without this side chain, a cavity with a diameter of 9.6 Å would form. Interestingly, searches with PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm) 3 reveal structural similarity to streptavidin (Fig. 3B). Superposition with a monomer of WT streptavidin from Streptomyces avidinii (WTStrep) resulted in an r.m.s.d. of 2.7 Å (89 residues). Strands β1–β4 of PvdP and WTStrep are similar in size (5–7 residues), but the following strands, β5–β8, of PvdP are shorter (6–9 residues compared with 10–13 amino acids in WTStrep), giving PvdP a more symmetrical appearance. However, because the BBD of PvdP lacks the biotin-binding pocket of streptavidin, it is not surprising that soaking or cocrystallization trials with biotin did not result in incorporation of the ligand. In fact, the position of both BBDs of the PvdP dimer relative to the active sites implies a structural role, similar to structurally related BBDs found in quinohemoprotein amine dehydrogenase (PDB entry code 1JMX; Ref. 24) and in erythrocruorin (PDB entry code 2GTL; Ref. 25), two heterooligomeric proteins with functions unrelated to PvdP (Fig. S2). Although the sequence identity to the BBD of PvdP is less than 20% for both of these proteins, their potential ligand-binding sites are also blocked with bulky hydrophobic amino acids, emphasizing their structural role.
Figure 3.
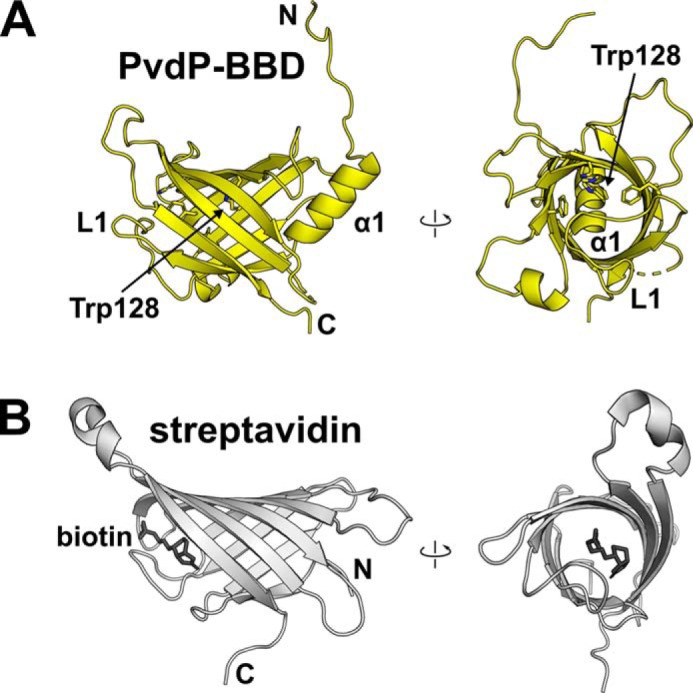
The N-terminal β-barrel domain of PvdP (PvdP-BBD) has structural similarity to streptavidin. A, two perpendicular views of PvdP-BBD. Access to the inside of the β-barrel is blocked by helix α1 and loop L1. The inside of the barrel is lined by the indicated aromatic residues. Removal of the side chain of Trp128 would generate a cavity with a diameter of almost 10 Å. B, biotin-bound streptavidin from S. avidinii (PDB entry code 3RY2; Ref. 23) shown from similar orientations as PvdP-BBD. Note that the hypothetical biotin-binding site in PvdP-BBD is blocked.
Because the similarity between BBD and streptavidin or the other mentioned proteins was not detected at the sequence level or by sophisticated structure prediction methods such as Phyre2 (26), we used different bioinformatics approaches to identify homologues and to learn about the potential function of this domain. Although searches with HHPred (27) in various databases as well as a search in CATH (28) returned only insignificant hits, a BLAST search (29) in the NCBI reference sequence database (30) excluding pseudomonads returned five proteins from γ-proteobacteria, namely from Enterobacter cloacae (SAJ31658.1), Cellvibrio japonicus (WP_012487482.1), Azotobacter vinelandii (WP_061289382.1), Stenotrophomonas rhizophila (KWW15420.1), and the unclassified γ-proteobacterium L18 (WP_027977676.1). Alignment with the complete sequence of PvdP (Fig. S3) shows that these proteins share large patches of conserved amino acids also beyond the BBD; i.e. they also contain a tyrosinase domain, implying that they may be involved in a similar biosynthetic pathway. Indeed, searches with PvdO, the other enzyme involved in PVD fluorophore maturation in P. aeruginosa (15), identified similar proteins in the genomic vicinity of two of these species (C. japonicus and A. vinelandii), suggesting that these strains could synthesize related compounds. However, with respect to PvdP, three of the proteins listed above lack an N-terminal periplasmic localization signal (Fig. S3), indicating that a potential siderophore biosynthesis must be organized differently in these organisms. Interestingly, the search also returned an uncharacterized fully identical but N-terminally truncated version of PvdP designated as coming from E. cloacae e403. Surprisingly, the genome of this bacterium seems to contain most PVD biosynthesis genes, and all of them share almost 100% identity to the respective proteins from P. aeruginosa PAO1. The N-terminal truncation of the PvdP-like gene comprises the first 83 amino acids and would also affect helix α1 and strands β1 and β2, which will likely impede the stability of the resulting protein. This may indicate that PVD biosynthesis is not functional in the respective isolate. In summary, although theses searches do not provide indications toward the function of the BBD, they show that there may be other γ-proteobacteria whose capacity to biosynthesize PVD-like siderophores has not been recognized.
