

Abstract
The influenza virus is a significant public health concern causing 250,000–500,000 deaths worldwide each year. Its ability to change quickly results in the potential for rapid generation of pandemic strains for which most individuals would have no antibody protection. This pandemic potential highlights the need for the continuous development of new drugs against influenza virus. As an essential component and well established virulence determinant, NS1 (nonstructural protein 1) of influenza virus is a highly prioritized target for the development of anti-influenza compounds. Here, we used NMR to determine that the NS1 effector domain (NS1ED) derived from the A/Brevig Mission/1/1918 (H1N1) strain of influenza (1918H1N1) binds to two previously described anti-influenza compounds A9 (JJ3297) and A22. We then used X-ray crystallography to determine the three-dimensional structure of the 1918H1N1 NS1ED. Furthermore, we mapped the A9/A22-binding site onto our 1918H1N1 NS1ED structure and determined that A9 and A22 interact with the NS1ED in the hydrophobic pocket known to facilitate binding to the 30-kDa subunit of the cleavage and polyadenylation specificity factor (CPSF30), suggesting that the two compounds likely attenuate influenza replication by inhibiting the NS1ED–CPSF30 interaction. Finally, our structure revealed that NS1ED could dimerize via an interface that we termed the α3–α3 dimer. Taken together, the findings presented here provide strong evidence for the mechanism of action of two anti-influenza compounds that target NS1 and contribute significant structural insights into NS1 that we hope will promote and inform the development and optimization of influenza therapies based on A9/A22.
Keywords: influenza, nuclear magnetic resonance (NMR), antiviral agent, protein structure, viral protein, viral replication, influenza virus, 1918 H1N1, 2013 H7N9, CPSF30, nonstructural protein 1, NS1
Introduction
Each year, the influenza virus circulates through human, avian, and mammal populations across the globe. Its ability to undergo antigenic shift via reassortment of its segmented genome renders its hosts constantly vulnerable to potentially pandemic strains. Every year unique strains of influenza cause seasonal outbreaks that result in over 200,000 hospitalizations (1) and ∼35,000 deaths (2) in the United States alone. Currently, the effort against influenza is primarily composed of preventative vaccines and antiviral drugs (3). Although these methods have been successful, high mutation rates among circulating strains are limiting the efficacy of currently approved therapeutics in both categories. For example, vaccination strategies have been effective in decreasing the number of infections and fatalities caused by influenza. However, rapid evolution of these viruses can lead to the appearance of novel strains that render current vaccine strategies ineffective (4, 5). This situation occurred in 2009 when it took several months to manufacture an effective vaccine to the pandemic H1N1 strain (termed “swine flu”) that infected ∼60 million individuals, resulting in more than 12,000 deaths worldwide (6). The severity of this situation highlights the need for effective antiviral treatments to impede the spread of novel strains for which vaccination is not a viable option.
Currently available antiviral strategies for the treatment of influenza infection include two distinct classes of antiviral compounds. Adamantanes, including amantadine and rimantadine, inhibit the activity of the influenza M2 ion channel, preventing release of the genome from the endosome. Additionally, neuraminidase inhibitors such as oseltamivir and zanamivir inhibit influenza replication by preventing release of viral particles from the infected cell membrane. Although these antivirals can be quite effective in preventing the replication and spread of influenza, viral resistance is an ongoing issue. Resistance to both classes of influenza antivirals has been reported, with high levels of such resistance rendering adamantanes ineffective (7). Viral mechanisms of resistance to oseltamivir have also been described in certain strains of the influenza A virus (IAV) 2 (8, 9). In fact, 98% of the 2009 seasonal H1N1 influenza strains were resistant to oseltamivir, a neuraminidase inhibitor, and over 90% of all circulating viruses in 2005 were resistant to amantadine, the M2 ion channel inhibitor (7, 10–15). This demonstrates the continual need for the development of novel antiviral compounds for use against potentially resistant strains.
One potential target for the development of novel anti-influenza compounds is NS1 (nonstructural protein 1). NS1 is a 215–237-amino acid protein encoded by the eighth segment of the influenza virus genome and is expressed at high levels during infection (16–18). The NS1 protein consists of two independently folding, functional domains: the RNA-binding domain (NS1RBD; residues 1–73) and the effector domain (NS1ED; residues 86–205). Its primary function is to counter the host innate immune response to infection primarily by antagonizing the type I interferon (IFN) response. NS1 accomplishes this by interacting with a multitude of host factors in different cellular compartments and at different times during infection. Some of these interactions include RIG-I (19, 20), TRIM-25 (21), PKR (22), PI3K (23), and CPSF30 (24–26). NS1's multifunctional role during infection makes it essential to the survival and adaptability of the influenza virus (27). Therefore, NS1 is a high-value target for the development of the antiviral compounds that could control the replication and spread of the influenza virus (28).
Interest in developing NS1 antagonists as anti-influenza inhibitors has increased in recent years (28). In 2006, the CPSF30 (the 30-kDa subunit of the cleavage and polyadenylation specificity factor)-binding site on the NS1ED was identified as a potentially effective target for antiviral drug development after a cell line stably expressing a small protein fragment derived from CPSF30 (F2F3) was shown to be highly resistant to the replication of influenza A virus (29). Additionally, these cells produced significantly more IFN-β mRNA compared with infected control cells (29). In 2009, a yeast-based screen was used to identify small molecules that target NS1 function with the goal of identifying potential inhibitors of influenza replication (30). Briefly, Ward et al. (31) demonstrated that expression of NS1 in Saccharomyces cerevisiae was toxic and led to a “slow growth” phenotype in the S. cerevisiae expressing the NS1. Using this phenotype, Basu et al. (30) created a yeast strain expressing NS1 from A/WSN/33 under control of the GAL1 promoter. Small molecules were screened for their ability to inhibit NS1 function, thereby rescuing growth kinetics back to WT levels. This screen was successful and led to the identification of four compounds shown to inhibit influenza replication in tissue culture (30). One of these small molecules (NSC125044) was subsequently used as the basis for synthesizing additional small molecules with the goal of increasing the efficacy of NSC125044 as an inhibitor of influenza replication. The two most promising compounds synthesized were A9 (JJ3297), with an EC50 of 0.8 μm (32), and A22, determined to be ∼10 times more potent than A9 (33). In particular, A22 is capable of inhibiting the replication of influenza by up to 3 orders of magnitude and its activity depends upon a functional IFN response pathway, consistent with its presumed function as an NS1 inhibitor (32, 33).
