

Abstract
Carbohydrate hydrolyzing α‐glucosidases are commonly found in microorganisms present in the human intestine microbiome. We have previously reported crystal structures of an α‐glucosidase from the human gut bacterium Blaubia (Ruminococcus) obeum (Ro‐αG1) and its substrate preference/specificity switch. This novel member of the GH31 family is a structural homolog of human intestinal maltase‐glucoamylase (MGAM) and sucrase–isomaltase (SI) with a highly conserved active site that is predicted to be common in Ro‐αG1 homologs among other species that colonize the human gut. In this report, we present structures of Ro‐αG1 in complex with the antidiabetic α‐glucosidase inhibitors voglibose, miglitol, and acarbose and supporting binding data. The in vitro binding of these antidiabetic drugs to Ro‐αG1 suggests the potential for unintended in vivo crossreaction of the α‐glucosidase inhibitors to bacterial α‐glucosidases that are present in gut microorganism communities. Moreover, analysis of these drug‐bound enzyme structures could benefit further antidiabetic drug development.
Keywords: α‐glucosidase, α‐glucosidase inhibitor, antidiabetic drug, voglibose, miglitol, acarbose, human gut microbiome, substrate/inhibitor selection
Short abstract
PDB Code(s): http://firstglance.jmol.org/fg.htm?mol=6C9X; http://firstglance.jmol.org/fg.htm?mol=6C9Z; http://firstglance.jmol.org/fg.htm?mol=6CA1; http://firstglance.jmol.org/fg.htm?mol=6CA3; http://firstglance.jmol.org/fg.htm?mol=3PHA
Introduction
Carbohydrate metabolism has regained interest recently because it is believed that the microbial communities living in the human gut greatly expand the range of complex carbohydrates available for conversion to simple sugars for energy.1, 2 It is also a focus of biomass–biofuel conversion research.3, 4 In bacterial genomes of the gut microbiota, the genes for carbohydrate transport and metabolism pathways are often significantly over‐represented and highly diverged (http://www.jgi.doe.gov).5, 6 This indicates the great adaptation of the enteric bacteria to a variety of carbohydrates present in diet of an omnivore such as man. Many of these dietary carbohydrates are being competed for with the host and are essential for the bacteria survival and growth. This allows bacterial species to colonize specific niches along the gastrointestinal tract and as result have impact on the physiology and pathology of the host.7, 8, 9
It is believed that human gut microbiota express many enzymes that have the ability to bind and metabolize a wide range of bioactive compounds. Drugs taken orally reach the gastrointestinal tract and are exposed to the intestinal microbiota. It has been shown that microbiota‐encoded enzymes are capable of transforming xenobiotics and can influence the pharmacokinetics of drugs taken orally and impact their bioavailability.10, 11, 12, 13 Therefore, the intestinal microbiota may have a significant impact on drug bioavailability and effectiveness. However, understanding of these processes and how they impact microbiome and change its composition is very limited.14
We have previously characterized the structure and function of an α‐glucosidase (Ro‐αG1) from the human gut bacterium Ruminococcus obeum 15 or Blautia obeum after reclassification.16 This novel member of the family 31 of glycoside hydrolases (GH31)17 has active site structurally and functionally resembling that of human intestinal maltase‐glucoamylase (MGAM),18 thermophilic archaeon Sulfolobus solfataricus α‐glucosidase MalA19 and the reported human intestinal sucrase–isomaltase (SI).20 However, the unique dimer structure of the exohydrolase Ro‐αG1 seems to limit its substrates to dietary maltose or isomaltose, and it is less active with longer maltose oligosaccharides.
α‐glucosidase inhibitors such as acarbose (a naturally occurring pseudotetrasaccharide), voglibose (also naturally occurring in Streptomyces) and miglitol (derived from 1‐deoxynojirimycin, isolated from plants) are the first‐line drugs in noninsulin‐dependent diabetes mellitus.21, 22 The inhibitors reversibly bind to MGAM and SI, inhibiting carbohydrate hydrolysis in the upper small intestine, and consequently retard the absorption of glucose into bloodstream and reduce postprandial hyperglycemia. Acarbose is poorly absorbed in the intestine and is mostly excreted in feces, while some is degraded by amylase and by the colonic microbiota.21, 23 The absorption of voglibose is similarly poor; its excretion is rapid with no metabolites measured.24 Miglitol, in contrast, is fully absorbed in the gut.25 There are very limited studies of the potential interactions of these α‐glucosidase inhibitors with enzymes present in intestinal bacteria or whether such interactions could create adverse gastrointestinal symptoms. It was reported that the acarbose, was a poor inhibitor of Ro‐αG1 in contrast to MGAM and SI.15 The locations of the two active sites of a Ro‐αG1 dimer seemingly restrict the access of longer linear substrates and inhibitors.15 There are many sequence orthologs of B. obeum Ro‐αG1 and other bacterial α‐glucosidases, present and abundant in the intestinal microbial community.26 These carbohydrate‐active enzymes may bind α‐glucosidase inhibitors and potentially reduce their efficacy. This inhibitory effect may also have an impact on the carbohydrate metabolism of gut microorganisms and may be a cause of microbial population fluctuations. The details of unintended interactions of biomedically important drugs with microorganisms living in our gastrointestinal tract are little understood, although the impact of xenobiotics on the physiology and gene expression of active gut microbiota is significant.12, 14 The characterization of the interaction of α‐glucosidase inhibitors with gut bacterial α‐glucosidases is an early step toward the understanding of this complex system. In this study, we characterized binding of voglibose, miglitol and acarbose inhibitors to Ro‐αG1 and determined crystal structures of their complexes with the enzyme. All of these inhibitors bind to the active site of Ro‐αG1. We also studied the interaction of these inhibitors with the Ro‐αG1 W169Y mutant that switches specificity of the enzyme from isomaltose to maltose. These data have revealed potential in vivo crossinteraction of human α‐glucosidase inhibitors with bacterial α‐glucosidases present in human intestinal microbiota. We also believe that these drug‐bound bacterial α‐glucosidase structures and the inhibitive activities of these drugs provide beneficial information for development of more potent and specific antidiabetic medicines.
