

Abstract
Viruses are the most abundant life form and infect practically all organisms. Consequently, these obligate parasites are a major cause of human suffering and economic loss. Rossmann‐like fold is the most populated fold among α/β‐folds in the Protein Data Bank and proteins containing Rossmann‐like fold constitute 22% of all known proteins 3D structures. Thus, analysis of viral proteins containing Rossmann‐like domains could provide an understanding of viral biology and evolution as well as could propose possible targets for antiviral therapy. We provide functional and evolutionary analysis of viral proteins containing a Rossmann‐like fold found in the evolutionary classification of protein domains (ECOD) database developed in our lab. We identified 81 protein families of bacterial, archeal, and eukaryotic viruses in light of their evolution‐based ECOD classification and Pfam taxonomy. We defined their functional significance using enzymatic EC number assignments as well as domain‐level family annotations.
Keywords: Rossmann‐like fold, viral proteins, evolution
Highlights
The Rossmann‐like fold is the most populated fold among α/β‐proteins in the Protein Data Bank. Current work is focused on evolutionary and functional features of viral protein families containing a minimal Rossmann like motif that can serve as potential antiviral targets. Obtained data outlines fast evolving viral proteins that could serve as drug targets as well as four protein families unique for viruses including proteins of Zika, dengue, and West Nile viruses.
Introduction
Rossmann‐like fold1, 2 is the most populated fold among α/β‐folds in the Protein Data Bank.3 It was first found in a wide range of nucleotide‐binding proteins that utilize diphosphate‐containing cofactors such as NAD(H). These structures included two sets of β‐α‐β‐α‐β units (321456 topology), forming a single parallel sheet flanked of a three layer α/β/α sandwich.4 An important structural feature of this fold includes a crossover observed between strands 3 and 4. This crossover creates a natural cavity that participates in the binding of the nucleotide ring.5 We can therefore define a minimal Rossmann‐like motif as a three‐layer α/β/α sandwich with at least three parallel β‐strands and a crossover between the second and third strands. In fact, protein structures containing this minimal defined unit constitute 22% of the Protein Data Bank (see “Materials and methods”). Rossmann domains are linked to a great variety of different catalytic domains and metabolic enzymes and can be found in different viruses.6
As an abundant life form that infects practically all organisms, viruses are a major cause of human suffering and economic loss. Previous studies showed that in marine, soil, and animal‐associated environments, the number of virus particles is typically 10–100 times greater than the number of cells.7 This so‐called “virosphere” is probably inclusive of every environment on the Earth, from the atmosphere to the deep biosphere.8 The viromes of the three domains of cellular life (bacteria, archea, and eukaryotes) are fundamentally different. In prokaryotes, most have double‐stranded DNA genomes, with a substantial minority of single‐stranded DNA viruses and only limited presence of RNA viruses. On the other hand, in eukaryotes, RNA viruses account for the majority of the virome diversity, although ssDNA and dsDNA viruses are common as well.9 Although several families of dsDNA viruses are represented in both bacteria and archea, no viruses are known to be shared by eukaryotes with any of the other two cellular domains, even at the family or order level.10 However, structural analyses of virion architecture and coat protein topology have revealed unexpected similarities, not visible in sequence comparisons, suggesting a common origin for viruses that infect hosts residing in different domains of life.11 Given the prevalence of Rossmann‐like folds in nature, their analysis in the viral structure proteome could provide an understanding of viral biology and evolution as well as could propose possible targets for antiviral therapy.
In this current work, we provide functional and evolutionary analysis of viral proteins containing a Rossmann‐like fold that can be found in the Evolutionary Classification of protein Domains (ECOD) database developed in our lab.12 ECOD is a hierarchical classification based on evolutionary concepts that consists of five levels: architecture (A), possible homology (X), homology (H), topology (T), and family (F).13 We identified and described protein families of bacterial, archeal, and eukaryotic viruses in the light of their classification in ECOD and defined their functional significance using enzymatic EC number assignments as well as domain‐level family annotations. 81 protein families were defined as viral proteins containing a minimal Rossmann fold motif. A few well‐populated viral folds tend to distribute across multiple host kingdoms, including P‐loop domains‐related (2004.1), Rossmann‐related (2003.1), and UDP‐glycosyltransferase/glycogen phosphorylase (2111.1). Alternatively, numerous fold types tend to be specific to their viral host kingdom, potentially explaining the difference in their viriomes and suggesting targets for therapy.
Results
Using the definition of a minimal Rossmann‐like folding unit that contains a three‐layer α/β/α sandwich with at least three parallel β‐strands and a crossover between the second and third strands, structures of 1427 viral Rossmann‐like domains were detected in the ECOD database. These domains were found in 512 (6.7% of all known viral protein structures) PDB structures and were assigned by ECOD to 81 protein family groups and 24 homology groups (Fig. 1). The structures represented gene products from 21 viral taxonomical families with host ranges from all kingdoms of life (http://prodata.swmed.edu/rossmann_fold/viruses/). 4 of 21 viral taxonomical families infect Bacteria, one infects Archea and eighteen infect Eukaryota (Fig. 1—green, violet, and orange colors, respectively). The biggest taxonomical family in terms of amount of proteins contains sixteen protein families from E.coli bacteriophage T4. Another two big taxonomical families are Eukaryote viruses from Poxviridae (8 families of vaccinia virus causing smallpox) and Reoviridae (9 protein families of Aquareovirus C, Rotavirus A, Cypovirus 1, and Bluetongue virus).
Figure 1.
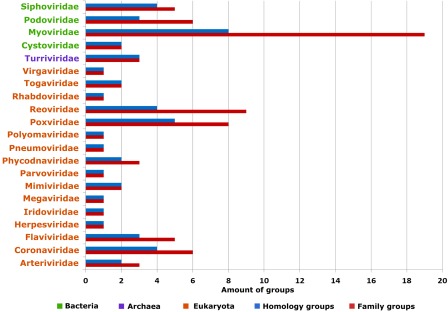
Amount of proteins groups in different viral families. Green color indicates virus families which parasite on Bacteria, violet—on Archea, orange—on Eukaryote. Red color indicates family groups, blue—homology groups.
Bacterial viruses
Phages are viruses that infect bacteria; their self‐replication depends on access to a bacterial host. The rising tide of antibiotic resistance coupled with the low rate of antibiotic discovery has revived interest in phages as antibacterial agents.14 Phages have been used not only to treat and prevent human bacterial infections but also to control plant diseases, detect pathogens, and assess food safety. E.coli bacteriophages are the most popular objects for these purposes,14 which perhaps explains their relative abundance of structures (Fig. 1). Our analysis detected 14 different bacterial virus structure topology types defined by ECOD T‐groups that contain a Rossmann‐like fold (Fig. 2, 12 topology groups shown). The largest “Rossmann‐related” homology group (2003.1) adopts a classical Rossmann‐like topology with additional β‐strands at the C‐terminal end for several families [Fig. 2(A,B)]. The tubulin family [Fig. 2(B)] of this homology group has a sheet topology 321456 representing a canonical GTP‐binding domain. Pseudomonas phiKZ‐like bacteriophages encode a group of related tubulin/FtsZ‐like proteins believed to be essential for the correct centering of replicated bacteriophage virions within the bacterial host.15 This phage protein classifies together with bacterial and archeal FtsZ proteins as well as eukaryotic tubulin alpha, beta, and gamma chains. The similar protein sequence and structure, in addition to the GTP‐dependent polymerization activities suggest all evolved from a common ancestor.
Figure 2.