C-terminal domain (TYD)
The C-terminal domain of PvdP is mostly α-helical with the exception of a small, solvent-exposed β-sheet (β9a and β9b). Its most prominent feature is a four-helix bundle (α5, α8, α13, and α16) that provides six histidine residues to form the active site (His216, His220, His271, His375, His379, and His432; Figs. 2, 4, and 5A). This arrangement is typical for type-3 copper proteins, and because PvdP displays tyrosinase activity (14), we refer to this domain as the TYD. Type-3 copper proteins contain two copper-binding sites termed CuA and CuB that are established by three histidines each, and it has recently been suggested that they have evolved into three subclasses that can be distinguished by the length of the sequences that separate the histidines in both copper-binding sites (31). Notably, with a His-X3-His-Xn-His-CuA motif (His216, His220, and His271) and a very long insertion between the second and third histidines of the CuB motif (His375, His379, and His432), PvdP seems to fall into the β-subclass of type-3 copper proteins (Fig. 4). This β-subclass has mainly been associated with arthropods, whereas bacterial enzymes in general belong to the α-subclass (α-subclass motifs are His-Xn-His-X8-His for the CuA site and His-X3-His-Xn-His for the CuB site, respectively). However, many other conserved β-subclass residues described previously (31) are not found in PvdP, suggesting that PvdP establishes a new, previously unrecognized subclass of type-3 copper proteins.
Figure 4.

Sequence alignment of the CuA and CuB sites of PvdP with other type-3 copper proteins of the α-subclass (HcOd, hemocyanin A-type, Octopus dofleini; AuSCg, plant aurone synthase, Coreopsis grandiflora; TyrSc, tyrosinase, S. castaneoglobisporus; TyrBm, tyrosinase, B. megaterium) and β-subclass (HCPi, hemocyanin A, Panulirus interruptus; HcLp, hemocyanin II, L. polyphemus; TyrDm, tyrosinase, D. melanogaster) as defined previously (31). Copper-coordinating histidines are shown in red, and similar residues are in blue (α-subclass only), gray (β-subclass only), or yellow (both α- and β-subclasses). Strictly conserved residues are shown in bold. Stars highlight two phenylalanines found in all type-3 copper proteins (Phe267 and Phe428 in PvdP). Filled circles mark every 10th amino acid of the top sequence.
Figure 5.

Details of the tyrosinase active site of PvdP. A, the TYD of PvdP contains a CuA and a CuB site, which were loaded with Zn2+ (gray spheres) in the complex with l-tyrosine determined here (thin black lines). In the apo structure, the placeholder residue Tyr531 occupies the binding site of the substrate's tyrosyl moiety, but autoxidation is hindered by holding the residue further away from the metal atoms (interaction with Gly417). Glu371 and Asp376 bind a water molecule that is implied in substrate deprotonation in other tyrosinases. Met270 and Met274 shield the active site from the solvent and could play a role in loading the enzyme with Cu2+. B, two representations of the molecular surface of PvdP in complex with l-tyrosine. The left side shows an electrostatic surface at ±10 kBT/e; the surface on the right has been colored according to the two chains of the PvdP homodimer. C, closeup of the l-tyrosine–binding site. Electrostatic potentials were calculated with APBS (46); A and C are cross-eyed stereoplots.
In addition to the six histidines, the TYD of PvdP contains several strictly to highly conserved type-3 copper protein residues such as phenylalanines four positions upstream of the third histidine in both copper-binding sites (CuA, Phe267/His271; CuB, Phe428/His432; Fig. 5A), which both point toward the active site. Other conserved residues are Pro426, which sits at the N-terminal end of helix α16 from the four-helix bundle and is probably required to provide a kink that leads to an almost 90° bend after helix α15 near the active site, and Asp436 in the middle of helix α16 where it interacts with the highly conserved Arg272 of helix α8 of the four-helix bundle.
A feature that sets PvdP truly apart from related proteins is the long insertions between the second and third histidines of both copper-binding sites. In PvdP, these sequences contain 50 (CuA) and 52 (CuB) amino acids, whereas they are much shorter in other tyrosinases (Fig. 4). For example, in the β-subclass tyrosinase from Drosophila melanogaster, they consist of only 24 (CuA) and 35 (CuB) amino acids. The extensions in PvdP lead to the formation of additional secondary structural elements such as the short helix α6 and the long helix α7 in the CuA site and to a long loop containing two short helices (α14 and α15) between helices α13 and α16 in the CuB site, respectively. Another unusually long sequence is located between the last histidine of the CuA (His271) and the first histidine of the CuB site (His375). This stretch includes helices α9–α12, and, with the exception of α12, all of these helices are involved in the interface with the BBD (Fig. 2B).