Although these compounds represent a significant step in the field of NS1 inhibitors, there is a pressing need to understand the specific mechanism by which these compounds function. To identify the mechanism of action of these inhibitors, we used an NMR technique known as chemical shift perturbation (CSP) (19, 34). This method has been used in the past to successfully identify binding interfaces between two proteins, as well as between proteins and small molecules. Here we present structural and biophysical evidence that both compounds interact with the NS1ED derived from the A/Brevig Mission/1/1918 (H1N1) strain of influenza (1918H1N1). Additionally, we show that this interaction is conserved in the NS1ED derived from the A/Shanghai/1/2013 (H7N9) strain of influenza (2013H7N9), suggesting that these compounds or others like them may hold broad-spectrum therapeutic potential. To provide structural context to the interaction between A22 and the NS1EDs, we solved a high-resolution structure of the NS1ED from the 1918H1N1 strain of influenza for residue specific mapping of our measured CSPs. This structure adopts a homologous fold when compared with NS1ED from other strains of influenza; however, our structure revealed a novel homodimer. By mapping the CSPs observed onto our newly solved structure of the 1918H1N1 NS1ED, we conclude that the A22 compound interacts with the known CPSF30-binding pocket. Our results provide a potential mechanism of action of these novel compounds and highlight their potential to be further developed as effective antiviral compounds.
Results
Although successful in identifying small molecules capable of inhibiting influenza replication, the mechanism of action for these compounds was never determined, thereby hindering efficient optimization by structure-based drug design. We sought to use an NMR technique known as CSP to determine the precise interface of interaction between each small molecule and NS1. We expressed both the NS1RBD (residues 1–73) and the NS1ED (residues 86–205) to determine the amenability of both domains for this type of NMR analysis. The 1H-15N heteronuclear single quantum coherence (HSQC) spectra show a dispersed single set of peaks, which is indicative of a well folded protein (Fig. 1). In both spectra, the expected number of amide resonances (excluding prolines) are visible in addition to amides from the side chains of glutamine and asparagine. Importantly, we introduced a previously described (35, 36) tryptophan to alanine mutation at amino acid 187 (W187A) in the effector domain construct to increase its solubility and prevent dimerization. Previous studies have shown that although this mutation inhibits dimerization, the overall fold of the monomeric unit of NS1ED is unaffected. All forthcoming references to the NS1ED constructs used in this study contain this mutation. These data indicate that the 1918H1N1 NS1RBD and NS1ED are amenable to analysis by NMR.
Figure 1.

1H-15N HSQC spectra of the 1918H1N1 NS1RBD and NS1ED acquired at 850 MHz. HSQC spectra of the 1918H1N1 NS1RBD (A) and NS1ED (B) show a dispersed, single set of peaks indicating well-folded proteins.
A9 and A22 show structural homology to a known binding partner of NS1
A close inspection of these compounds (Fig. 2) reveals an intriguing structural similarity to a fragment of CPSF30, a known binding partner of the NS1ED. CPSF30 is responsible for 3′ end processing of host mRNAs in the nucleus (24). By sequestering CPSF30, NS1 prevents maturation and nuclear export of host mRNAs, resulting in a global down-regulation of host gene expression (37). Previous studies have demonstrated that this interaction is necessary for NS1's ability to antagonize the host IFN response (24, 26, 37, 38). Each compound is composed of a phenyl ring along with a hydroxyphenyl group (A9) or an aminophenyl group (A22) that have extraordinary structural similarity with phenylalanines 98 and 102 of CPSF30. Analysis of the structure of the NS1ED bound to CPSF30 indicates that these phenylalanine side chains are buried within a hydrophobic pocket of the NS1ED (Fig. 2) (26). This led us to hypothesize that A9 and A22 block influenza viral replication and NS1 function by interfering with the interaction of NS1ED with CPSF30.
Figure 2.
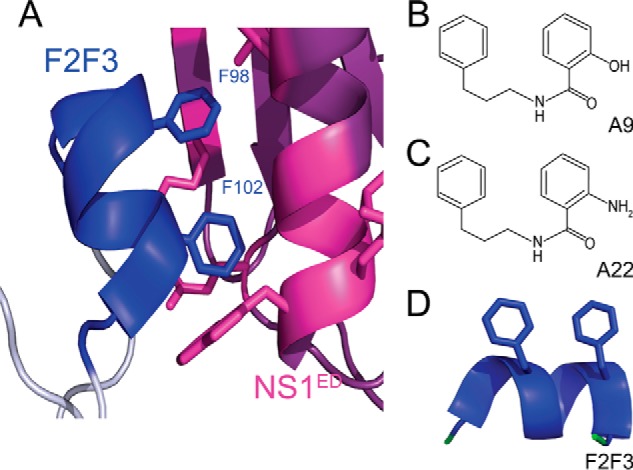
Structural homology between anti-influenza compounds and F2F3 of CPSF30. A, ribbon diagram of the NS1ED (purple) bound to the F2F3 region of CPSF30 (blue). The side chains of residues at the binding interface are shown in blue for CPSF30 and magenta for the NS1ED. B and C, chemical structures of the A9 (B) and A22 (C) anti-influenza compounds. D, residues of CPSF30 previously shown to bind the NS1ED with side chains of Phe-98 and Phe-102 indicated. The figure was generated using PDB code 2RHK.
Molecular docking was subsequently employed to determine whether this hypothesis was credible. Docking experiments were performed using Autodock Vina (39). For these experiments we used the NS1ED from the A/Puerto Rico/8/1934/H1N1 (PR8) strain of influenza and the A22 and A9 compounds modeled in Autodock Tools. We obtained binding energies for both A9 and A22 complexed with the PR8 NS1ED containing the W187A mutation (PDB code 3RVC) (Fig. 3). The docking results we obtained appeared to be in broad agreement with previously published docking data (40) (Fig. 3). The results of the docking models support our hypothesis that A9 and A22 bind to the NS1ED in the canonical CPSF30-binding region (Figs. 2 and 3). Importantly, we repeated the docking of A9 and A22 with Trp-187 modeled back into the PR8 NS1ED structure (data not shown). We found that Trp-187 participates in a π–π stacking with the phenyl ring of both small molecules. We note that our constructs, which require the Trp-187 mutation to make the NS1ED amenable to NMR, would theoretically exhibit decreased affinity for A9/A22 because of the absence of this π–π stacking.
Figure 3.

In silico docking of anti-influenza compounds. Both A9 (A) and A22 (B) were found to dock in the known CPSF30 binding pocket. The lowest binding free energies calculated by Autodock Vina are indicated for each compound. The figure was generated using PDB code 3RVC.