Results
Inhibition of Ro‐αG1 by voglibose, miglitol, and acarbose
The interaction of three widely used α‐glucosidase inhibitors with Ro‐αG1 was measured with a maltose hydrolysis assay. Voglibose is isolated from Streptomyces hygroscopius limoneus.27 This inhibitor comprises a validamine moiety and a 1,3‐propanediol group [Fig. 1(A)]. The validamine moiety mimics the nonreducing end glucose of a sugar substrate and blocks the −1 sugar‐binding subsite (–1 subsite) of α‐glucosidase. Miglitol is a pseudomonosaccharide derived from 1‐deoxynojirimycin and is one of the smallest α‐glucosidase inhibiton.28 The pseudomonosaccharide has a piperidine ring that is attached to a N‐hydroxyethyl chain [Fig. 1(A)]. Acarbose is a pseudotetrasaccharide, produced by Actinoplanes sp. SE 50/110.27 This compound is one of the largest α‐glucosidase inhibitors and includes a valienamine unit (ring A), a 4‐amino‐4,6‐dideoxy‐α‐d‐glucose unit (ring B) and two glucose units (rings C and D) joined through an α (1→4) maltose‐like linkage [Fig. 1(A)].
Figure 1.
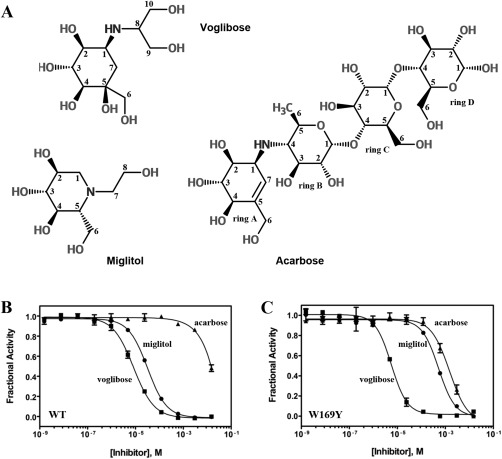
Selected inhibitors and their inhibition of Ro‐αG1. (A) Chemical Structures for voglibose, miglitol, and acarbose. Carbon atoms of each compound are numbered and labeled for the convenience of discussion. The four rings of acarbose are labeled in alphabetical order for the same purpose. (B), (C) Dose–response plots for inhibition of WT Ro‐αG1 and W169Y mutant, respectively, by voglibose (▪), miglitol (●) and acarbose (▲).
Voglibose is a good inhibitor of both wild type (WT) (IC50 = 8.3 μm) and the W169Y mutant (IC50 = 5.6 μm) of Ro‐αG1 [Table 1; Fig. 1(B,C)]. Miglitol is a moderate inhibitor of WT Ro‐αG1 (IC50 = 32.0 μm), but its inhibitory activity is significantly reduced towards the W169Y mutant (IC50 = 519.6 μm). Consistent with previous observations,15 acarbose barely inhibits WT Ro‐αG1 at the highest concentration of inhibitor used in the assay (15 mM), but it exhibits weak, although measurable, inhibition of the W169Y mutant. The Hill coefficients determined from fitting each dose‐response curve are approximately one, consistent with the structural observation that these inhibitors bind at the active site of Ro‐αG1.
Table 1.
Inhibitor Statistics
| WT | W169Y | |
|---|---|---|
| Voglibose | ||
| IC50 (µM) | 8.3 ± 1.6 | 5.6 ± 0.3 |
| Hill slope | 1.01 ± 0.08 | 1.31 ± 0.19 |
| Miglitol | ||
| IC50 (µM) | 32.0 ± 2.5 | 519.6 ± 28.5 |
| Hill slope | 1.09 ± 0.04 | 1.27 ± 0.16 |
| Acarbose | ||
| IC50 (µM) | n.d.a | 1483.0 ± 621.5 |
| Hill slope | n.d. | 1.14 ± 0.17 |
Not done; acarbose inhibition of the WT enzyme was incomplete at concentrations tested.
Structures of ro‐αG1 in complex with voglibose
Co‐crystallization of WT Ro‐αG1 in the presence of 10% (w/v) voglibose produced crystals of the same space group and similar unit cell dimensions as those of WT and D307A/isomaltose crystals (Table 2).15 There is one Ro‐αG1 dimer per asymmetric unit, and each active site is occupied by one voglibose which can be easily recognized in the excellent electron density maps [Fig. 2(A,B)]. A secondary structure matching (SSM) superposition of the WT dimer and the WT/Voglibose dimer results in a root mean square deviation (RMSD) value of 0.66 Å, therefore showing no significant intermonomer or interdomain conformational changes upon voglibose binding.
Table 2.
