

Abstract
Relaxin family peptide (RXFPs) 1‐4 receptors modulate the activity of cyclic adenosine monophosphate (cAMP) to produce a range of physiological functions. RXFP1 and RXFP2 increase cAMP via Gαs, whereas RXFP3 and RXFP4 inhibit cAMP via Gαi/o. RXFP1 also shows a delayed increase in cAMP downstream of Gαi3. In this study we have assessed whether the bioluminescence resonance energy transfer (BRET)‐based biosensor CAMYEL (cAMP sensor using YFP‐Epac‐Rluc), which allows real‐time measurement of cAMP activity in live cells, will aid in understanding ligand‐ and cell‐specific RXFP signaling. CAMYEL detected concentration‐dependent changes in cAMP activity at RXFP1‐4 in recombinant cell lines, using a variety of ligands with potencies comparable to those seen in conventional cAMP assays. We used RXFP2 and RXFP3 antagonists to demonstrate that CAMYEL detects dynamic changes in cAMP by reversing cAMP activation or inhibition respectively, with real‐time addition of antagonist after agonist stimulation. To demonstrate the utility of CAMYEL to detect cAMP activation in native cells expressing low levels of RXFP receptor, we cloned CAMYEL into a lentiviral vector and transduced THP‐1 cells, which express low levels of RXFP1. THP‐1 CAMYEL cells demonstrated robust cAMP activation in response to relaxin. However, the CAMYEL assay was unable to detect the Gαi3‐mediated phase of RXFP1 cAMP activation in PTX‐treated THP‐1 cells or HEK293A cells with knockout of Gαs. Our data demonstrate that cytoplasmically‐expressed CAMYEL efficiently detects real‐time cAMP activation by Gαs or inhibition by Gαi/o but may not detect cAMP generated in specific intracellular compartments such as that generated by Gαi3 upon RXFP1 activation.
Keywords: cAMP, CAMYEL, GPCR, relaxin, RXFP, signalling
Abbreviations
- AC
adenylate cyclase
- BRET
bioluminescence resonance energy transfer
- cAMP
cyclic adenosine monophosphate
- CAMYEL
cAMP sensor using YFP‐Epac‐Rluc
- DAPI
4′,6‐diamidino‐2‐phenylindole
- Epac
exchange protein activated by cAMP
- FACS
fluorescence‐activated cell sorting
- FRET
Förster/fluorescence resonance energy transfer
- GPCR
G protein‐coupled receptor
- INSL
insulin‐like peptide
- PDE
phosphodiesterase
- PI3K
phosphoinositide 3‐kinase
- PKA
protein kinase A
- PKC
protein kinase C
- PTX
pertussis toxin
- Rluc
Renilla luciferase
- RXFP
relaxin family peptide receptor
- TAMRA
5‐carboxytetramethylrhodamine
- YFP
yellow fluorescent protein
1. INTRODUCTION
G protein‐coupled receptors (GPCRs) are one of the largest families of proteins in the human genome, and regulate most aspects of human physiology. GPCRs initiate a variety of intracellular signaling cascades in response to a diverse range of ligands, by coupling to effectors including G proteins and β‐arrestins. Biased signaling is a key research topic in GPCR drug development, referring to the ability of different ligands to favor coupling of the receptor to particular effectors, leading to activation of only a subset of the receptor's signaling pathways.1 A related concept is system bias, whereby different cell types can show preferential coupling to particular downstream signaling pathways.2
The relaxin family of peptides target GPCRs to produce a broad array of physiological functions across a range of tissues including the reproductive system, cardiovascular system, connective tissue, gastrointestinal tract, and brain (reviewed in 3, 4). In humans, the relaxin family of peptides contains seven members, including H1 relaxin, H2 relaxin, H3 relaxin, insulin‐like peptide (INSL) 3, INSL4, INSL5, and INSL6. The actions of relaxin peptides are mediated by relaxin family peptide receptors (RXFPs) 1‐4.5 H2 relaxin and INSL3 are the cognate ligands for RXFP16 and RXFP2,7 and relaxin‐3 and INSL5 are the cognate ligands for RXFP38 and RXFP4,9 respectively. The receptors for INSL4 and INSL6 have not been identified.
One of the primary second messengers involved in RXFP receptor signaling is 3′,5′‐cyclic adenosine monophosphate (cAMP). Effectors of cAMP include protein kinase A (PKA), exchange protein directly activated by cAMP (Epac), and cyclic nucleotide‐gated ion channels. Activation of a Gαs‐coupled GPCR leads to synthesis of cAMP via adenylate cyclases (ACs), whereas activation of a Gαi/o‐coupled receptor leads to inhibition of ACs. RXFP1 and RXFP2 couple to Gαs to increase cAMP activity, which is negatively modulated by GαoB.6, 10 Unusually, RXFP1 also couples to Gαi3 to increase cAMP downstream of Gβγ, PI3K, PKC‐ζ, and AC5 in a number of cell types.10, 11, 12, 13 In contrast, RXFP3 and RXFP4 couple to Gαi/o proteins to inhibit the production of cAMP.14, 15 However, patterns of cAMP activity also depend on the cell type being stimulated. For example, fibroblasts that natively express RXFP1 show little or no increases in cAMP activity when stimulated with an RXFP1 agonist.16, 17, 18
Traditional assays for detecting cAMP activity do not easily measure the temporal aspects of signaling, but measuring the kinetics of signaling aids accurate detection of biased signaling.19 A real‐time assay for cAMP activity that can be used in native cells will therefore be valuable for understanding ligand‐ and cell‐specific effects of RXFP signaling, as well as for screening novel ligands acting at these receptors. Fortunately, there is a real‐time, genetically‐encoded bioluminescence resonance energy transfer (BRET)‐based biosensor for cAMP activity. BRET is the transfer of energy from an excited luciferase donor to a fluorophore acceptor, when they are in close proximity. CAMYEL (cAMP sensor using YFP‐Epac‐Rluc) is a unimolecular BRET‐based biosensor for cAMP activity, consisting of truncated and catalytically inactive human Epac1 sandwiched between Rluc (the donor) and a monomeric and circularly permuted form of the YFP citrine (the acceptor).20 When cAMP is not present, Epac adopts a “closed” conformation, where the donor and acceptor are in close proximity, producing a BRET signal. When cAMP binds to Epac, the donor and acceptor move farther apart, reducing BRET. These changes in BRET can be monitored in real time in live cells at 37°C.
In this study, we demonstrated the use and versatility of CAMYEL for detecting real‐time cAMP activity at RXFP1‐4 and developed and validated a CAMYEL lentiviral vector for the transduction of native cells expressing RXFP1. However, we were unable to detect the delayed phase of cAMP activity at RXFP1 mediated by Gαi3 suggesting that the CAMYEL sensor, when expressed in the cytoplasm, may not detect cAMP generated in specific intracellular compartments.
2. MATERIALS AND METHODS
2.1. Reagents and materials
DMEM, DMEM/F12, RPMI 1640, trypsin/EDTA, L‐glutamine, penicillin/streptomycin, DPBS, Gateway enzymes, Lipofectamine 2000 (Invitrogen); FBS (Scientifix); BSA, forskolin, HEPES, PTX (Sigma‐Aldrich); Viafect (Promega); coelenterazine h (Nanolight); CulturPlate96 (PerkinElmer); RXFP1 agonists: H2 relaxin and the small molecule ML29021; RXFP2 agonists: INSL3 and H2 relaxin; antagonist: INSL3 B‐chain dimer22; RXFP3 agonists: analogue 2 (H3 relaxin analogue)23, peptide 5 (single‐chain H3 relaxin analogue)24, R3/I59; antagonist: R3 B1‐22R25; RXFP4 agonists: INSL5 and analogue 13 (INSL5 analogue).26
2.2. Synthesis of H2 relaxin labeled with TAMRA
Individual A‐ and B‐chains of H2 relaxin with appropriate regioselective S‐protection were synthesised using a CEM Liberty peptide synthesiser.27 The amine‐reactive fluorophore 5(6)‐TAMRA succinimidyl ester (Anaspec) was attached to the N‐terminus of the A‐chain sequence using a manual coupling procedure. Following purification of the crude peptides stepwise formation of the three disulfide bonds was conducted as described.27 The final TAMRA‐labelled H2 relaxin was subjected to characterization by RP‐HPLC and MALDI‐TOF mass spectrometry to confirm its high purity and correct molecular mass (m/z calculated [M+Na]+, 6415.456, found 6419.70).
