

Abstract
Cytochrome c oxidase (CcO) is the terminal oxidase of cellular respiration, reducing O2 to water and pumping protons. X-ray structural features have suggested that CcO pumps protons via a mechanism involving electrostatic repulsions between pumping protons in the hydrogen-bond network of a proton-conducting pathway (the H-pathway) and net positive charges created upon oxidation of an iron site, heme a (Fea2+), for reduction of O2 at another iron site, heme a3 (Fea32+). The protons for pumping are transferred to the hydrogen-bond network from the N-side via the water channel of the H-pathway. Back-leakage of protons to the N-side is thought to be blocked by closure of the water channel. To experimentally test this, we examined X-ray structures of the azide-bound, oxidized bovine CcO and found that an azide derivative (N3−–Fea33+, CuB2+–N3−) induces a translational movement of the heme a3 plane. This was accompanied by opening of the water channel, revealing that Fea3 and the H-pathway are tightly coupled. The channel opening in the oxidized state is likely to induce back-leakage of pumping protons, which lowers the proton level in the hydrogen-bond network during enzymatic turnover. The proton level decrease weakens the electron affinity of Fea, if Fea electrostatically interacts with protons in the hydrogen-bond network. The previously reported azide-induced redox-potential decrease in Fea supports existence of the electrostatic interaction. In summary, our results indicate that the H-pathway is critical for CcO's proton-pumping function.
Keywords: enzyme mechanism, cytochrome c oxidase (complex IV), proton pump, mitochondrial membrane potential, X-ray crystallography, bioenergetics, heme, metalloenzyme, azide, copper
Introduction
Cytochrome c oxidase (CcO) 5 is the terminal oxidase of cellular respiration that reduces O2 to water, coupled with a proton-pumping process. The enzyme includes two redox-active metal sites as the low-potential sites (heme a (Fea) and CuA). These sites donate electrons to the O2 reduction site that comprises two high potential metal sites (heme a3 (Fea3) and CuB) (1, 2). Two proton-conducting pathways, designated as the K- and D-pathways as shown in Fig. 1, are located in the protein for connecting the O2 reduction site with the N-side surface of the CcO molecule. These pathways transfer protons for making water molecules after (or coupled with) donation of electrons to the O2 reduction site. The third proton-conducting pathway, the H-pathway, extends for proton pumping from the N-side surface to the P-side surface.
Figure 1.

X-ray structure of bovine heart CcO. The locations of the iron, copper, and magnesium atoms are shown by reddish brown, dark blue, and green balls, respectively. Small red balls are located at the positions of water molecules. Magenta structures denote hemes a and a3 as labeled. The positions of the central iron atoms, Fea and Fea3, of the hemes shown in reddish brown balls are indicated by arrows. The dark blue, red, and green portions of amino acids denote nitrogen, oxygen, and sulfur atoms, respectively. The carbon atoms of amino acid residues of subunits I and II are labeled by yellow and blue sticks, respectively. The possible pathways for O2, electrons, and protons for making water molecules and for pumping are denoted by gray, blue, and red curves, each with an arrowhead. The structure and location of the water exit pathway are still under debate. The inset is a schematic representation of the locations of the redox-active metal sites and the pathways for transportation of the substrates (O2, electrons, and protons) within the overall CcO structure. This figure is based on the X-ray diffraction data given in PDB 5B1A.
Extensive X-ray structural analyses of the system strongly suggest that proton pumping is driven by electrostatic repulsions between the protons transferred from the N-side by hydronium ions through a water channel, which forms part of the proton pumping pathway (the H-pathway) and the net positive charges created upon oxidation of heme a for reduction of O2 bound to heme a3. The directionality of the proton pump is provided by closure of the water channel after the pumping protons are collected (1, 3, 4). The water molecules (or the hydronium ions) are driven through the water channel by thermal motion of the protein moiety. Water cavities, where at least one mobile water molecule is stored, are located within the water channel. These water cavities accelerate the water exchange between the N-side and the hydrogen-bond network in the H-pathway that extends to the P-side, because the water molecules in the water cavities are mobile without significant interaction with the protein moiety. Extensive X-ray structural analyses thus far show that the water channel is closed by elimination of the largest cavity near the junction point with the hydrogen-bond network. The water channel opens only when the O2 reduction site comprising Fea3 and CuB is in the ligand-free fully reduced state and is closed in other oxidation states or in strong ligand (such as CO, NO and O2)-bound fully reduced states (5).
In contrast, proton-pumping mechanisms without involvement of the H-pathway have been proposed, based on mutational analyses of bacterial CcOs. Bovine CcO includes well-conserved residues, Glu242 and Asn98, near the upper end and halfway in the D-pathway, respectively, as described in Fig. 1. Bacterial E242Q mutant CcOs show neither the O2 reduction nor proton pumping, whereas bacterial N98D mutant CcOs reduce O2 without proton pumping. Based on these mutational results, it has been proposed that Gln242 abolishes transfer of protons for both pumping and O2 reduction, whereas Asp98 blocks only the proton-pumping transfer, at least, in bacterial CcOs (1–3). However, D51N mutation for bovine heart CcO abolishes proton-pumping function without decreasing the O2 reduction activity, confirming the critical role of the H-pathway in proton pumping of bovine CcO (6, 7), whereas Asp51 is not conserved in bacterial CcOs, and any mutation for the bacterial H-pathway residues shows no influence to both O2 reduction and pumping activities. One of the simplest interpretations for these mutational analysis results is that the mechanism of proton pumping of CcO is not completely conserved.
The proton/electron coupling mechanism of bovine CcO, proposed based on the static X-ray structural results, has been confirmed by mutational analyses of the function of the H-pathway, as described above (6, 7). However, the proposal has not been confirmed by experimental (or functional) analyses of the coupling process in the enzyme. For example, the X-ray structure of the hydrogen-bond network of the H-pathway, which is attached to heme a peripheral groups, strongly suggests that the electrostatic interactions between positively charged heme a and protons on the hydrogen-bond network drive proton-active transport (1–3, 7). However, the X-ray structural results themselves do not clearly show the existence of the electrostatic interactions sufficiently strong to promote active proton transfer. Thus, it is desirable to examine the effects of structural perturbation for the H-pathway on the enzymatic function of the bovine CcO.
Since the pioneering studies on respiration by Keilin and Hartree 78 years ago (8), azide has been known to be a potent inhibitor of CcO, which is nearly as strong as cyanide at low pH (9, 10). It was recognized that this inhibitor, which is obviously chemically different from O2, could be an excellent probe for investigating the function of the O2 reduction site of the enzyme. Extensive investigations of the interactions between azide and CcO are fairly limited, compared with investigations using CO and cyanide. One of the reasons is that the azide-induced absorption spectral changes of the oxidized CcO, as isolated from bovine heart, are very weak, and as a result, consistency among the reported results from different groups, which likely used different enzyme preparations, has tended to be fairly low (11–14).
All of the redox-active sites of the oxidized CcO as isolated from bovine heart are in the oxidized state (1, 15). Its reactivity with cyanide and its electron transfer and proton-pumping functions indicate that the fully oxidized form (or the “as prepared” form) is not directly involved in catalytic turnover (1). Thus, the preparation is designated as “resting oxidized” CcO. Although the physiological significance of this form remains controversial, it has been the most extensively investigated among the various forms and derivatives of CcO because of the availability of samples with high purity and yield (1, 2). Recently, it has been established that the resting oxidized form has a peroxide moiety bridged between Fea33+ and CuB2+ (16).
Recently, spectral changes in the absorption, which occur with binding of azide to the resting oxidized CcO, have been well-established using the CcO preparation purified by recrystallization. This investigation identifies a two-step binding of azide to the O2 reduction site revealed by accurately measuring the small spectral changes showing that CcO has two azide-binding sites with high and low affinities (Kd values of 78 μm and 42 mm, respectively) (17). Resonance Raman analyses showed that bound peroxide is removed by the initial azide binding to Fea33+ without influencing a spin state of heme a3. The absence of a spin-state transition provides much smaller spectral changes in absorbance upon binding of azide compared with the spectral changes that occur with cyanide binding accompanied by a high- to low-spin state transition (17, 18).