A22 interacts with the CPSF30-binding pocket of the 1918 NS1ED
To experimentally determine whether A9 and A22 interact with the NS1ED CPSF30-binding pocket, we used an NMR technique called chemical shift perturbation. Briefly, NMR chemical shifts are exquisitely sensitive to their surrounding chemical and electronic environments. This method relies on acquiring a 1H-15N HSQC spectrum of the NS1ED alone and in the presence of the ligand of interest. By identifying residues whose chemical shifts are perturbed upon the addition of a ligand, we can determine protein–ligand interaction sites with atomic resolution (34). This resolution is made possible by obtaining resonance assignments for the 1H-15N cross-peaks of the HSQC using three-dimensional NMR experiments such as the HNCACB and CBCA(CO)NH (Fig. 4). High quality triple-resonance spectra allowed backbone resonance assignments to be obtained for 99 of the 116 (85.3%) possible assigned residues (excluding prolines). The residues that we are unable to assign are likely in intermediate exchange, because the NS1ED is known to undergo dynamic motions on this timescale (41). Backbone resonances for the 1918H1N1 NS1ED have been deposited in the Biological Magnetic Resonance Bank (accession number 27539). Analysis of the 13Cα chemical shifts indicates that the 1918H1N1 NS1ED is composed of seven β-strands and three α-helices (42), which is consistent with the previously solved solution structure of the NS1ED derived from the Udorn1972 strain of influenza (43).
Figure 4.

1H-15N HSQC of the 1918H1N1 NS1ED with residue-specific assignments indicated.
Upon addition of the A22 compound, perturbations were observed for multiple residues when compared with the reference 1H-15N HSQC spectra of the 1918H1N1 NS1ED alone (Fig. 5). However, no perturbations were observed in the 1H-15N HSQC spectra of the 1918H1N1 NS1RBD upon addition of A22 (data not shown). The weighted 1H and 15N chemical shift differences observed in the 1918H1N1 NS1ED were plotted as a function of residue position. To ensure appreciable chemical shifts, we used an excess of the A22 compound relative to the concentration of the 1918H1N1 NS1ED (molar ratio of 5:1). We observed multiple statistically significant perturbations upon addition of A22 to the 1918 NS1ED (Fig. 5). The cutoff value used to determine statistically significant CSPs (2σ0corr) was calculated using an established method for discriminating between interacting and noninteracting residues (44). The amide resonances that showed the largest CSP (4σ0corr) included Arg-118, Val-111, and Gln-109, members of the β-2 and β-3 strands that make up the CPSF30-binding pocket. Phe-103, a residue upon which CPSF30 binding is contingent (26, 38, 45–48), also exhibited shifts above 4σ0corr. Other statistically significant CSPs exhibiting shifts above 4σ0corr include Lys-108, Gln-109, Lys-110, Ala-112, Gly-113, Ile-117, Met-119, Asp-120, Gln-121, Ala-122, Ile-123, Ile-160, Thr-170, Ala-177, Val-178, Val-180, Gly-183, Gly-184, Leu-185, Asn-188, and Asp-189, most of which are located within the β-2,3 strands or the α-2 helix that comprise the CPSF30-binding pocket (26). These results demonstrate that A22 indeed interacts with the 1918H1N1 NS1ED CPSF30-binding pocket, suggesting that this interaction may be the mechanism by which A22 inhibits NS1 function and IAV replication. Similar CSPs were observed upon the addition of A9, but the magnitude of these CSPs were lower (Fig. 5).
Figure 5.

NMR chemical shift perturbations reveal an interaction between the 1918H1N1 NS1ED and A9/A22. A and B, overlay of 1H-15N HSQC spectra of the 1918H1N1 NS1ED (black) and 1918H1N1 NS1ED after adding A9 (A) indicated in green and A22 (B) indicated in red. Chemical shift perturbations were quantified and plotted on a preresidue basis for A9 (A) and A22 (B). The dotted lines represent the calculated 2σ0corr and 4σ0corr cutoffs indicated. Residues experiencing shifts greater than 4σ0corr are in dark green (A9) or dark red (A22), whereas those experiencing shifts greater than 2σ0corr but less than 4σ0corr are indicated in light green or light red. Secondary structure predicted using Wishart analysis indicated above quantified chemical shifts for both A9 (A) and A22 (B).
A22 interacts with the A/Shanghai/1/2013 NS1ED in a similar manner
While demonstrating that the interaction between A22 and the NS1ED from a highly virulent strain such as the 1918H1N1 was important for understanding its anti-influenza properties, we also wanted to demonstrate its broad potential as an IAV inhibitor. Since 2013, a novel influenza virus of avian origin (2013H7N9) has circulated in China, resulting in over 1500 human infections with a 40% mortality rate (Centers for Disease Control and Prevention) (65). There are significant differences between the NS1 of this strain and the 1918H1N1 NS1 (Fig. 6), as well as most other NS1s. We therefore used the same methodology detailed above to identify the binding interface between the small molecule A22 and the NS1ED of the 2013H7N9 strain of influenza (83.4% identical to 1918). The construct used for the 2013 NS1ED studies included the same modification (the W187A mutation) that was used in the 1918 NS1ED construct. Backbone resonance assignments were obtained for 94 of the 115 (81.7%) possible assigned residues and have been deposited in the Biological Magnetic Resonance Bank (accession number 27541). Analysis of the 13Cα chemical shifts indicates that the 2013H7N9 NS1ED is composed of seven β-strands and three α-helices (42), identical to the secondary structural composition of the 1918H1N1 NS1ED.
Figure 6.

NMR chemical shift perturbations reveal an interaction between the 2013H7N9 NS1ED and A22. A, sequence alignment of the NS1ED derived from the 1918H1N1 and 2013H7N9 strains of influenza. B, 1H-15N HSQC spectra of the 2013H7N9 NS1ED with residue-specific assignments indicated. C, overlay of 1H-15N HSQC spectra of the 2013H7N9 NS1ED (black) and 2013H7N9 NS1ED after adding A22 indicated in red. D, chemical shift perturbations were quantified and plotted on a preresidue basis A22. The dotted lines represent the calculated 2σ0corr and 4σ0corr cutoffs indicated. Residues experiencing shifts greater than 4σ0corr are in dark blue (A22), whereas those experiencing shifts greater than 2σ0corr but less than 4σ0corr are indicated in light blue. Secondary structure predicted using Wishart Analysis indicated above quantified chemical shift plot.
Upon addition of the A22 to the 2013H7N9 NS1ED sample, perturbations for multiple residues were observed when comparing this 1H-15N HSQC spectra to the 2013H7N9 NS1ED alone. Interestingly, the CSPs observed for the 2013H7N9 NS1ED had significant overlap with resonances observed to undergo perturbations identified for the 1918H1N1 NS1ED (Fig. 6). Lys-108, Gln-109, Lys-110, Ile-117, Arg-118, Met-119, Asp-120, Gln-121, Ala-122, Ile-123, Ile-160, Ala-177, and Gly-184 experienced significant chemical shift perturbation as in 1918H1N1 NS1ED. Other statistically significant CSP included Leu-115, Glu-186, and Ala-187. This suggests that A22 can interact with NS1s from diverse strains of IAV and may provide broad-spectrum protection, highlighting its potential to be further developed as an effective antiviral compound.