Crystallographic Statistics
| Data collection | WT/voglibose | W169Y/voglibose | WT/miglitol | W169Y/miglitol | W169Y/acarbose | D307A/acarbose |
|---|---|---|---|---|---|---|
| Space group | P21 | P21 | P1 | P1 | P21 | P21 |
| Unit cell (Å, °) | a = 68.42, b = 124.3, c = 87.74, β = 108.5 | a = 67.75, b = 126.3, c = 88.19, β = 107.8 | a = 68.75, b = 70.06, c = 88.16, α = 111.3, β = 107.6, γ = 97.67 | a = 64.69, b = 70.83, c = 88.10, α = 111.3, β = 107.4, γ = 97.66 | a = 88.03, b = 125.9, c = 133.58, β = 107.7 | a = 67.95, b = 125.7, c = 87.97, β= 107.6 |
| MW Da (residue) | 77083 (663)a | 77083 (663)a | 77083 (663)a | 77083 (663)a | 77083 (663)a | 77083 (663)a |
| Mol (AU) | 2 | 2 | 2 | 2 | 4 | 2 |
| Wavelength (Å) | 0.9791 | 0.9792 | 0.9792 | 0.9792 | 0.9793 | 0.9792 |
| Resolution range (Å) | 29.89–1.46 | 32.3–2.10 | 31.5–1.95 | 31.7–1.74 | 42.4–2.18 | 41.4–2.00 |
| (Last resolution shell) | ||||||
| (1.49–1.46) | (2.14–2.10) | (1.98–1.95) | (1.77–1.74) | (2.22–2.18) | (2.00–2.03) | |
| Number of unique reflections | 226940 | 79048 | 94150 | 131158 | 143009 | 91803 |
| Redundancy | 4.2 (4.1)b | 4.0 (3.5)b | 1.9 (1.8)b | 2.0 (1.9)b | 3.6 (2.9)b | 3.4 (2.7)b |
| Completeness (%) | 100.0 (100.0)b | 97.6 (92.6)b | 96.6 (91.5)b | 96.2 (95.4)b | 98.8 (91.7)b | 96.0 (84.9)b |
| R merge (%) | 7.5 (62.5)b | 10.2 (59.0)b | 7.5 (42.4)b | 5.3 (59.4)b | 10.9 (58.0)b | 10.3 (58.5)b |
| I/σ(I) | 20.8 (1.9)b | 20.7 (1.9)b | 11.7 (1.7)b | 21.8 (1.5)b | 14.4 (1.4)b | 12,2 (1.2)b |
| Refinement | ||||||
| Resolution | 29.89–1.46 | 32.3–2.10 | 31.5–1.95 | 31.7–1.74 | 42.4–2.18 | 41.4–2.00 |
| Reflections (work/test) | 215511/11379 | 74931/3924 | 88434/4668 | 124532/6603 | 131921/6586 | 76870/4097 |
| R crystal/R free (%) | 14.2/16.2 | 19.2/24.6 | 15.2/20.1 | 15.3/19.1 | 20.0/26.5 | 17.1/22.5 |
| Rms deviation from ideal geometry Bond length (Å)/angle (°) | 0.006/1.151 | 0.006/1.011 | 0.006/1.051 | 0.006/1.042 | 0.008/1.086 | 0.007/1.028 |
| Protein atoms | 10898 | 10712 | 10855 | 10802 | 10644 | 10895 |
| Ligand/water/other atoms | 36/1493/44 | 36/263/0 | 28/806/12 | 28/1084/6 | 176/601/0 | 80/516/6 |
| Mean B value (Å2) (mainchain/sidechain) | 15.5/18.6 | 48.5/50.0 | 27.8/32.6 | 26.5/30.8 | 44.7/49.5 | 36.7/44.7 |
| Ramachandran plot statistic (%) | ||||||
| Residues in most favored regions, | 89.4 | 87.7 | 88.5 | 89.1 | 86.0 | 87.4 |
| in additional allowed regions, | 10.1 | 11.8 | 10.9 | 10.4 | 13.6 | 12.5 |
| in generously allowed regions, | 0.5 | 0.4 | 0.5 | 0.5 | 0.4 | 0.5 |
| in disallowed region | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB entry | 6C9X | 6C9Z | 6CA1 | 6CA3 | 3PHA | 3POC |
Not including cloning artifact.
(Last resolution shell).
Figure 2.

The structure of the Ro‐αG1/voglibose complex. (A) The ribbon diagram of a Ro‐αG1 dimer with voglibose bound at each active site. The two monomers of the dimer are colored in cyan and green, respectively. The two vogliboses are drawn in stick format. (B) The interaction pattern between one voglibose and its surrounding active site protein residues. The active site protein residues are drawn in stick format with carbon atoms colored in grey. The voglibose is drawn in thicker stick format with carbon atoms colored in yellow. The carbon atoms on the cyclohexane ring of voglibose are labeled in black. Hydrogen bonds are represented by magenta dashed lines. Water molecules involved in hydrogen bond network are drawn as small green spheres. The electron density map (2F o –F c) associated with the voglibose is represented in pink mesh at 1σ contour level. (C) The active site interaction pattern of an isomaltose in Ro‐αG1 D307A/isomaltose complex.15 The substrate‐binding mode is for comparison to inhibitor‐binding modes presented in this paper. The key residues at the active site (carbon atoms in yellow) and the substrate isomaltose (carbon atoms in cyan) are drawn in stick format. The carbon atoms on the pyranose ring of isomaltose at the −1 subsite are labeled. The three conserved water molecules involved in hydrogen bond network are drawn as small green spheres. The mutation D307A is highlighted in magenta.
At the −1 sugar‐binding subsite, the cyclohexane ring of voglibose adopts a chair conformation, similar to the glucopyranose ring of the nonreducing end glucose of isomaltose in the D307A/isomaltose complex [Fig. 2(B,C)]. The C2, C3, C4, and C6 hydroxyls of the validamine moiety of voglibose form hydrogen bonds to active site residues (D197, D420, and H478), similar to the equivalent hydroxyl groups of a glucose unit from the substrate.15 The extra C5 hydroxyl of voglibose does not form any hydrogen bonds to the surrounding protein residues. In fact, the indole ring of W169 beneath the cyclohexane ring tilts about 9° (not shown in figures) compared to the Ro‐αG1WT/miglitol structure as described later, seemingly to avoid close contact with the hydroxyl.
The (C1) amino group, that bridges the 1,3‐propanediol group at +1 subsite, forms a single hydrogen bond to the catalytic acid/base D420, a mimic of the interaction of the glycosidic oxygen of a substrate. The 1,3‐propanediol group forms multiple hydrogen bonds to residues D73 and R404 and involves a water‐mediated hydrogen bond network, therefore maintaining all major interactions of a substrate such as isomaltose at the +1 subsite with the surrounding protein residues.15
Compared to the D307/isomaltose complex, the three water molecules W(1), W(2), and W(3) at the active site are conserved. However, the hydrogen bond network associated with W(1) in WT/Voglibose is modified due to the lack of the hemiacetal oxygen O5 of a glucopyranose ring in the cyclohexane ring. The hydrogen bond network associated with W(3) (at the +1 subsite) is partially changed, too.