The IR analyses of azide binding to the enzyme in the five different overall oxidation states (i.e. 0 (resting oxidized) to the 4-electron reduced (the fully reduced) states) indicate that, in each oxidation state except for the fully reduced state, CcO shows sharp azide bands assignable to Fea33+–N3− or CuB2+–N3−, depending on the overall oxidation state. Based on these results, it has been proposed that the two metal sites in the O2 reduction site are occupied by azide simultaneously (12). However, the IR results do not preclude the possibility that a single azide molecule binds to either one of the two metal sites in the O2 reduction site. The X-ray structure of the azide-bound resting oxidized CcO has been determined at 2.9 Å resolution (PDB code 1OCZ). This structure shows a bridging ligand between the two metals in the O2 reduction site. This is inconsistent with the two sharp azide IR bands, which suggest terminal azide binding to a metal ion (19, 20). However, binding of single azide ion to the O2 reduction site is consistent with the narrow ligand binding space of the O2 reduction site. In fact, an early careful titration analysis indicates stoichiometric binding of a single azide ion to the resting oxidized bovine CcO (10). Furthermore, X-ray structural results thus far suggest that the site has the capacity to bind only a single diatomic molecule or ion, such as O2, CO, NO, and CN− (1).
To gain insights into the proton-pumping function of CcO as well as to clarify the structural basis of the inconsistent observations with regard to binding of azide to CcO, we reexamined the X-ray structure of the azide-bound resting oxidized CcO at significantly improved resolution (∼1.85 Å). The present results reveal that the azide-binding sites, Fea33+ and CuB2+, have an unexpectedly high tolerance for large ligands and that each can accept a terminal azide ion. The binding of two azide ions to the two metal centers induces opening of the water channel without changing the oxidation and spin states of the metal centers, which is uniquely detectable in the azide-bound state. The channel opening is highly likely to disturb the system for blockage of the water channel against the pumping proton back leaks to impair the normal enzymatic function, which is consistent with the proposal that the H-pathway is involved critically in the proton-pumping function.
Results
Two terminal azide-binding modes at 20 mm azide
The azide binding was confirmed by the visible spectral changes in the vicinity of the 650 nm shoulder as described previously (17). Fig. 2, A and B, shows the electron density maps of the O2 reduction site for the MR/DM map (see “X-ray structure determinations and refinements” under “Experimental procedures”) and the Fo − Fc map without azide ions, respectively, at 1.85 Å resolution. The structural refinement converged well for two azide ions, each coordinating to respective Fea3 and CuB, at the full occupancy, and the Fo − Fc map gave no significant residual electron density between the two metals (Fig. 2C). It is noteworthy that no other technique other than high-resolution X-ray crystallography is able to reveal the truly unexpected geometry of azide binding.
Figure 2.
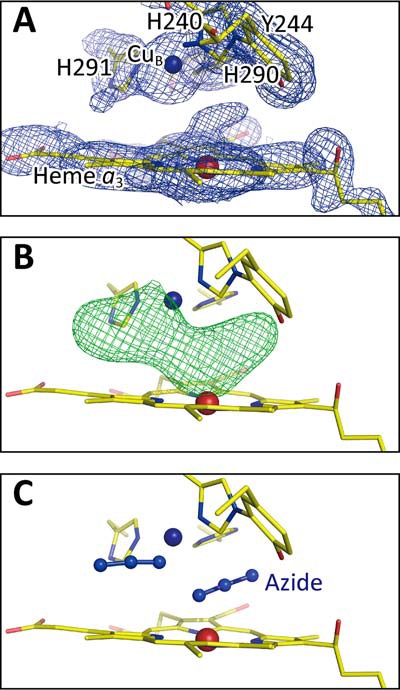
Electron density maps of the O2 reduction site of bovine heart resting oxidized CcO obtained from the crystals exposed to 20 mm azide at pH 5.7 for 4 days. A, MR/DM map of the O2 reduction site. B and C, the Fo − Fc map without and with azide ions, respectively, showing the residual densities (green mesh) at +3.0 σ. Significant residual density is not detectable in C.
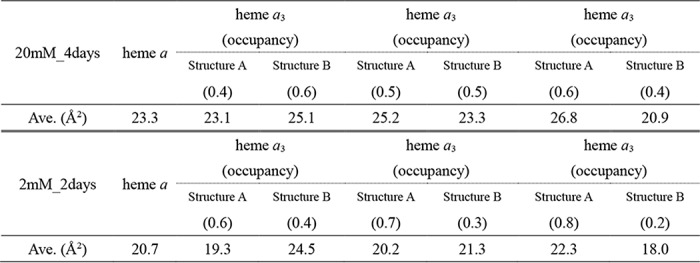
Whereas the refinement resulted in abnormal B-factors of heme a3, of which the averaged value is significantly higher than that of heme a (28.1 Å2 versus 23.3 Å2), in the Fo − Fc map, significant residual densities are detectable near ring C (in the PDB naming scheme) as shown in Fig. 3A. These results strongly suggest the existence of multiple structures. The two structures shown in Fig. 3B in equal occupancy with each other essentially eliminate the residual densities detectable near ring C in Fig. 3A and provide approximately equal B-factors (25.2 and 23.3 Å2, respectively) closely similar to that of heme a (23.3 Å2). The B-factors for structures A and B, calculated assuming the occupancy ratio of 0.4/0.6, are 23.1 and 25.1 Å2, respectively, which are also closely similar to that of heme a, as given in Table 1. The monotonous increase and decrease in the B-factors of structures A and B, respectively, with the occupancy increase in structure A (Table 1) indicate that a B-factor, 24.2 Å2, for both structures is attained at the occupancy ratio of 0.45:0.55. Thus, the reliability of the assessment of the occupancy ratio based on the average B-factors is higher than ±0.1. The two structures indicate a translational shift of the heme plane fixing the two propionate carboxyl groups and the terminal end of the hydroxy farnesyl ethyl group (Fig. 3B). The converged structure, assuming a single structure shown in Fig. 3A, is located between the two structures. The location of one of the structures (Fig. 3A, shown in magenta) is close to that of the resting oxidized form. In this paper, the azide-induced structure similar to that of the resting oxidized state is designated as structure A, and the other structure is designated as structure B throughout. The heme plane shift between the two structures (from magenta to light blue) is greater than the structural changes that occur upon complete reduction of resting oxidized CcO as given in Fig. 3B, although the change is in the same direction.
Figure 3.
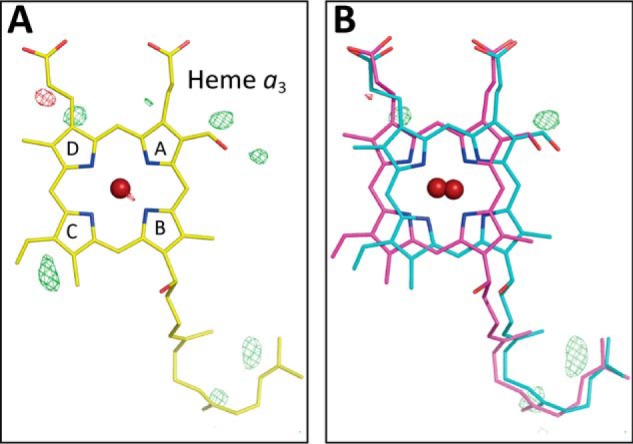
X-ray structure of heme a3 in the presence of 20 mm azide. A,Fo − Fc map determined assuming a single structure of heme a3. The residual densities are indicated with green mesh at +3.0 σ, and the converged structure is shown. B, Fo − Fc maps assuming the structures A and B of heme a3 (indicated by magenta and light blue sticks, respectively) in the 1:1 occupancy ratio. No significant residual density is detectable at the +3.0 σ level near the ring C.
Table 1.
Effects of structure occupancy on the B-factor of heme a3
Azide concentrations and exposure periods are indicated with the term “XX mM_Ydays.” For structure A, the nonshifted conformation is closely similar to the conformation of the resting oxidized state; structure B is the azide-induced shifted conformation. The averaged B-factors were calculated using the atoms of the composing porphyrin ring of heme a or heme a3.
