

Abstract
Mutations in the KRAS proto-oncogene are present in 50% of all colorectal cancers and are increasingly associated with chemotherapeutic resistance to frontline biologic drugs. Accumulating evidence indicates key roles for overactive KRAS mutations in the metabolic reprogramming from oxidative phosphorylation to aerobic glycolysis in cancer cells. Here, we sought to exploit the more negative membrane potential of cancer cell mitochondria as an untapped avenue for interfering with energy metabolism in KRAS variant–containing and KRAS WT colorectal cancer cells. Mitochondrial function, intracellular ATP levels, cellular uptake, energy sensor signaling, and functional effects on cancer cell proliferation were assayed. 3-Carboxyl proxyl nitroxide (Mito-CP) and Mito-Metformin, two mitochondria-targeted compounds, depleted intracellular ATP levels and persistently inhibited ATP-linked oxygen consumption in both KRAS WT and KRAS variant–containing colon cancer cells and had only limited effects on nontransformed intestinal epithelial cells. These anti-proliferative effects reflected the activation of AMP-activated protein kinase (AMPK) and the phosphorylation-mediated suppression of the mTOR target ribosomal protein S6 kinase B1 (RPS6KB1 or p70S6K). Moreover, Mito-CP and Mito-Metformin released Unc-51–like autophagy-activating kinase 1 (ULK1) from mTOR-mediated inhibition, affected mitochondrial morphology, and decreased mitochondrial membrane potential, all indicators of mitophagy. Pharmacological inhibition of the AMPK signaling cascade mitigated the anti-proliferative effects of Mito-CP and Mito-Metformin. This is the first demonstration that drugs selectively targeting mitochondria induce mitophagy in cancer cells. Targeting bioenergetic metabolism with mitochondria-targeted drugs to stimulate mitophagy provides an attractive approach for therapeutic intervention in KRAS WT and overactive mutant-expressing colon cancer.
Keywords: AMP-activated kinase (AMPK), cancer biology, cell growth, mitophagy, mitochondrial metabolism, colon cancer, KRAS proto-oncogene, cell proliferation, metformin
Introduction
Colorectal cancer is predicted to afflict 1 of every 20 people in the United States and is the third major cause of cancer with an estimated 100,000 new cases reported annually in the United States (1). With the identification of hereditary high-risk factors, modifiable lifestyle choices, and the increase in screening, the incidence rate of colon cancer has been on the decline. Nevertheless, with equal incidence between males and females, there were ∼50,000 colon cancer–associated mortalities projected in the United States for 2016 with another 700,000 worldwide (1). The first line of treatment for localized disease is surgical resection; perioperative chemotherapy and/or radiation therapy is often employed once the cancer has penetrated the bowel wall or spread to lymph nodes. Typically, the first-line chemotherapy for metastatic colon cancer is FOLFOX (5-FU, leucovorin, and oxaliplatin) or FOLFIRI (5-FU, leucovorin, and irinotecan). However, these treatments can be cytotoxic with pronounced side effects and off-target consequences. Alternative chemotherapies currently in development include recombinant fusion growth factor protein (aflibercept), tyrosine kinase inhibitor (regorafenib), and biologic monoclonal antibodies that block angiogenesis (bevacizumab) or target the epidermal growth factor receptor (cetuximab and panitumumab). A major caveat to these treatment regimens is that their efficacy is restricted to tumors that express WT KRAS protein. Unfortunately, KRAS is a common genetic mutation, accounting for ∼50% of colorectal cancer oncogenic mutations (2, 3). Although the treatment of WT KRAS colon cancer has advanced with the use of biologics, a recent report has attributed the emergence of oncogenic KRAS mutations as the cause of intrinsic or acquired cetuximab resistance, rendering previously cetuximab-sensitive cancer cells resistant to therapy and subsequent relapse (4). Thus, there remains a significant need for therapies with increased efficacy, reduced toxicity, and directed tumor specificity, particularly in the context of RAS oncogene expression.
Accumulating evidence suggests that constitutively active KRAS mutations aid the metabolic reprogramming of tumor cells involving a shift from oxidative phosphorylation (OXPHOS) 2 to aerobic glycolysis (the Warburg effect) (5). With the continuing demonstration that mitochondrial metabolism is a key player in cancer development, progression, and metastasis, it is rational to exploit this observation and explore mitochondria-targeted compounds as novel, untapped therapeutics. Indeed, we have shown that compounds targeted to the mitochondria induce anti-proliferative and cytotoxic effects in tumor cells without a significant impact on the surrounding normal cells (6–9). These novel mitochondria-targeted agents have been generated by chemically tethering an alkyl triphenylphosphonium cation (TPP+) moiety via an aliphatic linker chain to bioactive molecules (7, 8, 10–12). The lipophilic TPP+ cation facilitates the crossing of mitochondrial membrane, and the delocalized positively charged TPP+ moiety accumulates in the largely electronegative transmembrane potential of the mitochondria (13–16). Thus, the enhanced uptake and retention of TPP+-containing bioavailable compounds in tumor cell mitochondria has been posited to be the mechanism behind their selective proliferative inhibition (17–22). A number of these mitochondria-targeted molecules demonstrate considerable anti-tumor activity (8, 17, 18), however, the precise molecular mechanism(s) by which these drugs exert their anti-proliferative, anti-migratory, and anti-invasive effects remains poorly understood. Thus, we sought to determine the biomolecular pathways triggered by model mitochondria-targeted agents.
Mito-CP, a superoxide dismutase mimetic (3-carboxyl proxyl (CP) nitroxide), conjugated to TPP+, has demonstrated directed anti-proliferative and cytotoxic effects in regard to both pancreatic and breast cancer cells, without markedly affecting nontransformed cells (7, 8). Mito-CP, a potent disrupter of mitochondrial metabolism, in combination with 2-deoxyglucose (2-DG), an inhibitor of glycolysis, synergized to deplete intracellular ATP, reduce cell proliferation, and induce apoptosis of pancreatic tumor cells in vitro (7). Co-administration of Mito-CP and 2-DG led to significant tumor regression in a murine model of breast cancer (8). Anti-cancer effects of Mito-CP have also been shown in medullary thyroid cancer (23) and malignant mesothelioma (24). However, the mechanistic basis of these findings are not known. In addition to Mito-CP, we discovered that a TPP+-conjugated derivative of the FDA-approved type 2 diabetes drug Metformin, which we termed Mito-Met10, was 1000-fold more potent in inhibiting pancreatic cancer cell proliferation by impeding cell cycle progression, relative to the parental Metformin compound (6). Patients taking Metformin have a correlative lower risk of colorectal cancer (25, 26). Metformin is posited to inhibit the mitochondrial electron transport complex I and indirectly activates the AMP-activated protein kinase (AMPK) signaling cascade, leading to suppressed colon carcinoma proliferation and reduced polyp formation (27, 28). These results encouraged us to determine whether Metformin conjugated to TPP+ (Mito-Met10) might impact colon cancer cell dynamics. Here, the efficacy and biochemical mechanisms of Mito-CP and Mito-Met10 on colon cancer proliferation and bioenergetic metabolism were investigated. Both of these different agents restricted the ability of the tumor cells to cope with energetic stress. Examining a panel of both cell types, we found that KRAS WT colon cancer cells, as well as colon cancer cells with constitutively active KRAS, were exquisitely sensitive to both molecules as assessed by their impact upon cell proliferation. Mito-CP– and Mito-Met10–induced changes in mitochondrial bioenergetics activated AMPK signaling, concomitantly blocking mTOR-mediated proliferation and inducing mitophagy-like markers such as decreased mitochondrial membrane potential and disruption of cellular architecture. This study is the first to demonstrate the molecular mechanisms by which compounds engineered to localize within the mitochondria limit colon cancer proliferation and progression.