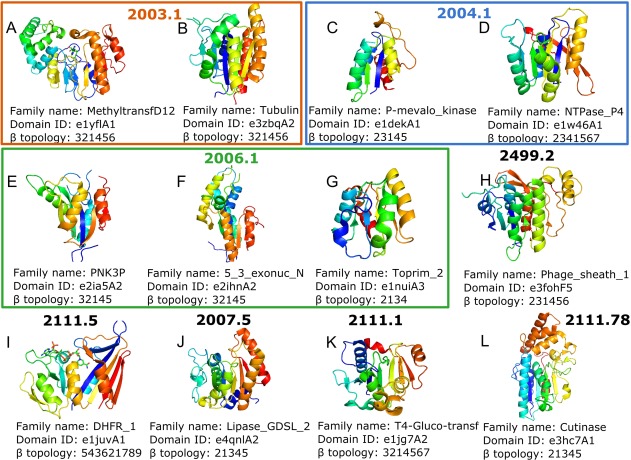
(A–L) Topology groups of phages proteins containing minimal Rossmann fold. Structure colored by rainbow. β topology is specified for middle β‐sheet only.
One family from the “P‐loop domains‐related” [2004.1, Fig. 2(C,D)] homology group, phosphomevalonate kinase [P‐mevalo_kinase, PMVK, Fig. 2(C)], contains a subfamily of phage T4 deoxynucleotide kinases that are somewhat distinct from the canonical animal enzymes catalyzing phosphorylation of 5‐phosphomevalonate into 5‐diphosphomevalonate, an essential step in isoprenoid biosynthesis. Given the idea that PMVK enzymes arose from nonorthologous gene displacement early in animal evolution,16 perhaps the phage system T4 deoxynucleotide kinases played a role in their alternate evolutionary origins. Another family member of this H‐group, the ATPase P4 from bacteriophage phi12 [NTPase_P4, Fig. 2(D)] contains motor proteins involved in the packaging of Pseudomonas phage phi‐12 genome into preformed capsids using ATP to drive translocation.17 The central P4 structure core, together with part of the C‐terminal region, forms a Rossmann‐type domain containing a twisted, eight‐stranded β‐sheet of mixed parallel and antiparallel topology flanked by five helices. Although the P4 family is limited to phage proteins, their presumed homologous relationship to other P‐loop structures suggests common evolutionary origin.18
The toprim‐like [Toprim_2, Fig. 2(G)] family belongs to “HAD domain‐related” homology group [2006.1, Fig. 2(E–G)] and is represented by DNA primase–helicase proteins.19 Phage T7 encodes DNA polymerase, primase, helicase, and single‐stranded DNA binding activities that catalyze the replication of double‐stranded phage DNA. Primase and helicase activities reside in a bifunctional primase–helicase protein that assembles into ring‐shaped hexamers.20 Given their essential role in phage replication, these viral toprim‐like proteins are predominantly found in bacteriophages and nucleocytoplasmic large DNA viruses.21 Two possible evolutionary scenarios have been proposed for primase–helicase proteins. The first involves fusion of primase and helicase genes, while the second considers the primase–helicase gene as an ancestor that underwent duplication and divergence followed by physical separation of primase and helicase functions.
The phage tail sheath protein domain [Phage_sheath_1, Fig. 2(H)] with the following common sheet topology (231456) contains proteins that play crucial roles in contracting the tail of bacteriophage T4 through the host outer membrane during infection.22 The phage tail sheath protein resembles the fold of the Type VI secretion system protein (T6SS) VipA/VipB heterodimer that intertwines to adopt the Rossmann‐like architecture.23 In fact, components of the T6SS system, which delivers effector proteins into a target cell, and the phage tail‐associated complex are thought to have arisen from a common ancestor.24
The T4‐glucosyltransferase family [Fig. 2(K)], limited to phage proteins, catalyzes the transfer of glucose from uridine diphosphoglucose to 5‐hydroxymethyl cytosine. The role of glycosylation in protecting the infecting viral DNA from host restriction enzymes has been reviewed.25 Comparison of the glucosyltransferase family fold to other families showed that it is completely embedded in the structure of glycogen phosphorylase. All nine α‐helices and 13 β‐strands of β‐glucosyltransferase match elements of glycogen phosphorylase in sequential order.26 The significance of the match is further supported by the fact that the first and second domains of the common core are architecturally distinct despite topographical similarity to the classical nucleotide‐binding fold. It appears paradoxical that the architecture of T4 β‐glucosyltransferase is simpler and therefore appears more primitive than that of glycogen phosphorylase, which is the older enzyme by phylogenetic arguments.26 The paradox may be resolved by either of two evolutionary models that differ in the point of divergence between glycogen phosphorylase and T4 β‐glucosyltransferase. The first model postulates that the gene for T4 β‐glucosyltransferase diverged rather recently from a fully evolved glycogen phosphorylase and evolution in T4 rapidly simplified the structure of the protein down to the essential catalytic core. In the second model, both descend along separate lineages from a very ancient common ancestor.
The N‐acetylmuramoyl‐l‐alanine amidase (Amidase_3) family contains viral proteins that exhibit high specificity towards the cell walls of their host bacteria. The core of these domains is formed by a twisted, six‐stranded β‐sheet flanked by six helices. β‐strands 4 and 5 are antiparallel, which makes the catalytic domain unique among the known Listeria phage endolysins.27 Based on their antimicrobial properties, endolysins from phages infecting Gram‐positive pathogens have recently attracted attention as potential therapeutic agents.28 Initial studies were successfully carried out with oral Streptococci in mice29 as well as with Bacillus anthracis in vitro.30 Also genetically modified lactic acid bacteria that are able to synthesize and secrete active Listeria phage endolysin were constructed to protect food fermentation products.31
Cutinase [Fig. 2(L)] belongs to the α/β‐hydrolase H‐group and is another big family that contains proteins from different domains of life. Cutinase is a serine esterase with the classical Ser, His, Asp triad of serine hydrolases. The structure of a cutinase shows an α/β sandwich organization similar to lysin B (LysB) enzymes. Viral LysB proteins are produced by mycobacteriophages to degrade the host peptidoglycan layer. The activity also circumvents a mycolic acid‐rich outer membrane covalently attached to the arabinogalactan–peptidoglycan complex in the wall of Mycobacterium.32 The five closest related structures of LysB are all cutinases, although there is no greater than 21% amino acid sequence identity with any of them. The LysB catalytic mechanism is expected to be similar to that for other serine esterases. Acquisition of LysB by mycobacteriophages throughout their evolution likely confers a substantial selective advantage over those without it by providing faster and more complete lysis.32
Archeal viruses
Like the bacterial and eukaryotic branches in the tree of life, the Archea are host to a multitude of viruses. However, compared to viruses infecting the domains Eukaryota and Bacteria, studies of viruses infecting the Archea are still in their infancy.33 The PDB database includes 305 domains in 55 structures of archeal‐specific viral nonchimeric proteins and only three domains in three structures were defined as Rossmann‐like (Fig. 3). In addition to these archea‐specific viruses, all types of dsDNA viruses known to infect bacteria can replicate in archea, including head‐tailed viruses (families Myoviridae, Siphoviridae, and Podoviridae), icosahedral viruses with internal lipid envelopes (families Sphaerolipoviridae and Turriviridae in archea; Tectiviridae and Corticoviridae in bacteria), and pleomorphic viruses (families Pleolipoviridae in archea and Plasmaviridae in bacteria).34 One example of an archeal‐infecting viral protein is the B116 protein (DUF1874 family) of Sulfolobus turreted icosahedral virus (STIV).35 While the 37 STIV open reading frames (ORFs) generally lack sequence similarity to other genes ORF B116 is common to the genomes of three additional hyperthermophilic Archeal viral families, the Rudiviridae, the Lipothrixviridae and the Bicaudaviridae. The B116 polypeptide folds to form a five‐stranded, predominantly parallel β‐sheet (topology 31452) lined on one side by three α‐helices, with strand β5 running antiparallel to the remaining strands. Interestingly, an intramolecular disulfide bond, contributed by Cys33 and Cys62, is also observed. This covalent link between the α1 and α2 helices is likely to enhance the thermostability of the B116 fold. Two copies of the B116 polypeptide are found in the asymmetric unit, giving rise to the homodimer and forming a larger 10‐stranded unclosed barrel, giving rise to a saddle shaped protein. Authors suggested that this unclosed barrel could be a place of nonspecific DNA binding in which the Rossmann‐like fold also could take part.35
Figure 3.