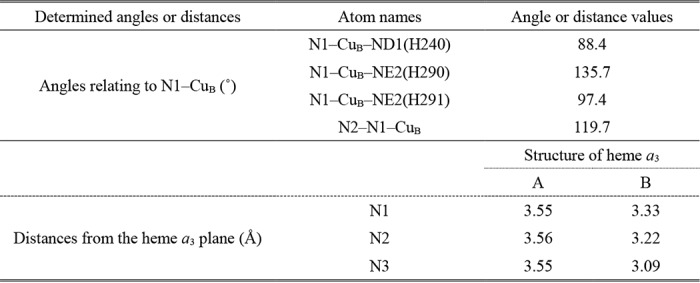
As shown in Fig. 4, A and B, azide 1 interacts with both pyrrole rings D of structure A and structure B with distances ∼3.5 and ∼3.3 Å, respectively. The geometries of azide 1 are summarized in Table 2. The strongest interaction of the terminally-bound azides to CcO is the coordination bond between Fea3 and one of the terminal nitrogen atoms of azide 2. The N–Fe–N angles of structure A and structure B are 80.9 and 105.5°, respectively. Furthermore, the terminal and central nitrogen atoms have close contacts with nitrogen atoms of pyrrole B of structure A with distances of 2.68 and 2.54 Å, respectively (Fig. 4C), and with those of pyrrole rings of B and D of structure B, with distances of 2.86 and 2.50 Å, respectively (Fig. 4D). The heme shift between structures A and B is accompanied by distortion of the Fea3–N coordination, which induces the multiple conformations in helix X as described below.
Figure 4.

Two modes of terminal azide bindings to Fea3 at 20 mm azide. A and B, atomic model of terminally-bound azide ions. Azide 1 and azide 2 in dark blue indicate azide ions bound to CuB and heme a3, respectively. Heme a3 planes giving structures A and B are drawn in magenta and light blue, respectively. C and D, geometry of bindings of azide 2 giving structures A and B, respectively, in the same color-code as in A and B.
Table 2.
Geometries of an azides bound to CuB, in the crystal equilibrated with 20 mm azide solution
Letters A and B indicate the structures A and B of heme a3, respectively. Other atomic names are given in Fig. 3. Heme a3 plane was calculated on the basis of FE, NA, NB, NC, and ND atoms in each heme a3 structure by geomcalc program (38).
Azide-induced conformational changes in helix X at 20 mm azide
The structural refinement for helix X with the conformation of the resting oxidized CcO, in which the water channel is closed, provides higher average B-factors of amino acid residues from 380 to 384 of the helix X (∼35 Å2) than those of the other residues of the helix (∼25 Å2). The Fo − Fc map provides significant positive and negative residual densities (Fig. 5A), suggesting a multiple conformation in the helix X closely similar to those observable when the open/closed conformation change in the water channel is induced upon ligand binding to the fully reduced ligand-free state or oxidation state changes of the fully reduced ligand-free state (5). Structural refinements were performed for two conformers of helix X with different occupancy ratios, each at 0.1 step. The multiple conformation with the two structures in equal occupancy (0.5:0.5) provides two conformers with closely similar values of B-factor as shown in Table 3 and the lowest residual electron densities among those obtained assuming various occupancy ratios (Fig. 5B).
Figure 5.

Fo − Fc maps of a segment (from Val380 to Gly384) of helix X. The residual electron densities are indicated with green mesh (+3.0 σ) and red mesh (−3.0 σ). A, determined assuming the conformation of the resting oxidized state (yellow). B, determined assuming the two conformations, one closely similar to that of the resting oxidized state (magenta) (conformer A) and the other obtained with 20 mm azide (light blue) (conformer B), in 1:1 occupancy ratio. C and D, water cavities located near the P-side end of the water channel in conformers A and B, detectable at 20 mm azide, respectively. The brown- and gray-dotted surfaces are determined by the program VOIDOO (39) using probes with a radius of 1.2 and 0.8 Å, respectively. E, cavities of the open conformation of the fully reduced ligand-free state, detectable in areas corresponding to those of C and D. The same color code as C and D was used to indicate the water-accessible surfaces. The dotted arrows denote possible locations of water channels. The locations of side chains of Ser382 and Met383 are indicated by red circles and blue ovals, respectively. The hydrogen bond between Met383 and the OH group of the hydroxy farnesyl ethyl group of heme a is indicated by a dotted line.
Table 3.
Effects of conformer occupancy on the B-factor of flexible amino acid residues in helix X at 20 mm azide
Conformer A is closely similar to the resting oxidized state. Conformer B is an azide-induced open conformation.
| Conformer | Occupancy | Val-380 | Leu-381 | Ser-382 | Met-383 | Gly-384 | Average |
|---|---|---|---|---|---|---|---|
| A | 0.4 | 21.0 | 19.5 | 22.8 | 23.8 | 25.7 | 22.6 |
| B | 0.6 | 30.7 | 30.0 | 32.0 | 29.6 | 28.0 | 30.1 |
| A | 0.5 | 24.7 | 23.3 | 27.6 | 28.2 | 27.1 | 26.2 |
| B | 0.5 | 27.3 | 25.5 | 28.7 | 25.8 | 26.7 | 26.8 |
| A | 0.6 | 27.9 | 26.6 | 31.9 | 32.3 | 28.3 | 29.4 |
| B | 0.4 | 23.4 | 20.7 | 24.5 | 21.6 | 24.8 | 23.0 |
One of the conformers (conformer A) is closely similar to that of the closed conformation, detectable in the resting oxidized state, in both of the main and side chains (Fig. 5C). The main-chain conformation of the other conformer (conformer B) (Fig. 5D) is closely similar to that of the open conformer of the ligand-free fully reduced state (Fig. 5E), whereas the side chain of Met383 (indicated with a light blue oval in Fig. 5D) folds in a bent conformation, definitely different from that of the open conformation of the ligand-free fully reduced state. This bent conformation is somewhat unusual and gives significantly higher B-factors compared with those of nearby residues. The unusual conformation of the Met383 side chain appears to be stabilized by a hydrogen bond formation with the hydroxyl group of the hydroxy farnesyl ethyl group of heme a, as marked by a dotted line in Fig. 5D. As shown in Fig. 5D, the side chain of Met383 eliminates one of the large cavities shown with brown cages detectable in the open conformation of the ligand-free fully reduced state (Fig. 5E). The position of the side chain of Ser382 marked by a red circle (Fig. 5D) is essentially identical to its position in the open conformation (Fig. 5E).
As shown in Fig. 5E, in the open conformation detectable in the ligand-free fully reduced state, the five water cavities located in the water channel near the junction point with the hydrogen-bond network (or the formyl group of heme a) contribute significantly to the accessibility of the water molecules from the N-side through the water channel, the location of which is marked by a dotted arrow in Fig. 5E. In conformer A, the side chain of Ser382, indicated with a red circle in Fig. 5C, eliminates two of the five cavities detectable in the ligand-free fully reduced state to essentially block the water channel as shown in Fig. 5C. In this conformation, an alternative water channel along the shortest distance between the two cavities near heme a and the one located closer to the N-side could be used as indicated with a dotted arrow in Fig. 5C. However, water exchange between these cavities is highly unlikely to occur at least within the physiological time scale (about 1 ms for the single turnover of the CcO reaction (1)), because even the water-accessible surfaces calculated with a probe radius of 0.8 Å (indicated by the gray-dotted surfaces) have no direct contact between these cavities. Thus, this conformation is designated as the “closed conformation.” This conformer is identical to the conformer observable in resting oxidized CcO within experimental accuracy.
The water-exchange efficiency difference between conformers A and B is obvious when comparing the water access surfaces calculated using a probe radius of 0.8 Å, as indicated by the gray-dotted surfaces. In conformer B, the water-accessibility surfaces of the two water cavities near heme a are more closely located with each other (Fig. 5D) compared with the corresponding surfaces in conformer A (Fig. 5C). Thus, the structure suggests that conformer B is unlikely to close the water channel as tightly as conformer A does. The conformation is designated as the azide-induced open conformation. Although Met383 eliminates the water cavity detectable in the open conformation (observable in the fully reduced ligand-free state (Fig. 5E)), the heme a3 migration induced by terminally-bound azide ions creates an alternative pathway for water exchange (Fig. 5D). Thus, 20 mm azide induces the closed/open conformational transition without changing the oxidation state of the O2 reduction site.