Results
Mito-CP and Mito-Met10 effectively inhibit colon cancer cell proliferation
Oncogenic KRAS drives metabolic reprogramming from mitochondrial (catabolic) to glycolytic (aerobic) energy production (the Warburg effect) (29). Indeed, Weinberg et al. have demonstrated that HCT116 cells shift their mitochondrial metabolism pathway to facilitate anaerobic glycolytic KRAS-induced anchorage-independent proliferation (5). The therapeutic potential of two potent mitochondria-targeted TPP+ biomolecules, Mito-Met10 and Mito-CP, was assessed using reductionist colon cancer models. Initially, HCT116 (KRASG13D) and HT-29 (WT KRAS) cells were seeded onto a 96-well plate and treated with increasing concentrations of Mito-CP (0–10 μm) or Mito-Met10 (0–100 μm). Cells were placed into an IncuCyte S3 and image acquisition started immediately to establish background proliferation. At day 1, cells were treated with titrated doses of Mito-CP or Mito-Met10 and images of each well were automatically acquired every 2 h for 5 days to allow us to assess cell confluence kinetics. The changes in percent confluency (% confluency), as a readout for proliferation, were monitored in real time. Both cell lines demonstrated a dose-dependent diminution in cell proliferation when treated with increasing concentrations of Mito-CP (Fig. 1, A and B) or Mito-Met10 (Fig. 1, C and D). Both cell lines exhibited an exquisite sensitivity to Mito-CP compared with Mito-Met10, with nearly 50-fold less Mito-CP required for a similar reduction in proliferation. HT-29 cells expressing WT KRAS were sensitive to low micromolar to nanomolar doses of both Mito-CP and Mito-Met10. HCT116 cells expressing constitutively active KRAS were somewhat more resistant to mitochondria-targeted drug (MTD) treatment with proliferation decreasing by 80% with 1 μm Mito-CP or 50 μm Mito-Met10 (Fig. 1, A and C). Additional oncogenic (SW480 and T84) (supporting Fig. S1, A–D) and WT (Caco2 BBE and HK 2–8) (supporting Fig. S1, E–H) KRAS-expressing cell lines were tested and validated the results that the MTDs exert their anti-proliferative effects independent of the KRAS allele (supporting Fig. S1). Furthermore, HCT116 (supporting Fig. S2, A and B) and HT-29 (supporting Fig. S2C) cells were treated with increasing doses of the parental compounds (carboxyl proxyl (CP) and Metformin) with 100- to 1000-fold more drug required to impart the anti-proliferative effects noted with the TPP+ conjugated moieties. Lastly, the TPP+ moiety alone had no effect on cell proliferation as HCT116 cells proliferate normally and reach confluency within 5 days after treatment with TPP+ (supporting Fig. S2D). Taken together, these data demonstrate that tethering known anti-oxidants or anti-diabetic molecules to a cationic moiety reduces colon cancer proliferation, independent of the KRAS protein.
Figure 1.

Mitochondria-targeted drugs inhibit colon cancer cell proliferation. A–D, HCT116 (A and C) and HT-29 (B and D) colon cancer cell lines were treated with increasing concentrations of Mito-CP (0–10 μm) or Mito-Met10 (0–100 μm). Images were acquired in real time every 2 h over a 6-day period using the IncuCyte S3 and representative phase contrast images are shown. Percent confluency (% confluency) was used as a readout for proliferation. A graphical representation of the dose response on cell confluence is expressed as a change in percentage of cell confluence. Values are mean ± S.E., n = 3; a two-way repeated measures ANOVA demonstrated p ≤ 0.0001.
Mito-CP and Mito-Met10 impact on mitochondria
To evaluate whether MTDs disrupted mitochondrial respiration, we first addressed the cellular uptake of the two drugs. A surrogate “normal” cell line (IEC6; nontransformed rat small intestine epithelia), along with the two colon cancer cell lines (HCT116 and HT-29) were treated with 0.5 μm Mito-CP or 25 μm Mito-Met10 for 24 h, and the cells processed for LC-MS/MS analysis. The colon cancer cells had a demonstrative increase in the uptake of both drugs when compared with the nontransformed cells (Fig. 2, A and B). To address the targeting of the MTDs to the mitochondria, HCT116 cells were treated 24 h and permeabilized to probe for complex I activity, which was significantly inhibited (Fig. 2C). Analysis of oxygen consumption over a 4-log range of drug concentrations revealed an IC50 = 0.5 ± 7.4 μm and 23 ± 8.7 μm for Mito-CP and Mito-Met10, respectively (Fig. 2C). This 46-fold difference in complex I activity correlates well with the anti-proliferative effects observed in Fig. 1. The diminished mitochondrial function was also reflected in the reduced levels of total ATP accumulated in Mito-CP– and Mito-Met10–treated cells (Fig. 2D). This dose-dependent decrease in ATP levels is consistent with Mito-CP and Mito-Met10 inhibition of complex I. Correlative with its weaker impact on complex I, Mito-Met10 had little overall effect on ATP levels in treated cells, suggesting its anti-proliferative effects reflect distinct signaling pathways relative to Mito-CP.
Figure 2.

The TPP+-biocompounds Mito-CP and Mito-Met10 impair mitochondrial bioenergetics. A and B, intracellular uptake of the compounds in nontransformed rat small intestine epithelial IEC6 cells, along with the two colon cancer cell lines, HCT116 and HT-29, was assessed after 24 h treatment with 0.5 μm Mito-CP (A) or 25 μm Mito-Met10 (B) using LC-MS/MS. C, complex I inhibition was addressed by treating HCT116 cells with increasing concentrations of Mito-Met10 (filled oval) or Mito-CP (empty rectangle) for 24 h. The mitochondrial complex I oxygen consumption (last oxygen consumption rate reading before succinate injection) is plotted against the drug concentrations. D, total cellular ATP levels were analyzed using a luciferase-based assay on HCT116 cells treated with increasing concentrations of the drugs (Mito-Met10 (filled oval) or Mito-CP (empty rectangle)) for 24 h. E and F, HCT116 mitochondrial respiration impairment after 24 h of treatment (Mito-CP (E) or Mito-Met10 (F)) was measured using the Seahorse XF analyzer. The OCR (pmol/min) was used as a marker for mitochondrial stress. Panels on the right are enlargements of the time course post–stress test drug additions (dashed boxes in E and F) and demarcate the sequential injections of oligomycin (Oligo) to measure ATP-linked OCR, FCCP to measure maximal OCR, and rotenone/antimycin A (Rot/AA) to measure nonmitochondrial respiration. * denotes p ≤ 0.05, ** denotes p ≤ 0.01, *** denotes p ≤ 0.001, **** denotes p ≤ 0.0001. n.s., not significant. Values are mean ± S.E. n = 4.
To gain a better understanding of MTD-mediated mitochondrial dysfunction, the oxygen consumption rate (OCR), as a readout of OXPHOS, and the extracellular acidification rate, as a surrogate marker for glycolysis, were measured in a Seahorse Bioscience extracellular flux analyzer. HCT116 cells were treated with increasing concentrations of drugs and a mitochondrial stress test was undertaken. In agreement with their ability to inhibit complex I, both compounds abrogated OCR in a dose-dependent manner, with Mito-CP (Fig. 2E) more potent than Mito-Met10 (Fig. 2F). Further, neither had an effect on extracellular acidification rate (data not shown), consistent with their ability to inhibit complex I. The mitochondrial respiration profile can be divided into five functional classes: basal respiration, ATP production, proton leak, maximal respiration, and spare capacity. These two compounds inhibit mitochondrial energy pathways through discrete mechanisms. All five aspects of mitochondrial function negatively impacted upon incubation with the two highest concentrations (1 and 10 μm) of Mito-CP, suggesting that Mito-CP treatment reduced the number of live cells and/or the number of mitochondria, rendering the cells unable to meet metabolic challenges (supporting Fig. S3, striped bars). For Mito-Met10, dysregulation of mitochondrial fitness was only noted at the highest concentration (100 μm) presenting with a significant decrease in maximum respiration and the reserve respiratory capacity (supporting Fig. S3, filled bars). This decrease in mitochondrial fitness and negative impact on bioenergetics is likely because of the directed targeting of the compounds to the mitochondria. Taken together our data show that MTDs influence cellular bioenergetic metabolism and proliferation.
AMPK signaling pathway is activated in MTD-treated colon cancer cells
To evaluate the molecular mechanism(s) regulating the Mito-CP and Mito-Met10 impact on colon cancer metabolism, we focused our attention on AMPK, a master energy sensor within the cell. AMPK, a strongly conserved eukaryotic Ser/Thr kinase, is rapidly activated when the intracellular ratio of AMP to ATP is elevated. The consequence of AMPK activation is to up-regulate ATP generating pathways while inhibiting ATP consuming functions of the cell (30). The heterotrimeric AMPK complex is composed of the AMPKα, AMPKβ, and AMPKγ subunits, and upon sensing increasing levels of intracellular AMP, the α subunit within the complex undergoes an allosteric change resulting in its activation via phosphorylation of the catalytic γ subunit at residue Thr-172 (31). Consistent with decreasing ATP levels, MTD treatment stimulated AMPK Thr-172 phosphorylation in a dose-dependent manner (Fig. 3A). As expected, Mito-CP and Mito-Met10 treatment activated AMPK at the same doses shown to inhibit proliferation and ATP levels.
Figure 3.