As described earlier, the indole ring of W169 tilts about 9 degrees to accommodate the C5 hydroxyl of voglibose in the WT complex. A tyrosine in the position of W169 is apparently less likely to form a close contact to the hydroxyl group, making it energetically favorable for inhibitor binding. This may overwhelm other factors resulting from the replacement as discussed later, and partially explains why voglibose has about a one‐third lower IC50 value with the W169Y mutant compared to the WT enzyme. No other significant changes of either overall structure or active site were observed between the WT/voglibose and W169Y/voglibose complexes. The rmsd value of a monomer‐to‐monomer superposition of the two complexes is less than 0.22Å.
Structures of Ro‐αG1 in complex with miglitol
Co‐crystallization of WT Ro‐αG1 with miglitol (10% (w/v) in crystallization buffer) produced crystals of similar forms as WT/voglibose (Table 2). In the structure, each active site of Ro‐αG1 is occupied by one miglitol fitted well into electron density maps [Fig. 3(A)]. No overall conformational change upon the inhibitor binding was observed.
Figure 3.

The structure of the Ro‐αG1/miglitol complex. (A) The interaction pattern between one miglitol and its surrounding active site protein residues. The active site protein residues are drawn in stick format with carbon atoms colored in grey. The miglitol is drawn in thicker stick format with carbon atoms colored in yellow. The carbon atoms on the cyclohexane ring of miglitol are labeled in black. Hydrogen bonds are represented by magenta dashed lines. Water molecules involved in hydrogen bond network are drawn as small green spheres. The electron density map (2F o –F c) associated with the mitlitol is represented in purple mesh at 1σ contour level. (B) The comparison of voglibose‐ and miglitol‐binding modes at the active site. The Ro‐αG1/voglibose and Ro‐αG1/miglitol complexes are superimposed (monomer to monomer) with an rmsd value of 0.33Å. All active site residues and inhibitors are drawn in stick format. The carbon atoms of protein residues and voglibose of the Ro‐αG1/voglibose complex are colored in cyan. The carbon atoms of protein residues and miglitol of the Ro‐αG1/miglitol complex are colored in cyan and red, respectively. The hydrogen bond between W271 and the N‐ethyl chain as discussed in the text is drawn in magenta dashed line. The conformations of active site key residues primarily involved in inhibitor binding are highly conserved. The cyclohexane ring of voglibose and the piperidine ring of miglitol are nearly overlapped. The conformational changes of some other key residues reflect their adjustments to accomodate different inhibitors.
Miglitol is the smallest α‐glucosidase inhibitor and occupies only the −1 subsite. Its piperidine ring also adopts a chair conformation like the bound substrate [Fig. 2(C)]. The C2, C3, C4, and C6 hydroxyls form hydrogen bonds or a hydrogen bond network at the active site similar to that observed in the D307/isomaltose and WT/voglibose complexes mentioned earlier. The hydroxyl of the N‐hydroethyl chain also forms a hydrogen bond to the Nɛ on the indole ring of W271. The indole ring of W271 has to rotate (Chi1 rotation) about 30° to make this interaction possible [Fig. 3(B)].
Due to the movement of W271 and the interaction of the piperidine ring nitrogen to the catalytic nucleophile D307, the hydrogen bond network associated with the conserved water W(1) is changed. At the +1 subsite, there is no direct hydrogen bond between miglitol and the surrounding protein residues. However, the conserved water W(3) and another water W(4) bridge multiple hydrogen bonds between the hydroxyl of the N‐hydroethyl chain to the residues of D73 and R404, which play important roles in substrate binding at the +1 subsite. The water W(4) is in a similar position as one of the hydroxyls of the 1,3‐propanediol of voglibose [Fig. 2(B)].
The overall mode of miglitol binding at the active site of Ro‐αG1 is similar to its binding to the N‐terminal domain of human intestinal brush‐border enzyme maltase‐glucoamylase (NtMGAM),29 a primary target of the drug. However, in the NtMGAM/miglitol complex, the hydroxyl of the N‐hydroethyl chain is not involved in any interaction with NtMGAM. Miglitol is also an inhibitor of human lysosomal acid α‐glucosidase (hGGA), which is critical for the degradation of glycogen to glucose in lysosomes. Mutations of the GGA gene cause glycogen storage disease type II (Pompe disease). A comparison of the unintended cross reaction product hGGA/miglitol complex and Ro‐αG1/miglitol shows almost identical conformation in their active site and the inhibitor bound.30
Miglitol also binds to the Ro‐αG1 W169Y mutant without significant structural alteration except for some variation in a few solvent molecules associated with the substitution. Since miglitol contributes no direct hydrogen bonds at the +1 subsite, a small structural variation at the −1 subsite could potentially cause a large change to its binding affinity. It is possible that the W‐>Y mutation reduced the interaction between the inhibitor and the flanking residue; so much so that it accounts for more than a 16‐fold increase in the IC50 value of miglitol with the W169Y mutant when compared with the WT enzyme.
Structures of Ro‐αG1 in complex with acarbose
Acarbose is the first α‐glucosidase inhibitor used for diabetes and is the most extensively studied. This antidiabetic drug also binds to many enzymes from all five kingdoms involved in glucose metabolism. Besides its targets, human intestinal MGAM and SI, it also binds to human pancreatic α‐amylase31 and glycogen branching enzyme (GBE1) (PDB ID: 5CLT).