X-ray structure of the azide-bound oxidized CcO at 2 mm azide
Effects of decreasing the azide concentration on the X-ray structure of the azide derivatives were analyzed for investigation of the mechanism of the two-step binding of azide to CcO revealed by spectroscopic analyses as described above (17). As shown in Fig. 6A, the Fo − Fc electron density at 2 mm azide at the σ level identical to that of Fig. 2B (the Fo − Fc electron density at 20 mm azide) indicates clear electron density with higher density at the central part than in the case of 20 mm azide. Structural refinement for a single bridging azide ion coexisting with the two partially occupied azide ions, each of which is terminally-bound as at 20 mm azide, in the occupancies of 0.7 (single) and 0.3 (double) was converged well and resulted in the lowest residual density in the Fo − Fc map as shown in Fig. 6B. Effects of the occupancy ratio on the average B-factor of the bound azides confirm that the above occupancy ratio provides the lowest and similar values of B-factor for the two structures as given in Table 4. The geometries of the azide bridging between Fea3 and CuB are given in Fig. 6C, which are consistent with the previously reported structure of azide derivatives prepared at the azide concentration significantly lower than 2 mm (19). Binding of three azide ions simultaneously to the O2 reduction site is not possible due to steric constraint. The two metal sites in the two azide-bound states do not have sufficient space to accept an additional single azide ion bridging between the two metals.
Figure 6.
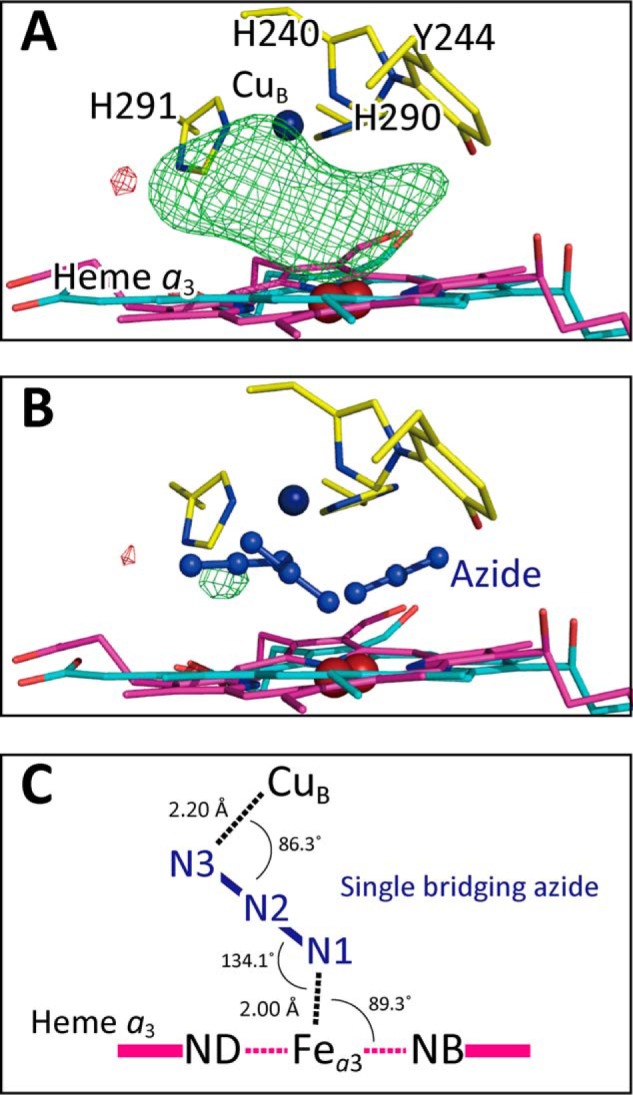
Fo − Fc maps of the O2 reduction site of bovine heart resting oxidized CcO obtained from crystals exposed to 2 mm azide. A, Fo − Fc map without azide ion, showing the residual densities (green mesh) at +3.0 σ identical to the σ level of Fig. 2B, B, Fo − Fc maps with two terminally-bound azide ions at Fea3 and CuB, each at 30% occupancy, and a single bridged azide ion, giving essentially no residual density, C, schematic representation of the geometry of the single bridging azide.
Table 4.
Effects of azide occupancy on the averaged B-factor of nitrogen atoms of azide ion at 2 mm
| Azide name | Occupancy | Averaged B-factor |
|---|---|---|
| Å2 | ||
| Single bridging azide | 0.9 | 26.8 |
| Terminal binding azide | 0.1 | 7.5 |
| Single bridging azide | 0.8 | 26.1 |
| Terminal binding azide | 0.2 | 15.6 |
| Single bridging azide | 0.7 | 24.3 |
| Terminal binding azide | 0.3 | 22.1 |
| Single bridging azide | 0.6 | 21.8 |
| Terminal binding azide | 0.4 | 25.2 |
| Single bridging azide | 0.5 | 19.7 |
| Terminal binding azide | 0.5 | 29.6 |
The structure of heme a3 at 2 mm azide determined assuming a single structure also shows an average B-factor higher than that of heme a and residual densities near ring C, suggesting a multiple structure. The refinement assuming two conformations with the same procedures as used for the analysis of the 20 mm azide structure provides two structures at 0.7/0.3 occupancy ratio. The dominant (0.7) and minor (0.3) structures are closely similar to those of structures A and B, observed at 20 mm azide, respectively (i.e. the nonshifted structure essentially identical to that of the resting oxidized CcO and the azide-induced shifted structure, respectively). Similarly, the helix X structure near Ser382 at 2 mm azide indicates coexistence of conformers A and B in a ratio of 0.8/0.2 as given in Table 5. If the state with two terminally-bound azide ions provides 1:1 multiple structures (structures A and B in heme a3 and conformers A and B in helix X as at 20 mm azide) and if the single azide bridging species provides essentially identical structures to those of the structure A in heme a3 and conformer A in helix X, the ratio for the single azide-bound state/the two azide-bound state of 7:3 provides the ratio of 8.5:1.5 for structures A to B in heme a3 and conformers A to B in helix X. The experimental results given in Table 5 for helix X and heme a3 structure (8:2 and 7:3, respectively) are consistent with the expected value within experimental error. Thus, the results suggest that the two azide ions terminally bound to Fea3 and CuB in the presence of 2 mm azide provide the two structures in both heme a3 and helix X as the two terminally-bound azide ions do at 20 mm.
Table 5.
Effects of azide on X-ray structures of resting oxidized CcO
The values indicate the occupancy of each multiple structure.
| N3− concentration | Exposure | O2 reduction site |
Heme a3 |
Helix X |
PDB ID | |||
|---|---|---|---|---|---|---|---|---|
| 1 N3− | 2 N3− | Structure A (nonshifted) | Structure B (shifted) | Conformer A (closed) | Conformer B (open) | |||
| 20 mm | 4 days | 0.0 | 1.0 | 0.5 | 0.5 | 0.5 | 0.5 | 5Z84 |
| 3 days | 0.0 | 1.0 | 0.5 | 0.5 | 0.5 | 0.5 | 5Z86 | |
| 2 days | 0.4 | 0.6 | 0.4 | 0.6 | 0.5 | 0.5 | 5ZCP | |
| 2 daysa | 0.0 | 1.0 | 0.5 | 0.5 | 0.5 | 0.5 | 5Z85 | |
| 2 mm | 2 daysa | 0.7 | 0.3 | 0.7 | 0.3 | 0.8 | 0.2 | 5ZCO |
a Different batches of crystals were used for these experiments.
Hydrogen-bond structures between Tyr244 and the OH group of the hydroxy farnesyl ethyl group of heme a3
In structure A of heme a3 at 2 mm azide (Fig. 7A), a hydrogen bond between Tyr244–OH and the OH group of the hydroxy farnesyl ethyl group of heme a3 is detectable, and a water molecule, W1, is hydrogen-bonded to Tyr244–OH. The structure is closely similar to that in the resting oxidized state in the absence of azide. The hydrogen bond is cleaved by the azide-induced heme a3 plane shift to introduce a water molecule, W2, in structure B, as shown in Fig. 7B. The occupancies of W1 and W2 at 2 mm azide, estimated with the procedure described under “Experimental procedures,” are 30 and 25%, respectively. The former is nearly identical to that of W1 detectable in the resting oxidized CcO in the absence of azide (about 30%). In contrast, the interactions of heme a3 with Tyr244 in structures A and B at 20 mm azide are essentially identical to those at 2 mm azide as given in Fig. 7. However, the occupancy of W2 is 65%, whereas W1 is undetectable at the 3 σ level. Therefore, the microenvironment of Tyr244 OH group at 2 mm azide, which receives W1 at 30% occupancy (Fig. 7A), is different from that in structure A at 20 mm azide, in which no significant W1 is detectable.