Activation of the AMPK signaling cascade concomitant with mTORC1 suppression is activated and induced mitophagy via ULK1. A, HCT116 cells were starved for glucose, glutamine, and serum for 6 h prior to a 30 min stimulation with increasing concentrations of Mito-Met10 or Mito-CP, or treated with DMSO (V, vehicle). Protein lysates (10 μg/lane) were resolved by SDS-PAGE and probed with antiserum directed toward phospho-Thr-172–AMPK (pAMPK), total AMPK, or GAPDH. Immunoblots were quantitated and represented graphically. B and C, HCT116 cells were treated with DMSO (V, vehicle) or increasing concentrations of Mito-Met10 or Mito-CP for 24 h. Proteins resolved by SDS-PAGE and transferred to PVDF were probed for phospho-Ser-792–Raptor (pRaptor), total Raptor, or GAPDH (B) or phospho-Thr-389–p70S6K, total p70S6K, or β-tubulin (C). * denotes p ≤ 0.05, ** denotes p ≤ 0.01, *** denotes p ≤ 0.001, **** denotes p ≤ 0.0001. Values are means ± S.E. n = 3.
mTORC1 and downstream targets are dysregulated
We next sought to determine whether AMPK up-regulation impacted the mTOR complex 1 (mTORC1), an essential intracellular master regulator of cell proliferation, that employs a network of molecular connections to regulate protein synthesis, cell proliferation, and autophagy (32). Additional molecular activators that sense survival cues and growth factor levels include the mTOR complex 2 (mTORC2) and the Ser/Thr protein kinase AKT. To assess if mTORC2/AKT had any role in the observed effects, HCT116 cell lysates were probed with an anti-AKT antibody. The levels of phospho-AKT (Ser-473), a modification that stimulates full AKT activity (33), were unchanged upon incubation with Mito-Met10 or Mito-CP (supporting Fig. S4), suggesting minimal to no involvement of AKT.
mTORC1 is a five-protein complex that can be distinguished from mTORC2 by the inclusion of Raptor, the regulatory-associated protein of mTOR. Raptor is a presumable nonenzymatic scaffold protein that dictates mTORC1 substrate specificity, localization, and complex assembly and is essential for the kinase activity of mTORC1 in response to energy level and mitochondrial uncoupling (34). When Raptor is complexed within mTORC1, it can be recognized and directly phosphorylated on Ser-722 and Ser-792 by AMPK (35). Phosphorylation of Raptor by AMPK redistributes its association to 14-3-3, an event correlative with the suppression of mTORC1 activity (35). HCT116 lysates probed with an anti–phospho-Ser-792 Raptor antibody demonstrated a significant increase in phospho-protein levels following with elevated MTD concentrations (Fig. 3B). In correlation with Gwinn et al., our data suggest that upon AMPK-mediated Raptor phosphorylation, mTORC1 is inactivated, likely reflecting energy stress imparted by MTD treatment (35).
mTORC1-mediated effects on protein synthesis and cell proliferation are facilitated through p70S6K, a major effector of cell proliferation and cell cycle progression (36, 37). MTD-treated HCT116 lysates were probed with a phospho-specific antibody that recognizes phospho-Thr-389–p70S6K, a direct substrate of mTORC1 (38). Indeed, Thr-389–p70S6K levels decreased in a dose-dependent manner (Fig. 3C). These data agree with prior reports that the presence of Raptor in mTORC1 is essential for phosphorylation of p70S6K at Thr-389 (39) and that phosphorylation of Thr-389 most closely correlates with p70 kinase activity in vivo (40). These observations raised the possibility that the observed anti-proliferative effects were because of induction of apoptosis. To test this idea, cell lysates from treated HCT116s were probed for pro–caspase 3 levels. Caspase 3, an executioner caspase, represents a terminal event in the intrinsic caspase 9–mediated apoptotic pathway. However, we found no change in pro–caspase 3 levels upon MTD stimulation (supporting Fig. S5, A and B), whereas gliotoxin, a potent inducer of apoptosis, shifted the levels of pro–caspase 3 (supporting Fig. S5, C and D). These data suggest that apoptosis is not a means by which Mito-CP and Mito-Met10 exert their anti-proliferative activity.
Activation of ULK1, an initiator of mitophagy
p70S6K is a moderator of protein synthesis which, and along with the autophagy-initiating kinase ULK1, has been used as a readout of autophagy induction (41). The autophagy cascade is known to be sensitive to the energy status of the cell (42). The energy sensor AMPK can activate ULK1 kinase by a coordinated cascade of events. In particular, AMPK has been indicated as a main upstream regulator of mTORC1-mediated autophagy suppression (43). Thus, if mTORC1 is in an active configuration, protein synthesis and cell proliferation are permitted, whereas autophagy is abrogated (44). Toward this end, mTORC1 directly phosphorylates ULK1 at Ser-757, thereby halting autophagy (44). However, activated AMPK can directly bind to and phosphorylate ULK1 at Ser-317, modifying ULK1 in such a manner by which the protein is constrained from contacting mTORC1, resulting in increased autophagy (44). To examine the phospho-ULK1 profile within treated HCT116 cells, total cell lysates were examined (Fig. 4, A and B). Comparing levels of p-Ser-757–ULK1 (Fig. 4A) to p-Ser-317–ULK1 (Fig. 4B) revealed a decrease in the autophagy inactivating form with a concomitant increase in the autophagy activating form of ULK1. The mechanism of action is independent of the KRAS allele in that stimulation of WT KRAS HT-29 cells demonstrated a similar decline in p-Ser-757–ULK1 levels (supporting Fig. S6, A and B). Collectively, these data begin to describe a molecular mechanism in which the low energy conditions evoked by MTDs trigger an AMPK–ULK1 signaling cascade that abrogates continued cancer cell proliferation, possibly through inducing mitophagy.
Figure 4.

MTD treatment shifts the phosphorylation status of ULK1 to promote mitophagy. A, HCT116 cells were treated with DMSO (V, vehicle) or increasing concentrations of Mito-Met10 or Mito-CP for 24 h. Protein lysates (10 μg/lane) were resolved by SDS-PAGE and probed with antiserum directed toward phospho-Ser-757–ULK1, total ULK1, or β-tubulin (A) or phospho-Ser-317–ULK1, total ULK1, or β-tubulin (B). Immunoblots were quantitated and represented graphically below each set of blots. ** denotes p ≤ 0.01. Values are mean ± S.E. n = 3. Immunoblot analyses of AKT analyzed concurrently with ULK are shown in supporting Fig. S4.
Mitochondrial structure and membrane potential (Δψm) are compromised in MTD-treated cells
To more thoroughly establish mitophagy as a mechanism for the anti-proliferative effects, we used transmission EM (TEM) to investigate the morphology of treated cells. HCT116 cells were harvested and processed for TEM 6 h post MTD addition. As presented in Fig. 5, we found a pronounced increase in vacuolization (E and H, arrowheads), ruptured mitochondria (F and I, black asterisk(s)), and even the appearance of double membranes associated with mitophagy (F and I, arrows) and autophagy (C, F, and I, white circles) following MTD treatment. Fig. 5C illustrates an autophagic vesicle observed in control cells; the pronounced structural changes seen in MTD-treated cells were largely absent in cancer cells treated with vehicle alone.
Figure 5.

Ultrastructural changes in mitochondria following MTD treatment. A–I, HCT116 cells were treated with Mito-CP (1 μm) or Mito-Met10 (50 μm) for 6 h prior to harvesting and processing by transmission EM. Representative images from vehicle (V)–treated (A–C), Mito-CP–treated (D–F), and Mito-Met10–treated cells (G–I) demonstrate the resulting changes in intracellular morphology upon MTD treatment. N represents the nucleus, white asterisk represents mitochondria with normal morphology, black asterisk represent mitochondria with abnormal morphology, white circles encircle autophagic vesicles, arrowheads visualize enlarged vacuoles, arrows represent mitophagy vesicles. The black box in D is enlarged in E, and the black box in E is enlarged in F. The black box in G is enlarged in H, and the black box in H is enlarged in I. Scale bar in A, D, E, G, and H represents 2 μm and in B, C, F, and I is 500 nm.
The TEM data strongly suggest that Mito-CP– or Mito-Met10 treatment damaged the mitochondria. To functionally quantitate the damage to the mitochondria, treated cells were stained with the membrane permeable, positively charged, TMRE (tetramethylrhodamine ethyl ester) red-orange dye. TMRE accumulates within mitochondria inversely proportional to the Δψm, with accumulation in metabolically active mitochondria, whereas depolarized, injured, or degraded mitochondria fail to sequester TMRE (45). As shown in Fig. 6, Mito-CP and Mito-Met10 potently and dose dependently disrupted mitochondrial membrane potential measured using flow cytometry or immunofluorescence. Mito-CP, at the 1 μm dose that blocked proliferation, reduced membrane potential greater than 75% in HCT116 (Fig. 6A) and HT-29 (Fig. 6C). As predicted, HCT116 (Fig. 6D) or HT-29 (Fig. 6F) cells treated with titrated doses of Mito-Met10 required higher levels to disrupt the mitochondrial membrane potential. Representative confocal microscopy images of Mito-CP (Fig. 6B) or Mito-Met10 (Fig. 6E) TMRE-labeled cells affirmed the loss of mitochondrial membrane potential. Together with the reduction in p-ULK and the structural mitochondrial abnormalities noted in MTD-treated cells, these data support the idea that Mito-CP, and to a lesser extent Mito-Met10, stimulate mitophagy, contributing to the proliferation inhibitory effects of these compounds in WT and oncogenic KRAS colon cancer cells.