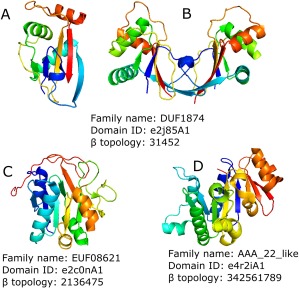
(A–D) Topology groups of Archeal viral proteins containing minimal Rossmann fold. Structure colored by rainbow. β topology is specified for middle β‐sheet only.
One more example from STIV is the A197 protein (EUF08621 family). The structure of the A197 monomer reveals a six‐stranded, predominantly parallel, α/β/α sandwich that is flanked by a four‐stranded antiparallel β‐sheet with an extended C terminus. Structure similarity data identified members of the glycosyltransferase (GT‐A) superfamily as the closest structural homologues of this protein. A197 is one of the smallest known glycosyltransferases, composed of only the core catalytic GT‐A fold and lacking additional functional domains. Thus, its structure may define the minimal components necessary for glycosyltransferase activity.36 The third example is the B204 protein (AAA_22_like family). It is an ATPase belonging to the P‐loop containing nucleoside triphosphate hydrolases H‐group that is thought to drive packaging of viral DNA during the replication process. The structure of STIV B204 is represented by a central nine‐stranded β‐sheet decorated with seven α‐helices. B204 contains a core Rossmann‐like fold (sheet topology 32451) followed by a β‐meander with a helical hairpin stemming from one of the loops. Related P‐loop ATPases with this identical topology also function to translocate DNA; including the bacterial conjugation protein TrwB that transfers bacterial DNA across membranes and between cells37 and the VirB4 ATPase of the bacterial type IV secretion system that mediates the transfer of proteins and DNA across bacterial membranes.38
Eukaryotic viruses
In contrast to bacteria and archea, eukaryota hosts numerous, diverse RNA viruses, retrotranscribing elements and retroviruses that typically integrate into the host genome.39 Our analysis detected 14 minimal Rossmann fold‐containing homology groups in virus that infect eukaryotes. The examples adopt 16 different topology types and contain 47 different families. By far the largest group of DNA viruses in eukaryotes consists of seven families of large and giant viruses (including mimiviruses, which have genomes in the megabase range).
The giant viruses of the family Mimiviridae are associated with a distinct class of satellite viruses, the virophages, which reproduce within viral “factories” inside their host protist cells and which depend on the latter for their replication.40 In our dataset Mimiviridae encodes two protein families. The glucose‐methanol‐choline oxidoreductase family, represented by the R135 protein, contains an N‐terminal FAD binding Rossmann‐related domain followed by a C‐terminal substrate recognition domain. The R135 oxidoreductase might participate in degrading the cell walls of their normal hosts, which include some lignin‐containing algae.41 The minimal Rossmann domain is composed of a five‐stranded parallel β‐sheet with the same β‐strand topology typical of a nucleotide‐binding fold. The second Mimiviridae protein family is represented by tyrosyl tRNA synthetases (TyrRS). These proteins have six‐stranded β‐sheet topology ordered 432561, with an antiparallel first strand. TyrRS shares 30% identity over 340 residues with the TyrRS of the hyperthermophilic Euryarchaeota Pyrococcus horikoshii, its closest known structural homologue. tRNA synthetases are pivotal in determining how the genetic code is translated in amino acids and in providing the substrate for protein synthesis. The discovery of four aminoacyl‐tRNA synthetases encoded in the genome of mimivirus together with a full set of translation initiation, elongation, and termination factors appeared to blur what was once a clear frontier between the cellular and viral world.42
Another eukaryotic infecting virus, vaccinia virus (Poxviridae), causes smallpox. The Poxviridae genome includes eight protein family groups with five different topology types. One of example is H3 envelop protein—an immunodominant antigen that is expressed late in infection and found as a membrane protein on the surface of virion particles. The nine‐stranded β‐sheet is made up of strands in the order 679584132 that is surrounded with helices on both sides, and all of the strands except 7 and 8 are oriented parallel to each other. The fold belongs to glycosyltransferases (GTs) of the GT‐A group.43 H3 is involved in attachment to the host cell, contributes to viral morphogenesis, and plays a role in infection.44 Since H3 is a major immune system target that is recognized by neutralizing antibodies, H3 is an important viral protein to be included in new vaccines.45 Another example is subunit D12 of vaccinia virus capping enzyme that executes all three steps in m7GpppRNA synthesis.46 The topology of the stimulatory subunit D12 reveals a class I N7‐methyl‐transferase (MT) like core, however, with a truncated S‐adenosyl‐homocysteine‐binding domain, consistent with its lack of MT activity. This enzyme has a completely unique mode of binding of the adenosine moiety of S‐adenosyl‐homocysteine, a feature that could be exploited for design of specific antipoxviral compound.47
Zika virus (Flaviviridae) include only one family with a minimal Rossmann structure, non‐structural protein NS1. The protein fold and domain arrangement of NS1 is virtually identical to dengue virus DENV2 protein and West Nile virus NS1 protein. Despite this overall similarity, the Zika virus NS1 crystal structure provides important new information about a domain containing minimal Rossmann motif flexible loop that is not visible in previous structures.48 The structure of the loop reveals an expanded surface permitting NS1 to associate with membranes during replication, to associate with immature virions during particle morphogenesis and to facilitate the interactions necessary for formation of the hexameric lipoprotein complex. The Zika virus, which has been implicated in an increase in neonatal microcephaly and Guillain–Barré syndrome, has spread rapidly through tropical regions of the world. NS1 plays crucial role in the Zika virus life cycle, being a multifunctional virulence factor.48
Reoviridae is a big viral family infecting fish, shellfish, crustacean species, insects, ruminants and human hosts. The Reoviridae family includes structure representatives from nine different protein families. Rotaviruses are the principal agents of infectious dehydrating diarrhea of infants and the cause of nearly a half‐million childhood deaths per year.49 They have a segmented, double stranded RNA (dsRNA) genome, packaged within a multi‐shelled virus particle. The outer protein layer of the virion, the molecular machinery for host‐cell binding and penetration, contains two protein components, VP4 and VP7.50 The domain containing minimal Rossmann motif of VP7 protein has five parallel β‐strands and is assigned to “Flavodoxin‐like” possible homology group. Rotavirus infection and parenteral immunization with virions both induce a strong VP7‐specific neutralizing antibody response. This protein is a principal target of protective antibodies.51
Taxonomic distribution of viral rossmann family representatives
We examined taxonomic distribution of sequences best hits of each family representative in order to define possible evolutionary relationships between viral and host proteins from the same family, which could be the result of Horizontal Gene Transfer (HGT) between virus and host organism. Looking at the similarity of the Rossmann motif ECOD families' sequences to those in the nonredundant sequence database, all Rossmann‐like fold families were divided into six groups according to appearance of BLAST hits from four taxonomy groups: A—Archea, B—Bacteria, E—Eukaryota and V—Viruses. The first group ‐ 31% (25 out of 81) of all protein families under study has universal BLAST hits from archea, Bacteria, Eukaryota and Viruses (“ABEV”, see two last columns at the online table: http://prodata.swmed.edu/rossmann_fold/viruses/). Half (13 out of 25, or 52%) of these families belong to double‐stranded DNA viruses (dsDNA), which can affect bacteria. BLAST score distributions allow us to assume that Horizontal Gene Transfer (HGT) took place between virus and host bacteria for 7 out of 13 protein families in this universal group (e3uj3X1, e3u5zE1, e2ocaA8, e2ia5B1, e1juvA1, e4ieeA2, e1xovA2). This assumption is based on high‐scoring bacterial hits being among viral hits in the BLAST score distributions. The second half of protein families with “ABEV” hits (12 out of 25, or 48%) belong to viruses that affect eukaryotes, including double‐stranded DNA and positive single‐stranded RNA ((+)ssRNA) viruses. Only one protein family seems to have evidence for HGT between virus and eukaryote host—vaccinia virus thymidine kinase (e2j87A3).