Active site
No metal ions were observed in the CuA or CuB sites of the apo structure of PvdP. Instead, the CuA site was occupied by a water molecule tetrahedrally coordinated by His216, His220, His271, and another water molecule. The entrance to the active site is blocked by Tyr531, which belongs to the short helix α19 close to the C terminus (Figs. 2B, 5A, and S5). Superimposition with the tyrosine complex of the Bacillus megaterium tyrosinase (PDB entry code 4P6R; Ref. 32) shows that Tyr531 occupies the substrate-binding site and hence acts as a placeholder, reminiscent of similar residues but from different regions in related polyphenoloxidases and hemocyanins (33) and again implicating that PvdP belongs to a new subclass of type-3 copper proteins. The hydroxyl group of Tyr531 points toward the CuA site and is located within van-der-Waals contact distance to Nϵ2-His220. The phenol ring of Tyr531 stacks with the imidazole group of His379 of the CuB site. The distance of the hydroxyl group toward the position of the metal ions at the CuA or CuB sites would be 3.6 and 3.9 Å as extrapolated from superimposition of PvdPapo on PvdPTyr.
The PVD precursor ferribactin (Fig. 1B) was not available to us. We therefore attempted soaking and cocrystallization with the surrogate substrates d- and l-tyrosine in the presence of CuSO4 but failed to obtain crystals. CuSO4 was therefore replaced by ZnCl2 because substitution of the cofactor Cu2+ by Zn2+ is known to lead to reduced activity of tyrosinases (32, 34), and we hypothesized that impeded turnover would support crystallization in the presence of substrates. Indeed, crystals with l-tyrosine could be obtained, but they were of lower quality than for the apoenzyme and belonged to a different crystal form (P21 with one PvdP dimer in the asymmetric unit), but single anomalous diffraction data collected at the zinc absorption edge (1.28 Å) clearly revealed the presence of Zn2+ ions in both the CuA and CuB sites (Fig. S4). The distance between the cations is 3.6 Å, and they are bridged by a water molecule at a distance of 2.0 Å toward each Zn2+. The most striking difference with respect to the apo structure is that not only the placeholder residue Tyr531 is displaced from the active site but also the structure surrounding it has become disordered such that residues 514–541 could not be traced in the electron density. This opens a large solvent-exposed and negatively charged pocket whose dimensions seem to reflect the size of the substrate molecule ferribactin (Fig. 5, B and C).
The placeholder position is occupied by poorly defined electron density that can be interpreted as MES from the crystallization buffer or as the surrogate substrate l-tyrosine. However, because superimposition with ligand complexes of related enzymes shows that tyrosine and derivatives bind in a similar fashion in these proteins, we interpreted the additional electron density as l-tyrosine (Figs. 5, A and C, and S4). The inferior quality of the electron density probably is a consequence of low solubility and weak binding of l-tyrosine, which is an order of magnitude smaller, has the opposite chirality, and is a zwitterion compared with the neutral d-tyrosyl moiety of the natural substrate ferribactin. This is also reflected in the fact that l-tyrosine, similar to d-tyrosine, is a relatively poor substrate of PvdP (Km values approximately 1 mm for l- and d-Tyr; see above).
Interestingly, the ligand-binding site consists mainly of residues from the tyrosinase domain, and only a small extended stretch of the first 10 amino acids from the N terminus of the second monomer lines parts of its perimeter (Fig. 5B). This again emphasizes the primarily structural function of the BBD but may also point toward a critical role of dimerization in PvdP.
Discussion
The structure analysis presented here reveals that PvdP possesses a novel two-domain architecture exclusively found in a small number of γ-proteobacterial species. Although the C-terminal domain has a core architecture commonly found in tyrosinases, the N-terminal domain is unique in primary sequence but resembles streptavidin in tertiary structure. Interestingly, sequence database searches identified one protein (GenBankTM accession number SAJ31658.1) that, with the exception of a deletion of the first 60 amino acids, is 100% identical to PvdP. The sequence of this protein was derived by whole-genome shotgun sequencing from a biological sample and was assigned as coming from E. cloacae strain e403. However, the high sequence identity makes the assignment to E. cloacae questionable, and indeed, other proteins from the same strain deposition are identical to P. aeruginosa proteins as well, corroborating this assumption. Conversely, the finding that searches with the PvdP sequence identified similar proteins in species not previously investigated for siderophore production such as C. japonicus, A. vinelandii, S. rhizophila, or γ-proteobacterium L18 shows that PVD production may be more widespread than anticipated.