Figure 6.

Mito-CP or Mito-Met10 treatment disrupts mitochondrial membrane potential. Colon cancer cell lines were concomitantly treated with DMSO as a vehicle control (V, black), FCCP (red), Mito-CP (green or blue, 0.5 μm or 1.0 μm, respectively) (HCT116 (A and B); HT-29 (C)), or Mito-Met10 (green, blue, or purple; 10 μm, 100 μm, and 250 μm, respectively) (HCT116 (D and E); HT-29 (F)) for 24 h. Mitochondria were stained with 200 nm TMRE and membrane potential was assessed by flow cytometry. A, C, D, and F, the top panels are representative TMRE staining histograms with a graphical depiction of the mean fluorescent intensity (MFI) presented below. B and E, visualization of mitochondria on a per cell basis was achieved by seeding HCT116 cells into glass-bottom dishes, treatment with drugs as above, stained with 100 nm TMRE and visualized at 60 x/1.6 by spinning disk confocal microscopy. Representative images are maximum projection of Z series acquired at 0.5 μm sections and are shown from the same multi-well experiment such that the vehicle control is the same image in B and E. * denotes p ≤ 0.05, ** denotes p ≤ 0.01. Values are mean ± S.E. n = 4.
Mito-CP and Mito-Met10 stimulate AMPK signaling to halt cancer cell proliferation
Pharmacological disruptions were used to explore the contributions of the AMPK signaling pathway in the MTD-mediated disruption of colon cancer cell proliferation. Compound C is a cell-permeable inhibitor of AMPK. Pretreatment of HCT116 cells prior to MTD treatment minimized the anti-proliferative effects of the MTDs (Fig. 7, A and B). Compound C pretreatment did not fully restore HCT116's proliferative capacity, suggesting the involvement of additional pathways. These data are the first to ascribe a role for cancer-selective mitochondria-targeted cations in the activation of AMPK-mediated signaling as an exquisite mechanism to arrest proliferation of KRAS WT and oncogenic cancer cells (Fig. 8).
Figure 7.

Pharmacological inhibition of the AMPK signaling pathway provides partial resistance to MTD treatment. A, HCT116 cells were treated (+) with 0.3 μm compound C (Cpd C) or remained untreated (−), for 48 h prior to incubation with DMSO (Vehicle), 50 μm Mito-Met10, or 1 μm Mito-CP. Cell images were acquired in real time on the IncuCyte S3 and percent confluency (% confluency) was used as a readout for proliferation after 3 days of MTD treatment. B, representative images of control and MTD-treated cells are shown. * denotes p ≤ 0.05, ** denotes p ≤ 0.01. Values are mean ± S.E. n = 3.
Figure 8.
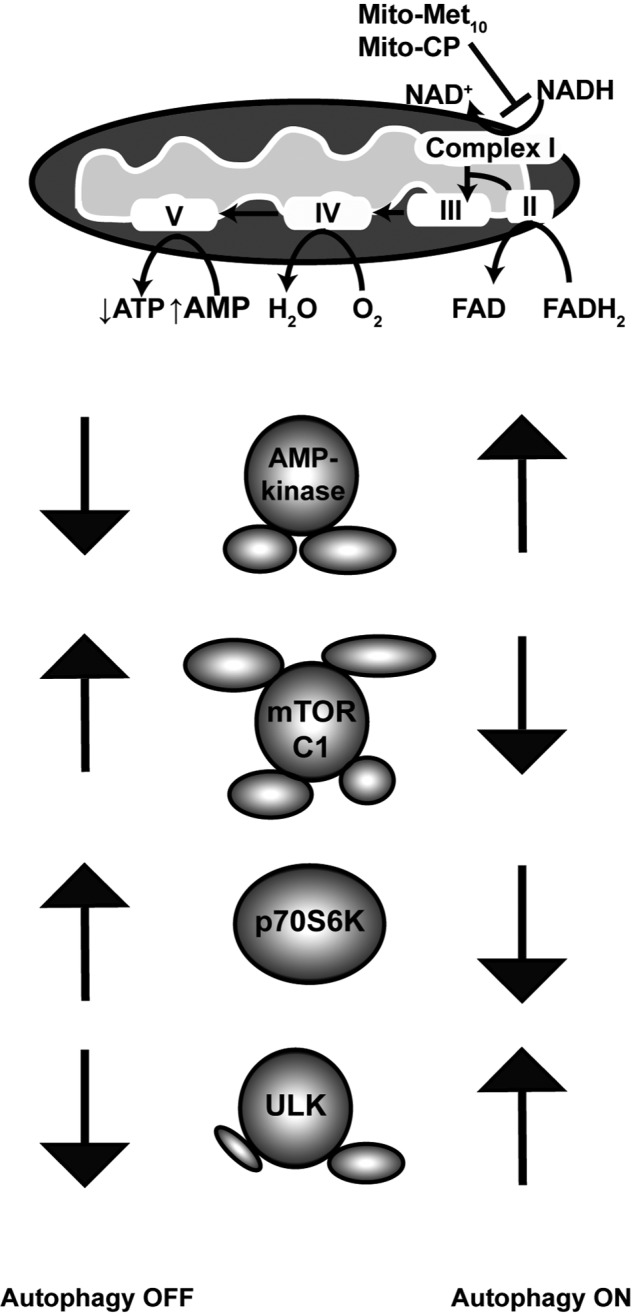
Schematic model a mitophagy mechanism of action of mitochondria-targeted drugs on colorectal cancer cells. Results from the analysis of two representative mitochondria-targeted drugs, Mito-CP and Mito-Met10, indicate selective uptake and localization within the mitochondria inhibits complex I activity, activating an AMPK-dependent signaling cascade which suppresses mTORC1, resulting in activation of the mitophagy regulator ULK1 and concomitant decreasing cell proliferation.
Discussion
Although increased screening for colorectal cancer has decreased the overall rate of incidence, current standard-of-care chemotherapies are restricted to tumors that express WT KRAS protein. With nearly half of all diagnosed colorectal cancers possessing oncogenic KRAS, there presents a significant need for new frontline biologic therapies with increased efficacy, reduced toxicity, and directed tumor specificity. In particular chemotherapies that target KRAS mutations or the functional consequences of that oncogenic mutation that account for lethal chemo-resistant forms of colon cancer are needed. The role of mitochondrial dynamics in cancer progression is increasingly appreciated for its multifaceted roles enhancing tumor cell proliferation, migration, and microenvironmental remodeling (46–48). We demonstrate here that disruption of complex I and mitochondrial bioenergetics potently arrested the proliferation capacity of colon cancer epithelial cells. HCT116 cells express oncogenic KRAS overactive mutations and are an aggressive cancer cell line that forms undifferentiated tumors, whereas the HT-29 cell line retains WT KRAS protein and presents with an intermediate differentiation profile (49). Although all colon cancer cell lines tested, irrespective of their KRAS allele, were sensitive to complex I–directed interventions, the anti-proliferative effect of our mitochondria-targeted compounds was selective for transformed cancers and had limited effects on normal epithelial cells (data not shown). As shown herein, independent of their KRAS genotype, colon cancer cells phenotypically respond to mitochondrial targeted drugs via AMPK activation/mTORC1 suppression signaling pathway. Our results demonstrate that the anti-neoplastic effects are two-pronged, with detrimental impacts on bioenergetics that restrict the ability of the tumor cell to cope with energetic stress, resulting in a cascade of interconnected AMPK signaling pathways that culminates in anti-tumor mitophagy.
Although KRAS mutations occur in several different types of cancer, they appear with increasing frequency in colorectal cancer, pancreatic cancer, and non-small cell lung cancer. One of the established outcomes of oncogenic KRAS is the reprogramming of cell metabolism (50). In nontransformed cells, WT KRAS is rapidly inactivated by guanine-exchange factor proteins into its GDP-bound form, whereas in cancer cells, KRAS remains predominantly GTP-bound, thereby functioning as a constitutively active protein. Metabolically, KRAS mutations decrease mitochondrial OXPHOS while increasing glycolysis. Oncogenic KRAS promotes glucose uptake through enhanced expression of GLUT1 that accelerates glycolytic activity (51). The KRAS oncogene also channels glucose in the pentose phosphate pathway. Signaling by GTP-bound KRAS results in the perpetual signaling of PI3K/AKT/mTOR, which functions to exacerbate cancer cell proliferation. Mito-CP and Mito-Met10 were effective in abrogating colon cancer energy production. The effect was largely restricted to mitochondrial, relative to aerobic, glycolytic metabolism. Following the mTORC2-mediated modification of phospho-Ser-473 AKT as a surrogate readout for mTORC2 involvement, we found little effect of Mito-CP or Mito-Met10 on AKT signaling, suggesting that although mTORC2 has been proposed to be in the center of cancer cell metabolic reprogramming (52), these MTD had limited impact on mTORC2 signaling. However, both compounds, with Mito-CP more effective than Mito-Met10, resulted in Raptor phosphorylation, correlative with repression of mTORC1 activity in accordance with previous studies (35). Further, decreased phosphorylation of the protein synthesis regulator p70S6K was consistent with the decrease in observed proliferation in treated colon cancer cells. The discrepancy in Raptor phosphorylation between Mito-CP and Mito-Met10 likely reflects that each compound activates divergent, and still as yet unknown, signaling mechanisms. This may also be reflected in the MTD's differing abilities to inhibit complex I activity.