Arcabose has been found to be a low affinity inhibitor of wild type Ro‐αG1 [Fig. 2(B); Table 1], which is consistent with earlier observations.15 Although the active sites of Ro‐αG1 and NtMGAM are conserved, only the active site of NtMGAM is wide open;18 therefore, we hypothesized that the difference in acarbose inhibition might be related to the lower accessibility of the inhibitor to the active site of Ro‐αG1 due to its unique hidden position under a four‐helix bundle [Fig. 2(A)].15
We first tried co‐crystallization of WT Ro‐αG1 by supplementing crystallization buffer with 10% (w/v) acarbose. Crystals were obtained and their structures were subsequently determined. However, no acarbose was found in these structures. Considering that the W169Y mutation switches the enzyme's substrate preference15 and the human NtMGAM that binds acarbose does have a tyrosine at the equivalent position of Ro‐αG1 W169,18 we co‐crystallized the W169Y mutant with acarbose and subsequently obtained a W169Y/acarbose complex structure (Table 2). In this structure, there are two parallel dimers residing in one asymmetric unit of the complex crystal. All four monomers are structurally ordered and the rmsd values of the pairwise SSM structural alignments of the four monomers range from 0.26 to 0.35 Å without gaps, suggesting all four monomers have the same structure. In the following descriptions and discussions, only one monomer (monomer A) and one dimer (the dimer from monomers A and B) will be used to describe the complex.
Each of the four monomers binds one acarbose at its active site [Fig. 4(A)]. The electron densities are well‐defined for the first two rings of the inhibitor, ring A and ring B [Figs. 1(A) and 4(B)]. The densities gradually become weaker towards the two glycone rings, ring C and ring D. In all four monomers, the bound acarbose is virtually in the same conformation. The overall B‐factor of acarbose and the corresponding conserved water molecules at each active site was higher (refined in full occupancies) than the surrounding protein atoms, indicating possible partial occupancy of the inhibitor. In fact, only in one monomer were all three conserved water molecules modeled and refined [Fig. 4(A)].
Figure 4.

The structure of the Ro‐αG1 W169Y/acarbose complex. (A) The interaction pattern between one acarbose and its surrounding active site protein residues. The active site protein residues are drawn in stick format with carbon atoms colored in gray. The acarbose is drawn in thicker stick format with carbon atoms colored in yellow. The carbon atoms on the valienamine ring of acarbose are labeled in black. Magenta dashed lines represent hydrogen bonds. The W169Y mutant is drawn in pink for highlighting. Water molecules involved in hydrogen bond network are drawn as small green spheres. (B) A simulated annealing omit 2F o–F c electron density map of one acarbose in the complex. The 2F o–F c map is drawn in gray mesh at 1σ contour level. The simulated annealing refinement with a starting temperature of 5000 K was performed with the acarbose being omitted.
Ring A of acarbose binds to the −1 subsite [Fig. 4(A)]. The unsaturated cyclitol unit is in a half‐chair conformation. The C2, C3, C4, and C6 hydroxyls form hydrogen bonds to Ro‐αG1 similarly to what is observed in other Ro‐αG1 complexes described above, particularly the Ro‐αG1/voglibose complex [Figs. 2(B,C) and 3(A)]. The catalytic acid/base D420 interacts with the C2 hydroxyl and the bridging nitrogen between rings A and B. At the +1 subsite, the interaction of ring B to Ro‐αG1 is primarily contributed by the two hydroxyls (C2 and C3 of ring B) to the residues D73 and R404 as well as the water W(3) mediated hydrogen bond network. At the +2 binding site, ring C does not form any hydrogen bond to Ro‐αG1. The electron density for ring D is quite smeared, suggesting multiple conformations of the ring, but the last glycone ring potentially forms a hydrogen bond to K348 or K422 (data not shown).
The overall acarbose binding is very similar in Ro‐αG1 W169Y/acarbose and NtMGAM/acarbose structures, particularly at the −1 and +1 subsites [Fig. 5(A)]. The lack of any interactions between the two glycone rings and Ro‐αG1 explains their conformational flexibility, and it may attribute to their low affinities to both NtMGAM and Ro‐αG1.
Figure 5.

Comparison of Inhibitor binding. (A) A comparison of acarbose binding at the active site in superimposed Ro‐αG1 W169Y/acarbose and NtMGAM/acarbose structures. The Cα trace of the W169Y peptide is drawn in green. The bound acarbose and some active site residues are drawn in stick format with carbon atoms colored in green. The W169Y mutant is highlighted in magenta while other residues are labeled in black. The Cα trace of NtMGAM peptide is drawn in orange. The bound acarbose and some equivalent active site residues are drawn in stick format with carbon atoms colored in orange. The labeling of each residue is in orange and mostly placed under the labeling of its corresponding residue of Ro‐αG1. (B) A structural superposition of Ro‐αG1 W169Y/acarbose and WT/voglibose complexes. The drawing of the W169Y/acarbose complex is the same as in (A). The labeling of the C6 atom of acarbose ring B is in red to highlight a potential close contact between the C6 methyl group of the ring and the W169 indole ring of WT Ro‐αG1. For WT/voglibose, the Cα trace is in grey. The voglibose and some active site residues are in stick format with carbon atoms colored in gray.
Additionally, the structure of a co‐crystal of D307A mutant with acarbose was also determined (Table 2), in which only the ring A of a bound acarbose at −1 subsite has well‐defined electron density while other three rings are disordered.
Discussion
Many host‐targeted medicines are metabolized by gastrointestinal microbiota, though only a few of them have been studied for their influence on these microorganisms.12, 13, 32, 33, 34, 35, 36 The molecular mechanisms underlying their interactions largely remain unanswered except for a few examples.37 Antidiabetic α‐glucosidase inhibitors that are not absorbed in the small intestine or not metabolized on their way to being excreted21, 23 could encounter an enormous number of gut bacteria during excretion, especially in the colon. This creates a chance for a potentially unintended cross‐interaction of the host‐targeted medicine with the α‐glucosidases of gut microbiota. The potential in vivo cross‐interaction is shown by the structures of bacterial Ro‐αG1 in complex with three common human α‐glucosidase inhibitors, voglibose, miglitol, and acarbose, presented here.