The second group—6% (5 out of 81) of Rossmann‐like fold proteins has BLAST hits from three taxonomical groups: Archea, Bacteria and Viruses (“ABV”). Two protein families infect bacteria (dsDNA) and three infect archea (dsRNA). All these protein families except one (e4r2iA1) have strong evidences for HGT between virus and host. The third group—16% (13 out of 81) has BLAST hits from three taxonomical groups: Bacteria, Eukaryota, and Viruses (“BEV”). Six protein families infect bacteria (dsDNA), and four of them have evidences of HGT (e3bgwF1, e3hc7A1, e5hd9A1, e2ihnA2). The rest infect eukaryotes and belong to double‐stranded DNA and positive single‐stranded RNA viruses. The fourth group—11% (9 out of 81) has BLAST hits from two taxonomical groups: Bacteria and Viruses (“BV”). Of the seven families that infect bacteria (dsDNA and dsRNA viruses), six have evidences for HGT (e4cu5B1, e4cu2A1, e1dekA1, e1y8zB2, e1y8zB1, e2ia5A2). The fifth smallest group—2.5% (2 out of 81) has BLAST hits from two taxonomical groups: Eukaryota and Viruses (“EV”). Both families belong to human vaccinia dsDNA virus with BLAST distributions having only low‐scoring eukaryotic hits. The biggest group—33% (27 out of 81) has BLAST hits limited to Viruses (“V”), with only four families infecting bacteria (dsDNA and dsRNA viruses) and the rest (23 families) infecting eukaryotes (dsRNA, (+) ssRNA, dsDNA, and (–) ssRNA viruses).
Distribution of viral protein fold types
Given the relatively high number of viral protein families that contain a Rossmann‐like fold (81 families), we sought to examine their evolutionary distributions among folds from the three major host kingdoms. Figure 4 highlights the protein family counts from bacteria (blue bar), archea (red bar), and eukaryota (green bar)—infecting viruses that fall within all distinct minimum Rossmann fold types. Importantly, each fold type includes protein families related by a common ancestor (ECOD H‐group). The fold types represented by more than one host kingdom tend to display relatively high family counts. These well‐populated folds include P‐loop domains‐related (2004.1), Rossmann‐related (2003.1), and UDP‐glycosyltransferase/glycogen phosphorylase (2111.1). Most of the other fold types include only one family representative, with 8 from eukaryotic hosts, 6 from bacterial hosts, and one from archeal hosts. The fold type nucleotide‐diphospho‐sugar transferases (2111.6) contains two families from eukaryote and one from archea, while the fold type SGNH hydrolase (2007.5) contains one family from bacteria and one from archea. This almost biphasic distribution, with one well‐populated universal fold set and one less populated unique fold set, suggests that despite the fundamental differences in the viriomes from the three domains of cellular life, they survive using a common set of folds. Furthermore, these well‐populated folds extend across all life forms, perhaps suggesting an ancient origin. Accordingly, the P‐loop domains‐like fold, whose nucleotide metabolic enzyme components are thought to form the origin of the protein world,52 also represent the most populated fold among viral genomes. Such examples of ancient viral proteins should be useful for understanding evolutionary relationships among members of this unique domain of life. Alternately, the unique fold types likely drive some of the differences observed in virus infecting different cellular forms of life. For example, the cystovirus bacteriophage phi12 encodes a unique P7 protein with a minimal Rossmann fold. Phi12 P7 is classified as its own unique X‐group in ECOD, implying a lack of evidence for homology to existing folds. P7 serves as a putative virion assembly cofactor thought to bind the unique three‐segmented double‐stranded cystovirus RNA genome.53 The avian coronavirus that infects chickens encodes another example (IBV Nsp2a) of a unique Rossmann‐like fold domain. The N‐terminal domain of Nsp2a adopts a Rossmann‐like sheet topology (2314), with β‐strand 2 being an insert to the core that is antiparallel to the rest. Although the function of Nsp2a is unknown, its lack of sequence/structure similarity to other proteins has suggested a role in host specificity.54
Figure 4.
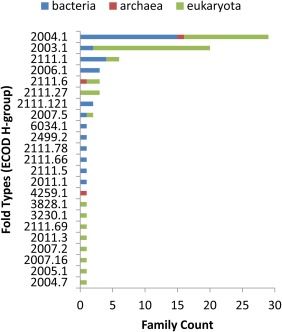
Homology groups distribution. Protein homology groups counts from bacteria—(blue bar), archea—(red bar), and eukaryote—(green bar) infecting viruses.
Viral helicases and other P‐loop domains‐related groups
Traditionally defined, helicases use energy derived from NTP hydrolysis to unwind double‐stranded nucleic acids.55 As such, they play roles in numerous cellular processes involving nucleic acids; including DNA replication and repair, transcription, translation, and RNA splicing and maturation, among others.56 Mechanistically, helicases can bind single‐stranded or double‐stranded nucleic acid; unwind RNA, DNA or hybrids, and translocate in both directions (3′–5′ or 5′–3′).57 Despite these functional distinctions, all helicases bind NTP using two structural elements formed by signature sequence motifs: a phosphate‐binding P‐loop (motif I/Walker A motif) and Mg2+ cofactor binding loop (motif II/Walker B). These motifs have helped classify P‐loop helicases into superfamilies (SF1–SF5) based on sequence, with the last family including also nonhelicase NTPases.58 Modular accessory domains or subdomains, including terminal extensions and insertions within the core, can also regulate helicase activity.58
By shuffling core and regulatory domains, nature has created a diverse range of cellular helicase machinery that also plays key roles in viral function. As such, the relative abundance of helicases in viral genomes has been used in part to assess picornaviral evolution.59 Their key roles in viral function also suggest helicases as novel targets for treating viral infections.58 In humans, several debilitating inherited disorders are linked to genetic defects in helicase genes, including Bloom's, Werner's, and Rothmund–Thomson's syndromes.60 The potential of helicases as antiviral drug targets has recently been reviewed.61
Among viral protein structures containing the minimal Rossmann fold, 14 protein families are known helicases (http://prodata.swmed.edu/rossmann_fold/viruses/). Helicase domains fall into two different homologs folds (H‐groups): P‐loop domains‐related (13 families) and HAD domain‐related (1 family).19 Given the number of P‐loop domains‐related representatives and their potential for informing viral evolution, we constructed a structure‐based tree of domains that contain P‐loop motifs (Fig. 5). The tree reproduces traditional sequence‐based classification:58 dividing the viral helicase domains into four superfamilies (SF1–SF4). Those in the SF1 and SF2 include a Rossmann‐like fold duplication, with the second domain helping form the DNA binding site and contributing a motif to the active site (i.e., motif IV in 3upuA3, not included in the tree).