Lipophilic cations known to inhibit tumor cell proliferation, including the conjugation of the lipophilic cationic TPP+ moiety to an antioxidant molecule and Metformin, are thought to act through selective accumulation into mitochondria and inhibition of mitochondrial respiration (8, 20, 53). The mitochondrial membrane potential of tumor cells is greater than nontransformed normal epithelial cells (54) and likely accounts for the observed cell selectivity. We and others have previously shown that Mito-CP has anti-proliferative effects in lung cancer, breast cancer, hepatic cell carcinoma, medullary thyroid cancer, mesothelioma, and pancreatic cancer (5, 7, 8, 55, 56). Further, a redox-inactive analog of Mito-CP, Mito-CP-Ac, similarly inhibited tumor cell proliferation via a mechanism independent of superoxide dismutation in mitochondria (7). The molecular and signaling mechanism(s) responsible for the enhanced anti-proliferative effects of mitochondria-targeted lipophilic compounds such as Mito-CP or Mito-Met10 has shown that these compounds trigger the major energy sensor kinase AMPK pathway. We have previously shown that pancreatic cancer cells respond to Mito-CP, administered in conjunction with the glycolytic inhibitor 2-DG, by transiently increasing active AMPK levels. Moreover, increased AMPK signaling led to a decrease in FOXM1 (Forkhead box protein M1) protein levels (7). That Mito-CP abrogated FOXM1 levels is wholly consistent with this cell cycle regulator participating in decreased cell proliferation in response to mitochondria-targeted drug treatment. Similarly, we observed that Mito-Met10 treatment of pancreatic cancer cells led to proliferation arrest via a complex I–mediated stimulation of AMPK activation and block in cell cycle progression (6). This decrease in cell proliferation was accompanied by a comparable decrease in FOXM1 transcription factor levels. Together, these data suggest that AMPK dampening of the cell cycle regulator FOXM1 is a key mechanism for the decreased cell proliferation. However, the blockade in cell cycle progression was not complete, suggesting additional mechanisms were participating in the anti-cancer cell proliferation effects of these mitochondria-targeted agents.
AMPK is one of two key signaling complexes, the other being the central cell-proliferation regulator mTORC1, that are tightly executed to regulate autophagy. Ultimately, these signaling pathways converge on regulating the phosphorylation status of ULK1, the central autophagy-initiating kinase. In normal, nontransformed cells, nutrient signals are often the major autophagy trigger. When nutrients are abundant, mTORC1 is intact and catalytically available to down-regulate autophagy via the phosphorylation of ULK1 at Ser-757, disrupting the ULK1/AMPK interaction (44). Under glucose starvation, activated AMPK directly phosphorylates Raptor at Ser-792. This posttranslational modification redirects Raptor by generating a binding site for the 14-3-3 complex, thus putative suppression of mTORC1 activity (35). Restructuring of mTORC1 by active AMPK leaves the ULK1 protein available for the autophagy-activating phosphorylation at Ser-317 by AMPK (44). In this report, we demonstrate that during normal nutrient conditions, perturbations in colon cancer metabolic bioenergetics activated an AMPK-mediated signaling cascade impeding cell proliferation. Blocking AMPK with the inhibitor compound C reversed the inhibition of cell proliferation induced by either Mito-CP or Mito-Met10. To uncover the mechanisms for the observed anti-proliferative effects in colon cancer we used an iterative approach to reveal that Mito-CP and Mito-Met10 directly activated ULK1 to stimulate an anti-tumor mitophagy program. Interestingly, we found that Mito-CP was a stronger activator of AMPK, Raptor, and ULK1 relative to Mito-Metformin. In contrast to these data from transformed pancreatic cancer cells, Mito-CP promoted cell survival through the activation of AMPK and autophagy (57) in normal nontransformed mouse embryonic fibroblasts, suggesting a role for autophagy in maintenance of healthy cells. This discrepancy suggests that these molecules have discrete binding partners or activate still as yet unknown signaling pathways to halt tumor progression.
Although the role of autophagy is dynamic and dependent on the cellular context, it should be noted that varying types of cancer will respond differently to the effects of the TPP+ conjugated compounds because of the fluctuation in the regulation and number of mitochondria (58). The role for autophagy in cancer progression or suppression, particularly in KRAS-dependent cancers, remains unclear, with reports ascribing anti-tumor or pro-tumor survival. Autophagy is an evolutionarily conserved catabolic process characterized by cellular auto-digestion in the recycling of the cellular building blocks and the removal of dysfunctional or damaged organelles (59, 60). This recycling of resources ensures continued homeostatic balance and conserves energy in times of stress. Mitophagy, the sequestration and degradation of damaged mitochondria via autophagy (61), fulfills a vital function in normal nontransformed cells through the reduction in levels of reactive oxygen species (ROS) maintaining genomic stability thus limiting carcinogenesis (62). In this capacity, autophagy is believed to be largely tumor-suppressive. However, current dogma posits dual, conflicting roles for autophagy in cancer progression, with reports supporting anti-tumor as well as pro-tumor effects (63). This dichotomy is contextually and temporally dependent. In advanced stages of cancer, activation of the autophagic program facilitates proliferation under metabolic strain (ischemic or hypoxic conditions) (64, 65). In a prior report, a ubiquinone conjugated to TPP+ induced an autophagic-mediated proliferation arrest of metastatic breast cancer cells (66). With this report, we have determined that Mito-CP and Mito-Met10, potently halt colorectal cancer cell proliferation through the activation of an AMPK-repressive mTORC1-mitophagy signaling module. Although basal levels of autophagy in the colon cancer cells remained low in nutrient-rich normoxic conditions, we were able to induce a mitophagy/autophagy response with Mito-CP and Mito-Met10. However, following disruption of mitochondrial function, MTDs induced autophagy with a concomitant arrest in cell proliferation. Overall, our results are in concordance with other studies in which autophagy displays anti-oncogenic properties by suppressing transformed colon cancer cell proliferation (67–69). Our data reveal that selective disruption of the function of cancer cell mitochondria may be an effective and transitional therapeutic. The penetrance of the undruggable oncogenic KRAS allele is a serious and growing clinical issue with the imminent need for therapeutic agents whose mechanisms of action are not biased by the KRAS signaling pathway. The work here provides a framework upon which continued generation and characterization of mitochondria-targeted compounds will aid in the quest for better and safer chemotherapeutics. The potential activation of an anti-tumor mitophagy by Mito-CP and Mito-Met, or even Metformin, suggests new avenues for chemotherapeutic intervention.
Experimental procedures
Cell culture
HCT116 (CCL-247), T84 (CCL-248), HT-29 (HTB-38), and C2BBe1 (Caco2; CRL-2102) human colorectal carcinoma cells were purchased from ATCC (Manassas, VA) and maintained as described previously (70). SW480 and HK2–8 carcinoma cells (a generous gift from Dr. Guan Chen, Medical College of Wisconsin) were maintained as described (71). HCT116, T84, and SW480 cells express an overactive mutant KRAS protein, and HT-29, Caco2, and HK2–8 cells express WT KRAS. Normal nontransformed IEC-6 cells, isolated from rat small intestine tissue fragments and that retain a normal karyotype, were obtained from the ATCC (CRL-1592) and cultured as described previously (72, 73).
Reagents
Mito-CP and Mito-Met10 were synthesized as described previously (6, 19). 3-Carboxy-PROXYL, Metformin, and compound C were from purchased from Sigma-Aldrich. Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) was obtained from Agilent Technologies (Santa Clara, CA). Tetramethylrhodamine ethyl ester (TMRE) was purchased from Thermo Fisher Scientific. Gliotoxin was purchased from Merck KGaA (Darmstadt, Germany).