PvdP is only the third bacterial tyrosinase whose structure has been determined. The other two representatives are tyrosinases from Streptomyces castaneoglobisporus (TyrSC; PDB entry code 1WXC; Ref. 35) and B. megaterium (TyrBM; PDB entry code 3NM8; Ref. 36), but they are only relatively distantly related to PvdP. In fact, sequence analysis places PvdP closer to arthropod rather than bacterial tyrosinases (31), albeit still with large dissimilarities at the sequence level (Fig. 4). The evolutionary distance of PvdP may also be responsible for the failure to discover related structures of the C-terminal domain with default parameters in the structure similarity search program PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm).3 Such relatives could be identified with DALI (37), clearly revealing similarities to other type-3 copper proteins such as hemocyanins, arylphorins, and phenoloxidases in addition to tyrosinases. According to this analysis, the closest homologue to the tyrosinase domain of PvdP is the type-3 copper protein domain of a prophenolxidase from Manduca sexta (PDB entry code 3HHS; Ref. 38), which aligns with an r.m.s.d. of 3.9 Å over 177 residues (Z-score = 11.3). However, the overall structure of this protein is grossly different from PvdP, and the sequence identity within the aligned structural elements is only 14%.
The large evolutionary distance of PvdP to related proteins offers an opportunity to re-evaluate the importance of several sequence motifs that have been identified as having key roles in other tyrosinases (for a recent review, see Ref. 39). These enzymes execute a complicated reaction cycle that involves different redox states of the two copper atoms and can eventually lead to loss of the metal due to side reactions that generate Cu0 atoms. This may hint at generally rather weak metal affinity to enable enzyme reactivation by copper reloading and could explain why we observed the apoprotein in a metal-free state. At the same time, low metal affinity would require evolving means for recharging the enzyme with Cu2+, and indeed, several such mechanisms are discussed for different types of type-3 copper proteins. For example, the role of two structurally neighboring methionines in supplying the protein with Cu2+ has been demonstrated in TyrBM (Met61 and Met184 in TyrBM; Ref. 40). These residues are not conserved in PvdP, but interestingly, a similar solvent-exposed methionine motif (Met270 and Met274 in PvdP) that shields the active site and may hence serve as a copper load port can be found at another location in PvdP, suggesting a similar role that deserves attention in future studies (Fig. 5A). Another highly conserved methionine that may be involved in H2O2 scavenging via sulfur oxidation (39), Met215 in TyrBM, is replaced by a leucine in PvdP (Leu416), which may reflect the fact that P. aeruginosa has other very effective detoxification systems for reactive oxygen species (41).
Mechanistic studies indicate that the phenol group of the substrate has to be deprotonated to initiate binding to the CuA site of tyrosinases. Experimental and in silico evidence suggests that this is achieved by a conserved glutamate/asparagine dyad (Glu195 and Asn205 in TyrBM) that binds a water molecule to perform the deprotonation (32). In PvdP, the glutamic acid is conserved (Glu371), but we found an aspartic acid (Asp376) instead of asparagine. Nevertheless, a water molecule bound in a similar place as in other tyrosinases can be observed in the better resolved apo structure, indicating that PvdP applies a similar mechanism for substrate activation (Fig. 5A).
An interesting feature of most type-3 copper proteins is the presence of a placeholder residue that occupies the substrate-binding site in the resting state of the enzyme and needs to be displaced via structural rearrangements or by proteolytic cleavage to activate the enzyme. PvdP provides a new placeholder motif to the type-3 copper protein family by bearing the placeholder residue Tyr531 in an α-helix within its flexible C terminus as indicated by high B-factors and the partial absence of traceable electron density. The fact that the enzyme was active in in vitro assays both toward the model substrate tyrosine and toward ferribactin (14) indicates that PvdP does not undergo proteolytic activation but rather uses displacement of the last 30 amino acids (beyond Phe512 at the C terminus of helix α18) to provide a binding interface for the large PVD precursor molecule. In the apo structure, Tyr531 binds the active center at a position in which the hydroxo group of the side chain is 1.4 Å more distant to the CuA site than in the complex with the tyrosine ligand. This probably avoids autoxidation of Tyr531 and is achieved by locating Tyr531 in a short η-helix (Pro529–Arg533) that interacts with Gly417 at its N terminus, thus avoiding further slipping of Tyr531 into the active center (Fig. 5A).
The finding that the N-terminal domain adopts a streptavidin-like fold that could not be predicted from its sequence was a surprise to us. The current analysis of the structure suggests that it takes a structural rather than a functional role by establishing contacts with the tyrosinase domain of the second chain of the PvdP homodimer, leading to an unprecedented overall structure within the type-3 copper protein family. This family is known to contain largely different quaternary structural arrangements, reaching from monomers such as TyrBM to large complexes consisting of up to 48 chains as in the case of hemocyanin from the horseshoe crab Limulus polyphemus (42). The diversity of these structures probably reflects a long evolutionary history of this protein family, explaining why PvdP deviates in so many details from previously studied tyrosinases and establishes a previously unrecognized subclass of type-3 copper enzymes.
In summary, the data presented here unravel the structural basis of the activity of PvdP in pyoverdin biosynthesis. The finding that PvdP, although keeping essential residues involved in the chemistry catalyzed by tyrosinases, replaces several sequence motifs involved in mechanisms not directly associated with catalysis provides deeper insight into this protein family and may also serve as a starting point for the structure-guided development of PvdP-specific inhibitors against disease inflicted by P. aeruginosa. Toward this, the large body of known natural and synthetic tyrosinase inhibitors (43) should provide leads into the chemical nature of such compounds.