In addition, we used chemical shift perturbation to determine the Kd of the interactions between A22 and both the 1918H1N1 and 2013H7N9 NS1EDs (49). The measured Kd values for A22 binding were 425 ± 225 and 475 ± 218 μm for the 1918H1N1 and 2013H7N9 NS1EDs, respectively (Fig. 7). These values suggest a relatively weak interaction, yet this was expected because of the potential effects of the necessary W187A mutation discussed previously.
Figure 7.

Disassociation constants for the 1918H1N1 NS1ED–A22 and 2013H7N9 NS1ED–A22 interactions. A and B, representative binding isotherms and CSPs used to calculate the Kd of the interactions between A22 and both the 1918H1N1 (A) and 2013H7N9 (B) NS1EDs.
High-resolution crystal structure of the 1918H1N1 NS1ED
To provide structural context for the interaction discovered herein, we solved a 1.9 Å resolution crystal structure of the 1918 NS1ED (Table 1). Our structure has the typical fold associated with previously solved NS1EDs from different strains of influenza (35, 36, 50–52). Specifically, there are three α-helices and seven β-strands that form a twisted, antiparallel β-sheet that surrounds a long, central α-helix (α2) (Fig. 8). Atomic coordinates and structure factors have been deposited in the PDB (code 6DGK). Interestingly, the asymmetric unit of the crystal structure contains two monomers (Fig. 8). This was unexpected because we incorporated a W187A mutation into our expression construct to prevent dimerization of the protein. This is a common modification made for structural studies of the NS1ED because the dimerization of the WT protein can present solubility issues. Interestingly, the interaction between the monomers in the asymmetric unit of our structure has not been observed in any previous NS1ED structures (Fig. 9). Other structures have displayed the “helix–helix” dimer (52, 53) and the less common “strand–strand” dimer (51). However, dimerization of the NS1ED in our structure appears to be dependent on the burial of Trp-203 in the hydrophobic pocket formed by the α2 and α3 helices and β7 strand of the opposing monomer (Figs. 8 and 9). Because Trp-203 is on α3, we have termed this novel NS1ED dimeric interaction the “α3–α3 dimer.” To further characterize this dimeric interaction, we calculated the buried surface area for each dimeric interface using the PDBe Protein Interfaces, Surfaces, and Assemblies (PISA) web service (54). We determined that the buried surface area for the α3–α3 dimer (638.0 Å2) is comparable with the helix–helix dimer (596.8 Å2) and the strand–strand dimer (629.7 Å2). Further studies aimed at determining the presence of this potential dimeric interaction in solution are needed to understand any functional consequences during infection.
Table 1.
X-ray diffraction and refinement statistics
The values in parentheses correspond to the highest resolution data shell.
| Parameter | 1918H1N1 NS1ED WT W187A |
|---|---|
| Wavelength | 1.00 |
| Resolution range | 29.67–1.9 (1.968–1.9) |
| Space group | P 21 21 21 |
| Unit cell | 32.8, 69.7, 105.0 90 90 90 |
| Total reflections | 138,275 |
| Unique reflections | 19,479 (1905) |
| Multiplicity | 7.1 |
| Completeness (%) | 98.71 (98.45) |
| Mean I/σ(I) | 54.9 (8.9) |
| Wilson B factor | 24.95 |
| Rmerge | 0.060 (0.247) |
| Rmeas | 0.065 (0.266) |
| Rpim | 0.025 (0.099) |
| CC½ | 0.992 |
| Reflections used in refinement | 19,471 (1903) |
| Reflections used in Rfree | 1452 (143) |
| Rwork | 0.1697 (0.2167) |
| Rfree | 0.2123 (0.2967) |
| Number of non-hydrogen atoms | 2094 |
| Macromolecules | 1886 |
| Solvent | 208 |
| Protein residues | 240 |
| RMS bonds | 0.006 |
| RMS angles | 0.80 |
| Ramachandran favored (%) | 97.03 |
| Ramachandran outliers (%) | 2.97 |
| Rotamer outliers (%) | 0.00 |
| Clashscore | 3.16 |
| Average B factor | 37.28 |
| Macromolecules | 36.73 |
| Solvent | 42.29 |
| Number of TLS groups | 19 |
Figure 8.
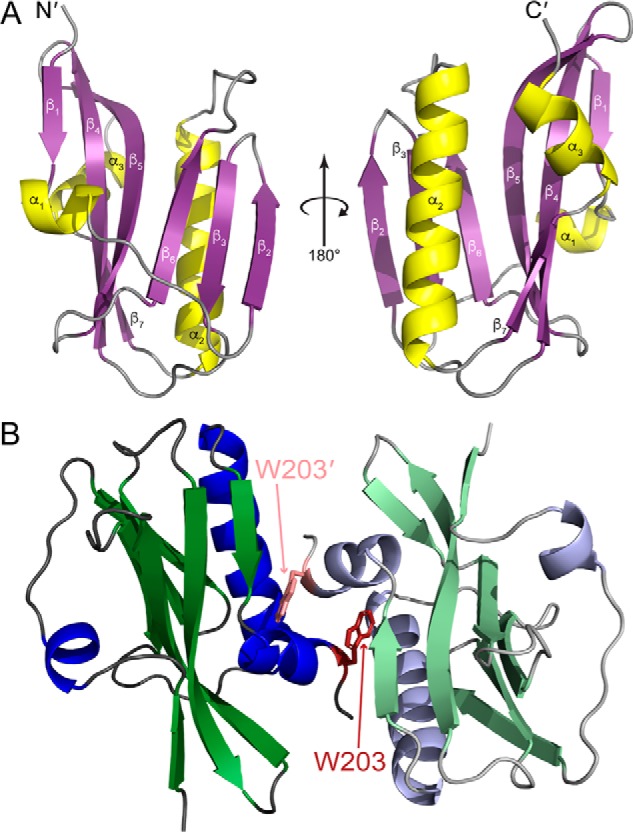
High-resolution crystal structure of the 1918H1N1 NS1ED. A, ribbon diagram of the 1918H1N1 NS1ED (PDB code 6DGK) with the secondary structure labeled (β-strands in purple and α-helices in yellow). B, dimer found in the asymmetric unit of the 1918H1N1 NS1ED crystal structure. Tryptophan 203 (indicated in red and pink) is buried in the hydrophobic pocket formed by the α2 helix, α3 helix, and β7 strand of the opposing monomer.
Figure 9.

The three proposed dimerization interfaces of the NS1ED. The 187 backbone and side chain (Trp for Udorn1972 and PR81934, and Ala for 1918H1N1) are indicated by orange sticks, whereas the Trp-203 backbone and side chain are indicated in red. The interface solvent-accessible surface area is indicated for each dimeric structure. A, the α3–α3 dimer found in the 1918H1N1 NS1ED crystal structure (PDB code 6DGK). B, the helix–helix dimer found in the Udorn1972 NS1ED crystal structure (PDB code 3EE9). C, the strand–strand dimer found in the PR81934 NS1ED crystal structure (PDB code 2GX9).