Cell proliferation
Cells (2 × 103) were plated into a 96-well plate and allowed to settle overnight. Cells were left untreated, or incubated in the presence of compound C (0.3 μm) for 48 h, after which time, increasing concentrations of Mito-Met10 and Mito-CP were added. Cells were placed into either an IncuCyte ZOOM or IncuCyte S3 Live-Cell Imaging System (Essen Bioscience, Ann Arbor, MI). Live cell images were acquired via the IncuCyte Analyzer at 2- to 3-h intervals over 5–7 days to provide real-time cellular confluence data based on segmentation of high-definition phase-contrast images. The IncuCyte S3 analysis software package allows for the post-acquisition assessment of phase object confluence (%) for all/any select images acquired during the time course of the dose experiments. The cell proliferation is expressed as an increase in percentage of confluence (6, 74).
Changes in mitochondrial bioenergetics and complex I inhibition
HCT116 cells (2 × 104) were plated into Seahorse XF96 well plates (Agilent Technologies, Santa Clara, CA), allowed to adhere for 4 h, and treated with a Mito-Met10 or Mito-CP dose curve. Measurement of the mitochondrial respiratory complex I in permeabilized cells was performed according to the manufacturer's directions and as described previously (74–76). Briefly, after a 24-h incubation with drugs, intact cells were permeabilized with 1 nm plasma membrane permeabilizer (Agilent Technologies) immediately before the OCR measurement was taken on a Seahorse Bioscience XF96 Extracellular Flux Analyzer. OCR was measured as a readout of mitochondrial function and was assessed by injecting oligomycin (1 μg/ml) to inhibit ATP synthase, FCCP (50 μm) to uncouple the mitochondria, and rotenone (1 μm) and antimycin A (10 μm) as inhibitors of complex I and complex III, respectively. The oxygen consumption derived from mitochondrial complex I or complex II activity was measured before and after addition of different substrates to mitochondria, e.g. pyruvate/malate for complex I and succinate for complex II activity. Rotenone, malonate, and antimycin A were used as specific inhibitors of mitochondrial complex I, II, and III activities, respectively.
Total ATP measurements
HCT116 cells (2 × 104) were plated into a 96-well plate and treated with increasing concentrations of Mito-CP or Mito-Met10 for 24 h. Intracellular ATP levels were determined in cell lysates using a luciferase-based assay per manufacturer's directions (Sigma Aldrich) as described previously (6).
Quantification of intracellular accumulation of MTDs by LC-MS/MS
Cells (1 × 106) were incubated with Mito-CP or Mito-Met10 for 24 h. Extraction of Mito-CP or Mito-Met10 was described previously, but without butylated hydroxytoluene (6). LC-MS/MS analyses were performed using a Kinetex Phenyl-Hexyl column (50 mm × 2.1 mm, 1.7 μm, Phenomenex) equilibrated with water:acetonitrile mixture (4:1) containing 0.1% formic acid. Compounds were eluted by increasing the content of acetonitrile from 20 to 100% over 4 min and detected using the MRM mode.
Protein harvesting and immunoblot analysis
Cells (1.25 × 106) were treated for 18 h with Mito-Met10 or Mito-CP and lysed in a modified RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.25% v/v sodium deoxycholate, 1% v/v Nonidet P-40, 0.1% v/v SDS, 1 mm EDTA, 10 mm sodium orthovanadate, 40 mm β-glycerol phosphate, 20 mm sodium fluoride). Lysates were quantified, normalized for protein concentration, and 10 μg protein per lane size separated using reducing SDS-PAGE. Proteins were electrotransferred to PVDF membranes (Millipore) and probed using primary and horseradish peroxidase–conjugated secondary antibodies. Primary antibodies purchased from Cell Signaling Technology (Danvers, MA) include phospho-Thr-172 AMPK (40H9), total AMPK (D5A2), phospho-Ser-792 Raptor (2083), total Raptor (24C12), phospho-AKT (Ser-473), total AKT, phospho-Thr-389 p70S6K (108D2), total p70S6K, phospho-Ser-757 ULK1 (D706U), phospho-Ser-317 ULK1, total ULK1 (D8H5), caspase 3, and GAPDH were used at the manufacturer's recommended dilutions. Beta-tubulin (E7) anti-sera was obtained from Developmental Studies Hybridoma Bank (University of Iowa). Proteins were visualized by chemiluminescence and quantified by densitometric analysis using the FluorChem HD2 from Cell Biosciences (Santa Clara, CA). For AMPK, cells were starved of glucose, glutamine, and serum for 6 h prior to a 30-min stimulation with titrated doses of Mito-Met10 or Mito-CP.
TMRE staining to assess mitochondrial integrity
Flow cytometry analysis
Cells (5 × 105) were treated with FCCP (20 μm) for 15 min or increasing concentrations of Mito-Met10 or Mito-CP for 24 h. TMRE (200 nm) was added for 30 min prior to harvesting for analysis on a BD-LSR II flow cytometer (BD Biosciences); data were analyzed using FlowJo (FlowJo, LLC, Ashland, OR).
Live cell confocal imaging
Cells (1 × 105) were plated on 35 mm No. 1.5 cover glass (0.16–0.19 mm) dishes and treated with FCCP (20 μm) for 15 min or increasing concentrations of Mito-Met10 or Mito-CP for 24 h. TMRE (100 nm) was added for 30 min, washed, and quenched by incubation of HEPES containing media prior to visualization. Live-cell images were acquired with a Spinning Disc Confocal system configured with an IX83 inverted microscope (Olympus America, Center Valley, PA) equipped with an electron-multiplying charge-coupled device camera (Hamamatsu Corporation, Bridgewater, NJ) and a silicone oil immersion 60×/1.6 numerical aperture (Olympus) objective lens driven by MetaMorph software.
Transmission electron microscopy
1 × 106 HCT116 cells were treated with vehicle (DMSO), Mito-Met10 (50 μm), or Mito-CP (1 μm) for 6 h. Cells were washed, fixed in situ with 4% paraformaldehyde + 2% glutaraldehyde in 0.1 m sodium cacodylate buffer for 10 min at room temp. Cells were gently scraped from the culture dishes, pelleted, washed with 3 × 10 min rinses in 0.1 m cacodylate buffer and postfixed in potassium ferricyanide reduced 1% osmium tetroxide for 2 h on ice. Cell pellets were processed into epoxy resin (EMBed 812). 60 nm sections were stained with uranyl acetate and lead citrate and imaged on a Hitachi H600 TEM.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0 (La Jolla, CA). Paired analyses were calculated using a Student's t test and a Dunnett post hoc analysis to identify pair-wise differences between experimental and control groups. Multiple comparisons between groups were analyzed using a one-way or two-way repeated measures analysis of variance (ANOVA). Values provided represent mean ± S.E. Statistical significance was defined as p ≤ 0.05 and denoted with the appropriate asterisks in the figure legends.
Author contributions
K. A. B., J. V. W., R. B. H., B. K., and M. B. D. conceptualization; K. A. B., R. B. H., A. M., B. K., and M. B. D. resources; K. A. B., J. V. W., and R. B. H. data curation; K. A. B., J. V. W., R. B. H., A. M., B. K., and M. B. D. formal analysis; K. A. B., R. B. H., A. M., B. K., and M. B. D. supervision; B. K. and M. B. D. funding acquisition; K. A. B., J. V. W., R. B. H., B. K., and M. B. D. investigation; K. A. B., J. V. W., R. B. H., A. M., and B. K. methodology; K. A. B., J. V. W., R. B. H., and M. B. D. writing-original draft; K. A. B., B. K., and M. B. D. project administration; K. A. B., J. V. W., R. B. H., A. M., B. K., and M. B. D. writing-review and editing.
Supplementary Material
Acknowledgments
We thank members of the Dwinell lab for their insight and scholarly contributions to this work. We are particularly indebted to Donna McAllister, Olivia Koehn, Melissa Whyte, Megan Harwig, Elizabeth English, and Clive Wells for their technical assistance.
This work was supported by grants from the NCI, National Institutes of Health Grants U01 CA178960 (to M. B. D. and B. K.) and R01 CA152810 (to B. K.), the Medical College of Wisconsin Cancer Center (to M. B. D. and B. K.), the Advancing a Healthier Wisconsin Endowment (to M. B. D.), and the Bobbie Nick Voss Charitable Foundation continuing philanthropic donations (to M. B. D.). M. B. D. has ownership interests in Protein Foundry, LLC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting Figs. S1–S6.
- OXPHOS
- oxidative phosphorylation
- Mito-CP
- 3-carboxyl proxyl nitroxide
- CP
- carboxyl proxyl
- 2-DG
- 2-deoxyglucose
- AMPK
- AMP-activated protein kinase
- OCR
- oxygen consumption rate
- TEM
- transmission EM
- TMRE
- tetramethylrhodamine ethyl ester
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- MTD
- mitochondria-targeted drug.