Because of the high sequence and structural identity of the active sites of human α‐glucosidases (MGAM and SI) and gut bacterial α‐glucosidases such as Ro‐αG1, gut bacterial α‐glucosidases can process dietary carbohydrates (e.g., maltose and isomaltose) as well as be effectively inhibited by α‐glucosidase inhibitors with comparable strengths.38, 39 The nearly identical binding modes (of miglitol and acarbose) to Ro‐αG1 and human NtMGAM suggest that most of the currently used α‐glucosidase inhibitors can inhibit gut bacterial α‐glucosidases (e.g., Ro‐αG1) [Fig. 2(A)]. Though the NtMGAM/voglibose structure has not been reported, we believe the binding mode of voglibose to NtMGAM or other domains of MGAM and SI will be very similar to that observed in Ro‐αG1/voglibose [Fig. 2(B)].
Though the active site is highly conserved, its location and any changes in nearby residues may affect the access and specificity of these α‐glucosidases to inhibitors. Comparing the W169Y/acarbose and NtMGAM/acarbose structures, acarbose binding is nearly identical at the −1 and +1 subsites, while the conformation of the two glycone rings is different and flexible due to lack of interaction with the enzymes. The active site of NtMGAM is wide open for access of the substrate/inhibitor. The active site of Ro‐αG1, in contrast, is hidden under a helix‐bundle. The two active sites of a Ro‐αG1 dimer face each other and are separated by about 37 Å, which is measured by the distance between the O4 atoms (ring A) of two bound acarboses. The length of one acarbose is about 19 Å (not fully stretched). It is clear that a long inhibitor like acarbose will not easily approach an active site of Ro‐αG1, as indicated by IC50. Even a bound acarbose at one active site can create steric hindrance for another acarbose to reach and block the active site in the opposite subunit. This may be one of reasons that acarbose is not a good inhibitor of Ro‐αG1.15 However, small inhibitors like voglibose and miglitol will be much less influenced by such a steric limitation.
The 6‐membered rings of voglibose, miglitol and acarbose that contribute to inhibitor binding at the −1 subsite are different in their chemical composition. However, their largely similar conformations and the four hydroxyls (C2, C3, C4, and C6) that they commonly possess are the determinants of binding. At the −1 subsite, the conformations of Ro‐αG1 residues including D197, D449, H478, and acid/base D420, which are involved in forming hydrogen bonds to these hydroxyls, are quite conserved. Meanwhile, the residues including W169, W271, and nucleophile D307 are seemingly more or less adjustable in their positions in order to avoid close contact (in the case of voglibose binding) or form a new hydrogen bond (in the case of miglitol binding) upon inhibitor docking. The mutant D307A increased Ro‐αG1 binding to acarbose in contrast to WT even though the improvement is very limited and only the ring A of the inhibitor is visible in the structure.
We demonstrated earlier that Ro‐αG1 prefers isomaltose over maltose because of a potential clash of the indole ring of W169 with the O6 atom of the maltose glycone binding at the +1 subsite.15 Replacing the tryptophan with a tyrosine relieves the congestion and switches the substrate specificity of Ro‐αG1 to maltose. Because the W169Y mutant, unlike the WT, is able to form a complex with acarbose, the W169Y mutation apparently has increased the affinity for acarbose. The low affinity of the WT Ro‐αG1 for acarbose, we believe, similarly results from a potential close contact of the W169 indole ring to, in this case, ring B's methyl group (C6) of acarbose at the +1 subsite [Fig. 5(B)]. A sidechain movement that the W169 may have to help accommodate ring B at the +1 subsite seems to be limited. Therefore, a W‐>Y mutation removes the potential congestion, leading to an increased affinity to acarbose. There are naturally occurring α‐glucosidases with W‐>Y substitution and they are also present in intestinal microbiota.
The reviving of an inhibitor's binding by a mutation as described above also suggests that the moiety of an inhibitor at the +1 subsite plays a subtle role in determining the affinity of an inhibitor. Miglitol is regarded as a pseudomonosaccharide, but it is not simply an analog of a monosaccharide. It has an N‐hydroxyethyl chain that forms water‐bridged hydrogen bonds to residues D73 and R404, which primarily interact with the moiety of the inhibitor at the +1 subsite [Figs. 2(B), 3(A), and 4(A)]. Since Ro‐αG1 does not form a complex with glucose even in the presence of elevated concentration of the monosaccharide in crystallization buffer, we believe that a simple glucose analog will not bind and inhibit Ro‐αG1 either. The N‐hydroxyethyl chain of miglitol could be critical in the binding of the inhibitor.
The study of the human gut microbiome has been expanding rapidly; however, the structural and functional characterization of the key molecules of these microorganisms is still in its infancy. Here, we have presented the structures and functions of the α‐glucosidase Ro‐αG1 from a species of Blaubia, a genus of the prominent Firmicutes division. The bacterial α‐glucosidase, a representative of many sequence homologs, can process dietary carbohydrates and interact with human α‐glucosidase drugs in vitro. The potential unintended crossinteraction in vivo may influence microbial population and community dynamics. Though its consequent physiological and/or pathological impact to the host is unknown and remain to be further explored, we believe for the purpose of? novel antidiabetic drug development, the understanding of such interaction between an α‐glucosidase inhibitor and its potential receptor(s) from gut microbial community could be an important part of exploration.
Materials and Methods
Crystallization of Ro‐αG1 or its mutants in the presence of inhibitors
The cloning of wild type Ro‐αG1 into the pMCSG19 vector and the construction of site‐direct mutant W169Y using the Polymerase Incomplete Primer Extension (PIPE) method40 have been reported earlier.15 The wild type and W169Y mutant were over‐expressed in E. coli pRK1037 and purified using Ni‐affinity chromatography as described earlier.41 The purified proteins, concentrated to 28–61 mg/mL, were crystallized in the presence of 10% (w/v) voglibose, miglitol, or acarbose under the same condition as that of the wild type (0.1M Bis–Tris pH 5.5, 25%(w/v) PEG3350) at 4°C.15 All crystals were cryo‐protected directly in liquid nitrogen prior to X‐ray diffraction data collection.