Experimental procedures
Chemicals and reagents
All chemicals were from Sigma-Aldrich unless otherwise indicated. Molecular biology reagents were purchased from Fermentas.
Cloning
The PvdP gene of P. aeruginosa UCBPP-PA14 (PA14_33740) was cloned without the predicted signal sequence (PvdP(26-544)) into a pOPINB plasmid (44) using a touchdown PCR protocol for gene amplification (forward primer, 5-ggaagtgctgtttcagggtaccgacgagggcgccctgtacgg-3; reverse primer, 5-gatgtttaaactggtctagaaagcttagtccgccttcaccgggcg-3). Cloning was done using the sequence- and ligation-independent method (SLIC) and KpnI/HindIII restriction sites for vector opening. Initial transformations were plated on SOC agar with kanamycin using ultracompetent E. coli Omnimax (Thermo Fisher Scientific). The full construct contained an N-terminal His6 tag followed by a PreScission protease cleavage site before PvdP(26–544).
Expression and protein purification
PvdP expression was achieved in E. coli Rosetta2(DE3) (Novagen) using 1 liter of SOC medium supplemented with 30 mg/liter kanamycin at 37 °C at 130 rpm. Induction with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside was started when cell density reached A600 nm 0.6–0.8 at which point the temperature was decreased to 20 °C. Cells were harvested after 20 h of incubation. Selenomethionine (SeMet)-containing PvdP was obtained as follows. Precultures were grown in 100 ml of LB including 30 mg/liter kanamycin and incubated overnight at 37 °C. 20 ml of the culture were harvested, washed twice with M9 minimal medium without antibiotic, and used for starting 1-liter cultures of M9 medium including kanamycin. After reaching an A600 nm of 0.5, an amino acid mixture (100 mg/liter Lys, Phe, and Thr and 50 mg/liter Ile, Leu, and Val) was supplied. After a 0.5-h incubation at 20 °C, 60 mg of SeMet powder and 0.1 mm isopropyl 1-thio-β-d-galactopyranoside were added. Cultures were shaken at 130 rpm for 24 h before harvesting. For purification of protein for crystallization experiments, 2 (native) or 4 liters (SeMet) of cell culture were used. Purification started with a 5-ml HisTrap FF column (GE Healthcare) and buffer A (50 mm Tris/HCl, pH 8.0, 0.1 m NaCl) versus buffer B (50 mm Tris/HCl, pH 8.0, 0.1 m NaCl, 0.5 m imidazole) on an ÄKTApurifier system (GE Healthcare). Because cleavage of the tag was not successful, His6-tagged PvdP was directly run on an Superdex S75 26/600 gel filtration column (GE Healthcare) using buffer A. Using this buffer system, PvdP reversibly aggregated when concentrated to more than 12 mg/ml.
Enzyme kinetic measurements
Enzyme kinetic parameters for l- and d-tyrosine were determined with a colorimetric assay detecting the generation of dopachrome at 475 nm (ϵ = 3600 m−1 cm−1; Ref. 14) in an Evolution 260 UV-visible spectrophotometer (Thermo Fisher Scientific) thermostated at 303.15 K. The substrate concentration was varied between 0 and 4 mm by mixing the required ratios of buffer (50 mm CHES, pH 9, 0.25 mm CuSO4) containing no or 4 mm substrate to 1 ml in 1-cm plastic cuvettes. The reaction was initiated by adding 126 μg of His6-tagged PvdP and then followed for 300 s in 10-s intervals. All measurements were performed in triplicates and evaluated with the Enzyme Kinetics Module in SigmaPlot (Systat Software, Inc.) using a simple Michaelis–Menten model.
Crystallization
PvdP was crystallized with the sitting drop vapor diffusion method in 96-well format using Intelli-Plates (Art Robbins Instruments) at 293.15 K. Native PvdP concentrations ranged from 4 to 12 mg/ml, and promising crystals were identified with a precipitant consisting of 20% PEG 3350 and 0.18 m ammonium citrate. These crystals could only be optimized by microseeding (1:10 to 1:1000 diluted seed stock). Final crystals were small thin plates and often contained defects. For SeMet-protein, conditions had to be rescreened, resulting in 25% PEG 3350, 0.2 m NaCl, 0.1 m Tris/HCl, pH 8.4, as the precipitant. Reliability of crystal growth was again enhanced using microseeding. For ligand-bound PvdP, the buffer was changed to 50 mm CHES, pH 9.0, prior to crystallization (14), which resulted in better solubility of PvdP. Crystals grew in 0.86 m (NH4)2SO4, 0.1 m MES, pH 5.5. Cocrystallization was used to obtain ligand complexes. PvdP was incubated with 0.5 mm ZnCl2 and 1 mm l-tyrosine for 0.5 h on ice before setting up crystallization experiments. Prior to flash cooling in liquid nitrogen, crystals were washed in precipitant supplemented with 10% (2R,3R)-2,3-butanediol and 0.5 mm l-tyrosine, 0.3 mm ZnCl2 in the case of cocrystallized PvdP.