Figure 5.
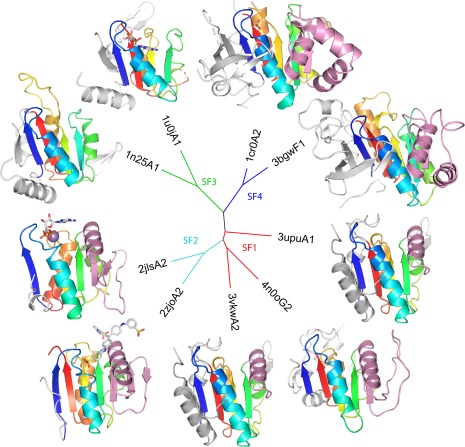
A tree of viral helicases. Structure‐based distances between representative P‐loop domains from ECOD family viral helicases were estimated using DaliZ scores. Nodes of the tree are labeled by PDB and colored according to superfamily. Structures are colored in rainbow according to the core Rossmann‐like topology common to all viral helicase domains. Terminal extentions (white) and insertions (pink) decorate the core fold. Where present, active site molecules are in stick.
All the catalytic viral helicase domains include a core topology of four strands (order 1432) sandwiched by a helix on one side and two helices on the other. The common core binds nucleotide using the Walker A (following core strand 1) and Walker B (following strand 3) motifs. The structure‐based tree correctly defines superfamilies based on terminal extensions and insertions to the core, with SF1 having an insertion (Fig. 6, pink cartoon) that extends one side of the β‐sheet by a strand and a C‐terminal extension that extends the other side. SF2 domains have a longer insertion than SF1 that extends the sheet by two strands. SF3 have a similar C‐terminal extension as SF1 but lack the insertion, while SF4 includes both a longer C‐terminal extension that adopts a four‐stranded b‐meander and a longer insertion that extends the sheet and has an additional helical subdomain. Given the ability of the viral P‐loop helicase domains with minimal Rossmann folds to recapitulate traditional sequence‐based classification, these domains might provide useful for further analysis of viral evolution.
Figure 6.
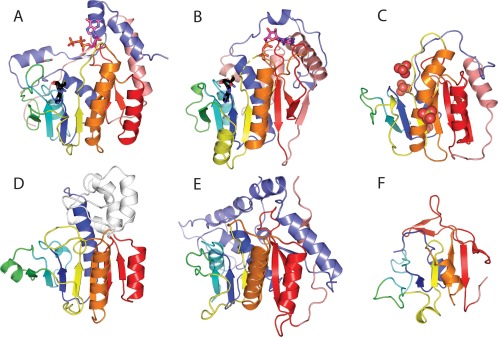
Viral methyltransferase (MT) domains. The core MT topology (sheet order 3214576) is colored by α/β units in rainbow from N‐terminus (blue) to C‐terminus (red), with the last antiparallel strand that marks the fold in red. N‐terminal (slate) and C‐terminal (salmon) extensions and insertions (white) to the core MT fold are colored. (A) Slow evolving viral FtsJ family MT from Dengue virus NS5 [4v0r] highlights typical AdoMet (black stick) and cap (magenta stick) binding. (B) Fast evolving Vaccinia virus V39 MT [1v39] retains similar AdoMet binding but diverges in cap binding. (C) Fast evolving inactive Alphavirus P32 MT domain [4gua] has deteriorated around the AdoMet site, yet binds RNA (indicated by SO4 spheres). (D) Duplicated inactive MT domain in transcribing cytoplasmic polyhedrosis virus [3jay] binds AdoMet and alters capsid conformation to ultimately activate (E) functional MT domain. (F) MT‐like domain from SARS virus NSP15 [2h85] has significantly deteriorated AdoMet binding site, yet retains the antiparallel strand (red).
Viral genomes possess several additional types of P‐loops domain‐related homologs that do not function as helicases. Their activities include terminase or viral nucleic acid packaging that couples NTP hydrolysis to directional motion along nucleic acids, thymidine and other deoxynucleotide kinases providing DNA precursors for synthesis in the host cytoplasm, polynucleotide kinase functioning in nucleic acid repair, and a recombinase promoting strand exchange. These additional structures bring the total number of P‐loop domains‐related families to 27 (removed incorrect dynein domains), which represents almost one third of the existing Rossmann‐like fold domains in viruses.
Discussion
Looking at similarity to known protein sequences, about one third of the Rossmann motif ECOD families are limited to viral sequences (27 out of 81, or 33%). Most of these viral‐specific protein families exhibit characteristics of fast evolution, with their sequences being distinct from homologs (24 out of 27, or 89%). Interestingly, most of these fast evolving families infect eukaryotes (21 out of 24, or 87%), with many of these classified as diverse methyltransferase domains (12 families). Viral methyltransferase domains tend to function in mRNA 5' cap biosynthesis. One viral methyltransferase example that has not rapidly diverged from its FtsJ counterparts in other kingdoms (i.e., “ABVE”) illustrates a typical methyltransferase fold bound to AdoMet substrate and cap [Fig. 6(A)]. Alternately, a fast evolving structure of the vaccinia virus protein VP39 highlights the typical methyltransferase binding sites for AdoMet substrate [Fig. 6(B), black stick] with diversity arising from extensions at the termini, a replacement of the C‐terminal helix with a strand, and a unique cap binding pocket [Fig. 6(B), magenta stick] that allows for sensing substrate methylation status.62 Evolution of viral methyltransferases has been discussed, with the unique sequence features arising from their invention of alternate capping pathways, their intimate interactions with additional viral enzymes functioning in the process, and their inactivation as methyltransferases.63 Such inactive domains have transformed into RNA‐binding modules64 displaying significant deterioration surrounding the AdoMet binding site [Fig. 6(C)] or allosteric modulators of RNA processing65 that function together with a duplicated methyltransferase domain [Fig. 6(D,E), respectively]. Interestingly, classified viral methyltransferase domains from SARS NSP15 and PRRSV NSP11 endoribonucleases have a largely degraded N‐terminus where AdoMet substrate usually binds; however, the distinguishing C‐terminal β‐hairpin that marks the methyltransferase fold remains intact [Fig. 6(F)]. The function of this domain, like other viral structure additions, appears to be in oligomerization. Given the uniqueness of the folds with respect to host methyltransferases, the rapidly diverging viral methyltransferases might serve as targets for therapy. Indeed, Zika virus NSP5, which includes an FtsJ family methyltransferase domain, shows promise for drug design.66
Alternatively, to the majority of fast evolving viral Rossmann domains, only a few of them (3 out of 27, or 11%) appear as being unique to virus. These belong to virus with diverse hosts, with one infecting bacteria (Pseudomonas phage core protein P7) and two infecting eukaryotic hosts (IBV Nsp2a and Zika virus NS1). The IBV Nsp2a N‐terminus contains a minimal Rossmann‐like motif with an α/β insertion following the first helix that forms an antiparallel interaction with the crossover strand 2 [Fig. 7(A)]. While NSP2 is one of the first proteins to be translated and processed in the IBV life cycle, its function remains unknown. Zika virus NS1 also adopts a minimal Rossmann‐like fold. However, NS1 replaces the C‐terminal helix addition with an N‐terminal helix addition. It also has an insertion in the same position as Nsp2a, but the β‐strand forms a parallel interaction with strand 2 [Fig. 7(B)]. The function of Zika virus NS1 remains unclear. The unique properties of these apparent viral‐specific proteins, which could have arisen from degradation of more complete Rossmann domains, preclude functional inference from their structure.