2.3. Cell culture
Cell culture media was supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% L‐glutamine (complete media). Human embryonic kidney (HEK) 293T and 293A cells were grown in complete Dulbecco's modified Eagle medium (DMEM), Chinese hamster ovary (CHO‐K1) cells in complete DMEM/F12, and THP‐1 cells in complete RPMI 1640 medium (ATCC modification). All cells were cultured at 37°C with 5% CO2. Experiments were conducted in HEK293T cells stably expressing RXFP1 (HEK‐RXFP1), RXFP2 (HEK‐RXFP2), or RXFP3 (HEK‐RXFP3), THP‐1 cells endogenously expressing RXFP1, CHO‐K1 (FlpIn) cells stably expressing RXFP3 (CHO‐RXFP3),28 and CHO‐K1 cells stably expressing RXFP4 (CHO‐RXFP4).29 Experiments were also conducted in HEK293A parental cells and HEK293A cells that do not express the Gαs subunit (ΔGαs).30
2.4. Real‐time BRET assays for cAMP activity in live cells
Adherent cells were seeded on six‐well plates and were transfected the following day with CAMYEL in the pcDNA3.1/His vector,20 using Lipofectamine 2000 (HEK) or Viafect (CHO), according to the manufacturers’ instructions. HEK293A cells were also transfected with RXFP1 or the Gαs subunit in the pcDNA3.1/Zeo vector. The next day, cells were detached resuspended in complete media containing 25 mmol L−1 HEPES but no phenol red, reseeded into opaque white 96‐well CulturPlates and grown overnight. Non‐adherent THP‐1 cells stably expressing CAMYEL (see below) were plated directly onto CulturPlates at 80 000 (PTX assays) or 120 000 cells per well in complete RPMI 1640 containing 25 mmol L−1 HEPES but no phenol red. For all Gαi/o inhibition assays, PTX was added to wells at a final concentration of 100 ng mL−1 at least 16 hours before ligand stimulations. Peptides were diluted in complete phenol red free media and ML290 was diluted in DMSO. Media for cells stimulated with ML290 was also replaced with 0.5% FBS media before the assay.
Light emissions from the luciferase (Rluc) and YFP (citrine) were measured using a POLARstar Omega plate reader (BMG Labtech) at 37°C. Cells were pre‐incubated with the luciferase substrate coelenterazine h (Nanolight) at a final concentration of 5 μmol L−1 for 5 minutes to establish a baseline, followed by addition of vehicle or ligand. For Gαi/o assays, indicated concentrations of forskolin were added after a pre‐incubation of at least four minutes with agonist. Emissions were measured simultaneously at the wavelength windows 475/30 nm (Rluc) and 535/30 nm (citrine). Ligand‐induced BRET ratio was calculated by subtracting the ratio of citrine to Rluc emissions for vehicle‐treated wells from the same ratio for ligand‐treated cells.31 For CAMYEL, a decrease in ligand‐induced BRET ratio corresponds to an increase in cAMP, so negative values were converted to positive values and vice versa.
2.5. Analysis of BRET data
cAMP data were analysed using GraphPad Prism 7. Data represent the mean ± standard error of the mean (SEM) of at least three independent experiments. For time‐courses, data were normalized as a percentage of the maximum forskolin response, and were plotted against time, with the final pre‐incubation reading displayed at the zero‐time point (time of vehicle/ligand addition). Concentration‐response curves were generated by fitting non‐linear regressions to data from indicated time points, which generated pEC50 and E max values. For Gαs data, the curves were expressed as a percentage of maximum forskolin response, whereas for Gαi/o data, the curves were expressed as a percentage of the forskolin response at the same time point. Statistical significance was determined using an unpaired t‐test or one‐way ANOVA.
2.6. Molecular cloning of the CAMYEL biosensor into lentiviral vector
A lentiviral vector containing the CAMYEL sequence under the control of the Ef1α promoter was cloned using Gateway technology (Invitrogen). The CAMYEL‐coding sequence was flanked with Gateway‐compatible attB5 and attB2 sites and amplified by polymerase chain reaction (PCR) using the forward primer 5′ GGGGACAACTTTGTATACAAAAGTTGACCATGATCACTCTCGGCATGGAC 3′ and reverse primer 5′ GGGGACCACTTTGTACAAGAAAGCTGGGTATTACTGCTCGTTCTTCAGCACTC 3′. The PCR product was gel purified and cloned into the pDONR 221 P5‐P2 vector using BP Clonase II, to create the pENTR L5‐L2 CAMYEL entry clone. The entry clone was sequenced using Sanger sequencing to confirm the sequence was correct. Our pENTR L1‐R5 Ef1α entry clone was cloned along with the pENTR L5‐L2 CAMYEL entry clone into the pLenti X1 Zeo DEST vector32 using LR Clonase II, to create the pLenti X1 Ef1α CAMYEL expression clone.
2.7. Production of the THP‐1 CAMYEL stable cell lines
For transduction of THP‐1 suspension cells with CAMYEL lentivirus, a spinoculation protocol using purified lentivirus was followed. HEK293T cells were seeded on five 20 cm2 dishes at 12 million cells per dish. The following day, cells were transfected with the pLenti X1 Ef1α CAMYEL lentiviral vector and the lentiviral packaging and envelope plasmids pMDL, pRSV‐Rev, and pCMV‐VSV‐G, using Lipofectamine 2000. After 2 days, media containing secreted lentivirus was harvested and spun at 2000 RPM for 5 minutes, and supernatant containing lentivirus was filtered through a 0.45 μm filter and spun at 20 000 RPM at 4°C for 2.5 hours to concentrate the virus. Supernatant was removed and virus was resuspended in 50 μL of cold PBS per tube and stored at −80°C. 400 000 THP‐1 cells in 2 mL of cell culture medium were transduced with 2 μL of purified lentivirus and 4 μg mL−1 hexadimethrine bromide (Polybrene) for 20 minutes at room temperature, then centrifuged for 30 minutes at 800 rpm. The cell pellet was resuspended in media and cultured for 3 days before fluorescence‐activated cell sorting (FACS) analysis using a Becton Dickinson FACS Aria III to select viable cells that expressed CAMYEL (based on fluorescence of the YFP) and sort them into three expression levels (low, medium, and high). DAPI was used to stain dead cells, which were detected by laser excitation at 405 nm and emission at 450/40 nm, and were removed by gating. From the population of live single cells, YFP fluorescence was measured by laser excitation at 488 nm and emission at 530/30 nm.
2.8. Flow cytometry analysis and fluorescent ligand binding assay
A sample of the same HEK293A parental and ΔGαs cells that were co‐transfected with CAMYEL and RXFP1 and used for BRET assays, along with untransfected control cells, were detached from a plate and analysed using flow cytometry to confirm co‐expression of CAMYEL (by YFP expression) and RXFP1 (by fluorescent ligand binding), using a Beckman Coulter CytoFLEX S. To show specific RXFP1 binding cells were washed and resuspended in DPBS containing 1% bovine serum albumin (BSA) and incubated either alone, with TAMRA‐labeled H2 relaxin (20 nmol L−1), or with TAMRA‐labeled H2 relaxin and an excess of unlabeled H2 relaxin (2 μmol L−1), for 30 minutes at room temperature. Unbound relaxin was removed by centrifugation, and cells were resuspended in cold 1% BSA DPBS containing DAPI, and used for flow cytometry. Live cells expressing YFP and TAMRA were isolated as above, but with a few differences. DAPI emission was measured at 450/45 nm, YFP emission was measured at 525/40 nm, and TAMRA was measured by laser excitation at 561 nm and emission at 585/42. A quadrant gate was used to show there was a subpopulation of cells expressing YFP and binding TAMRA‐labeled relaxin. Data were analysed using FlowJo 10.
3. RESULTS
3.1. Detection of cAMP activity at RXFP1
To establish the range of intracellular cAMP activity that can be detected using CAMYEL HEK‐RXFP1 cells transiently transfected with CAMYEL were stimulated with a range of concentrations of forskolin, a diterpene that directly activates adenylate cyclase.33 HEK‐RXFP1 cells showed fast, concentration‐dependent increases in cAMP activity that peaked at about 5‐10 minutes and were sustained over 45 minutes (Figure 1A). A concentration‐response curve generated for 45 minutes showed a potency (pEC50) of 6.77 ± 0.07 (Figure 1B) and a maximal response at 10 μmol L−1 forskolin.
Figure 1.
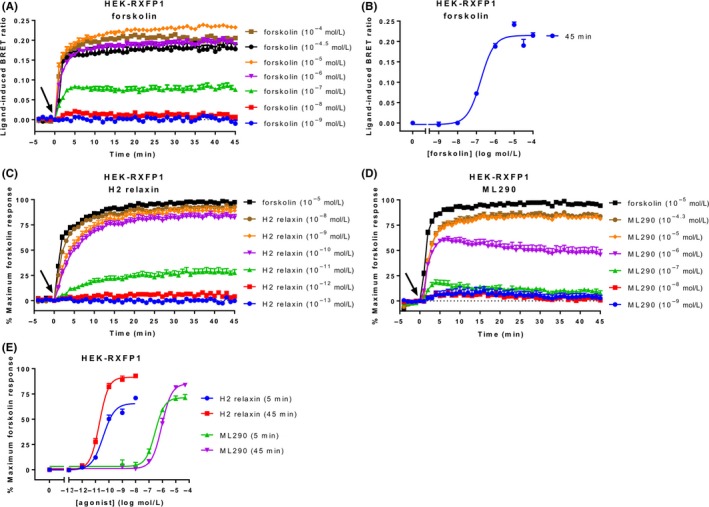
Ligand‐induced cAMP activity in HEK‐RXFP1 cells. HEK293T cells stably expressing RXFP1 were stimulated with vehicle, forskolin, or an RXFP1 agonist for 45 minutes. (A) Time‐course of forskolin‐induced cAMP activity. (B) Concentration‐response curve for forskolin at 45 minutes. Time‐course for (C) H2 relaxin‐ and (D) ML290‐induced cAMP activity. (E) Concentration‐response curves for H2 relaxin and ML290 at 5 and 45 minutes. Time of ligand or vehicle addition is represented by an arrow. Data represent mean ± SEM of three independent experiments
After establishing that CAMYEL can detect concentration‐dependent increases in cAMP activity in HEK‐RXFP1 cells, we investigated agonist activation. Stimulation with the native ligand H2 relaxin or ML29021 showed concentration‐dependent cAMP increases that also plateaued by 5‐10 minutes and were maintained for 45 minutes (Figure 1C and D). The responses approached or reached the maximum forskolin response, suggesting they are approaching the detection limit of the sensor. Notably, sub‐maximal concentrations of ML290 showed slow decreases in cAMP activity after peaking at about 5 minutes. Concentration‐response curves were generated for five and 45 minutes to compare ligand efficacy and potency values over time (Figure 1E, Table 1). Ligand efficacy was greater at 45 minutes compared to 5 minutes and was similar for both ligands. However, the potency of H2 relaxin was >10 000 times higher than that of ML290 at both time points (P < 0.01).