Data collection
X-ray diffraction data were collected at 100 K at the PETRAIII (Deutsches Elektronen-Synchrotron (DESY), Hamburg, Germany), BESSYII (Berlin, Germany), and SLS (Paul Scherrer Institute, Villigen, Switzerland) synchrotrons. For PvdPapo and PvdPTyr, data from single crystals were collected on beamline P11 (PETRAIII) or beamline 14.2 (BESSYII) using 3600 nonoverlapping frames of 0.1°. Data were indexed and integrated with XDS (45) and then submitted to the STARANISO server (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi)3 to calculate the ellipsoidal resolution limit, setting 1.5 I/σ(I) as the lowest acceptable signal. Because no search model for molecular replacement was available, PvdPSeMet was used for phasing. To improve the anomalous signal, multiple SeMet-containing crystals were measured, and data sets were tested for scalability using XSCALE (45). Data of three crystals were sufficiently similar to be merged: of crystal I, four wedges (χ-rotation of 0°, 10°, 20°, and 30°) were measured and scaled to 10 wedges from crystal II (seven data sets at χ 0°, 5°, 10°, 15°, 20°, 25°, and 30° and three data with χ-/ϕ-rotation of 7°/10°, 17°/10°, and 27°/10°). These data were collected at the SLS on beamline PXIII at a wavelength of 0.978 Å. However, more data were required to obtain initial phases, which were contributed by crystal III measured at PETRAIII beamline P11 at a wavelength of 0.979 Å. Scaling all of these SeMet data with XSCALE increased redundancy to 49-fold and led to usable anomalous signal to 3.5-Å resolution. After data reduction, SeMet data were converted using XDSCONV (45).
Structure solution and refinement
Structure solution and refinement were carried out using programs from the Phenix software suite (47). Initial phases were obtained with the HySS subroutine of Autosol using the scaled and merged SeMet data and an apo-PvdP data set to 4.0 Å. The output was then used for Autobuild and Buccaneer from the CCP4 suite (48, 49). Both programs calculated different parts of the structure and still contained missing or misplaced connections, which were then curated manually. The initial model was used for phasing the PvdPapo and PvdPTyr data. Iterative refinement was done with phenix.refine after manual inspection using Coot (50). All figures were prepared with PyMOL (51). Coordinates and diffraction data have been deposited in the PDB (52) with entry codes 6EYS for PvdPapo and 6EYV for PvdPTyr.
Author contributions
J. P. and W. B. conceptualization; J. P., J. R., and W. B. formal analysis; J. P. and W. B. validation; J. P. investigation; J. P., J. R., and W. B. visualization; J. P., J. R., and W. B. methodology; J. P., J. R., and W. B. writing-original draft; J. P. and W. B. writing-review and editing; W. B. resources; W. B. supervision; W. B. project administration.
Supplementary Material
Acknowledgments
We thank the synchrotrons BESSYII (Berlin/Germany) (53), PETRAIII (Hamburg/Germany) (54), and SLS (Villigen/Switzerland) for access to facilities and beamline staff for support.
The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S5.
The atomic coordinates and structure factors (codes 6EYS and 6EYV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- PVD
- pyoverdine
- PDB
- Protein Data Bank
- SLS
- Swiss Light Source
- TEV
- tobacco etch virus protease
- BBD
- β-barrel domain
- TYD
- tyrosinase domain
- r.m.s.d.
- root mean square deviation
- SOC
- super optimal broth with catabolite repression
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
References
- 1. Meyer J.-M., Gruffaz C., Raharinosy V., Bezverbnaya I., Schäfer M., and Budzikiewicz H. (2008) Siderotyping of fluorescent Pseudomonas: molecular mass determination by mass spectrometry as a powerful pyoverdine siderotyping method. Biometals 21, 259–271 10.1007/s10534-007-9115-6 [DOI] [PubMed] [Google Scholar]
- 2. Budzikiewicz H. (2004) Siderophores of the Pseudomonadaceae sensu stricto (fluorescent and non-fluorescent Pseudomonas spp.), in Progress in the Chemistry of Organic Natural Products, pp. 81–237, Springer, Vienna, Austria: [DOI] [PubMed] [Google Scholar]
- 3. Albrecht-Gary A.-M., Blanc S., Rochel N., Ocaktan A. Z., and Abdallah M. A. (1994) Bacterial iron transport: coordination properties of pyoverdin PaA, a peptidic siderophore of Pseudomonas aeruginosa. Inorg. Chem. 33, 6391–6402 10.1021/ic00104a059 [DOI] [Google Scholar]
- 4. Meyer J.-M., Stintzi A., De Vos D., Cornelis P., Tappe R., Taraz K., and Budzikiewicz H. (1997) Use of siderophores to type pseudomonads: the three Pseudomonas aeruginosa pyoverdine systems. Microbiology 143, 35–43 10.1099/00221287-143-1-35 [DOI] [PubMed] [Google Scholar]
- 5. Schalk I. J., and Guillon L. (2013) Pyoverdine biosynthesis and secretion in Pseudomonas aeruginosa: implications for metal homeostasis. Environ. Microbiol. 15, 1661–1673 10.1111/1462-2920.12013 [DOI] [PubMed] [Google Scholar]