Figure 7.
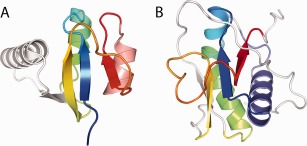
Novel viral Rossmann‐like motifs. Two viral‐specific families retain minimal Rossmann‐like motifs colored in rainbow from N‐terminus (blue) to C‐terminus (red). (A) The N‐terminal domain from IBV Nsp2a [3ld1] has a unique β/α insertion (white) after the first helix forming an antiparallel interaction with strand 2 and an additional C‐terminal helix (salmon). (B) The N‐terminal domain from Flavivirus NS1 includes an N‐terminal helix (slate) and a different insertion (white) in the same position as in IBV Nsp2a, but forming a parallel interaction with strand 2.
The remaining families include BLAST hits from other domains of life (54 out of 81, or 66%). These could either represent proteins of ancient origin or proteins that have been horizontally transferred (HGT) between viral genomes and their hosts. 51% (28 out of 54) of these families infect bacteria, 44% (23 out of 54) infect eukaryotes and 5% (3 out of 54) infect archea. This nearly equal distribution suggests the potential for viral protein families that derive from ancient origin, as the prevalence of HGT stems from bacterial origins.67 Although evidence does exist for viral acquisition of eukaryotic proteins68 (i.e., from HGT, not ancient origins), these viral families of eukaryotic hosts that perform universal functions serve as a potential examples of viral proteins with ancient origins.
Many of these more universal proteins serve as helicases, which provide important functions to all domains of life. Rossmann‐like fold containing viral helicases are divided into four superfamilies (SF1–SF4), according to traditional sequence‐based classification58 and our classification of the P‐loop containing domains (Fig. 5). The SF3 helicases, whose viral examples belong to universal families, are thought to have been present at the last universal common ancestor stage as in virus‐like “selfish” replicons.69 Key role of these proteins in viral function also suggest helicases as novel targets for treating viral infections.
Only one family appears as forming a homology group that is unique and distant from the others—resolvase protein family (PF00239). Resolvases or recombinases are proteins that cause conserved DNA rearrangements, interact with short sequences in the DNA, bring two sites together in a synapse and then catalyze strand exchange so that the DNA is cleaved and religated to opposite partners.70 There are several known types of recombinases but only serine recombinases or resolvase/invertase family proteins contain a Rossmann fold motif. This family has emerged from studies of phages, prophages, and transposons from predominantly Gram‐positive bacteria and consists of three groups. The structural and evolutionary differences imply that an ancestral catalytic domain has fused to unrelated sequences to result in a family of structurally and functionally diverse proteins. Thus, the modular nature of the serine recombinases resembles that in other recombination enzymes, such as the tyrosine integrases and the DDE superfamily of transposases.71 Interestingly, serine recombinases have significant sequence similarity to the poxvirus F16 protein whose function is still unclear, but it is proposed that this protein may affect signaling functions of the nucleoli and it is unlikely to have serine recombinase activity.72 Thus, the most parsimonious evolutionary scenario of these orthologs involves acquisition of a serine recombinase gene by the ancestor of poxviruses from a transposon or a bacteriophage.72
Materials and Methods
The minimal Rossmann fold motif was defined as a three‐layer α/β/α sandwich motif with a crossover between elements III and V, which contained three parallel β‐strands as a middle layer and three variations of the crossover element IV (Fig. 8). Element IV can be represented as α‐helix [Fig. 8(A)], β‐strand [Fig. 8(B)] or linker [Fig. 8(C)]. Element II was represented only as a helix since it forms the active site of most of Rossmann fold proteins.
Figure 8.
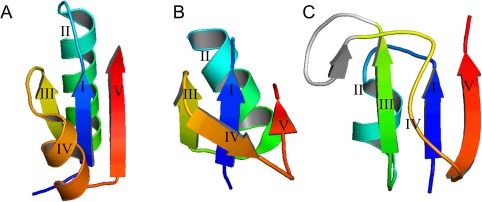
Minimal Rossmann fold motifs. (A–C) Examples of minimal Rossmann fold motifs. Secondary structure is colored by rainbow from blue (N‐terminal part) to red (C‐terminal part).
For the motif search, we used the ProSMoS program developed in our lab.73 We generated a database of PDB domains (ECOD database version: develop159/20161205) with each represented by a secondary structure element (SSE) interaction matrix describing the interactions (parallel or antiparallel) and hydrogen‐bonding between the secondary structure elements of the PDB structure. This database was generated using PALSSE.74 The structure consensuses of minimal Rossmann fold proteins were represented as query matrices. Query matrices specified the number and types of secondary structure elements in the motif under consideration, the hydrogen bonding and parallel or antiparallel relationships between its elements and also minimum and maximum length of the three component b‐strands. Then we used query matrixes as input for ProSMoS program. False positives were removed by visual inspection. Domains were considered to belong to Rossmann‐like fold only when minimal Rossmann fold motif in them formed the structural core of the protein domain.
A full list of protein families and their characteristics can be found online: http://prodata.swmed.edu/rossmann_fold/viruses/. We generated functional information for each PDB representative included in the online table from several databases. Virus taxonomy is from the latest report of the International Committee for Taxonomy of Viruses (ICTV). Topology and evolutionary information are from ECOD. The protein name and structure are from the Protein Data Bank.3 General functional descriptions are from the Pfam database.75 Finally, enzyme function in the form of an EC number—is from the KEGG Enzyme database.76
The helicase tree was built from structure‐based distances of ECOD domains (2004.1.1) with helicase EC function using the FITCH program from PHYLIP package77 (available from: http://evolution.genetics.washington.edu/phylip.html) with global rearrangements. Distances between representative structures were estimated by transforming Dali Z scores78 from pairwise superpositions using the following equation: Distance12 = ln[DaliZ12/minimum(DaliZ11, DaliZ22)].
For each PDB family representative sequence from the online table (http://prodata.swmed.edu/rossmann_fold/viruses/), a search against the NCBI non‐redundant protein sequence database (National Center for Biotechnology Information, NIH, Bethesda, MD) was performed using BLAST.79 Settings of BLAST search were used as follows: number of hits—5,000, e‐value cutoff—0.01. The rest settings were set on default. All hits were sorted in four big taxonomical groups: A—Archea, B—Bacteria, E—Eukaryota, V—Viruses, and were plotted as distributions against BLAST score. These plots can be accessed through the online table (http://prodata.swmed.edu/rossmann_fold/viruses/), see column “BLAST distribution plot”. The last column of the online table entitled “BLAST hits kingdoms” defines taxonomical groups from the distribution plot.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Funding Information
National Institutes of Health, Grant Number: GM094575 and GM127390; Welch Foundation, Grant Number(s): I‐1505 to N.V.G.
Contributor Information
Kirill E. Medvedev, Email: Kirill.Medvedev@UTSouthwestern.edu.