References
- 1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society, Atlanta, GA [Google Scholar]
- 2. Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., and Der C. J. (2014) Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 13, 828–851 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Roock W., Claes B., Bernasconi D., De Schutter J., Biesmans B., Fountzilas G., Kalogeras K. T., Kotoula V., Papamichael D., Laurent-Puig P., Penault-Llorca F., Rougier P., Vincenzi B., Santini D., Tonini G., et al. (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 11, 753–762 10.1016/S1470-2045(10)70130-3 [DOI] [PubMed] [Google Scholar]
- 4. Misale S., Yaeger R., Hobor S., Scala E., Janakiraman M., Liska D., Valtorta E., Schiavo R., Buscarino M., Siravegna G., Bencardino K., Cercek A., Chen C. T., Veronese S., Zanon C., et al. (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536 10.1038/nature11156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R., and Chandel N. S. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng G., Zielonka J., Ouari O., Lopez M., McAllister D., Boyle K., Barrios C. S., Weber J. J., Johnson B. D., Hardy M., Dwinell M. B., and Kalyanaraman B. (2016) Mitochondria-targeted analogues of metformin exhibit enhanced anti-proliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. 76, 3904–3915 10.1158/0008-5472.CAN-15-2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng G., Zielonka J., McAllister D., Hardy M., Ouari O., Joseph J., Dwinell M. B., and Kalyanaraman B. (2015) Antiproliferative effects of mitochondria-targeted cationic antioxidants and analogs: Role of mitochondrial bioenergetics and energy-sensing mechanism. Cancer Lett. 365, 96–106 10.1016/j.canlet.2015.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng G., Zielonka J., Dranka B. P., McAllister D., Mackinnon A. C. Jr., Joseph J., and Kalyanaraman B. (2012) Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 72, 2634–2644 10.1158/0008-5472.CAN-11-3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zielonka J., Joseph J., Sikora A., Hardy M., Ouari O., Vasquez-Vivar J., Cheng G., Lopez M., and Kalyanaraman B. (2017) Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 117, 10043–10120 10.1021/acs.chemrev.7b00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chandran K., Aggarwal D., Migrino R. Q., Joseph J., McAllister D., Konorev E. A., Antholine W. E., Zielonka J., Srinivasan S., Avadhani N. G., and Kalyanaraman B. (2009) Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q. Biophys. J. 96, 1388–1398 10.1016/j.bpj.2008.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mukhopadhyay P., Horváth B., Zsengellér Z., Zielonka J., Tanchian G., Holovac E., Kechrid M., Patel V., Stillman I. E., Parikh S. M., Joseph J., Kalyanaraman B., and Pacher P. (2012) Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 52, 497–506 10.1016/j.freeradbiomed.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith R. A., Adlam V. J., Blaikie F. H., Manas A. R., Porteous C. M., James A. M., Ross M. F., Logan A., Cochemé H. M., Trnka J., Prime T. A., Abakumova I., Jones B. A., Filipovska A., and Murphy M. P. (2008) Mitochondria-targeted antioxidants in the treatment of disease. Ann. N.Y. Acad. Sci. 1147, 105–111 10.1196/annals.1427.003 [DOI] [PubMed] [Google Scholar]
- 13. Kelso G. F., Porteous C. M., Coulter C. V., Hughes G., Porteous W. K., Ledgerwood E. C., Smith R. A., and Murphy M. P. (2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596 10.1074/jbc.M009093200 [DOI] [PubMed] [Google Scholar]
- 14. Smith R. A., Porteous C. M., Coulter C. V., and Murphy M. P. (1999) Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 263, 709–716 10.1046/j.1432-1327.1999.00543.x [DOI] [PubMed] [Google Scholar]
- 15. Smith R. A., Porteous C. M., Gane A. M., and Murphy M. P. (2003) Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 5407–5412 10.1073/pnas.0931245100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Callahan J., and Kopecek J. (2006) Semitelechelic HPMA copolymers functionalized with triphenylphosphonium as drug carriers for membrane transduction and mitochondrial localization. Biomacromolecules 7, 2347–2356 10.1021/bm060336m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dong L. F., Jameson V. J., Tilly D., Prochazka L., Rohlena J., Valis K., Truksa J., Zobalova R., Mahdavian E., Kluckova K., Stantic M., Stursa J., Freeman R., Witting P. K., Norberg E., et al. (2011) Mitochondrial targeting of α-tocopheryl succinate enhances its pro-apoptotic efficacy: A new paradigm for effective cancer therapy. Free Radic. Biol. Med. 50, 1546–1555 10.1016/j.freeradbiomed.2011.02.032 [DOI] [PubMed] [Google Scholar]
- 18. Millard M., Pathania D., Shabaik Y., Taheri L., Deng J., and Neamati N. (2010) Preclinical evaluation of novel triphenylphosphonium salts with broad-spectrum activity. PLoS One 5, e13131 10.1371/journal.pone.0013131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dhanasekaran A., Kotamraju S., Karunakaran C., Kalivendi S. V., Thomas S., Joseph J., and Kalyanaraman B. (2005) Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial superoxide. Free Radic. Biol. Med. 39, 567–583 10.1016/j.freeradbiomed.2005.04.016 [DOI] [PubMed] [Google Scholar]
- 20. Modica-Napolitano J. S., and Aprille J. R. (2001) Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Delivery Rev. 49, 63–70 10.1016/S0169-409X(01)00125-9 [DOI] [PubMed] [Google Scholar]
- 21. Rodríguez-Enríquez S., Hernández-Esquivel L., Marín-Hernández A., Dong L. F., Akporiaye E. T., Neuzil J., Ralph S. J., and Moreno-Sánchez R. (2012) Molecular mechanism for the selective impairment of cancer mitochondrial function by a mitochondrially targeted vitamin E analogue. Biochim. Biophys. Acta 1817, 1597–1607 10.1016/j.bbabio.2012.05.005 [DOI] [PubMed] [Google Scholar]
- 22. Nadakavukaren K. K., Nadakavukaren J. J., and Chen L. B. (1985) Increased rhodamine 123 uptake by carcinoma cells. Cancer Res. 45, 6093–6099 [PubMed] [Google Scholar]
- 23. Starenki D., and Park J. I. (2013) Mitochondria-targeted nitroxide, Mito-CP, suppresses medullary thyroid carcinoma cell survival in vitro and in vivo. J. Clin. Endocrinol. Metab. 98, 1529–1540 10.1210/jc.2012-3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murphy M. P. (1997) Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 15, 326–330 10.1016/S0167-7799(97)01068-8 [DOI] [PubMed] [Google Scholar]
- 25. Higurashi T., Takahashi H., Endo H., Hosono K., Yamada E., Ohkubo H., Sakai E., Uchiyama T., Hata Y., Fujisawa N., Uchiyama S., Ezuka A., Nagase H., Kessoku T., Matsuhashi N., Yamanaka S., Inayama Y., Morita S., and Nakajima A. (2012) Metformin efficacy and safety for colorectal polyps: A double-blind randomized controlled trial. BMC Cancer 12, 118 10.1186/1471-2407-12-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Z. J., Zheng Z. J., Kan H., Song Y., Cui W., Zhao G., and Kip K. E. (2011) Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 34, 2323–2328 10.2337/dc11-0512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomimoto A., Endo H., Sugiyama M., Fujisawa T., Hosono K., Takahashi H., Nakajima N., Nagashima Y., Wada K., Nakagama H., and Nakajima A. (2008) Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 99, 2136–2141 10.1111/j.1349-7006.2008.00933.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hosono K., Endo H., Takahashi H., Sugiyama M., Uchiyama T., Suzuki K., Nozaki Y., Yoneda K., Fujita K., Yoneda M., Inamori M., Tomatsu A., Chihara T., Shimpo K., Nakagama H., and Nakajima A. (2010) Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol. Carcinog. 49, 662–671 10.1002/mc.20637 [DOI] [PubMed] [Google Scholar]
- 29. Summerhayes I. C., Lampidis T. J., Bernal S. D., Nadakavukaren J. J., Nadakavukaren K. K., Shepherd E. L., and Chen L. B. (1982) Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 79, 5292–5296 10.1073/pnas.79.17.5292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kahn B. B., Alquier T., Carling D., and Hardie D. G. (2005) AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25 10.1016/j.cmet.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 31. Inoki K., Zhu T., and Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]
- 32. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hemmings B. A., and Restuccia D. F. (2012) PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 4, a011189 10.1101/cshperspect.a011189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., and Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 10.1016/S0092-8674(02)00808-5 [DOI] [PubMed] [Google Scholar]
- 35. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., and Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dufner A., and Thomas G. (1999) Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res. 253, 100–109 10.1006/excr.1999.4683 [DOI] [PubMed] [Google Scholar]
- 37. Pullen N., and Thomas G. (1997) The modular phosphorylation and activation of p70s6k. FEBS Lett. 410, 78–82 10.1016/S0014-5793(97)00323-2 [DOI] [PubMed] [Google Scholar]