Table 1.
Efficacy and potency of agonists acting at RXFP1
| Time point | H2 relaxin | ML290 | |
|---|---|---|---|
| E max (% maximum forskolin) | 5 minutes | 65.65 ± 2.21 | 71.32 ± 2.02 |
| 45 minutes | 91.58 ± 1.84* | 85.22 ± 2.72** | |
| pEC50 | 5 minutes | 10.43 ± 0.08 | 6.54 ± 0.08*** |
| 45 minutes | 10.71 ± 0.05 | 6.05 ± 0.06*** |
HEK‐RXFP1 cells were stimulated for 45 minutes with ML290 or H2 relaxin. The efficacy (E max) and potency (pEC50) were determined at 5 and 45 minutes. Efficacy is expressed as a percentage of the maximum response to forskolin (10 μmol L−1). Data represent mean ± SEM of three independent experiments. *P < 0.01 compared to 5 minutes; **P < 0.05 compared to 5 minutes; ***P < 0.01 compared to H2 relaxin (unpaired t‐tests).
3.2. Detection of cAMP activity at RXFP2
A forskolin time‐course in HEK‐RXFP2 cells showed a similar profile to that seen in HEK‐RXFP1 cells, with rapid cAMP activity increases that became stable after 5‐10 minutes and were sustained for 45 minutes (Figure 2A). A concentration‐response curve for forskolin at 45 minutes showed a potency (pEC50) of 5.92 ± 0.08 (Figure 2B), which was nearly ten times lower than that seen in the HEK‐RXFP1 cells (P < 0.01), despite being isolated from the same parental cell line.
Figure 2.
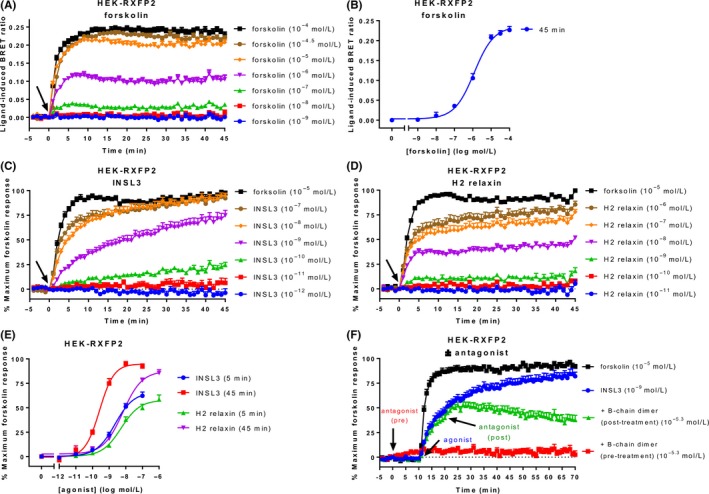
Ligand‐induced cAMP activity in HEK‐RXFP2 cells. HEK293T cells stably expressing RXFP2 were stimulated with ligands for 45 minutes. (A) Time‐course of forskolin‐induced cAMP activity. (B) Concentration‐response curve for forskolin at 45 minutes. Time‐course for (C) INSL3‐ and (D) H2 relaxin‐induced cAMP activity. (E) Concentration‐response curves for INSL3 and H2 relaxin at 5 and 45 minutes. (F) Blocking and reversibility of the CAMYEL signal was demonstrated by adding an RXFP2 antagonist (INSL3 B‐chain dimer) 10 minutes before or after agonist addition. Time of ligand or vehicle addition is represented by an arrow. Data represent mean ± SEM of three independent experiments
Stimulation of HEK‐RXFP2 cells with the native ligand INSL3 or H2 relaxin (which also activates RXFP2) induced concentration‐dependent increases in cAMP activity which approached the forskolin maximum response more slowly than agonists in HEK‐RXFP1 cells (Figure 2C and D). The two ligands showed different temporal patterns of cAMP activity which was most evident at sub‐maximal concentrations where INSL3 showed slow, sustained increases, whereas equivalent concentrations of H2 relaxin produced fast cAMP increases to 5 minutes which were then sustained. These differences were evident in concentration‐response curves generated at 5 and 45 minutes whereby the potency of INSL3 increased nearly 10‐fold between 5 and 45 minutes (P < 0.01), whereas the potency of relaxin stayed relatively stable for the duration of the time‐course (Figure 2E, Table 2).
Table 2.
Efficacy and potency of agonists acting at RXFP2
| Time point | INSL3 | H2 relaxin | |
|---|---|---|---|
| E max (% maximum forskolin) | 5 minutes | 65.92 ± 3.94 | 57.79 ± 3.02 |
| 45 minutes | 94.84 ± 2.89 | 87.53 ± 3.25 | |
| pEC50 | 5 minutes | 8.65 ± 0.12 | 8.28 ± 0.13 |
| 45 minutes | 9.58 ± 0.08* | 8.18 ± 0.09** |
HEK‐RXFP2 cells were stimulated for 45 minutes with INSL3 or H2 relaxin. The efficacy (E max) and potency (pEC50) were determined at 5 and 45 minutes. Efficacy is expressed as a percentage of the maximum response to forskolin (10 μmol L−1). Data represent mean ± SEM of three independent experiments. *P < 0.01 compared to 5 minutes; **P < 0.01 compared to INSL3 (unpaired t‐tests).
As stimulation of HEK‐RXFP1 and HEK‐RXFP2 both demonstrate rapid and sustained increases in cAMP, it is possible that the CAMYEL sensor is not dynamic and does not easily reverse the conformational change that occurs when cAMP binds. To simply assess whether the sensor was able to respond to rapid changes in cAMP we utilized a specific receptor antagonist. As there is no relevant RXFP1 antagonist we utilized a well‐characterized RXFP2 antagonist INSL3 B‐chain dimer.22 Blockade of RXFP2 by the antagonist 10 minutes before adding the agonist INSL3 abolished the ability of INSL3 to increase cAMP activity (Figure 2F). Importantly, addition of antagonist 10 minutes after addition of INSL3 resulted in real‐time reversal of the BRET signal highlighting the CAMYEL sensor is able to respond to dynamic changes in cAMP activity.
3.3. Inhibition of cAMP activity at RXFP3
We further validated the use of the CAMYEL assay for the Gαi/o‐coupled RXFP receptors, starting with RXFP3. As Gαi/o‐coupled receptors inhibit adenylate cyclase to reduce cAMP activity, we stimulated cells with forskolin and measured inhibition of forskolin‐stimulated cAMP. CHO‐RXFP3 cells transiently expressing CAMYEL were stimulated with either analogue 2 or peptide 5 for 4 minutes, before forskolin was added. 10 μmol L−1 forskolin was used, as it is the approximate EC50 for cAMP response in these cells (Figure S1), and has been used previously for measuring cAMP inhibition using CAMYEL in CHO cells at other GPCRs.19, 34 The two agonists showed similar, concentration‐dependent cAMP inhibition profiles (Figure 3A and B). Complete or near‐complete inhibition was reached by higher concentrations of agonist (100 nmol L−1‐10 μmol L−1) at about 20‐25 minutes, before cAMP activity started to slowly increase again. Inhibitory concentration‐response curves were generated for 10 and 45 minutes, expressed as a percentage of forskolin at each time point (Figure 3C, Table 3). Notably, ligand efficacy was not different at 10 or 45 minutes however, peptide 5 demonstrated greater potency at 45 minutes than at 10 minutes and was more potent than analogue 2 only at 45 minutes (P < 0.05).
Figure 3.