Figure 7.
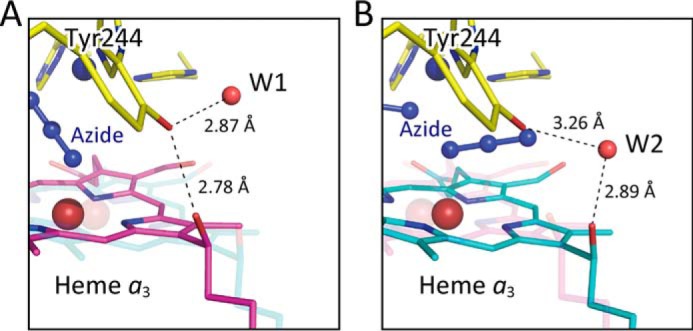
Hydrogen bond system between the hydroxy farnesyl ethyl group and Tyr244 covalently linked to one of the imidazoles coordinated to CuB at 2 mm azide. A, one of the structures detectable at 2 mm azide, closely similar to that detectable in the resting oxidized state showing the direct hydrogen bonding between the two OH groups of Tyr244 and hydroxy farnesyl ethyl group (structure A). The Tyr244–OH group has a fixed water molecule (W1) hydrogen-bonded. B, (structure B) another structure detectable at 2 mm azide showing a large translational shift of heme a3 plane from that of structure A. The direct hydrogen bond between the two OH groups is replaced by a bridging water molecule (W2) hydrogen-bonded to the two OH groups.
The azide-induced hydrogen bond break as given in Fig. 7 is not detectable with the heme a3 shift induced by reduction of heme a3 of bovine CcO (21). However, it has been reported that reduction of fully oxidized bacterial CcO induces loss of a hydrogen bond, which is very similar to the hydrogen bond loss observed upon addition of a high concentration of azide to the resting oxidized bovine CcO (22).
Saturation of the terminal azide-binding sites in the resting oxidized CcO crystals
The two azide terminally-bound CcO crystals were prepared by exposing resting oxidized CcO crystals to 20 mm azide for 3 days at 4 °C as described under “Experimental procedures.” Complete saturation of the azide-binding metal sites is not attained by exposure of CcO to 20 mm azide for 2 days, whereas elongation of the exposure time up to 4 days does not significantly change the X-ray crystal structures (Table 5). Thus, in contrast to the binding kinetics for the second (low affinity) azide-binding site in solution (faster than the time scale of minutes) (17), 3 days of exposure at 4 °C is necessary for saturation of the two terminal azide-binding sites. The discrepancy is likely to be due to restriction of flexibility in the O2 transfer pathway (23) induced by the crystal packing. As typically shown in the results obtained by a 2-day exposure to 20 mm azide (Table 5), the process of azide binding to the metal sites does not tightly synchronize with the progress of the structural changes in the heme a3 and helix X. The azide saturation period was found to be somewhat dependent on the CcO preparation batch. Some CcO batches provided the crystals, which require 2 days for saturation of the two terminal azide-binding sites (Table 5).
Even at the highest azide concentration employed (20 mm) in this work, no azide ion was detectable on the transmembrane surface of the CcO molecule where azide binding has been proposed based on the X-ray structure of the azide-bound bovine heart CcO at 2.9 Å resolution (19). In the present structure, a fairly stable fatty acid tail of a triacylglycerol molecule occupies the electron density previously assigned to an azide ion.
Discussion
Reactions of azide with the resting oxidized CcO
The present results indicate that there is unexpectedly high flexibility in the O2 reduction site that allows the site to accept two triatomic ligands. The reported two terminal azide IR bands (12) are likely due to the two terminally-bound azide ions in the present X-ray structure.
The azide ion bridging between the two metals in the O2 reduction site detectable in the X-ray structure at 2 mm azide does not provide any detectable band in the azide stretch band region (2100–2000 cm−1). The results suggest that bridging between the two metals decreases the polarization of the azide molecule significantly, which weakens the IR band intensity. The B-factors of nitrogen atoms of the azide ion bridging between the two metals in the present X-ray structures are roughly identical with those of the terminally-bound azide ions. Thus, the bridging between the two metals does not increase the diversity of orientation of the bound azide. (The diversity increase in orientation of metal-bound ligand broadens the IR band to weaken the maximal band intensity.) Thus, we propose that the main reason for weakening of the azide band is the polarity decrease upon bridging between the two metals.
The terminal binding of two azide ions simultaneously to Fea3 and CuB was observed also at 2 mm azide as described above, and the occupancies of the two terminally-bound azide ions are identical to each other irrespective of the azide concentration down to concentrations that are insufficient to saturate the two terminal binding sites. Thus, the terminally-bound azide at Fea33+ is as stable as the terminally-bound azide at CuB2+. Alternatively, it is possible that any single azide terminally bound at either one of the two metals is much less stable than the azide bridging between the two metals.
Earlier extensive absorption spectral analysis for azide binding to the resting oxidized CcO (17) reveals a two-step absorbance change with an increase in the azide concentration, in which each step is induced by a single azide ion. The high-affinity binding eliminates a resonance Raman band assignable to the O–O stretch band of the peroxide bound to the resting oxidized CcO, leaving a band assignable to the ν16 porphyrin mode of the high-spin heme a3. This porphyrin mode band is not influenced by increasing the azide concentration up to 100 mm sufficient for saturating the low-affinity (second) azide-binding site. Based on these spectral results, it has been proposed that the high-affinity binding terminally to Fea33+ eliminates the peroxide bridging between Fea33+ and CuB2+ in the resting oxidized CcO, whereas both Fea33+ and CuB2+ sites are fully occupied by terminally-bound azide ions at high-azide concentrations (17).
The present X-ray structural results show that the high-affinity azide binding provides a bridging azide between Fea33+ and CuB2+, in contrast to the above proposed structure. In general, an azide ion terminally bound to a ferric heme stabilizes the low-spin state, because the azide ion is a strong ligand to ferric heme. However, the bridging azide could promote the high-spin state in heme a3. The present X-ray structure is consistent with the resonance Raman results showing that the high-affinity azide binding provides the high-spin heme a3. Furthermore, the resonance Raman and X-ray structural results at a high-azide concentration suggest that the azide at CuB2+ in the two azide-bound CcOs stabilizes the high-spin state in the terminally-bound azide derivative of the ferric heme a3.
The present results giving a binding mode ratio of 7 (bridging):3 (terminal) at 2 mm azide indicate that the Kd value of the second step of the azide binding (formation of the two-azide–bound form from the one-azide–bound form) is 4.6 mm. This Kd value obtained at pH 5.7 is consistent with the reported Kd value of 42 mm (17) for the low-affinity site determined by absorption spectral analyses at pH 7.4 in solution, because the reported pH dependence of Kd indicates that the Kd value determined at pH 5.7 is about 1 order of magnitude less than the Kd value determined at neutral pH (10). This consistency in the azide-binding affinity between solution and crystalline states suggests that the structure of the O2 reduction site in the physiological conditions (i.e. in solution state) is essentially preserved in the crystalline state, although the O2 transfer pathway to the O2 reduction site is significantly perturbed to decrease the azide-binding rate as described above.
Structural coupling between the O2 reduction site and the H-pathway
The present X-ray structural analyses reveal that the second step of the azide binding to the resting oxidized CcO is accompanied by the azide-binding mode transition from the bridging to the terminal, which is undetectable by the absorption and Raman spectroscopic analyses. A simultaneous two azide binding directly to the resting oxidized state is unlikely because the spectral changes in absorbance for the azide binding to the low-affinity site (the second step) is consistent with an equilibrium, CcO–N3− + N3− = CcO–(N3−)2 (17). Thus, the bridging/terminal-binding mode transition induces the closed/open transition in the water channel without changing the oxidation and spin states of the O2 reduction site, mediated by the translational migration of heme a3 plane.