X‐ray diffraction and structure determination
The X‐ray diffraction data were obtained at the 19‐ID beamline of the Structural Biology Center at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect.42 The intensities were integrated and scaled with the HKL3000 suite43 (Table 2). All structures were solved by molecular replacement using the program MolRep44 with the Ro‐αG1 wild type structure (PDB ID: 3N04) as a search template (Table 2). The model rebuilding of the enzyme and the model building of voglibose or miglitol or acarbose were performed using the program COOT.45 All refinements including TLS refinement46 were carried out with the program Phenix.Refine47 (Table 2).
Inhibitor assays
Maltose hydrolysis in the presence of the inhibitors voglibose, miglitol and acarbose (all from Sigma‐Aldrich, St. Louis, MO) was determined by the glucose oxidase/peroxidase method as described earlier in (15). Reactions were carried out in duplicate by incubating 100 nM wild type (WT) enzyme or 20 nM W169Y with 30 mM maltose for 60 min at 37°C (in 50 mM MES, 100 mM NaCl, pH 6.5). Inhibitor concentrations ranged from 1.54 µM to 15 mM. Reactions were quenched for 3 min at 95°C, and then glucose was quantitated with the Glucose (GO) Assay Kit (Sigma‐Aldrich, St. Louis MO). Contaminating maltase activity in the GO reagent was inhibited by 1 mM acarbose as described in (10). None of the inhibitors interfered with glucose quantitation (not shown). Data analysis was performed using GraphPad Prism 5.04 for Windows (GraphPad, San Diego, CA; http://www.graphpad.com). The data were fit to the built‐in equation for log(inhibitor) versus response curve with variable Hill slope. Standard deviations for IC50 values were calculated from the fits to the individual replicate data sets.
ACKNOWLEDGMENTS
The authors wish to thank members of the Structural Biology Center at Argonne National Laboratory for their help with data collection at the 19‐ID beamline.
The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE‐AC02‐06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid‐up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
REFERENCES
- 1. Turnbaugh PJ, Ley RE, Hamady M, Fraser‐Liggett CM, Knight R, Gordon JI (2007) The human microbiome project. Nature 449:804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke‐Reed P, Zakhari S, Read J, Watson B, Guyer M (2009) The NIH Human Microbiome Project. Genome Res 19:2317–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Jr. , Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T (2006) The path forward for biofuels and biomaterials. Science 311:484–489. [DOI] [PubMed] [Google Scholar]
- 4. Pauly M, Keegstra K (2008) Cell‐wall carbohydrates and their modification as a resource for biofuels. Plant J 54:559–568. [DOI] [PubMed] [Google Scholar]
- 5. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI (2003) A genomic view of the human–Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076. [DOI] [PubMed] [Google Scholar]
- 6. Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, Taylor TD, Noguchi H, Mori H, Ogura Y, Ehrlich DS, Itoh K, Takagi T, Sakaki Y, Hayashi T, Hattori M (2007) Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 14:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blaut M, Clavel T (2007) Metabolic diversity of the intestinal microbiota: implications for health and disease. J Nutr 137:751S–755S. [DOI] [PubMed] [Google Scholar]
- 8. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. [DOI] [PubMed] [Google Scholar]
- 9. Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, Corthesy I, Macé K, Chou CJ (2008) Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. Faseb J 22:2416–2426. [DOI] [PubMed] [Google Scholar]
- 10. Jourova L, Anzenbacher P, Anzenbacherova E (2016) Human gut microbiota plays a role in the metabolism of drugs. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 160:317–326. [DOI] [PubMed] [Google Scholar]
- 11. Stojancevic M, Bojic G, Salami HA, Mikov M (2014) The influence of intestinal tract and probiotics on the date of orally administered drugs. Curr Issues Mol Biol 16:55–68. [PubMed] [Google Scholar]
- 12. Maurice CF, Haiser HJ, Turnbaugh PJ (2013) Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoo HH, Kim IS, Yoo DH, Kim DH (2016) Effects of orally administered antibiotics on the bioavailability of amlodipine: gut microbiota‐mediated drug interaction. J Hypertens 34:156–162. [DOI] [PubMed] [Google Scholar]
- 14. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira‐Silva S, Gudmundsdottir V, Pedersen HK, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Costea PI, Kultima JR, Li J, Jorgensen T, Levenez F, Dore J, Meta HITc, Nielsen HB, Brunak S, Raes J, Hansen T, Wang J, Ehrlich SD, Bork P, Pedersen O (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528:262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan K, Tesar C, Wilton R, Keigher L, Babnigg G, Joachimiak A (2010) Novel {alpha}‐glucosidase from human gut microbiome: substrate specificities and their switch. Faseb J 24:3939–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lawson PA, Finegold SM (2015) Reclassification of Ruminococcus obeum as Blautia obeum comb. nov. Int J Syst Evol Microbiol 65:789–793. [DOI] [PubMed] [Google Scholar]
- 17. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate‐Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sim L, Quezada‐Calvillo R, Sterchi EE, Nichols BL, Rose DR (2008) Human intestinal maltase‐glucoamylase: crystal structure of the N‐terminal catalytic subunit and basis of inhibition and substrate specificity. J Mol Biol 375:782–792. [DOI] [PubMed] [Google Scholar]