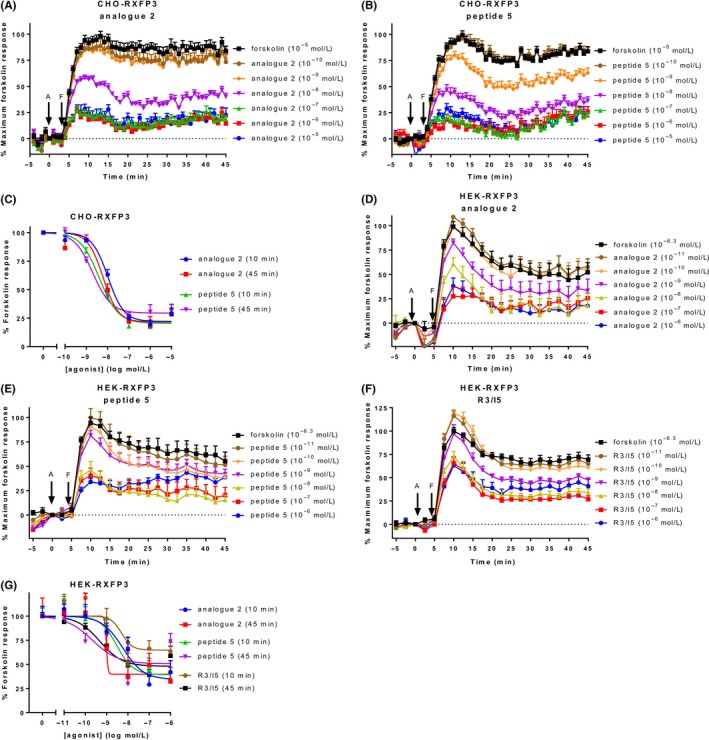
Ligand‐induced cAMP activity in cells expressing RXFP3. CHO‐K1 and HEK293T cells stably expressing RXFP3 (CHO‐RXFP3 and HEK‐RXFP3) were stimulated with vehicle, forskolin, and an RXFP3 agonist for 45 minutes. CHO‐RXFP3 cells were stimulated with (A) analogue 2 or (B) peptide 5, with addition of forskolin (10 μmol L−1) after 4 minutes. (C) Concentration‐response curves were generated for CHO‐RXFP3 at 10 and 45 minutes. HEK‐RXFP3 cells were stimulated with (D) analogue 2, (E) peptide 5, or (C) R3/I5, with addition of forskolin (500 nmol L−1) after five minutes. (F) Concentration‐response curves were generated for HEK‐RXFP3 cells at 10 and 45 minutes. Time of agonist or vehicle (A) and forskolin or vehicle (F) addition are represented by arrows. Concentration‐response data is expressed as a percentage of forskolin for the indicated time point. Data represent mean ± SEM of three independent experiments
Table 3.
Efficacy and potency of agonists acting at RXFP3 in CHO‐K1 cells
| Time point | Analogue 2 | Peptide 5 | |
|---|---|---|---|
| E max (% forskolin) (bottom) | 10 minutes | 22.06 ± 2.54 | 20.42 ± 2.79 |
| 45 minutes | 21.58 ± 3.93 | 29.4 ± 2.68 | |
| pEC50 | 10 minutes | 7.98 ± 0.07 | 8.39 ± 1.00 |
| 45 minutes | 8.21 ± 0.12 | 8.70 ± 0.11* |
CHO‐RXFP3 cells were stimulated with agonist for 45 minutes, with addition of forskolin four minutes after agonist addition. The efficacy (E max) and potency (pEC50) were determined at 10 and 45 minutes. Efficacy is expressed as a percentage of the response to forskolin (10 μmol L−1). Data represent mean ± SEM of three independent experiments. *P < 0.05 compared to analogue 2 (unpaired t‐test).
As our lab also had HEK‐RXFP3 cells, they were also tested for agonist‐induced cAMP inhibition together with another agonist, R3/I5. Forskolin is more potent in HEK293T than CHO‐K1 cells, so we used a lower concentration (500 nmol L−1, ~EC50) to allow detection of agonist‐induced inhibition (Figure S2). All agonists showed concentration‐dependent inhibition of forskolin‐stimulated cAMP, which reached maximum inhibition after about 15‐20 minutes and, unlike in CHO‐RXFP3 cells, were sustained for the duration of the time‐course (Figure 3D‐F). All the ligands demonstrated lower efficacy than in CHO‐RXFP3 cells especially R3/I5 which had significantly lower efficacy than the other ligands but only at 10 minutes (Figure 3G, Table 4). Importantly this ligand is not a partial agonist of cAMP activation9 and the data may reflect the strong potency of forskolin in HEK cells and the R3/I5 data stimulations being performed at a later time to the other agonists. Notably R3/I5 potency was the same as the other ligands as expected from previous data although the data from analogue 2 at 45 minutes was not of sufficient quality to generate accurate curve fits in Prism.
Table 4.
Efficacy and potency of agonists acting at RXFP3 in HEK293T cells
| Time point | Analogue 2 | Peptide 5 | R3/I5 | |
|---|---|---|---|---|
| E max (% forskolin) (bottom) | 10 minutes | 34.27 ± 9.834 | 39.18 ± 7.80 | 64.77 ± 6.23 |
| 45 minutes | 39.72 ± 10.82 | 50.57 ± 9.44 | 47.97 ± 4.61 | |
| pEC50 | 10 minutes | 8.24 ± 0.36 | 8.52 ± 0.32 | 8.30 ± 0.43 |
| 45 minutes | Ambiguous | 9.68 ± 0.65 | 9.32 ± 0.26 |
HEK‐RXFP3 cells were stimulated with agonist for 45 minutes, with addition of forskolin 5 minutes after agonist addition. The efficacy (E max) and potency (pEC50) were determined at 10 and 45 minutes. Efficacy is expressed as a percentage of the response to forskolin (10 μmol L−1). Data represent mean ± SEM of three independent experiments.
3.4. Modulation of CAMYEL signal using Gαi/o inhibition and antagonist
We also demonstrated the ability to inhibit or reverse the BRET signal at a Gαi/o receptor, in HEK‐RXFP3 cells. Treatment with the Gαi/o inhibitor pertussis toxin (PTX) (100 ng mL−1; 20 hours) abolished the ability of R3/I5 to inhibit forskolin‐induced cAMP activity at RXFP3 (Figure 4A). Treatment with the antagonist R3 B1‐22R (10 μmol L−1) after stimulation with agonist (30 minutes) rapidly reversed the BRET signal (Figure 4B), again demonstrating that we are able to measure dynamic cAMP changes using the CAMYEL sensor.
Figure 4.
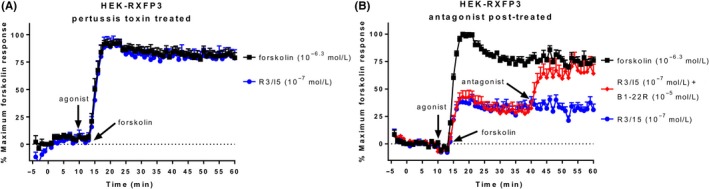
Blocking and reversing the CAMYEL signal in HEK‐RXFP3 cells. (A) Pre‐treatment of HEK‐RXFP3 cells with the Gαi/o inhibitor pertussis toxin (100 ng mL−1; 20 hours) abolished R3/I5‐induced inhibition of cAMP activity. (B) Treatment with the RXFP3 antagonist R3 B1‐22R after stimulation with R3/I5 (30 minutes) and forskolin showed reversal of the R3/I5 response in real time. Time of ligand or vehicle addition is represented by an arrow. Data represent the mean ± SEM of three independent experiments
3.5. Inhibition of cAMP activity at RXFP4
Finally, in our initial characterisation of CAMYEL in recombinant cells, we tested the ability of agonists to inhibit cAMP activity at RXFP4. We only had CHO‐K1 cells stably expressing RXFP4 (CHO‐RXFP4) available, and tested the native ligand INSL5 and analogue 13.26 As with CHO‐RXFP3, cells were pre‐incubated for 4 minutes with agonist, before adding forskolin at a concentration of 10 μmol L−1, which is the approximate EC50 for forskolin in these cells (Figure S3). The two agonists showed concentration‐dependent decreases in cAMP activity, which plateaued after about 30 minutes (Figure 5A and B). Concentration‐response curves were generated for 10 and 45 minutes, expressed as a percentage of forskolin activity at the same time point (Figure 5C). At 45 minutes, the efficacy of INSL5 was greater than that of analogue 13 but this was not statistically significant (P > 0.05) (Table 5). The lower efficacy of the ligands seen in these cells compared to ligands in CHO‐RXFP3 cells may be related to the lower expression of RXFP4 in the CHO‐RXFP4 cells. The potency of INSL5 (8.58 ± 0.16) was not significantly higher than analogue 13 (7.75 ± 0.49) (P > 0.05).
Figure 5.
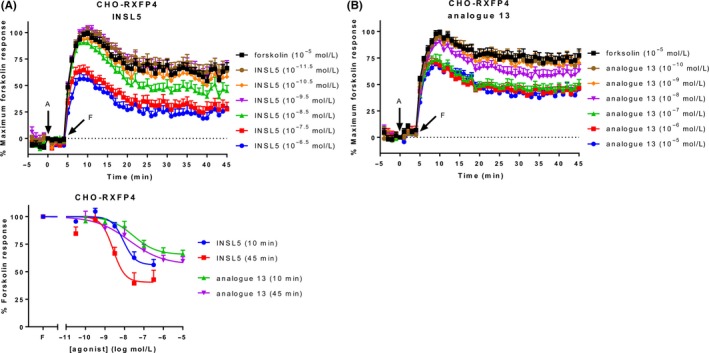
Ligand‐induced cAMP activity in CHO‐RXFP4 cells. CHO‐K1 cells stably expressing RXFP4 (CHO‐RXFP4) were stimulated with vehicle, forskolin, and an RXFP4 agonist for 45 minutes, with addition of forskolin (10 μmol L−1) 4 minutes after agonist addition. Time‐courses were generated for (A) INSL5 and (B) analogue 13. (C) Concentration‐response curves were generated for both agonists at 10 and 45 minutes. Time of agonist or vehicle (A) and forskolin or vehicle (F) addition are represented by arrows. Concentration‐response data is expressed as a percentage of forskolin for the indicated time point. Data represent mean ± SEM of three independent experiments
Table 5.