The conformation of the water channel is perturbed also by the coordination state change in Fea3 in the fully reduced state of the O2 reduction site (i.e. release of CO from CcO-CO) to give an open conformation in the water channel, the structure of which is significantly different from the azide-induced open conformation (1, 5). The structural coupling is also mediated by the translational movement of heme a3 plane (4). These structural couplings between the O2 reduction site and the H-pathway support the critical involvement of the H-pathway as the proton pump system, driven by O2 reduction in the O2 reduction site.
Possible inhibition mechanism by the azide binding to the low-affinity site
It has been proposed that protons for pumping are collected into a Mg2+-containing water cluster through the water channel of the H-pathway in the open state, which is attained when the O2 reduction site is in the fully reduced state (4). The water channel is closed upon O2 binding to CuB after the four protons were collected into the Mg2+-containing water cluster (21). The collected protons are transferred to the hydrogen-bond network of the H-pathway to be pumped to the P-side electrostatically driven by net positive charges created on heme a upon electron transfer to the O2 reduction site (4). The water channel closure determines the direction of the proton pump by blockage of the proton back-leak to the N-side. Thus, the opening of the water channel in the fully oxidized state upon azide binding to the low-affinity site would critically decrease the efficiency in the proton pump.
The channel opening is also likely to lower the level of protons to be pumped into the hydrogen-bond network under turnover conditions due to spontaneous proton leakage to the N-side. The proton level decrease is likely to decrease the electron affinity of heme a under the electrostatic influence from the hydrogen-bond network of the H-pathway. Then, the electron flow from CuA via heme a to the azide-bound Fea33+ would be decreased. It is well-known that azide does not bind to Fea32+ (12). Therefore, the suppression of the electron flow would increase the level of Fea33+–N3− under turnover conditions to enhance the azide inhibition.
The electron affinity decrease in heme a is consistent with the reported redox potential decrease induced by azide at high concentration sufficient for saturating the low-affinity site (24, 25). Similarly, CO binding to Fea32+, accompanied by the channel closure, increases the potential (25). These results support the existence of the electrostatic influence of heme a to the hydrogen-bond network of the H-pathway. However, structural changes induced by the azide binding other than the channel opening is also likely to influence the redox potential of heme a. In fact, cyanide binding to Fea33+ decreases the redox potential without opening the channel (25), although the resolution (2.0 Å) of the crystal structure of cyanide-bound oxidized CcO (PDB code 3X2Q) is not sufficiently high for concluding that the water channel is completely closed. Moreover, the present X-ray structural analyses for the azide-bound CcO at 20 mm indicate that no significant structural change other than the channel opening is likely to influence the redox potential of heme a.
Alternative proton-pumping mechanisms
The present results support the critical involvement of the H-pathway in the proton pump function of bovine CcO. However, it has been proposed based on the mutational and simulation analyses of bacterial CcOs as mentioned above that bacterial CcOs pump protons without using the H-pathway. In these mechanisms, both pumping and water-forming protons are transferred through a single proton-conducting pathway (either the D-pathway or the K-pathway) (1–3). In contrast, in the proton pump mechanism of bovine CcO, the proton pump pathway (H-pathway) is completely separated from the pathways for the water-forming protons. The difference should not be ignored, because the separation is prerequisite for the efficient energy transduction in CcO (26).
Recently, it has been proposed that the H-pathway does not function as the proton-pumping system by a simulation analysis using X-ray structural data of bovine CcO determined at 1.8 Å resolution (5), because access of the water molecules from the N-side to the water channel of the H-pathway is essentially impossible (27). His413 located near the entrance of the water channel, in the protonated state, stimulates penetration of water molecules from the N-side and forms a water wire through which protons are transferred to the hydrogen-bond network of the H-pathway. The residue in the deprotonated (or neutral) state strongly suppresses the water accumulation and disconnects the water wire. The latter simulation result, showing 5–6 water molecules in the channel region, is consistent with the X-ray structure at 1.8 Å resolution (5). However, 12 water molecules are resolved in the channel region in the X-ray structure at 1.6 Å resolution (4). These water molecules include all those proposed by stimulation assuming that His413 is protonated (27). These results indicate that the X-ray structure at 1.8 Å resolution (5) is not sufficiently high for identification of all the water molecules in the water channel detectable at 1.6 Å resolution (4). Reevaluations are desirable for the simulation analysis (27).
Experimental procedures
Preparation of azide-bound resting oxidized CcO crystals
The resting oxidized bovine heart CcO crystals were prepared as described before (15). Crystals were frozen at 90 K in 40 mm sodium phosphate buffer, pH 5.7, 0.2% decyl maltoside, 8% PEG 4000, 40% ethylene glycol, and 2–20 mm sodium azide. The final medium composition was attained by a 48 stepwise manual exchange from the initial medium composed of 40 mm sodium phosphate buffer, pH 6.5, 0.2% decyl maltoside, 1% PEG 4000, and 2% ethylene glycol in which the crystals are stable at 4 °C. The stepwise exchange of the medium was performed typically within 3 days (i.e. 20, 20, and 8 steps in the 1st, 2nd, and 3rd days, respectively). The azide concentration was gradually increased by adding increasing concentrations of azide into the media for the stepwise exchange up to the final concentration in the 1st day. The final azide concentration was retained over the following 2 days. Thus, the crystals were exposed for 2 days with azide at the final concentration. For attainment of a longer exposure time, the stepwise exchange was performed over a longer duration, but the azide concentration was increased to the final concentration within the 1st day. For example, a longer exposure was performed in the stepwise exchange in 5 days (12, 9, 9, 10, and 8 steps on each of the five consecutive days, respectively) concomitantly with a stepwise increase in azide concentration to the final concentration within the 1st day. In this paper, the azide exposure period (days) after attaining the final concentration is designated as the exposure time. Immediately after finishing the 48-step medium exchange simultaneously with azide exposure, the crystals were frozen at 90 K.
X-ray diffraction experiments
Crystals for the X-ray diffraction experiments were prepared under four different conditions with respect to azide concentration and soaking period. All X-ray experiments were carried out at beamline BL44XU/SPring-8 equipped with an MAR300HE CCD detector. Crystals with sizes ∼700 × 700 × 200 μm were used in the diffraction experiments. In a representative series of diffraction measurements, the thin edge of a crystal was aligned parallel to the X-ray beam at a rotation angle of 0.0°. The wavelength was 0.9 Å, the photon number at the sample position was 4.0 × 1011 photons/s, and the crystal was shot with X-rays in a helium gas stream at 50 K and translated by 10 μm after each shot to reduce radiation damage. Other experimental conditions for low-resolution data collection were X-ray beam cross-section of 20 μm (vertical) × 20 μm (horizontal) at the crystal, a camera distance of 431 mm, exposure period of 1.0 s, and an oscillation angle of 1.0°. Conditions employed for high-resolution data were X-ray beam cross-section of 50 μm (vertical) × 30 μm (horizontal) at the crystal, a camera distance of 230 mm, exposure period of 3.0 s, and an oscillation angle of 0.5°. Crystalline absorption spectra were measured before and after the X-ray diffraction experiment with the spectrophotometer system as described before (28). The full occupancy of azide ions observed in the present X-ray structural results suggests that no significant structural damage in the azide-binding site is induced by X-ray irradiation, although the X-ray exposure in the present X-ray diffraction experimental conditions provides slight spectral changes assignable to heme a reduction. A total of 5–11 crystals were used for acquisition of the full data sets at 1.65 to 1.85 Å. Data processing and scaling were carried out using HKL2000 and SCALEPACK (29). A total of 678–851 images were successfully processed and scaled. The structure factor amplitude (|F|) was calculated using the CCP4 program TRUNCATE (30, 31). Other statistics obtained from the intensity data are provided in Table S1.
X-ray structural determinations and refinements
Four types of crystals, exposed to 2 mm azide for 2 days (2mm_2days), to 20 mm azide for 2 days (20mm_2days), to 20 mm azide for 3 days (20mm_3days), and to 20 mm azide for 4 days (20mm_4days), prepared as described above (Table 5), have essentially identical cell dimensions within less than 0.2% deviation, indicating that all crystals are isomorphous with each other at the present resolution. The same procedures applied for the previous structural analyses of CcO crystals crystallized at pH 5.7 (4) were followed in the structural determinations. Initial phase angles of structure factors up to 4.0 Å resolution were calculated by the MR method (32) using the fully oxidized structure, previously determined at 1.5 Å resolution (PDB code 5B1A) (4). The phases were extended to each highest resolution by density modification (33) coupled with noncrystallographic symmetry (NCS) averaging (34, 35) using the CCP4 program DM (36). The resultant phase angles (αMR/DM) were used to calculate the electron-density map (MR/DM map) with Fourier coefficients |F| exp(iαMR/DM), where |F| is the observed structure factor amplitude.