Nick V. Grishin, Email: grishin@chop.swmed.edu
References
- 1. Aravind L, Anantharaman V, Koonin EV (2002) Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP‐ATPase nucleotide‐binding domains: Implications for protein evolution in the RNA world. Proteins 48:1–14. [DOI] [PubMed] [Google Scholar]
- 2. Aravind L, de Souza RF, Iyer LM (2010) Predicted class‐I aminoacyl tRNA synthetase‐like proteins in non‐ribosomal peptide synthesis. Biol Direct 5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, Westbrook J (2000) The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol 7:957–959. [DOI] [PubMed] [Google Scholar]
- 4. Burroughs AM, Iyer LM, Aravind L (2009) Natural history of the E1‐like superfamily: Implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins 75:895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rossmann MG, Moras D, Olsen KW (1974) Chemical and biological evolution of a nucleotide‐binding protein. Nature 250:194–199. [DOI] [PubMed] [Google Scholar]
- 6. Bashton M, Chothia C (2002) The geometry of domain combination in proteins. J Mol Biol 315:927–939. [DOI] [PubMed] [Google Scholar]
- 7. Edwards RA, Rohwer F (2005) Viral metagenomics. Nat Rev Microbiol 3:504–510. [DOI] [PubMed] [Google Scholar]
- 8. Suttle CA (2007) Marine viruses: Major players in the global ecosystem. Nat Rev Microbiol 5:801–812. [DOI] [PubMed] [Google Scholar]
- 9. Koonin EV, Dolja VV, Krupovic M (2015) Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 479–480:2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. King AM, Lefkowitz E, Adams MJ, Carstens EB, editors (2011) Virus taxonomy: Ninth report of the International Committee on Taxonomy of Viruses. Amsterdam: Elsevier. [Google Scholar]
- 11. Bamford DH, Grimes JM, Stuart DI (2005) What does structure tell us about virus evolution? Curr Opin Struct Biol 15:655–663. [DOI] [PubMed] [Google Scholar]
- 12. Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim BH, Grishin NV (2014) ECOD: An evolutionary classification of protein domains. PLoS Comp Biol 10:e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng H, Liao Y, Schaeffer RD, Grishin NV (2015) Manual classification strategies in the ECOD database. Proteins 83:1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pires DP, Cleto S, Sillankorva S, Azeredo J, Lu TK (2016) Genetically engineered phages: A review of advances over the last decade. Microbiol Mol Biol Rev 80:523–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aylett CH, Izoré T, Amos LA, Löwe J (2013) Structure of the tubulin/FtsZ‐like protein TubZ from Pseudomonas bacteriophage ΦKZ. J Mol Biol 425:2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Houten SM, Waterham HR (2001) Nonorthologous gene displacement of phosphomevalonate kinase. Mol Genet Metab 72:273–276. [DOI] [PubMed] [Google Scholar]
- 17. Mancini EJ, Kainov DE, Grimes JM, Tuma R, Bamford DH, Stuart DI (2004) Atomic snapshots of an RNA packaging motor reveal conformational changes linking ATP hydrolysis to RNA translocation. Cell 118:743–755. [DOI] [PubMed] [Google Scholar]
- 18. Iyer LM, Makarova KS, Koonin EV, Aravind L (2004) Comparative genomics of the FtsK–HerA superfamily of pumping ATPases: Implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res 32:5260–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burroughs AM, Allen KN, Dunaway‐Mariano D, Aravind L (2006) Evolutionary genomics of the HAD superfamily: Understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol 361:1003–1034. [DOI] [PubMed] [Google Scholar]
- 20. Kato M, Ito T, Wagner G, Richardson CC, Ellenberger T (2003) Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol Cell 11:1349–1360. [DOI] [PubMed] [Google Scholar]
- 21. Gupta A, Patil S, Vijayakumar R, Kondabagil K (2017) The polyphyletic origins of primase–helicase bifunctional proteins. J Mol Evol 85:188–204. [DOI] [PubMed] [Google Scholar]
- 22. Aksyuk AA, Leiman PG, Kurochkina LP, Shneider MM, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG (2009) The tail sheath structure of bacteriophage T4: A molecular machine for infecting bacteria. EMBO J 28:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kudryashev M, Wang RY, Brackmann M, Scherer S, Maier T, Baker D, DiMaio F, Stahlberg H, Egelman EH, Basler M (2015) Structure of the type VI secretion system contractile sheath. Cell 160:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ (2009) Type VI secretion apparatus and phage tail‐associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci USA 106:4154–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moréra S, Larivière L, Kurzeck J, Aschke‐Sonnenborn U, Freemont PS, Janin J, Rüger W (2001) High resolution crystal structures of T4 phage β‐glucosyltransferase: Induced fit and effect of substrate and metal binding. J Mol Biol 311:569–577. [DOI] [PubMed] [Google Scholar]
- 26. Holm L, Sander C (1995) Evolutionary link between glycogen phosphorylase and a DNA modifying enzyme. EMBO J 14:1287–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Korndörfer IP, Danzer J, Schmelcher M, Zimmer M, Skerra A, Loessner MJ (2006) The crystal structure of the bacteriophage PSA endolysin reveals a unique fold responsible for specific recognition of Listeria cell walls. J Mol Biol 364:678–689. [DOI] [PubMed] [Google Scholar]
- 28. Fischetti VA (2005) Bacteriophage lytic enzymes: Novel anti‐infectives. Trends Microbiol 13:491–496. [DOI] [PubMed] [Google Scholar]
- 29. Loeffler JM, Nelson D, Fischetti VA (2001) Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 294:2170–2172. [DOI] [PubMed] [Google Scholar]
- 30. Schuch R, Nelson D, Fischetti VA (2002) A bacteriolytic agent that detects and kills Bacillus anthracis . Nature 418:884–889. [DOI] [PubMed] [Google Scholar]
- 31. Gaeng S, Scherer S, Neve H, Loessner MJ (2000) Gene cloning and expression and secretion of Listeria monocytogenes bacteriophage‐lytic enzymes in Lactococcus lactis . Appl Environ Microbiol 66:2951–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Payne K, Sun Q, Sacchettini J, Hatfull GF (2009) Mycobacteriophage lysin B is a novel mycolylarabinoga‐lactan esterase. Mol Microbiol 73:367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prangishvili D (2013) The wonderful world of archaeal viruses. Annu Rev Microbiol 67:565–585. [DOI] [PubMed] [Google Scholar]
- 34. Prangishvili D (2015) Archaeal viruses: Living fossils of the ancient virosphere? Ann New York Acad Sci 1341:35–40. [DOI] [PubMed] [Google Scholar]
- 35. Larson ET, Eilers BJ, Reiter D, Ortmann AC, Young MJ, Lawrence CM (2007) A new DNA binding protein highly conserved in diverse crenarchaeal viruses. Virology 363:387–396. [DOI] [PubMed] [Google Scholar]
- 36. Larson ET, Reiter D, Young M, Lawrence CM (2006) Structure of A197 from Sulfolobus turreted icosahedral virus: A crenarchaeal viral glycosyltransferase exhibiting the GT‐A fold. J Virol 80:7636–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gomis‐Rüth FX, Moncalián G, Pérez‐Luque R, González A, Cabezón E, de la Cruz F, Coll M (2001) The bacterial conjugation protein TrwB resembles ring helicases and F1‐ATPase. Nature 409:637–641. [DOI] [PubMed] [Google Scholar]
- 38. Walldén K, Williams R, Yan J, Lian PW, Wang L, Thalassinos K, Orlova EV, Waksman G (2012) Structure of the VirB4 ATPase, alone and bound to the core complex of a type IV secretion system. Proc Natl Acad Sci USA 109:11348–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krupovic M, Koonin EV (2015) Polintons: A hotbed of eukaryotic virus, transposon and plasmid evolution. Nat Rev Microbiol 13:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krupovic M, Cvirkaite‐Krupovic V (2011) Virophages or satellite viruses? Nat Rev Microbiol 9:762–763. [DOI] [PubMed] [Google Scholar]