We mapped perturbations that were greater than 4σ0corr onto our newly solved structure of the 1918H1N1 NS1ED (Fig. 10). This analysis revealed that A9 and A22 bind to the 1918H1N1 NS1ED in the same location as the F2F3 portion of CPSF30 (26). This suggests that the mechanism by which A22 inhibits influenza replication is by interfering with the interaction between NS1 and CPSF30. Furthermore, mapping our 2013H7N9 NS1ED/A22 CSP data onto the 1918H1N1 NS1ED crystal structure indicates a high degree of overlap with the 1918H1N1 NS1ED CSP data, indicating that A22 also binds the 2013H7N9 NS1ED in the CPSF30-binding pocket. Taken together, these findings suggest that A22 could serve as a broad-spectrum inhibitor of multiple strains of influenza.
Figure 10.
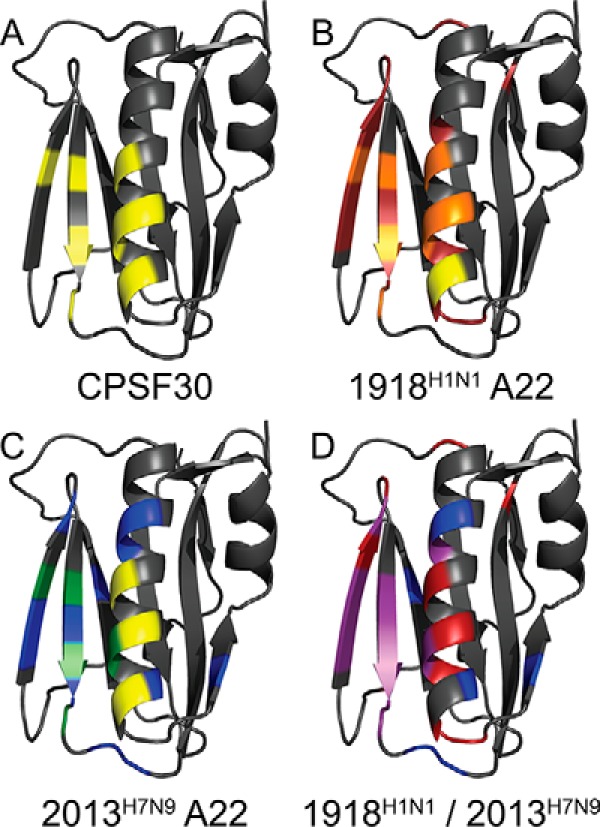
CSPs induced upon addition of the compound reveal that A22 binds in the CPSF30-binding pocket of the NS1ED. A, residues known to play a role in CPSF30 binding are yellow. B, perturbations measured for 1918H1N1 NS1ED upon addition of A22 are mapped onto the 1918H1N1 NS1ED crystal structure. Residues with shifts greater than 4σ0corr are indicated in red, residues known to play a role in CPSF30 binding in yellow, and residues with shifts greater than 4σ0corr and known to play a role in CFSP30 binding are indicated in orange. C, perturbations measured for 2013H7N9 NS1ED upon addition of A22 are mapped onto the 1918H1N1 NS1ED crystal structure. Residues with shifts greater than 4σ0corr are indicated in blue, residues known to play a role in CPSF30 binding in yellow, and residues with shifts greater than 4σ0corrand known to play a role in CFSP30 binding are indicated in green. D, overlay of perturbations found for 1918H1N1 NS1ED and 2013H7N9 NS1ED upon addition of A22 mapped onto the 1918H1N1 NS1ED crystal structure. Perturbations found in both strains are indicated in purple.
Discussion
Here we have identified a potential mechanism by which two anti-influenza compounds inhibit the activity of NS1 and thereby inhibit the replication of IAV. We show that A9 (JJ3297) and A22 bind to the CPSF30-binding pocket of the NS1ED from both the 1918H1N1 and the 2013H7N9 strains of IAV. This binding event could obstruct NS1's interaction with host CPSF30, an interaction responsible for NS1's ability to block global host gene expression including genes involved in the host interferon response. In addition, we have solved a 1.9 Å crystal structure of the 1918 NS1ED and identified a novel interface through which dimerization of this domain could occur. This finding reinforces the importance of this interaction as a target for anti-influenza drug development (28, 29) and highlights the potential for further optimization of A9 and A22. Furthermore, our results suggest that the efficacy of these compounds could be broadly specific against many strains of IAV.
The significant efficacy with which A9 and A22 inhibited the replication of IAV in vitro was originally demonstrated using the A/Puerto Rico/8/1934 H1N1 (PR8) mouse adapted strain of IAV (33). Interestingly, PR8 NS1 contains two mutations (F103L and M106I) that decrease CPSF30 binding affinity and is thus unable to properly control host gene expression (37, 55). This could suggest that even the relatively low levels of CPSF30 binding retained by PR8 NS1 are sufficient to support the replication of the virus. Additionally, CPSF30 binding by NS1 may impart some other unknown benefit to the replication of IAV such as proper subcellular localization or increased association with host RNAs. Moreover, there may be other NS1–host interactions that are dependent upon the same pocket in which CPSF30 binds. A previous study has identified Trp-187, a residue buried within the CPSF30-binding pocket of the dimerized NS1ED, as an important residue contributing to NS1's ability to bind dsRNA (43). NS1's dsRNA-binding activity is essential for shielding IAV from the activation of 2′-5′ oligo(A) synthetase/RNase L pathway (56). Interestingly, it is this pathway on upon which the anti-influenza activity of A9 was shown to be dependent (32). This highlights a potentially alternative or complementary mechanism for the anti-influenza activity of A9 and A22 and provides a foundation for the design of new antivirals that could inhibit similar pathways.
Full-length NS1 exists as a dimer in solution, stabilized by a strong self-association of its N-terminal RBD (43, 50). However, the nature of dimerization of the effector domain in solution is not completely understood. Some studies have suggested that the helix-helix dimerization of the NS1ED occurs at high protein concentrations and can lead to oligomerization of full-length NS1 (41, 43), but the affinity and specificity of this dimerization in a biological setting is not established. It is possible that the NS1ED samples multiple dimeric interfaces during infection and that the α3–α3 dimer that we observe could be involved. In fact, previous crystal structures and 19F NMR data have demonstrated conformational heterogeneity in the orientation of the helix–helix interface, suggesting that NS1ED dimerization is dynamic in nature (35, 41).
In summary, we have identified the NS1ED-binding interface for two potent anti-influenza compounds and confirmed their interaction with the NS1ED from two divergent strains of IAV. We further identified this interface as the CPSF30-binding pocket, an important functional interface for the NS1ED. Finally, we solved a crystal structure of the 1918H1N1 NS1ED and identified a novel potential dimerization interface within the NS1ED. Together, these data shed light on important questions regarding the continued development of new anti-influenza antiviral drugs and the structural characteristics driving the function of NS1.