Bulk solvent correction and anisotropic scaling of the observed and calculated structure factor amplitudes and TLS parameters were incorporated into the refinement calculation using REFMAC5 (37). The anisotropic B-factors for the iron, copper, and zinc atoms were imposed on the calculated structure factors. Each monomer of two NCS-related monomers was assigned to a single TLS group in the refinement. The crystal structure was refined under NCS restraints between two monomers. Ligands in the O2 reduction center, water molecules, ethylene glycol molecules, lipids, and detergents were located in an Fo − Fc map composed with the phases calculated with atomic parameters of protein atoms and cofactors.
Each Fo − Fc map was found to have significant electron density corresponding to azide ions. In the 20mm_4days map, two azide ions each terminally bound to Fea3 and CuB, respectively, were accommodated each with full occupancy of 1.0. In the 2mm_2days map, an azide group bridged between Fea3 and CuB. An additional Fo − Fc map was calculated by including the bridging azide group with occupancy of 1.0 to detect significant residual densities at the same sites as those of 20mm_4days. Because the bridging azide ions could not coexist with the terminally-bound azide ions, both types of azide ions were included in the refinement with their partial occupancies. The occupancies of azide ions were determined by inspecting the residual densities of the Fo − Fc map in each iteration of the refinement at a different occupancy of each 0.1 step. The structures of azide ions in the O2 reduction center of the other crystals were conducted by following these two cases.
When heme a3 structures were refined from the oxidized form determined at 1.5 Å resolution (PDB code 5B1A), they were converged to different locations from each other, and each of the average B-factor value of heme a3 was significantly higher than those of heme a of the same crystal. Further refinements for heme a3 were conducted with multiple structures. One structure was the same heme a3 structure as the resting oxidized form, and the other was a structure shifted further than that of the reduced form (PDB code 5B1B). Convergence of each structural refinement with respect to heme a3 was confirmed by inspecting B-factor values of heme a3 and the residual electron density at heme a3.
Significant residual density was found to exist at residues 380–383 of helix X in each Fo − Fc map, and B-factors of atoms of residues 380–383 were found to be higher than those of other atoms of helix X. The structures of residues 380–383 were refined by imposing multiple conformations. One conformation was the same structure as the oxidized form, and the other conformation was that of the reduced form. Convergency of each refinement with respect to helix X was confirmed as in the case of heme a3.
Although the Fo − Fc electron density of the water molecule, W1, hydrogen-bonded to Tyr244–OH was detected in 2mm_2days map, it was not detected in 20mm_4days map at the 3 σ level. The occupancies of the water molecule (W2) hydrogen-bonded to both Tyr244–OH and the OH group of the hydroxy farnesyl ethyl group of heme a3 and W1 were determined by inspecting each B-factor. To minimize the difference of the B-factors between W1 and Tyr244–OH, each refinement was performed with changing the occupancy of W1 in 5% step. In the case for W2, the differences between the B-factor values of W2 and the average of the B-factor values of Tyr244–OH and the hydroxy farnesyl ethyl OH group of heme a3 were examined for optimizing the occupancy value. Statistics of the refinements for these azide derivatives of CcO are summarized in Table S2.
Author contributions
A. S., E. Y., K. M., T. T., and S. Y. conceptualization; A. S., K. H., H. T., N. Y., K. S.-I., E. Y., K. M., T. T., and S. Y. resources; A. S., K. H., H. T., N. Y., K. S.-I., E. Y., K. M., T. T., and S. Y. data curation; A. S., N. Y., E. Y., and T. T. software; A. S., K. H., H. T., N. Y., E. Y., T. T., and S. Y. formal analysis; A. S., K. S.-I., E. Y., K. M., T. T., and S. Y. supervision; A. S., T. T., and S. Y. funding acquisition; A. S., K. H., H. T., N. Y., E. Y., K. M., and T. T. validation; A. S., K. H., H. T., N. Y., K. S.-I., E. Y., K. M., T. T., and S. Y. investigation; A. S., K. H., H. T., N. Y., E. Y., K. M., and T. T. visualization; A. S., H. T., N. Y., K. S.-I., E. Y., T. T., and S. Y. methodology; A. S., T. T., and S. Y. writing-original draft; T. T. and S. Y. project administration.
Supplementary Material
Acknowledgments
Diffraction data were collected in the SPring-8 Facility at BL44XU under proposals 2013A6500, 2013B6500, 2014A6500, 2014B6500, 2015A6500, 2015B6500, 2016A6500, and 2016B6500 from Spring-8. MAR300HE was financially supported by Academia Sinica and National Synchrotron Radiation Research Center. We thank Rika Inaoka and Ichiro Nakadai for preliminary experiments for X-ray structural analyses of azide-bound CcO.
This work was supported by Grants-in-aid for the Global Center of Excellence Program (to S. Y.), for Scientific Research (A) 2247012 (to S. Y.), Photon and Quantum Basic Research Coordinated Development Program (to T. T., S. Y., and K. S.-I.), each provided by the Japanese Ministry of Education, Culture, Sports, Science and Technology, JSPS KAKENHI Grants 22370060 (to K. M.), 26291033 (to S. Y.), 15K18493 (to A. S.), and 12101577 (to T. T.) by JST, CREST, and by Targeted Proteins Research Program. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S2.
- CcO
- cytochrome c oxidase
- heme a and heme a3
- low and high-spin heme A molecules of CcO, respectively
- Fea and Fea3
- iron ions of heme a and heme a3, respectively
- CuA and CuB
- low and high potential copper sites of CcO
- H-pathway
- the proton-pumping pathway of animal CcO
- PDB
- Protein Data Bank
- MR
- molecular replacement
- DM
- density modification
- NCS
- noncrystallographic symmetry.