- 41. Klose T, Herbst DA, Zhu H, Max JP, Kenttämaa HI, Rossmann MG (2015) A mimivirus enzyme that participates in viral entry. Structure 23:1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abergel C, Rudinger‐Thirion J, Giegé R, Claverie JM (2007) Virus‐encoded aminoacyl‐tRNA synthetases: Structural and functional characterization of mimivirus TyrRS and MetRS. J Virol 81:12406–12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singh K, Gittis AG, Gitti RK, Ostazeski SA, Su HP, Garboczi DN (2016) The vaccinia virus H3 envelope protein, a major target of neutralizing antibodies, exhibits a glycosyltransferase fold and binds UDP–glucose. J Virol 90:5020–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin CL, Chung CS, Heine HG, Chang W (2000) Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo . J Virol 74:3353–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davies DH, McCausland MM, Valdez C, Huynh D, Hernandez JE, Mu Y, Hirst S, Villarreal L, Felgner PL, Crotty S (2005) Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J Virol 79:11724–11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kyrieleis OJ, Chang J, de la Peña M, Shuman S, Cusack S (2014) Crystal structure of vaccinia virus mRNA capping enzyme provides insights into the mechanism and evolution of the capping apparatus. Structure 22:452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De la Peña M, Kyrieleis OJ, Cusack S (2007) Structural insights into the mechanism and evolution of the vaccinia virus mRNA cap N7 methyl‐transferase. EMBO J 26:4913–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brown WC, Akey DL, Konwerski JR, Tarrasch JT, Skiniotis G, Kuhn RJ, Smith JL (2016) Extended surface for membrane association in Zika virus NS1 structure. Nat Struct Mol Biol 23:865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parashar UD, Gibson CJ, Bresee JS, Glass RI (2006) Rotavirus and severe childhood diarrhea. Emerg Infect Dis 12:304–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC (2011) Atomic model of an infectious rotavirus particle. EMBO J 30:408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aoki ST, Settembre EC, Trask SD, Greenberg HB, Harrison SC, Dormitzer PR (2009) Structure of rotavirus outer‐layer protein VP7 bound with a neutralizing Fab. Science 324:1444–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Caetano‐Anollés G, Wang M, Caetano‐Anollés D, Mittenthal JE (2009) The origin, evolution and structure of the protein world. Biochem J 417:621–637. [DOI] [PubMed] [Google Scholar]
- 53. Eryilmaz E, Benach J, Su M, Seetharaman J, Dutta K, Wei H, Gottlieb P, Hunt JF, Ghose R (2008) Structure and dynamics of the P7 protein from the bacteriophage ϕ12. J Mol Biol 382:402–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu K, Ming Z, Li Y, Chen C, Bao Z, Ren Z, Liu B, Tao W, Rao Z, Lou Z (2012) Purification, crystallization and preliminary X‐ray analysis of nonstructural protein 2 (nsp2) from avian infectious bronchitis virus. Acta Crystallogr 68:716–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soultanas P, Wigley DB (2001) Unwinding the ‘Gordian knot’ of helicase action. Trends Biochem Sci 26:47–54. [DOI] [PubMed] [Google Scholar]
- 56. James JA, Aggarwal AK, Linden RM, Escalante CR (2004) Structure of adeno‐associated virus type 2 Rep40–ADP complex: Insight into nucleotide recognition and catalysis by superfamily 3 helicases. Proc Natl Acad Sci USA 101:12455–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kwong AD, Rao BG, Jeang KT (2005) Viral and cellular RNA helicases as antiviral targets. Nat Rev Drug Disc 4:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gorbalenya AE, Koonin EV (1993) Helicases: Amino acid sequence comparisons and structure–function relationships. Curr Opin Struct Biol 3:419–429. [Google Scholar]
- 59. Koonin EV, Wolf YI, Nagasaki K, Dolja VV (2008) The Big Bang of picorna‐like virus evolution antedates the radiation of eukaryotic supergroups. Nat Rev Microbiol 6:925–939. [DOI] [PubMed] [Google Scholar]
- 60. Nakayama H (2002) RecQ family helicases: Roles as tumor suppressor proteins. Oncogene 21:9008. [DOI] [PubMed] [Google Scholar]
- 61. Lou Z, Sun Y, Rao Z (2014) Current progress in antiviral strategies. Trends Pharmacol Sci 35:86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hodel AE, Gershon PD, Quiocho FA (1998) Structural basis for sequence‐nonspecific recognition of 5′‐capped mRNA by a cap‐modifying enzyme. Mol Cell 1:443–447. [DOI] [PubMed] [Google Scholar]
- 63. Byszewska M, Śmietański M, Purta E, Bujnicki JM (2014) RNA methyltransferases involved in 5′ cap biosynthesis. RNA Biol 11:1597–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shin G, Yost SA, Miller MT, Elrod EJ, Grakoui A, Marcotrigiano J (2012) Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc Natl Acad Sci USA 109:16534–16539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yu X, Jiang J, Sun J, Zhou ZH (2015) A putative ATPase mediates RNA transcription and capping in a dsRNA virus. Elife 4:e07901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang B, Thurmond S, Hai R, Song J (2018) Structure and function of Zika virus NS5 protein: Perspectives for drug design. Cell Mol Life Sci 75:1723–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Koonin EV (2016) Horizontal gene transfer: Essentiality and evolvability in prokaryotes, and roles in evolutionary transitions. F1000 Res 5:1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rappoport N, Linial M (2012) Viral proteins acquired from a host converge to simplified domain architectures. PLOS Comput Biol 8:e1002364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Iyer LM, Leipe DD, Koonin EV, Aravind L (2004) Evolutionary history and higher order classification of AAA + ATPases. J Struct Biol 146:11–31. [DOI] [PubMed] [Google Scholar]
- 70. Smith M, Thorpe HM (2002) Diversity in the serine recombinases. Mol Microbiol 44:299–307. [DOI] [PubMed] [Google Scholar]
- 71. Haren L, Ton‐Hoang B, Chandler M (1999) Integrating DNA: Transposases and retroviral integrases. Annu Rev Microbiol 53:245–281. [DOI] [PubMed] [Google Scholar]
- 72. Senkevich TG, Koonin EV, Moss B (2011) Vaccinia virus F16 protein, a predicted catalytically inactive member of the prokaryotic serine recombinase superfamily, is targeted to nucleoli. Virology 417:334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shi S, Zhong Y, Majumdar I, Sri Krishna S, Grishin NV (2007) Searching for three‐dimensional secondary structural patterns in proteins with ProSMoS. Bioinformatics 23:1331–1338. [DOI] [PubMed] [Google Scholar]
- 74. Majumdar I, Krishna SS, Grishin NV (2005) PALSSE: A program to delineate linear secondary structural elements from protein structures. BMC Bioinform 6:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador‐Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aoki KF, Kanehisa M (2005) Using the KEGG database resource. Curr Protoc Bioinform 1.12.1–1.12.54. [DOI] [PubMed] [Google Scholar]
- 77. Felsenstein J (2010) PHYLIP (Phylogeny Inference Package) version 3.69. Seattle: University of Washington. [Google Scholar]
- 78. Holm L, Laakso LM (2016) Dali server update. Nucleic Acids Res 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI‐BLAST: A new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]