Efficacy and potency of agonists acting at RXFP4
| Time point | INSL5 | Analogue 13 | |
|---|---|---|---|
| E max (% forskolin) (bottom) | 10 minutes | 56.16 ± 3.92 | 65.51 ± 4.01 |
| 45 minutes | 40.42 ± 5.91 | 56.52 ± 7.49 | |
| pEC50 | 10 minutes | 8.05 ± 0.17 | 7.55 ± 0.28 |
| 45 minutes | 8.58 ± 0.16 | 7.75 ± 0.49 |
CHO‐RXFP4 cells were stimulated with agonist for 45 minutes, with addition of forskolin 4 minutes after agonist addition. The efficacy (E max) and potency (pEC50) were determined at 10 and 45 minutes. Efficacy is expressed as a percentage of the response to forskolin (10 μmol L−1). Data represent mean ± SEM of three independent experiments.
3.6. Investigation of RXFP1 signaling in a native expressing cell line using CAMYEL
So far, we have shown that CAMYEL can detect cAMP activity with high sensitivity in cells that recombinantly overexpress the receptor of interest. However, we have not demonstrated its use in cells that natively express the receptor at lower levels. To enable the use of CAMYEL in all types of mammalian cells, we cloned CAMYEL into the pLenti X1 Zeo DEST vector under control of the constitutive Ef1α promoter using Gateway cloning, to create the pLenti X1 Ef1α CAMYEL expression clone (see Methods). We showed that transfection of this vector allows robust expression of CAMYEL (data not shown) and therefore produced lentivirus for the transduction of cells.
Human monocytic leukaemia THP‐1 cells have commonly been used for detecting relaxin induced cAMP activity even though they have low endogenous expression of RXFP1 (~275 receptors per cell35), in contrast to HEK‐RXFP1 cells, which have ~50 000‐100 000 receptors per cell.36 As THP‐1 cells are a non‐adherent cell line we used a spinoculation protocol involving purified lentivirus to transduce the cells, followed by sorting using FACS into populations expressing low, medium, or high levels of CAMYEL (Figure 6). The “high” CAMYEL population was initially tested for concentration‐dependent increases in forskolin‐induced cAMP activity (Figure 7A). Forskolin produced rapid dose‐dependent increases in cAMP with a maximum response at 100 μmol L−1 and a higher potency at 5 minutes (5.29 ± 0.03) than at 45 minutes (4.93 ± 0.05) (P < 0.01) (Figure 7B).
Figure 6.
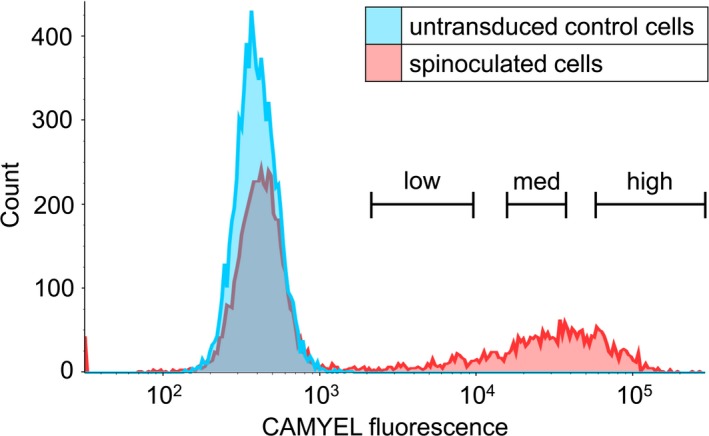
Representation of fluorescence‐activated cell sorting (FACS) of transduced THP‐1 CAMYEL cells. THP‐1 cells were transduced by spinoculation with CAMYEL lentivirus and were sorted to remove untransduced cells. Single, live cells were isolated, and sorted by expression of YFP into low, medium, and high levels of CAMYEL expression, based on YFP fluorescence. Cells with YFP expression that was no greater than control THP‐1 cells were removed
Figure 7.
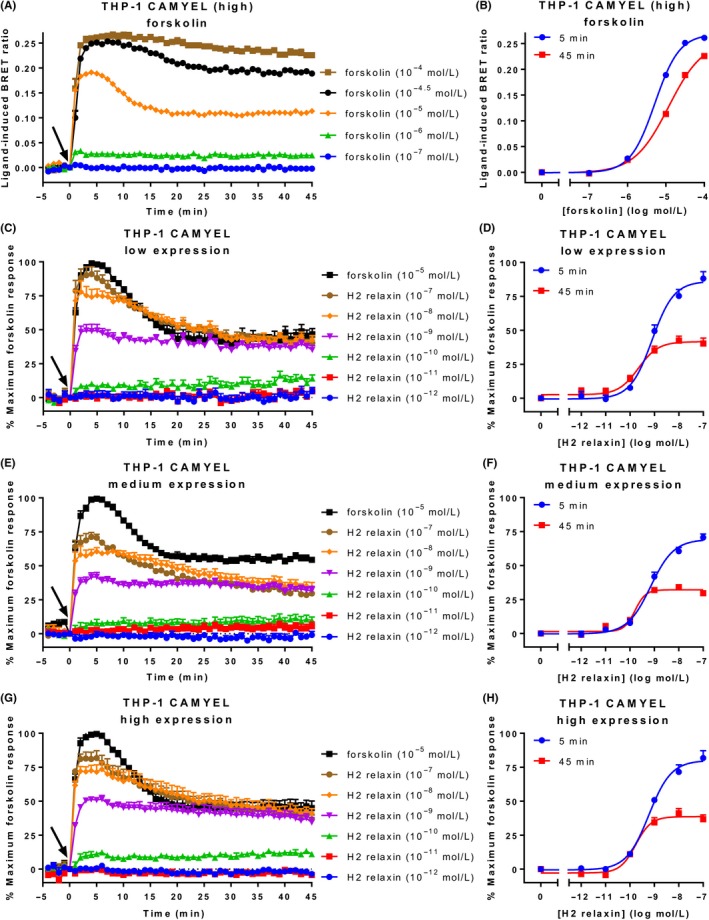
Ligand‐induced cAMP activity in THP‐1 CAMYEL cells. THP‐1 cells that endogenously express RXFP1 and stably express high, medium, or low levels of CAMYEL (THP‐1 CAMYEL) were stimulated with vehicle, forskolin, or H2 relaxin for 45 minutes. (A) Time‐course for forskolin activity in high expression cells. (B) Concentration‐response curves for forskolin at 5 and 45 minutes. (C, E, G) Time‐courses for H2 relaxin‐induced cAMP activation in the low‐, medium‐, and high‐expressing cells. (D, F, H) Concentration‐response curves for H2 relaxin at 5 and 45 minutes in the low‐, medium‐, and high‐expressing cells. Time of ligand or vehicle addition is represented by an arrow. Data represent mean ± SEM of three independent experiments
Stimulation of the three sorted THP‐1 CAMYEL populations with H2 relaxin for 45 minutes revealed robust concentration‐dependent increases in cAMP activity (Figure 7C, E, G). The higher concentrations of H2 relaxin (10 and 100 nmol L−1) showed more transient responses than seen in HEK‐RXFP1 cells, as cAMP reached a maximum by 5 minutes before declining to less than half of the maximum forskolin response. Notably, the response to H2 relaxin did not approach or reach the maximal detectable response by the CAMYEL in THP‐1 cells (Figure 7A). Concentration‐response curves were generated for 5 and 45 minutes to compare the three populations (Figure 7D, F, H). Notably, there was no difference in H2 relaxin efficacy or potency between the cells, indicating the level of CAMYEL had no effect on the response to H2 relaxin (Table 6). Ligand efficacy was greater at 5 minutes as expected from the shape of the response curves, but ligand potency was higher at 45 minutes (P < 0.05).
Table 6.
Efficacy and potency of H2 relaxin in the THP‐1 CAMYEL stable cell line
| Time point | Low CAMYEL | Medium CAMYEL | High CAMYEL | |
|---|---|---|---|---|
| E max (% maximum forskolin) | 5 minutes | 86.11 ± 2.23 | 69.29 ± 2.52 | 80.02 ± 2.93 |
| 45 minutes | 41.61 ± 2.29* | 32.15 ± 1.87* | 38.64 ± 1.77* | |
| pEC50 | 5 minutes | 9.10 ± 0.09 | 9.15 ± 0.08 | 9.23 ± 0.08 |
| 45 minutes | 9.67 ± 0.15* | 9.80 ± 0.21* | 9.77 ± 0.10* |
THP‐1 CAMYEL cells sorted into three levels (high, medium, low) of CAMYEL expression were stimulated with H2 relaxin for 45 minutes. The efficacy (E max) and potency (pEC50) was determined for each expression level at 5 and 45 minutes. Efficacy is expressed as a percentage of the maximum response to forskolin (10 μmol L−1), with vehicle expressed as 0%. Data represent mean ± SEM of three independent experiments. *P < 0.05 compared to 5 minutes (unpaired t‐test).