Experimental procedures
Cloning, mutagenesis, protein expression, and purification
The effector domains of the A/Brevig Mission/1/1918 H1N1 (1918 NS1ED) and A/Shanghai/1/2013 H7N9 (2013 NS1ED) NS1 gene were subcloned into a His6-SMT3 fusion T7 expression vector (pE-SUMO; Life Sensors) using primers designed to amplify residues 86–205 of full-length 1918 and 2013 NS1 plasmids (Biomatik). Site-directed mutagenesis was then performed using primers designed to introduce the W187A mutation into each plasmid. This modification was made to increase protein solubility for NMR and X-ray crystallography. These expression plasmids were transformed into DE3 Star BL21 Escherichia coli cells and grown in minimal medium supplemented with 15NH4Cl and/or d-glucose (U-13C6-99%) depending on the desired labeling scheme. NS1ED cultures were induced at an A600 0.6 with 1 mm isopropyl β-d-1-thiogalactopyranoside for 48 h at 16 °C. The cells were lysed in buffer containing 20 mm HEPES, 500 mm NaCl, 15 mm imidazole, 5 mm DTT, and 0.25 mg/ml lysozyme (lysis buffer) via sonication. Clarified lysates were loaded onto a cOmplete His-tag purification column (Roche), and protein was eluted using lysis buffer with 250 mm imidazole. The His6-SMT3 tag was then removed by the recombinant SUMO protease, leaving no extraneous residues on the N terminus. Finally, the samples were re-loaded onto the cOmplete His-tag purification columns to separate the cleaved tag from the desired protein. Buffer exchange was accomplished via size exclusion chromatography using the Hi-Prep 26/60 Sephacryl S-100 HR (GE). Typical yields were 20 mg/liter with >99% purity, as verified by SDS-PAGE (data not shown).
X-ray crystallography and structure determination
The purified 1918 NS1ED was subjected to numerous commercially available crystallization screens. Diffraction quality crystals were obtained using hanging-drop vapor diffusion in a solution (pH 6.8) of 30% PEG (molecular weight 3350) and 0.2 m diammonium tartrate. Crystals were cryo-cooled in the same conditions. X-ray diffraction data were collected with native protein crystals at the Advance Photon Source, SER-Cat Beamline 22-ID. Raw intensity data were processed with the HKL2000 software package (57). The initial electron density map was generated via molecular replacement using the previously solved structure of the PR8 NS1ED (PDB code 3RVC) with PHASER (58). The structure was then iteratively refined with PHENIX (59) and Coot (60). All figures were created using PyMOL (61).
NMR spectroscopy
NMR experiments were carried out at 25 °C using Bruker Avance III spectrometers equipped with TCI cryoprobes operating at 600 and 850 MHz 1H frequencies. All NMR data were processed using NMRPipe (62) and analyzed using NMRView (63) and CARA compiled on Linux workstations. Backbone 1H, 15N, and 13C resonances were assigned using standard triple-resonance assignment experiments: HNCACB and CBCA(CO)NH (64). CSPs were assessed using 1H-15N HSQC spectra. For these experiments, we collected 1H-15N HSQC spectra from the NS1ED of interest in the unbound state and in the presence of A9 or A22. When comparing the two spectra, the changes in chemical shift resonances were calculated using weighted combination of chemical shifts given by the following equation.
| (Eq. 1) |
The cutoff value used to determine statistically significant chemical shift differences (2σ0corr) was calculated using an established method determined to result in less than 4.5% false positives when discriminating between interacting and noninteracting residues. All NMR experiments were performed in following buffer: 20 mm NaPO4, 150 mm NaCl, 1 mm tris(2-carboxyethyl) phosphine, 1% glycerol, 0.03% sodium azide, 5% deuterium oxide (pH 6.8). The protein concentration in all CSP experiments was 0.1 mm. 5% DMSO was added to all buffers used in CSP experiments to ensure solubility of small molecules. Isotherms were generated using the titration analysis tool in NMRViewJ.
Molecular docking/modeling
Docking experiments were performed using Autodock Vina. The NS1ED from the A/Puerto Rico/8/1934/H1N1 (PR8) strain of influenza (PDB code 3RVC) and the A22 and A9 compounds were modeled in Autodock Tools (with six rotatable bonds each). The search space was defined as a 20 × 20 × 20 Å box centered on the CPSF30 binding site of the NS1ED derived from PR8 (no residues were designated as flexible). The nine lowest energy solutions were then analyzed, and figures were generated in PyMOL using the lowest energy solution. Autodock Vina employs a strategy of initiating multiple runs starting from random ligand conformations. Free energy of binding calculations are performed using the intermolecular component of the lowest-scoring conformation as calculated via AutoDock Vina's scoring function (39).
Author contributions
A. B. K. and C. M. P. conceptualization; A. B. K., A. S. J., S. M. F., T. J. G., and C. M. P. data curation; A. B. K., A. S. J., S. M. F., T. J. G., and C. M. P. formal analysis; A. B. K. and C. M. P. validation; A. B. K. and C. M. P. investigation; A. B. K., T. J. G., and C. M. P. methodology; A. B. K. and C. M. P. writing-original draft; A. B. K., A. S. J., and C. M. P. writing-review and editing; C. M. P. supervision; C. M. P. funding acquisition; C. M. P. visualization; C. M. P. project administration.
This work was supported by National Institutes of Health Grants R01 AI1346931 (to C. M. P.) and R01 AI116738 (to T. J. G). This work was supported in part by NCI, National Institutes of Health Grants 1P30 CA-13148 and 1P30 CA-13148, National Center for Research Resources Grant 1S10 RR022994-01A1 and by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 6DGK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IAV
- influenza A virus
- RBD
- RNA-binding domain
- IFN
- interferon
- CSP
- chemical shift perturbation
- HSQC
- heteronuclear single quantum coherence
- PDB
- Protein Data Bank
- SUMO
- small ubiquitin-like modifier.