References
- 1. Yoshikawa S., and Shimada A. (2015) Reaction mechanism of cytochrome c oxidase. Chem. Rev. 115, 1936–1989 10.1021/cr500266a [DOI] [PubMed] [Google Scholar]
- 2. Yoshikawa S., Muramoto K., and Shinzawa-Itoh K. (2011) Proton-pumping mechanism of cytochrome c oxidase. Annu. Rev. Biophys. 40, 205–223 10.1146/annurev-biophys-042910-155341 [DOI] [PubMed] [Google Scholar]
- 3. Rich P. R., and Maréchal A. (2013) Functions of the hydrophilic channels in protonmotive cytochrome c oxidase. J. R. Soc. Interface 10, 20130183 10.1098/rsif.2013.0183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yano N., Muramoto K., Shimada A., Takemura S., Baba J., Fujisawa H., Mochizuki M., Shinzawa-Itoh K., Yamashita E., Tsukihara T., and Yoshikawa S. (2016) The Mg2+-containing water cluster of mammalian cytochrome c oxidase collects four pumping proton equivalents in each catalytic cycle. J. Biol. Chem. 291, 23882–23894 10.1074/jbc.M115.711770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muramoto K., Ohta K., Shinzawa-Itoh K., Kanda K., Taniguchi M., Nabekura H., Yamashita E., Tsukihara T., and Yoshikawa S. (2010) Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. U.S.A. 107, 7740–7745 10.1073/pnas.0910410107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimokata K., Katayama Y., Murayama H., Suematsu M., Tsukihara T., Muramoto K., Aoyama H., Yoshikawa S., and Shimada H. (2007) The proton pumping pathway of bovine heart cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 4200–4205 10.1073/pnas.0611627104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsukihara T., Shimokata K., Katayama Y., Shimada H., Muramoto K., Aoyama H., Mochizuki M., Shinzawa-Itoh K., Yamashita E., Yao M., Ishimura Y., and Yoshikawa S. (2003) The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl. Acad. Sci. U.S.A. 100, 15304–15309 10.1073/pnas.2635097100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keilin D., and Hartree E. F. (1939) Cytochrome and cytochrome oxidase. Proc. R. Soc. Lond. B Biol. Sci. 127, 847 10.1098/rspb.1939.0016 [DOI] [Google Scholar]
- 9. Yoshikawa S., and Orii Y. (1972) The inhibition mechanism of the cytochrome oxidase reaction II classification of inhibitors based on their modes of action. J. Biochem. 71, 859–872 10.1093/oxfordjournals.jbchem.a129835 [DOI] [PubMed] [Google Scholar]
- 10. Wever R., Muijsers A. O., van Gelder B. F., Bakker E. P., and van Buuren K. J. (1973) Biochemical and biophysical studies on cytochrome c oxidase. XI. Reaction with azide. Biochim. Biophys. Acta 325, 1–7 10.1016/0005-2728(73)90144-8 [DOI] [PubMed] [Google Scholar]
- 11. Wever R., Muijsers A. O., and Van Gelder B. F. (1973) Biochemical and biophysical studies on cytochrome c oxidase. XII. Kinetics of azide binding. Biochim. Biophys. Acta-Bioenerg. 325, 8–15 10.1016/0005-2728(73)90145-X [DOI] [PubMed] [Google Scholar]
- 12. Yoshikawa S., and Caughey W. S. (1992) Infrared evidence of azide binding to iron, copper, and non-metal sites in heart cytochrome c oxidase. J. Biol. Chem. 267, 9757–9766 [PubMed] [Google Scholar]
- 13. Li W., and Palmer G. (1993) Spectroscopic characterization of the interaction of azide and thiocyanate with the binuclear center of cytochrome oxidase: evidence for multiple ligand sites. Biochemistry 32, 1833–1843 10.1021/bi00058a018 [DOI] [PubMed] [Google Scholar]
- 14. Vamvouka M., Müller W., Ludwig B., and Varotsis C. (1999) Fourier transform infrared and resonance Raman studies of the interaction of azide with cytochrome c oxidase from Paracoccus denitrificans. J. Phys. Chem. B. 103, 3030–3034 10.1021/jp984589o [DOI] [Google Scholar]
- 15. Mochizuki M., Aoyama H., Shinzawa-Itoh K., Usui T., Tsukihara T., and Yoshikawa S. (1999) Quantitative reevaluation of the redox-active sites of crystalline bovine heart cytochrome c oxidase. J. Biol. Chem. 274, 33403–33411 10.1074/jbc.274.47.33403 [DOI] [PubMed] [Google Scholar]
- 16. Hirata K., Shinzawa-Itoh K., Yano N., Takemura S., Kato K., Hatanaka M., Muramoto K., Kawahara T., Tsukihara T., Yamashita E., Tono K., Ueno G., Hikima T., Murakami H., Inubushi Y., et al. (2014) Determination of damage-free crystal structure of an X-ray-sensitive protein using an XFEL. Nat. Methods 11, 734–736 10.1038/nmeth.2962 [DOI] [PubMed] [Google Scholar]
- 17. Hikita M., Yamamoto A., Shinzawa-Itoh K., Ogura T., and Yoshikawa S. (2015) Stepwise binding of two azide ions to the O2 reduction site of bovine heart cytochrome c oxidase shown by resonance Raman analyses. Chem. Lett. 44, 1142–1144 10.1246/cl.150384 [DOI] [Google Scholar]
- 18. Sakaguchi M., Shinzawa-Itoh K., Yoshikawa S., and Ogura T. (2010) A resonance Raman band assignable to the O–O stretching mode in the resting oxidized state of bovine heart cytochrome c oxidase. J. Bioenerg. Biomembr. 42, 241–243 10.1007/s10863-010-9282-y [DOI] [PubMed] [Google Scholar]
- 19. Fei M. J., Yamashita E., Inoue N., Yao M., Yamaguchi H., Tsukihara T., Shinzawa-Itoh K., Nakashima R., and Yoshikawa S. (2000) X-ray structure of azide-bound fully oxidized cytochrome c oxidase from bovine heart at 2.9 A resolution. Acta Crystallogr. D Biol. Crystallogr. 56, 529–535 10.1107/S0907444900002213 [DOI] [PubMed] [Google Scholar]
- 20. Iwata S., Ostermeier C., Ludwig B., and Michel H. (1995) Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 376, 660–669 10.1038/376660a0 [DOI] [PubMed] [Google Scholar]
- 21. Shimada A., Kubo M., Baba S., Yamashita K., Hirata K., Ueno G., Nomura T., Kimura T., Shinzawa-Itoh K., Baba J., Hatano K., Eto Y., Miyamoto A., Murakami H., Kumasaka T., et al. (2017) A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci. Adv. 3, e1603042 10.1126/sciadv.1603042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qin L., Liu J., Mills D. A., Proshlyakov D. A., Hiser C., and Ferguson-Miller S. (2009) Redox-dependent conformational changes in cytochrome c oxidase suggest a gating mechanism for proton uptake. Biochemistry 48, 5121–5130 10.1021/bi9001387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shinzawa-Itoh K., Aoyama H., Muramoto K., Terada H., Kurauchi T., Tadehara Y., Yamasaki A., Sugimura T., Kurono S., Tsujimoto K., Mizushima T., Yamashita E., Tsukihara T., and Yoshikawa S. (2007) Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 26, 1713–1725 10.1038/sj.emboj.7601618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilson D. F., Lindsay J. G., and Brocklehurst E. S. (1972) Heme–heme interaction in cytochrome oxidase. Biochim. Biophys. Acta 256, 277–286 10.1016/0005-2728(72)90058-8 [DOI] [PubMed] [Google Scholar]
- 25. Wikström M. K., Harmon H. J., Ingledew W. J., and Chance B. (1976) A re-evaluation of the spectral, potentiometric and energy-linked properties of cytochrome c oxidase in mitochondria. FEBS Lett. 65, 259–277 10.1016/0014-5793(76)80127-5 [DOI] [PubMed] [Google Scholar]
- 26. Williams R. J. (1995) Bioenergetics. Purpose of proton pathways. Nature. 376, 643 10.1038/376643a0 [DOI] [PubMed] [Google Scholar]
- 27. Sharma V., Jambrina P. G., Kaukonen M., Rosta E., and Rich P. R. (2017) Insights into functions of the H channel of cytochrome c oxidase from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 114, E10339–E10348 10.1073/pnas.1708628114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aoyama H., Muramoto K., Shinzawa-Itoh K., Hirata K., Yamashita E., Tsukihara T., Ogura T., and Yoshikawa S. (2009) A peroxide bridge between Fe and Cu ions in the O2 reduction site of fully oxidized cytochrome c oxidase could suppress the proton pump. Proc. Natl. Acad. Sci. U.S.A. 106, 2165–2169 10.1073/pnas.0806391106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otwinowski Z., and Minor W. (1997) Macromolecular crystallography, Part A. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 30. French S., and Wilson K. (1978) On the treatment of negative intensity observations. Acta Cryst. A 34, 517–525 10.1107/S0567739478001114 [DOI] [Google Scholar]
- 31. Weiss M. S. (2001) Global indicators of X-ray data quality. J. Appl. Crystallogr. 34, 130–135 10.1107/S0021889800018227 [DOI] [Google Scholar]
- 32. Rossmann M. G., and Blow D. M. (1962) The detection of sub-units within the crystallographic asymmetric unit. Acta Crystallogr. 15, 24–31 10.1107/S0365110X62000067 [DOI] [Google Scholar]
- 33. Wang B. C. (1985) Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 115, 90–112 10.1016/0076-6879(85)15009-3 [DOI] [PubMed] [Google Scholar]
- 34. Bricogne G. (1974) Geometric sources of redundancy in intensity data and their use for phase determination. Acta Crystallogr. Sect. A 30, 395–405 10.1107/S0567739474010722 [DOI] [Google Scholar]
- 35. Bricogne G. (1976) Methods and programs for direct-space exploitation of geometric redundancies. Acta Crystallogr. Sect. A. 32, 832–847 10.1107/S0567739476001691 [DOI] [Google Scholar]
- 36. Cowtan K. (1994) DM: an automated procedure for phase improvement by density modification. Jt. CCP4 ESF-EACBM Newsl. Protein Crystallogr. 31, 34–38 [Google Scholar]
- 37. Winn M. D., Isupov M. N., and Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 10.1107/S0907444900014736 [DOI] [PubMed] [Google Scholar]
- 38. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kleywegt G. J., and Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50, 178–185 10.1107/S0907444993011333 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.