Relaxin‐stimulated intracellular cAMP accumulation through RXFP1 has been shown to be modulated by three G proteins. Immediate increases in cAMP are mediated by Gαs and negatively modulated by GαoB,10 whereas there is a delayed increase in cAMP activity after ~15 minutes mediated by Gαi3 in a number of cell types, including HEK‐RXFP1 and THP‐1.10, 12, 13 However, using the CAMYEL assay we have not detected a biphasic increase in cAMP activity at RXFP1. To investigate further, we tested the effect of treatment with PTX (100 ng mL−1; 16‐18 hours) on H2 relaxin‐induced cAMP activity over time in THP‐1 CAMYEL cells (Figure 8A and B). Comparing the H2 relaxin responses as a percentage of the forskolin response at 5 minutes showed a trend toward higher cAMP activity in PTX‐treated cells at higher concentrations of H2 relaxin, suggesting that PTX is inhibiting the GαoB inhibition, but this was not significant (P > 0.05) (Figure 8C). At 45 minutes, there was no difference between treated and untreated cells (Figure 8D), suggesting we are not seeing a delayed Gαi3‐mediated enhancement of cAMP.
Figure 8.
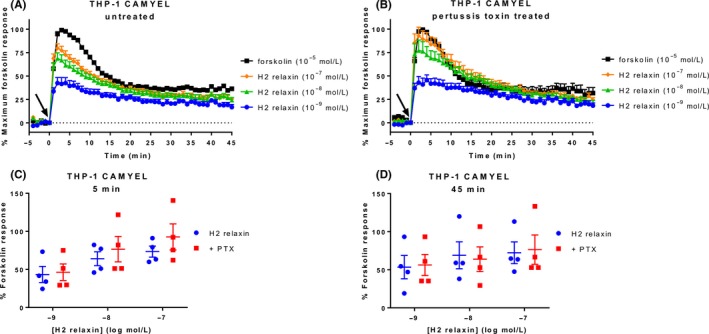
Effect of pertussis toxin on cAMP activity in THP‐1 CAMYEL cells. THP‐1 CAMYEL (high expression) were either (A) untreated or (B) treated with pertussis toxin (100 ng mL−1; 16‐18 hours), and stimulated with vehicle, forskolin, or H2 relaxin for 45 minutes. Treated and untreated cells were compared at (C) 5 and (D) 45 minutes. Time of ligand or vehicle addition is expressed by an arrow. Data in (C) and (D) are expressed as a percentage of the forskolin response at the same time point. Data represent the mean ± SEM of four independent experiments
3.7. Dissecting RXFP1 cAMP activity using HEK293A ΔGαs cells
To further investigate this potential lack of Gαi3 cAMP effect we used novel HEK293A cells with CRISPR/Cas9‐mediated knockout of Gαs members (GNAS and GNAL genes).30 We first produced forskolin concentration‐response data for the HEK293A parental and Gαs knockout (ΔGαs) cells. HEK293A parental cells showed robust increases in cAMP activity that were very similar to those seen earlier in HEK293T cells expressing RXFP1 or RXFP2 (Figure 9A). However, the ΔGαs cells showed very weak cAMP activity, requiring relatively high concentrations of forskolin (>10 μmol L−1) to detect a cAMP increase, which was only transient (Figure 9B). As forskolin has a lower affinity for adenylate cyclase in the absence of Gαs,37 we transfected Gαs into these cells to show that forskolin potency could be rescued by re‐expression of Gαs (Figure 9C). At 45 minutes, the potency of forskolin in HEK293A parental cells was 5.91 ± 0.04. In ΔGαs cells with re‐expression of Gαs, the potency was 7.1 ± 1.29 (Figure 9D).
Figure 9.
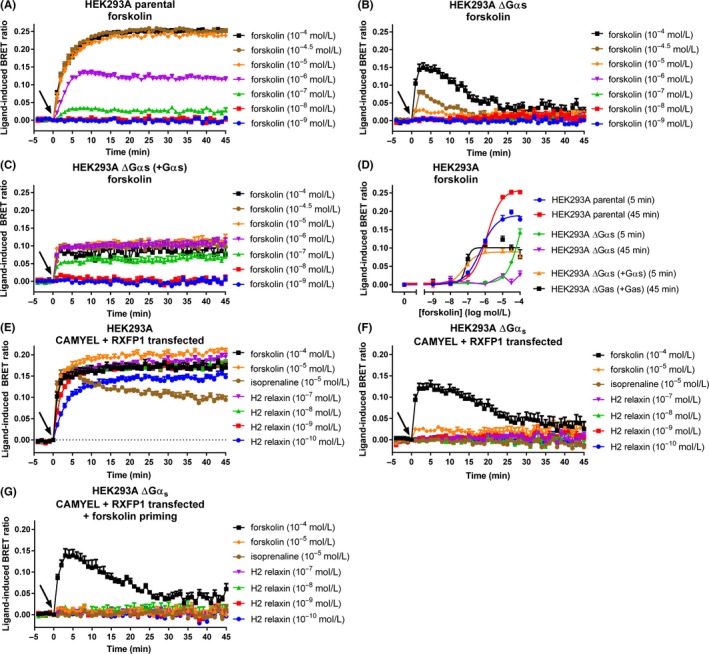
cAMP activity in HEK293A parental and Gαs knockout (ΔGαs) cells. (A) HEK293A parental cells, (B) HEK293A ΔGαs cells, and (C) HEK293A ΔGαs cells with Gαs transfected in, were transfected with CAMYEL and stimulated with vehicle or forskolin for 45 minutes. (D) Concentration‐response curves were generated for forskolin at 5 and 45 minutes. (E) HEK293A parental and (F) HEK293A ΔGαs cells were co‐transfected with CAMYEL and RXFP1, and were stimulated with vehicle, forskolin, isoprenaline, or H2 relaxin for 45 minutes. (G) HEK293A ΔGαs cells were primed with forskolin (33 μmol L−1) to compensate for the loss of Gαs, before adding forskolin, isoprenaline, or H2 relaxin. Time of ligand or vehicle addition is represented by an arrow. Data represent mean ± SEM of three independent experiments
Parental HEK293A and ΔGαs cells were then transiently co‐transfected with CAMYEL and RXFP1 to detect the Gαi3‐mediated phase of cAMP activity. Isoprenaline (10 μmol L−1), which targets Gαs‐coupled β‐adrenoceptors endogenous to HEK293 cells, was used as a control for these experiments to confirm lack of Gαs activity. As expected, the response to isoprenaline was robust in the parental cells (Figure 9E), but was abolished in the ΔGαs cells (Figure 9F). Similarly, the response to H2 relaxin (100 pmol L−1‐100 nmol L−1) was robust in parental cells, but was completely abolished in the ΔGαs cells. To test whether pre‐activation of adenylate cyclase is required to detect the delayed Gαi3‐mediated phase of cAMP activity, we primed the ΔGαs cells with forskolin (33 μmol L−1) to compensate for the loss of Gαs, before adding the ligands (Figure 9G). However, no response to H2 relaxin was detectable. Additionally, we used flow cytometry to confirm that a subpopulation of cells from the same transfected population used in the assay were co‐expressing CAMYEL (by YFP expression of the sensor) and RXFP1 (by binding of fluorescent TAMRA‐H2 relaxin) (Figure S4), demonstrating that the lack of response is not due to CAMYEL and RXFP1 not being present in the same cells.
4. DISCUSSION
Biased signaling is the ability of a ligand to selectively activate particular signaling pathways downstream of a GPCR in the same cell type, compared to the endogenous ligand. Biased signaling has been shown to occur at relaxin family peptide receptors. For example, at RXFP1, H2 relaxin promotes ERK1/2 activation but ML290 does not.16 Relatedly, system bias has been shown, particularly for RXFP1. For example, HEK‐RXFP1 cells signal via cAMP but not cGMP, whereas fibroblasts that natively express RXFP1 signal via cGMP but not cAMP, and human vascular cells that express RXFP1 signal through both.16 Measuring the temporal aspects of signaling facilitates accurate detection of biased ligands,19 and the use of native cells will facilitate understanding of how relaxin family peptide receptors signal in different cell backgrounds. Therefore, in this study, we set out to demonstrate the use of the BRET‐based biosensor CAMYEL for real‐time cAMP signaling at relaxin family peptide receptors, initially in recombinant cells, but with the goal of detecting cAMP in native cells.
RXFP receptor signaling has typically been measured in HEK293T or CHO‐K1 cells expressing the receptor of interest, using traditional end point cAMP assays, such as a cAMP response element (CRE) reporter gene assay, or accumulation kits including AlphaScreen and Homogeneous Time Resolved Fluorescence (HTRF). However, the CRE reporter gene assay provides a readout of total downstream activity from a combination of signaling pathways, including cAMP/PKA, Ca2+/CaM, Ras/ERK, and PI3K/Akt,38 and is therefore inappropriate for detecting bias. Although kit‐based assays directly measure cAMP, they are laborious and involve stimulations in the presence of phosphodiesterase inhibitors to stop the degradation of cAMP, and therefore do not reveal the kinetics of cAMP activity. We have demonstrated that CAMYEL is a sensitive and robust alternative for detecting real‐time cAMP activity in cells in the absence of phosphodiesterase inhibitors. Notably, we were able to detect both Gαs‐mediated cAMP activation and Gαi/o‐mediated cAMP inhibition for all RXFP receptors and multiple cell types, including recombinant and native cells. At the Gαs‐coupled RXFP1 and RXFP2 stably expressed in HEK293T cells, we showed sustained concentration‐dependent increases in cAMP activity that differed over time between the agonists. At RXFP3 and RXFP4 expressed in CHO‐K1 or HEK293T cells, we showed sustained concentration‐dependent inhibition of forskolin‐stimulated cAMP activity, with more similar patterns of cAMP inhibition induced by agonists. Agonists at the four receptors also showed similar potencies to those already seen using traditional cAMP end point assays.