References
- 1. Thompson W. W., Comanor L., and Shay D. K. (2006) Epidemiology of seasonal influenza: use of surveillance data and statistical models to estimate the burden of disease. J. Infect. Dis. 2, S82–S91 10.1086/507558 [DOI] [PubMed] [Google Scholar]
- 2. Molinari N. A., Ortega-Sanchez I. R., Messonnier M. L., Thompson W. W., Wortley P. M., Weintraub E., and Bridges C. B. (2007) The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25, 5086–5096 10.1016/j.vaccine.2007.03.046 [DOI] [PubMed] [Google Scholar]
- 3. Beigel J., and Bray M. (2008) Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res. 78, 91–102 10.1016/j.antiviral.2008.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zost S. J., Parkhouse K., Gumina M. E., Kim K., Diaz Perez S., Wilson P. C., Treanor J. J., Sant A. J., Cobey S., and Hensley S. E. (2017) Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. U.S.A. 114, 12578–12583 10.1073/pnas.1712377114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu N. C., Zost S. J., Thompson A. J., Oyen D., Nycholat C. M., McBride R., Paulson J. C., Hensley S. E., and Wilson I. A. (2017) A structural explanation for the low effectiveness of the seasonal influenza H3N2 vaccine. PLoS Pathog. 13, e1006682 10.1371/journal.ppat.1006682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang Y., Sugimoto J. D., Halloran M. E., Basta N. E., Chao D. L., Matrajt L., Potter G., Kenah E., and Longini I. M. Jr. (2009) The transmissibility and control of pandemic influenza A (H1N1) virus. Science 326, 729–733 10.1126/science.1177373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bright R. A., Shay D. K., Shu B., Cox N. J., and Klimov A. I. (2006) Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 295, 891–894 10.1001/jama.295.8.joc60020 [DOI] [PubMed] [Google Scholar]
- 8. Kwon J. J., Choi W. S., Jeong J. H., Kim E. H., Lee O. J., Yoon S. W., Hwang J., Webby R. J., Govorkova E. A., Choi Y. K., Baek Y. H., and Song M. S. (2018) An I436N substitution confers resistance of influenza A(H1N1)pdm09 viruses to multiple neuraminidase inhibitors without affecting viral fitness. J. Gen. Virol. 99, 292–302 10.1099/jgv.0.001029 [DOI] [PubMed] [Google Scholar]
- 9. Trebbien R., Christiansen C. B., and Fischer T. K. (2018) Antiviral resistance due to deletion in the neuraminidase gene and defective interfering-like viral polymerase basic 2 RNA of influenza A virus subtype H3N2. J. Clin. Virol. 102, 1–6 10.1016/j.jcv.2018.02.005 [DOI] [PubMed] [Google Scholar]
- 10. de St Maurice A., and Halasa N. (2017) Immunization and treatment updates: 2016–2017 influenza season. Pediatr. Transpl. 21, 12884 10.1111/petr.12884 [DOI] [PubMed] [Google Scholar]
- 11. Abed Y., Goyette N., and Boivin G. (2005) Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. Antimicrob. Agents Chemother. 49, 556–559 10.1128/AAC.49.2.556-559.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lackenby A., Hungnes O., Dudman S. G., Meijer A., Paget W. J., Hay A. J., and Zambon M. C. (2008) Emergence of resistance to oseltamivir among influenza A(H1N1) viruses in Europe. Euro Surveill. 13, 8026 [DOI] [PubMed] [Google Scholar]
- 13. Sheu T. G., Deyde V. M., Okomo-Adhiambo M., Garten R. J., Xu X., Bright R. A., Butler E. N., Wallis T. R., Klimov A. I., and Gubareva L. V. (2008) Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 52, 3284–3292 10.1128/AAC.00555-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nelson M. I., Simonsen L., Viboud C., Miller M. A., and Holmes E. C. (2009) The origin and global emergence of adamantane resistant A/H3N2 influenza viruses. Virology 388, 270–278 10.1016/j.virol.2009.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lackenby A., Moran Gilad J., Pebody R., Miah S., Calatayud L., Bolotin S., Vipond I., Muir P., Guiver M., McMenamin J., Reynolds A., Moore C., Gunson R., Thompson C., Galiano M., et al. (2011) Continued emergence and changing epidemiology of oseltamivir-resistant influenza A(H1N1)2009 virus, United Kingdom, winter 2010/11. Euro Surveill. 16, 19784 [PubMed] [Google Scholar]
- 16. Hale B. G., Randall R. E., Ortín J., and Jackson D. (2008) The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89, 2359–2376 10.1099/vir.0.2008/004606-0 [DOI] [PubMed] [Google Scholar]
- 17. Ayllon J., and García-Sastre A. (2015) The NS1 protein: a multitasking virulence factor. Curr. Top. Microbiol. Immunol. 386, 73–107 [DOI] [PubMed] [Google Scholar]
- 18. Krug R. M. (2015) Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 12, 1–6 10.1016/j.coviro.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jureka A. S., Kleinpeter A. B., Cornilescu G., Cornilescu C. C., and Petit C. M. (2015) Structural basis for a novel interaction between the NS1 protein derived from the 1918 influenza virus and structure RIG-I. Structure 23, 2001–2010 10.1016/j.str.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pichlmair A., Schulz O., Tan C. P., Näslund T. I., Liljeström P., Weber F., and Reis e Sousa C. (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 21. Gack M. U., Albrecht R. A., Urano T., Inn K. S., Huang I. C., Carnero E., Farzan M., Inoue S., Jung J. U., and García-Sastre A. (2009) Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449 10.1016/j.chom.2009.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li S., Min J.-Y., Krug R. M., and Sen G. C. (2006) Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349, 13–21 10.1016/j.virol.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 23. Hale B. G., Jackson D., Chen Y. H., Lamb R. A., and Randall R. E. (2006) Influenza A virus NS1 protein binds p85β and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 14194–14199 10.1073/pnas.0606109103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noah D. L., Twu K. Y., and Krug R. M. (2003) Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology 307, 386–395 10.1016/S0042-6822(02)00127-7 [DOI] [PubMed] [Google Scholar]
- 25. Nemeroff M. E., Barabino S. M., Li Y., Keller W., and Krug R. M. (1998) Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell 1, 991–1000 10.1016/S1097-2765(00)80099-4 [DOI] [PubMed] [Google Scholar]
- 26. Das K., Ma L. C., Xiao R., Radvansky B., Aramini J., Zhao L., Marklund J., Kuo R. L., Twu K. Y., Arnold E., Krug R. M., and Montelione G. T. (2008) Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. U.S.A. 105, 13093–13098 10.1073/pnas.0805213105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. García-Sastre A., Egorov A., Matassov D., Brandt S., Levy D. E., Durbin J. E., Palese P., and Muster T. (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252, 324–330 10.1006/viro.1998.9508 [DOI] [PubMed] [Google Scholar]
- 28. Engel D. A. (2013) The influenza virus NS1 protein as a therapeutic target. Antiviral Res. 99, 409–416 10.1016/j.antiviral.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Twu K. Y., Noah D. L., Rao P., Kuo R. L., and Krug R. M. (2006) The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J. Virol. 80, 3957–3965 10.1128/JVI.80.8.3957-3965.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Basu D., Walkiewicz M. P., Frieman M., Baric R. S., Auble D. T., and Engel D. A. (2009) Novel influenza virus NS1 antagonists block replication and restore innate immune function. J. Virol. 83, 1881–1891 10.1128/JVI.01805-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ward A. C., Azad A. A., and Macreadie I. G. (1994) Expression and characterisation of the influenza A virus non-structural protein NS1 in yeast. Arch. Virol. 138, 299–314 10.1007/BF01379133 [DOI] [PubMed] [Google Scholar]