- 19. Ernst HA, Lo Leggio L, Willemoës M, Leonard G, Blum P, Larsen S (2006) Structure of the Sulfolobus solfataricus alpha‐glucosidase: implications for domain conservation and substrate recognition in GH31. J Mol Biol 358:1106–1124. [DOI] [PubMed] [Google Scholar]
- 20. Sim L, Willemsma C, Mohan S, Naim HY, Pinto BM, Rose DR (2010) Structural basis for substrate selectivity in human maltase‐glucoamylase and sucrase‐isomaltase N‐terminal domains. J Biol Chem 285:17763–17770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Standl E, Schnell O (2012) Alpha‐glucosidase inhibitors 2012 ‐ cardiovascular considerations and trial evaluation. Diab Vasc Dis Res 9:163–169. [DOI] [PubMed] [Google Scholar]
- 22. Derosa G, Maffioli P (2012) Efficacy and safety profile evaluation of acarbose alone and in association with other antidiabetic drugs: a systematic review. Clin Ther 34:1221–1236. [DOI] [PubMed] [Google Scholar]
- 23. Desai A, Tandon N (2007) Management of type 2 diabetes mellitus with oral antihyperglycaemic therapy. Natl Med J India 20:192–198. [PubMed] [Google Scholar]
- 24. Goke B, Fuder H, Wieckhorst G, Theiss U, Stridde E, Littke T, Kleist P, Arnold R, Lucker PW (1995) Voglibose (AO‐128) is an efficient alpha‐glucosidase inhibitor and mobilizes the endogenous GLP‐1 reserve. Digestion 56:493–501. [DOI] [PubMed] [Google Scholar]
- 25. Standl E, Schernthaner G, Rybka J, Hanefeld M, Raptis SA, Naditch L (2001) Improved glycaemic control with miglitol in inadequately‐controlled type 2 diabetics. Diabet Res Clin Pract 51:205–213. [DOI] [PubMed] [Google Scholar]
- 26. Lozupone CA, Hamady M, Cantarel BL, Coutinho PM, Henrissat B, Gordon JI, Knight R (2008) The convergence of carbohydrate active gene repertoires in human gut microbes. Proc Natl Acad Sci USA 105:15076–15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flatt PM, Mahmud T (2007) Biosynthesis of aminocyclitol‐aminoglycoside antibiotics and related compounds. Nat Prod Rep 24:358–392. [DOI] [PubMed] [Google Scholar]
- 28. Kingma PJ, Menheere PP, Sels JP, Nieuwenhuijzen Kruseman AC (1992) alpha‐Glucosidase inhibition by miglitol in NIDDM patients. Diabet Care 15:478–483. [DOI] [PubMed] [Google Scholar]
- 29. Sim L, Jayakanthan K, Mohan S, Nasi R, Johnston BD, Pinto BM, Rose DR (2010) New glucosidase inhibitors from an ayurvedic herbal treatment for type 2 diabetes: structures and inhibition of human intestinal maltase‐glucoamylase with compounds from Salacia reticulata . Biochemistry 49:443–451. [DOI] [PubMed] [Google Scholar]
- 30. Roig‐Zamboni V, Cobucci‐Ponzano B, Iacono R, Ferrara MC, Germany S, Bourne Y, Parenti G, Moracci M, Sulzenbacher G (2017) Structure of human lysosomal acid alpha‐glucosidase—a guide for the treatment of Pompe disease. Nat Commun 8:1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brayer GD, Sidhu G, Maurus R, Rydberg EH, Braun C, Wang YL, Nguyen NT, Overall CH, Withers SG (2000) Subsite mapping of the human pancreatic alpha‐amylase active site through structural, kinetic, and mutagenesis techniques. Biochemistry 39:4778–4791. [DOI] [PubMed] [Google Scholar]
- 32. Sousa T, Paterson R, Moore V, Carlsson A, Abrahamsson B, Basit AW (2008) The gastrointestinal microbiota as a site for the biotransformation of drugs. Int J Pharm 363:1–25. [DOI] [PubMed] [Google Scholar]
- 33. Yoo DH, Kim IS, Van Le TK, Jung IH, Yoo HH, Kim DH (2014) Gut microbiota‐mediated drug interactions between lovastatin and antibiotics. Drug Metab Dispos 42:1508–1513. [DOI] [PubMed] [Google Scholar]
- 34. Saad R, Rizkallah MR, Aziz RK (2012) Gut pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut‐associated microbes. Gut Pathog 4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H, He J, Jia W (2016) The influence of gut microbiota on drug metabolism and toxicity. Expert Opin Drug Metab Toxicol 12:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kang MJ, Kim HG, Kim JS, Oh DG, Um YJ, Seo CS, Han JW, Cho HJ, Kim GH, Jeong TC, Jeong HG (2013) The effect of gut microbiota on drug metabolism. Expert Opin Drug Metab Toxicol 9:1295–1308. [DOI] [PubMed] [Google Scholar]
- 37. Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, Redinbo MR (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330:831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuriyama C, Kamiyama O, Ikeda K, Sanae F, Kato A, Adachi I, Imahori T, Takahata H, Okamoto T, Asano N (2008) In vitro inhibition of glycogen‐degrading enzymes and glycosidases by six‐membered sugar mimics and their evaluation in cell cultures. Bioorg Med Chem 16:7330–7336. [DOI] [PubMed] [Google Scholar]
- 39. Natori Y, Imahori T, Murakami K, Yoshimura Y, Nakagawa S, Kato A, Adachi I, Takahata H (2011) The synthesis and biological evaluation of 1‐C‐alkyl‐l‐arabinoiminofuranoses, a novel class of alpha‐glucosidase inhibitors. Bioorg Med Chem Lett 21:738–741. [DOI] [PubMed] [Google Scholar]
- 40. Klock HE, Lesley SA (2009) The polymerase incomplete primer extension (PIPE) method applied to high‐throughput cloning and site‐directed mutagenesis. Methods Mol Biol 498:91–103. [DOI] [PubMed] [Google Scholar]
- 41. Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A (2004) Automation of protein purification for structural genomics. J Struct Funct Genomics 5:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat 13:30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr 62:859–866. [DOI] [PubMed] [Google Scholar]
- 44. Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr 66:22–25. [DOI] [PubMed] [Google Scholar]
- 45. Emsley P, Cowtan K (2004) Coot: Model‐building tools for molecular graphics. Acta Crystallogr 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 46. Painter J, Merritt EA (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr 62:439–450. [DOI] [PubMed] [Google Scholar]
- 47. Afonine PV, Grosse‐Kunstleve RW, Adams PD (2005) CCP4 Newsl 42:contribution 8.