- 6. Meneely K. M., Barr E. W., Bollinger J. M. Jr., and Lamb A. L. (2009) Kinetic mechanism of ornithine hydroxylase (PvdA) from Pseudomonas aeruginosa: substrate triggering of O2 addition but not flavin reduction. Biochemistry 48, 4371–4376 10.1021/bi900442z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McMorran B. J., Shanta Kumara H. M., Sullivan K., and Lamont I. L. (2001) Involvement of a transformylase enzyme in siderophore synthesis in Pseudomonas aeruginosa. Microbiology 147, 1517–1524 10.1099/00221287-147-6-1517 [DOI] [PubMed] [Google Scholar]
- 8. Vandenende C. S., Vlasschaert M., and Seah S. Y. (2004) Functional characterization of an sminotransferase tequired for pyoverdine diderophore biosynthesis in Pseudomonas aeruginosa PAO1. J. Bacteriol. 186, 5596–5602 10.1128/JB.186.17.5596-5602.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yeterian E., Martin L. W., Guillon L., Journet L., Lamont I. L., and Schalk I. J. (2010) Synthesis of the siderophore pyoverdine in Pseudomonas aeruginosa involves a periplasmic maturation. Amino Acids 38, 1447–1459 10.1007/s00726-009-0358-0 [DOI] [PubMed] [Google Scholar]
- 10. Ochsner U. A., Snyder A., Vasil A. I., and Vasil M. L. (2002) Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 8312–8317 10.1073/pnas.082238299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamont I. L., and Martin L. W. (2003) Identification and characterization of novel pyoverdine synthesis genes in Pseudomonas aeruginosa. Microbiology 149, 833–842 10.1099/mic.0.26085-0 [DOI] [PubMed] [Google Scholar]
- 12. Lewenza S., Gardy J. L., Brinkman F. S., and Hancock R. E. (2005) Genome-wide identification of Pseudomonas aeruginosa exported proteins using a consensus computational strategy combined with a laboratory-based PhoA fusion screen. Genome Res. 15, 321–329 10.1101/gr.3257305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drake E. J., and Gulick A. M. (2011) Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6, 1277–1286 10.1021/cb2002973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nadal-Jimenez P., Koch G., Reis C. R., Muntendam R., Raj H., Jeronimus-Stratingh C. M., Cool R. H., and Quax W. J. (2014) PvdP is a tyrosinase that drives maturation of the pyoverdine chromophore in Pseudomonas aeruginosa. J. Bacteriol. 196, 2681–2690 10.1128/JB.01376-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ringel M. T., Dräger G., and Brüser T. (2018) PvdO is required for the oxidation of dihydropyoverdine as the last step of fluorophore formation in Pseudomonas fluorescens. J. Biol. Chem. 293, 2330–2341 10.1074/jbc.RA117.000121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drake E. J., and Gulick A. M. (2016) 1.2 Å resolution crystal structure of the periplasmic aminotransferase PvdN from Pseudomonas aeruginosa. Acta Crystallogr. F Struct. Biol. Commun. 72, 403–408 10.1107/S2053230X16006257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ringel M. T., Dräger G., and Brüser T. (2016) PvdN enzyme catalyzes a periplasmic pyoverdine modification. J. Biol. Chem. 291, 23929–23938 10.1074/jbc.M116.755611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Voulhoux R., Filloux A., and Schalk I. J. (2006) Pyoverdine-mediated iron uptake in Pseudomonas aeruginosa: the Tat system is required for PvdN but not for FpvA transport. J. Bacteriol. 188, 3317–3323 10.1128/JB.188.9.3317-3323.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan Z., Gao F., Bai G., Xia H., Gu L., and Xu S. (2017) Crystal structure of PvdO from Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 484, 195–201 10.1016/j.bbrc.2016.12.181 [DOI] [PubMed] [Google Scholar]
- 20. Dorrestein P. C., Poole K., and Begley T. P. (2003) Formation of the chromophore of the pyoverdine siderophores by an oxidative cascade. Org. Lett. 5, 2215–2217 10.1021/ol034531e [DOI] [PubMed] [Google Scholar]
- 21. Turner K. H., Everett J., Trivedi U., Rumbaugh K. P., and Whiteley M. (2014) Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 10, e1004518 10.1371/journal.pgen.1004518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 23. Le Trong I., Wang Z., Hyre D. E., Lybrand T. P., Stayton P. S., and Stenkamp R. E. (2011) Streptavidin and its biotin complex at atomic resolution. Acta Crystallogr. D Biol. Crystallogr. 67, 813–821 10.1107/S0907444911027806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Satoh A., Kim J.-K., Miyahara I., Devreese B., Vandenberghe I., Hacisalihoglu A., Okajima T., Kuroda S., Adachi O., Duine J. A., Van Beeumen J., Tanizawa K., and Hirotsu K. (2002) Crystal structure of quinohemoprotein amine dehydrogenase from Pseudomonas putida. Identification of a novel quinone cofactor encaged by multiple thioether cross-bridges. J. Biol. Chem. 277, 2830–2834 10.1074/jbc.M109090200 [DOI] [PubMed] [Google Scholar]