- 32. Walkiewicz M. P., Basu D., Jablonski J. J., Geysen H. M., and Engel D. A. (2011) Novel inhibitor of influenza non-structural protein 1 blocks multi-cycle replication in an RNase L-dependent manner. J. Gen. Virol. 92, 60–70 10.1099/vir.0.025015-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jablonski J. J., Basu D., Engel D. A., and Geysen H. M. (2012) Design, synthesis, and evaluation of novel small molecule inhibitors of the influenza virus protein NS1. Bioorg. Med. Chem. 20, 487–497 10.1016/j.bmc.2011.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foster M. P., Wuttke D. S., Clemens K. R., Jahnke W., Radhakrishnan I., Tennant L., Reymond M., Chung J., and Wright P. E. (1998) Chemical shift as a probe of molecular interfaces: NMR studies of DNA binding by the three amino-terminal zinc finger domains from transcription factor IIIA. J. Biomol. NMR 12, 51–71 10.1023/A:1008290631575 [DOI] [PubMed] [Google Scholar]
- 35. Kerry P. S., Ayllon J., Taylor M. A., Hass C., Lewis A., García-Sastre A., Randall R. E., Hale B. G., and Russell R. J. (2011) A transient homotypic interaction model for the influenza A virus NS1 protein effector domain. PLoS One 6, e17946 10.1371/journal.pone.0017946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia S., and Robertus J. D. (2010) X-ray structures of NS1 effector domain mutants. Arch. Biochem. Biophys. 494, 198–204 10.1016/j.abb.2009.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hale B. G., Steel J., Medina R. A., Manicassamy B., Ye J., Hickman D., Hai R., Schmolke M., Lowen A. C., Perez D. R., and García-Sastre A. (2010) Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J. Virol. 84, 6909–6922 10.1128/JVI.00081-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dankar S. K., Miranda E., Forbes N. E., Pelchat M., Tavassoli A., Selman M., Ping J., Jia J., and Brown E. G. (2013) Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol. J. 10, 243 10.1186/1743-422X-10-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang L., Zhao J., Ding G., Li X., and Liu H. (2012) Screening of potent inhibitor of H1N1 influenza NS1 CPSF30 binding pocket by molecular docking. Adv. Infect. Dis. 2, 92–96 10.4236/aid.2012.24015 [DOI] [Google Scholar]
- 41. Aramini J. M., Hamilton K., Ma L. C., Swapna G. V. T., Leonard P. G., Ladbury J. E., Krug R. M., and Montelione G. T. (2014) 19F NMR reveals multiple conformations at the dimer interface of the nonstructural protein 1 effector domain from influenza A virus. Structure 22, 515–525 10.1016/j.str.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wishart D. S., and Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 43. Aramini J. M., Ma L. C., Zhou L., Schauder C. M., Hamilton K., Amer B. R., Mack T. R., Lee H. W., Ciccosanti C. T., Zhao L., Xiao R., Krug R. M., and Montelione G. T. (2011) Dimer interface of the effector domain of non-structural protein 1 from influenza A virus: an interface with multiple functions. J. Biol. Chem. 286, 26050–26060 10.1074/jbc.M111.248765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schumann F. H., Riepl H., Maurer T., Gronwald W., Neidig K. P., and Kalbitzer H. R. (2007) Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275–289 10.1007/s10858-007-9197-z [DOI] [PubMed] [Google Scholar]
- 45. Ayllon J., Domingues P., Rajsbaum R., Miorin L., Schmolke M., Hale B. G., and García-Sastre A. (2014) A single amino acid substitution in the novel H7N9 influenza A virus NS1 protein increases CPSF30 binding and virulence. J. Virol. 88, 12146–12151 10.1128/JVI.01567-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Twu K. Y., Kuo R. L., Marklund J., and Krug R. M. (2007) The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 81, 8112–8121 10.1128/JVI.00006-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Spesock A., Malur M., Hossain M. J., Chen L. M., Njaa B. L., Davis C. T., Lipatov A. S., York I. A., Krug R. M., and Donis R. O. (2011) The virulence of 1997 H5N1 influenza viruses in the mouse model is increased by correcting a defect in their NS1 proteins. J. Virol. 85, 7048–7058 10.1128/JVI.00417-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forbes N., Selman M., Pelchat M., Jia J. J., Stintzi A., and Brown E. G. (2013) Identification of adaptive mutations in the influenza A virus non-structural 1 gene that increase cytoplasmic localization and differentially regulate host gene expression. PLoS One 8, e84673 10.1371/journal.pone.0084673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 10.1016/j.pnmrs.2013.02.001 [DOI] [PubMed] [Google Scholar]
- 50. Kerry P. S., Long E., Taylor M. A., and Russell R. J. (2011) Conservation of a crystallographic interface suggests a role for β-sheet augmentation in influenza virus NS1 multifunctionality. Acta Crystallogr. F Struct. Biol. Crystallogr. Commun. 67, 858–861 10.1107/S1744309111019312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bornholdt Z. A., and Prasad B. V. (2006) X-ray structure of influenza virus NS1 effector domain. Nat. Struct. Mol. Biol. 13, 559–560 10.1038/nsmb1099 [DOI] [PubMed] [Google Scholar]
- 52. Xia S., Monzingo A. F., and Robertus J. D. (2009) Structure of NS1A effector domain from the influenza A/Udorn/72 virus. Acta Crystallogr. D Biol. Crystallogr. 65, 11–17 10.1107/S0907444908032186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hale B. G., Barclay W. S., Randall R. E., and Russell R. J. (2008) Structure of an avian influenza A virus NS1 protein effector domain. Virology 378, 1–5 10.1016/j.virol.2008.05.026 [DOI] [PubMed] [Google Scholar]
- 54. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 55. Kochs G., García-Sastre A., and Martínez-Sobrido L. (2007) Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81, 7011–7021 10.1128/JVI.02581-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Min J. Y., and Krug R. M. (2006) The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 7100–7105 10.1073/pnas.0602184103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 58. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Delano W. L. (2008) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto [Google Scholar]
- 62. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 63. Johnson B. A., and Blevins R. A. (1994) NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 64. Muhandiram D. R., and Kay L. E. (1994) Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Res. Series B 103, 203–216 10.1006/jmrb.1994.1032 [DOI] [Google Scholar]
- 65. Kile J. C., Ren R., Liu L., Greene C. M., Roguski K., Iuliano A. D., Jang Y, Jones J., Thor S., Song Y., Zhou S., Trock S. C., Dugan V., Wentworth D. E., Levine M. Z., Uyeki T. M., et al. (2017) Update: Increase in human infections with novel Asian Lineage Avian Influenza A(H7N9) viruses during the Fifth Epidemic — China, October 1, 2016–August 7, 2017. MMWR Morb, Mortal. Wkly. Rep. 66, 928–932 10.15585/mmwr.mm6635a2 [DOI] [PMC free article] [PubMed] [Google Scholar]