The sustained cAMP activity seen at RXFP1 and RXFP2 is consistent with the finding that these receptors lack β‐arrestin‐mediated desensitization and show poor internalization in response to ligand stimulation.39 However, we also saw sustained cAMP inhibition at RXFP3 (particularly in HEK293T cells) and RXFP4, which have been shown to interact with β‐arrestins and internalize after stimulation with their endogenous ligands.14, 15 Although the original characterization of CAMYEL in RAW 264.7 murine macrophage cells showed that CAMYEL can report substantial and rapid decreases in BRET signal,20 studies on some GPCRs, including the cannabinoid 1,40 dopamine D2L,19 and mu‐opioid34 receptors, have also shown relatively sustained changes in CAMYEL signal over extended periods of time. However, most studies using CAMYEL do not show time‐course data and some include phosphodiesterase inhibitors in the assay to stop the breakdown of cAMP. As sustained changes in the BRET signal have been observed in the present study and other studies, it was thought possible that cAMP does not easily dissociate from the sensor, or that the sensor does not easily reverse its conformation and therefore does not precisely detect real‐time fluctuations in cAMP levels. Therefore, we demonstrated reversibility of the BRET signal at RXFP2 and RXFP3 by adding an antagonist after pre‐incubation with agonist, which both showed real‐time reversal of the signal. These experiments demonstrated that the sensor is dynamic and able to detect changes in BRET signal reflective of changes in cAMP levels, over extended periods of time.
Targeting RXFP receptors with drugs has potential for a range of therapeutic applications (reviewed in 4). Much effort has therefore been put into designing analogues of relaxin family peptides, including agonists and antagonists used in the present study.41 In particular, RXFP1 is a promising therapeutic target for cardiovascular diseases and fibrosis. Novel ligands acting at RXFP1, including peptide analogues (e.g., 42) and small molecules,21 have been screened in THP‐1 cells due to the robust cAMP activation by relaxin in these cells. Our newly‐developed THP‐1 CAMYEL cell line will be a valuable tool for drug discovery targeting RXFP1, as it is sensitive and able to detect concentration‐dependent increases in cAMP activity over time in the absence of phosphodiesterase inhibitors. Importantly, the assay showed a similar potency of H2 relaxin to that observed using traditional cAMP assays such as enzyme immunoassay43 and HTRF cAMP assays.42 The THP‐1 CAMYEL assay also streamlines the process of screening ligands, as these suspension cells can be plated out on the day of the assay, and no extra steps for lysis or addition of detection reagents are required. Additionally, while recombinant HEK‐RXFP1 cells showed cAMP responses that reached or closely approached the ceiling set by forskolin, the native THP‐1 CAMYEL cells showed that the level of agonist‐induced cAMP activity was clearly within the dynamic range of the assay. The sorted THP‐1 CAMYEL cells also showed that the level of biosensor present in the cells did not affect the observed potency of H2 relaxin, suggesting that the expression level of CAMYEL is of little importance and does not need to be tightly controlled. The successful use of CAMYEL in a cell line that has low RXFP1 expression also suggests that the lentiviral CAMYEL construct can be utilized to transduce other native cells, including human primary cells, in order to understand RXFP1 signaling in the context of human diseases.
RXFP1 has been shown to couple to at least three G proteins to modulate cAMP activity in some human cells. Gαs is the primary mediator of cAMP, and its actions are negatively modulated by GαoB. 10 Further increases in cAMP activity have also been shown to occur in a number of cell types, including THP‐1,13 HEK‐RXFP1,10 and some human primary vascular cell types44 downstream of Gαi3, Gβγ, PI3K, PKCζ, and adenylate cyclase 5.10, 12, 13 Coupling of RXFP1 to Gαi3 has also been confirmed by BRET protein‐protein interaction studies,16 and is dependent on localization of RXFP1 and Gαi/o in membrane raft microdomains.45 This finding suggests that cAMP activity at RXFP1 is compartmentalized, which is consistent with the finding that in HEK‐RXFP1 cells, Gαs‐induced cAMP activity affects CRE transcription, whereas Gαi3‐induced cAMP activity affects NFκB transcription.46 In the present study, PTX inhibition in THP‐1 CAMYEL cells demonstrated a potential inhibition by GαoB at 5 minutes which was not significant but no effect at any other time point indicated a complete absence of a Gαi3 effect. We then used HEK293A cells that do not express the Gαs subunit to further investigate. Although the potency of forskolin was diminished in these cells, re‐expression of Gαs restored the potency of forskolin, showing that the diminished forskolin response was not an off‐target effect of the gene editing in these cells. However, stimulation of these cells with H2 relaxin when RXFP1 and CAMYEL were co‐expressed did not show any increases in cAMP activity, including when adenylate cyclase was primed using forskolin to compensate for the loss of Gαs. Therefore, we have been unable to reproduce the Gαi3‐mediated increase in cAMP activity at RXFP1 using the CAMYEL assay. It is therefore possible that there is a localized pool of cAMP that CAMYEL is unable to detect. The original paper that described CAMYEL20 suggested that CAMYEL is expressed throughout the cytoplasm, as it uses a cytosolic mutant of Epac1.47 Further studies have shown cytosolic distribution of CAMYEL via confocal microscopy and western blot.48 We have confirmed this using confocal microscopy in HEK293T cells stably expressing CAMYEL (data not shown). Importantly, there is precedent for the inability of cytosolic biosensors to detect localized hotspots of cAMP activity. For example, in GH3B6 cells treated with vasoactive intestinal peptide, FRET‐based biosensors for cAMP activity located around the cytosol or localized to the plasma membrane reported robust cAMP increases that were not detected by an AC8‐localized sensor.49 In contrast, increases in cAMP activity after stimulation with thyrotropin‐releasing hormone were detected by the AC8‐localized sensor but not the cytosolic or membrane forms.49
In summary, CAMYEL is a valuable tool for the direct detection of real‐time cAMP activity at the relaxin family peptide receptors 1‐4. We have shown robust cAMP activation and inhibition at Gαs‐ and Gαi/o‐coupled receptors when CAMYEL was transfected into recombinant cell lines, and have also shown robust cAMP activity in a cell line that expresses low native levels of RXFP1, when CAMYEL was delivered virally. We have also detected differences in cAMP activity between different ligands and cell types, which can be modulated by antagonists and an inhibitor. However, we could not replicate the biphasic cAMP response at RXFP1 using this assay, which may suggest compartmentalization of cAMP activity at RXFP1, consistent with previous findings. The development of a lentiviral CAMYEL construct will allow the transduction of any mammalian cell for the real‐time analysis of cAMP signaling.
DISCLOSURES
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
ACKNOWLEDGEMENTS
This research was supported by National Health and Medical Research Council of Australia project grants [1100676] and [1043750] (RADB) and the Victorian Government Operational Infrastructure Support Program. AI was funded by Japan Agency for Medical Research and Development (AMED) Grant Number JP17gm5910013 and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 17K08264. RADB is supported by an NHMRC Research Fellowship. The authors thank Tania Ferraro and Sharon Layfield for technical assistance; Dr Michelle Halls and Dr Meritxell Canals (Monash Institute of Pharmacological Sciences, Parkville, Victoria, Australia) for helpful suggestions on the manuscript and provision of the CAMYEL plasmid, respectively; and V. Jameson and J. Kie (MBC Flow Cytometry Facility) for assistance with FACS.
Valkovic AL, Leckey MB, Whitehead AR, et al. Real‐time examination of cAMP activity at relaxin family peptide receptors using a BRET‐based biosensor. Pharmacol Res Perspect. 2018;e00432 10.1002/prp2.432
Contributor Information
Martina Kocan, Email: martina.kocan@florey.edu.au.
Ross A. D. Bathgate, Email: bathgate@florey.edu.au.