- 25. Royer W. E. Jr., Sharma H., Strand K., Knapp J. E., and Bhyravbhatla B. (2006) Lumbricus erythrocruorin at 3.5 Å resolution: architecture of a megadalton respiratory complex. Structure 14, 1167–1177 10.1016/j.str.2006.05.011 [DOI] [PubMed] [Google Scholar]
- 26. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alva V., Nam S.-Z., Söding J., and Lupas A. N. (2016) The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 44, W410–W415 10.1093/nar/gkw348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dawson N. L., Lewis T. E., Das S., Lees J. G., Lee D., Ashford P., Orengo C. A., and Sillitoe I. (2017) CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 45, D289–D295 10.1093/nar/gkw1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aguilera F., McDougall C., and Degnan B. M. (2013) Origin, evolution and classification of type-3 copper proteins: lineage-specific gene expansions and losses across the Metazoa. BMC Evol. Biol. 13, 96 10.1186/1471-2148-13-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldfeder M., Kanteev M., Isaschar-Ovdat S., Adir N., and Fishman A. (2014) Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 5, 4505 10.1038/ncomms5505 [DOI] [PubMed] [Google Scholar]
- 33. Decker H., Schweikardt T., Nillius D., Salzbrunn U., Jaenicke E., and Tuczek F. (2007) Similar enzyme activation and catalysis in hemocyanins and tyrosinases. Gene. 398, 183–191 10.1016/j.gene.2007.02.051 [DOI] [PubMed] [Google Scholar]
- 34. Han H.-Y., Zou H.-C., Jeon J.-Y., Wang Y.-J., Xu W.-A., Yang J.-M., and Park Y.-D. (2007) The inhibition kinetics and thermodynamic changes of tyrosinase via the zinc ion. Biochim. Biophys. Acta 1774, 822–827 10.1016/j.bbapap.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 35. Matoba Y., Kumagai T., Yamamoto A., Yoshitsu H., and Sugiyama M. (2006) Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 281, 8981–8990 10.1074/jbc.M509785200 [DOI] [PubMed] [Google Scholar]
- 36. Sendovski M., Kanteev M., Ben-Yosef V. S., Adir N., and Fishman A. (2011) First structures of an active bacterial tyrosinase reveal copper plasticity. J. Mol. Biol. 405, 227–237 10.1016/j.jmb.2010.10.048 [DOI] [PubMed] [Google Scholar]
- 37. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., Wang Y., Jiang H., and Deng J. (2009) Crystal structure of Manduca sexta prophenoloxidase provides insights into the mechanism of type 3 copper enzymes. Proc. Natl. Acad. Sci. U.S.A. 106, 17002–17006 10.1073/pnas.0906095106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanteev M., Goldfeder M., and Fishman A. (2015) Structure-function correlations in tyrosinases. Protein Sci. 24, 1360–1369 10.1002/pro.2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanteev M., Goldfeder M., Chojnacki M., Adir N., and Fishman A. (2013) The mechanism of copper uptake by tyrosinase from Bacillus megaterium. J. Biol. Inorg. Chem. 18, 895–903 10.1007/s00775-013-1034-0 [DOI] [PubMed] [Google Scholar]
- 41. Hassett D. J., Charniga L., Bean K., Ohman D. E., and Cohen M. S. (1992) Response of Pseudomonas aeruginosa to pyocyanin: mechanisms of resistance, antioxidant defenses, and demonstration of a manganese-cofactored superoxide dismutase. Infect. Immun. 60, 328–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van Holde K. E., and Miller K. I. (1995) Hemocyanins. Adv. Protein Chem. 47, 1–81 10.1016/S0065-3233(08)60545-8 [DOI] [PubMed] [Google Scholar]
- 43. Lee S. Y., Baek N., and Nam T. (2016) Natural, semisynthetic and synthetic tyrosinase inhibitors. J. Enzyme Inhib. Med. Chem. 31, 1–13 10.3109/14756366.2015.1004058 [DOI] [PubMed] [Google Scholar]
- 44. Berrow N. S., Alderton D., Sainsbury S., Nettleship J., Assenberg R., Rahman N., Stuart D. I., and Owens R. J. (2007) A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 35, e45 10.1093/nar/gkm047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 10.1107/S0907444906022116 [DOI] [PubMed] [Google Scholar]
- 49. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.8.6.2, Schrödinger, LLC, New York [Google Scholar]
- 52. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., and Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 10.1107/S0909049512006395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burkhardt A., Pakendorf T., Reime B., Meyer J., Fischer P., Stübe N., Panneerselvam S., Lorbeer O., Stachnik K., Warmer M., Rödig P., Göries D., and Meents A. (2016) Status of the crystallography beamlines at PETRA III. Eur. Phys. J. Plus 131, 56 [Google Scholar]
- 55. Kleywegt G. J. (1997) Validation of protein models from Cα coordinates alone. J. Mol. Biol. 273, 371–376 10.1006/jmbi.1997.1309 [DOI] [PubMed] [Google Scholar]
- 56. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bond C. S. (2003) TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19, 311–312 10.1093/bioinformatics/19.2.311 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.