- 38. Pearson R. B., Dennis P. B., Han J. W., Williamson N. A., Kozma S. C., Wettenhall R. E., and Thomas G. (1995) The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 14, 5279–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hara K., Maruki Y., Long X., Yoshino K., Oshiro N., Hidayat S., Tokunaga C., Avruch J., and Yonezawa K. (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 10.1016/S0092-8674(02)00833-4 [DOI] [PubMed] [Google Scholar]
- 40. Weng Q. P., Kozlowski M., Belham C., Zhang A., Comb M. J., and Avruch J. (1998) Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J. Biol. Chem. 273, 16621–16629 10.1074/jbc.273.26.16621 [DOI] [PubMed] [Google Scholar]
- 41. Neufeld T. P. (2010) TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell Biol. 22, 157–168 10.1016/j.ceb.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alers S., Löffler A. S., Wesselborg S., and Stork B. (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 32, 2–11 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tchevkina E., and Komelkov A. (2012) Protein phosphorylation as a key mechanism of mTORC1/2 signaling pathways. In Protein Phosphorylation in Human Health (Huang C., ed), pp. 1–50, IntechOpen, London, UK: 10.5772/2944 [DOI] [Google Scholar]
- 44. Kim J., Kundu M., Viollet B., and Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perry S. W., Norman J. P., Barbieri J., Brown E. B., and Gelbard H. A. (2011) Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 50, 98–115 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kashatus J. A., Nascimento A., Myers L. J., Sher A., Byrne F. L., Hoehn K. L., Counter C. M., and Kashatus D. F. (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 57, 537–551 10.1016/j.molcel.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Senft D., and Ronai Z. A. (2016) Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 39, 43–52 10.1016/j.ceb.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Serasinghe M. N., Wieder S. Y., Renault T. T., Elkholi R., Asciolla J. J., Yao J. L., Jabado O., Hoehn K., Kageyama Y., Sesaki H., and Chipuk J. E. (2015) Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 57, 521–536 10.1016/j.molcel.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yeung T. M., Gandhi S. C., Wilding J. L., Muschel R., and Bodmer W. F. (2010) Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. U.S.A. 107, 3722–3727 10.1073/pnas.0915135107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ying H., Kimmelman A. C., Lyssiotis C. A., Hua S., Chu G. C., Fletcher-Sananikone E., Locasale J. W., Son J., Zhang H., Coloff J. L., Yan H., Wang W., Chen S., Viale A., Zheng H., et al. (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bryant K. L., Mancias J. D., Kimmelman A. C., and Der C. J. (2014) KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 39, 91–100 10.1016/j.tibs.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Masui K., Cavenee W. K., and Mischel P. S. (2014) mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab. 25, 364–373 10.1016/j.tem.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cheng G., Zielonka J., McAllister D. M., Mackinnon A. C. Jr., Joseph J., Dwinell M. B., and Kalyanaraman B. (2013) Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer 13, 285 10.1186/1471-2407-13-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Beckham T. H., Lu P., Jones E. E., Marrison T., Lewis C. S., Cheng J. C., Ramshesh V. K., Beeson G., Beeson C. C., Drake R. R., Bielawska A., Bielawski J., Szulc Z. M., Ogretmen B., Norris J. S., and Liu X. (2013) LCL124, a cationic analog of ceramide, selectively induces pancreatic cancer cell death by accumulating in mitochondria. J. Pharmacol. Exp. Ther. 344, 167–178 10.1124/jpet.112.199216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cunniff B., Benson K., Stumpff J., Newick K., Held P., Taatjes D., Joseph J., Kalyanaraman B., and Heintz N. H. (2013) Mitochondrial-targeted nitroxides disrupt mitochondrial architecture and inhibit expression of peroxiredoxin 3 and FOXM1 in malignant mesothelioma cells. J. Cell Physiol. 228, 835–845 10.1002/jcp.24232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dilip A., Cheng G., Joseph J., Kunnimalaiyaan S., Kalyanaraman B., Kunnimalaiyaan M., and Gamblin T. C. (2013) Mitochondria-targeted antioxidant and glycolysis inhibition: Synergistic therapy in hepatocellular carcinoma. Anticancer Drugs 24, 881–888 10.1097/CAD.0b013e32836442c6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao B., Qiang L., Joseph J., Kalyanaraman B., Viollet B., and He Y. Y. (2016) Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis. 3, 82–87 10.1016/j.gendis.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 10.1016/j.freeradbiomed.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. He C., and Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stipanuk M. H. (2009) Macroautophagy and its role in nutrient homeostasis. Nutr. Rev. 67, 677–689 10.1111/j.1753-4887.2009.00252.x [DOI] [PubMed] [Google Scholar]
- 61. Youle R. J., and Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Czarny P., Pawlowska E., Bialkowska-Warzecha J., Kaarniranta K., and Blasiak J. (2015) Autophagy in DNA damage response. Int. J. Mol. Sci. 16, 2641–2662 10.3390/ijms16022641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Burada F., Nicoli E. R., Ciurea M. E., Uscatu D. C., Ioana M., and Gheonea D. I. (2015) Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 7, 271–284 10.4251/wjgo.v7.i11.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Galluzzi L., Pietrocola F., Bravo-San Pedro J. M., Amaravadi R. K., Baehrecke E. H., Cecconi F., Codogno P., Debnath J., Gewirtz D. A., Karantza V., Kimmelman A., Kumar S., Levine B., Maiuri M. C., Martin S. J., et al. (2015) Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 10.15252/embj.201490784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Panda P. K., Mukhopadhyay S., Das D. N., Sinha N., Naik P. P., and Bhutia S. K. (2015) Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin. Cell Dev. Biol. 39, 43–55 10.1016/j.semcdb.2015.02.013 [DOI] [PubMed] [Google Scholar]
- 66. Rao V. A., Klein S. R., Bonar S. J., Zielonka J., Mizuno N., Dickey J. S., Keller P. W., Joseph J., Kalyanaraman B., and Shacter E. (2010) The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 285, 34447–34459 10.1074/jbc.M110.133579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ellington A. A., Berhow M., and Singletary K. W. (2005) Induction of macroautophagy in human colon cancer cells by soybean B-group triterpenoid saponins. Carcinogenesis 26, 159–167 10.1093/carcin/bgh297 [DOI] [PubMed] [Google Scholar]
- 68. Psahoulia F. H., Moumtzi S., Roberts M. L., Sasazuki T., Shirasawa S., and Pintzas A. (2007) Quercetin mediates preferential degradation of oncogenic Ras and causes autophagy in Ha-RAS-transformed human colon cells. Carcinogenesis 28, 1021–1031 10.1093/carcin/bgl232 [DOI] [PubMed] [Google Scholar]
- 69. Patel B. B., Sengupta R., Qazi S., Vachhani H., Yu Y., Rishi A. K., and Majumdar A. P. (2008) Curcumin enhances the effects of 5-fluorouracil and oxaliplatin in mediating growth inhibition of colon cancer cells by modulating EGFR and IGF-1R. Int. J. Cancer 122, 267–273 10.1002/ijc.23097 [DOI] [PubMed] [Google Scholar]
- 70. Wendt M. K., Johanesen P. A., Kang-Decker N., Binion D. G., Shah V., and Dwinell M. B. (2006) Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene 25, 4986–4997 10.1038/sj.onc.1209505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yin N., Lepp A., Ji Y., Mortensen M., Hou S., Qi X. M., Myers C. R., and Chen G. (2017) The K-Ras effector p38γ MAPK confers intrinsic resistance to tyrosine kinase inhibitors by stimulating EGFR transcription and EGFR dephosphorylation. J. Biol. Chem. 292, 15070–15079 10.1074/jbc.M117.779488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Smith J. M., Johanesen P. A., Wendt M. K., Binion D. G., and Dwinell M. B. (2005) CXCL12 activation of CXCR4 regulates mucosal host defense through stimulation of epithelial cell migration and promotion of intestinal barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G316–G326 10.1152/ajpgi.00208.2004 [DOI] [PubMed] [Google Scholar]
- 73. Quaroni A., Wands J., Trelstad R. L., and Isselbacher K. J. (1979) Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J. Cell Biol. 80, 248–265 10.1083/jcb.80.2.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cheng G., Zielonka J., McAllister D., Tsai S., Dwinell M. B., and Kalyanaraman B. (2014) Profiling and targeting of cellular bioenergetics: Inhibition of pancreatic cancer cell proliferation. Br. J. Cancer 111, 85–93 10.1038/bjc.2014.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Divakaruni A. S., Wiley S. E., Rogers G. W., Andreyev A. Y., Petrosyan S., Loviscach M., Wall E. A., Yadava N., Heuck A. P., Ferrick D. A., Henry R. R., McDonald W. G., Colca J. R., Simon M. I., Ciaraldi T. P., and Murphy A. N. (2013) Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. U.S.A. 110, 5422–5427 10.1073/pnas.1303360110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Salabei J. K., Gibb A. A., and Hill B. G. (2014) Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 9, 421–438 10.1038/nprot.2014.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.