References
- 1. Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven‐transmembrane receptors. Nat Rev Drug Discov. 2010;9:373‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17:243‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bathgate RA, Kocan M, Scott DJ, et al. The relaxin receptor as a therapeutic target – perspectives from evolution and drug targeting. Pharmacol Ther. 2018;187:114‐132. [DOI] [PubMed] [Google Scholar]
- 4. Bathgate RAD, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405‐480. [DOI] [PubMed] [Google Scholar]
- 5. Bathgate RA, Ivell R, Sanborn BM, Sherwood OD, Summers RJ. International Union of Pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol Rev. 2006;58:7‐31. [DOI] [PubMed] [Google Scholar]
- 6. Hsu SY, Nakabayashi K, Nishi S, et al. Activation of orphan receptors by the hormone relaxin. Science. 2002;295:671‐674. [DOI] [PubMed] [Google Scholar]
- 7. Kumagai J, Hsu SY, Matsumi H, et al. INSL3/Leydig insulin‐like peptide activates the LGR8 receptor important in testis descent. J Biol Chem. 2002;277:31283‐31286. [DOI] [PubMed] [Google Scholar]
- 8. Liu C, Eriste E, Sutton S, et al. Identification of relaxin‐3/INSL7 as an endogenous ligand for the orphan G‐protein‐coupled receptor GPCR135. J Biol Chem. 2003;278:50754‐50764. [DOI] [PubMed] [Google Scholar]
- 9. Liu C, Kuei C, Sutton S, et al. INSL5 is a high affinity specific agonist for GPCR142 (GPR100). J Biol Chem. 2005;280:292‐300. [DOI] [PubMed] [Google Scholar]
- 10. Halls ML, Bathgate RAD, Summers RJ. Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol Pharmacol. 2006;70:214‐226. [DOI] [PubMed] [Google Scholar]
- 11. Nguyen BT, Dessauer CW. Relaxin stimulates cAMP production in MCF‐7 cells upon overexpression of type V adenylyl cyclase. Ann N Y Acad Sci. 2005a;1041:296‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen BT, Dessauer CW. Relaxin stimulates protein kinase C ζ translocation: requirement for cyclic adenosine 3′,5′‐monophosphate production. Mol Endocrinol. 2005b;19:1012‐1023. [DOI] [PubMed] [Google Scholar]
- 13. Nguyen BT, Yang L, Sanborn BM, Dessauer CW. Phosphoinositide 3‐kinase activity is required for biphasic stimulation of cyclic adenosine 3′,5′‐monophosphate by relaxin. Mol Endocrinol. 2003;17:1075‐1084. [DOI] [PubMed] [Google Scholar]
- 14. Ang SY, Hutchinson DS, Patil N, et al. Signal transduction pathways activated by insulin‐like peptide 5 at the relaxin family peptide RXFP4 receptor. Br J Pharmacol. 2016;174:1077‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kocan M, Sarwar M, Hossain MA, Wade JD, Summers RJ. Signalling profiles of H3 relaxin, H2 relaxin and R3(BΔ23‐27)R/I5 acting at the relaxin family peptide receptor 3 (RXFP3). Br J Pharmacol. 2014;171:2827‐2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kocan M, Sarwar M, Ang SY, et al. ML290 is a biased allosteric agonist at the relaxin receptor RXFP1. Sci Rep. 2017;7:2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mookerjee I, Hewitson TD, Halls ML, et al. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009;23:1219‐1229. [DOI] [PubMed] [Google Scholar]
- 18. Samuel CS, Unemori EN, Mookerjee I, et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125‐4133. [DOI] [PubMed] [Google Scholar]
- 19. Klein Herenbrink C, Sykes DA, Donthamsetti P, et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. 2016;7:10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang LI, Collins J, Davis R, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1‐phosphate/G13 pathway. J Biol Chem. 2007;282:10576‐10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao J, Huang Z, Chen CZ, et al. Identification and optimization of small‐molecule agonists of the human relaxin hormone receptor RXFP1. Nat Commun. 2013;4:1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shabanpoor F, Zhang S, Hughes RA, et al. Design and development of analogues of dimers of insulin‐like peptide 3 B‐chain as high‐affinity antagonists of the RXFP2 receptor. Biopolymers. 2011;96:81‐87. [DOI] [PubMed] [Google Scholar]
- 23. Shabanpoor F, Hossain M, Ryan P, et al. Minimization of relaxin‐3 leading to high affinity analogues with increased selectivity for relaxin‐family peptide 3 receptor (RXFP3) over RXFP1. J Med Chem. 2012;55:1671‐1681. [DOI] [PubMed] [Google Scholar]
- 24. Hojo K, Hossain MA, Tailhades J, et al. Development of a single‐chain peptide agonist of the relaxin‐3 receptor RXFP3 using hydrocarbon stapling. J Med Chem. 2016;59:7445‐7456. [DOI] [PubMed] [Google Scholar]
- 25. Haugaard‐Kedström L, Shabanpoor F, Hossain M, et al. Design, synthesis and characterization of a single‐chain peptide antagonist for the relaxin‐3 receptor RXFP3. J Am Chem Soc. 2011;133:4965‐4974. [DOI] [PubMed] [Google Scholar]
- 26. Patil NA, Hughes RA, Rosengren KJ, et al. Engineering of a novel simplified human insulin‐like peptide 5 agonist. J Med Chem. 2016;59:2118‐2125. [DOI] [PubMed] [Google Scholar]
- 27. Hossain MA, Bathgate RAD, Kong CK, et al. Synthesis, conformation, and activity of human insulin‐like peptide 5 (INSL5). ChemBioChem. 2008;9:1816‐1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Westhuizen ET, Werry TD, Sexton PM, Summers RJ. The relaxin family peptide receptor 3 activates extracellular signal‐regulated kinase 1/2 through a protein kinase C‐dependent mechanism. Mol Pharmacol. 2007;71:1618‐1629. [DOI] [PubMed] [Google Scholar]
- 29. Belgi A, Bathgate RAD, Kocan M, et al. Minimum active structure of insulin‐like peptide 5. J Med Chem. 2013;56:9509‐9516. [DOI] [PubMed] [Google Scholar]
- 30. Stallaert W, van der Westhuizen ET, Schönegge A‐M, et al. Purinergic receptor transactivation by the β2‐adrenergic receptor increases intracellular Ca2 + in nonexcitable cells. Mol Pharmacol. 2017;91:533‐544. [DOI] [PubMed] [Google Scholar]
- 31. Kocan M, See HB, Seeber RM, Eidne KA, Pfleger KDG. Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein‐coupled receptors in live cells. J Biomol Screen. 2008;13:888‐898. [DOI] [PubMed] [Google Scholar]
- 32. Campeau E, Ruhl VE, Rodier F, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009;4:e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci. 1981;78:3363‐3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson GL, Lane JR, Coudrat T, Sexton PM, Christopoulos A, Canals M. Systematic analysis of factors influencing observations of biased agonism at the mu‐opioid receptor. Biochem Pharmacol. 2016;113:70‐87. [DOI] [PubMed] [Google Scholar]
- 35. Parsell DA, Mak JY, Amento EP, Unemori EN. Relaxin binds to and elicits a response from cells of the human monocytic cell line, THP‐1. J Biol Chem. 1996;271:27936‐27941. [DOI] [PubMed] [Google Scholar]
- 36. Sudo S, Kumagai J, Nishi S, et al. H3 relaxin is a specific ligand for LGR7 and activates the receptor by interacting with both the ectodomain and the exoloop 2. J Biol Chem. 2003;278:7855‐7862. [DOI] [PubMed] [Google Scholar]
- 37. Dessauer CW, Scully TT, Gilman AG. Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem. 1997;272:22272‐22277. [DOI] [PubMed] [Google Scholar]
- 38. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605‐623. [DOI] [PubMed] [Google Scholar]
- 39. Callander GE, Thomas WG, Bathgate RAD. Prolonged RXFP1 and RXFP2 signaling can be explained by poor internalization and a lack of β‐arrestin recruitment. AJP Cell Physiol. 2009;296:C1058‐C1066. [DOI] [PubMed] [Google Scholar]
- 40. Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M. Real‐time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol. 2013;170:893‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hossain MA, Bathgate RAD. Challenges in the design of insulin and relaxin/insulin‐like peptide mimetics. Bioorg Med Chem. 2018;26:2827‐2841. [DOI] [PubMed] [Google Scholar]
- 42. Hossain MA, Kocan M, Yao ST, et al. A single‐chain derivative of the relaxin hormone is a functionally selective agonist of the G protein‐coupled receptor, RXFP1. Chem Sci. 2016;7:3805‐3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bathgate RAD, Samuel CS, Burazin TCD, et al. Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene: novel members of the relaxin peptide family. J Biol Chem. 2002;277:1148‐1157. [DOI] [PubMed] [Google Scholar]
- 44. Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ. Serelaxin‐mediated signal transduction in human vascular cells: bell‐shaped concentration‐response curves reflect differential coupling to G proteins. Br J Pharmacol. 2015;172:1005‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Halls ML, van der Westhuizen ET, Wade JD, Evans BA, Bathgate RAD, Summers RJ. Relaxin family peptide receptor (RXFP1) coupling to Gαi3 involves the C‐terminal Arg752 and localization within membrane raft microdomains. Mol Pharmacol. 2009;75:415‐428. [DOI] [PubMed] [Google Scholar]
- 46. Halls ML, Bathgate RAD, Summers RJ. Comparison of signaling pathways activated by the relaxin family peptide receptors, RXFP1 and RXFP2, using reporter genes. J Pharmacol Exp Ther. 2007;320:281‐290. [DOI] [PubMed] [Google Scholar]
- 47. Ponsioen B, Zhao J, Riedl J, et al. Detecting cAMP‐induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matthiesen K, Nielsen J. Cyclic AMP control measured in two compartments in HEK293 cells: phosphodiesterase KM is more important than phosphodiesterase localization. PLoS ONE. 2011;6:e24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wachten S, Masada N, Ayling L‐J, et al. Distinct pools of cAMP centre on different isoforms of adenylyl cyclase in pituitary‐derived GH3B6 cells. J Cell Sci. 2010;123:95‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials