

Summary
Neutral zinc alkoxide complexes show high activity toward the ring-opening polymerization of cyclic esters and carbonates, to generate biodegradable plastics applicable in several areas. Herein, we use a ferrocene-chelating heteroscorpionate complex in redox-switchable polymerization reactions, and we show that it is a moderately active catalyst for the ring-opening polymerization of L-lactide, ɛ-caprolactone, trimethylene carbonate, and δ-valerolactone. Uniquely for this type of catalyst, the oxidized complex has a similar polymerization activity as the corresponding reduced compound, but displays significantly different rates of reaction in the case of trimethylene carbonate and δ-valerolactone. Investigations of the oxidized compound suggest the presence of an organic radical rather than an Fe(III) complex. Electronic structure and density functional theory (DFT) calculations were performed to support the proposed electronic states of the catalytic complex and to help explain the observed reactivity differences. The catalyst was also compared with a monomeric phenoxide complex to show the influence of the phosphine-zinc interaction on catalytic properties.
Subject Areas: Chemistry, Inorganic Chemistry, Catalysis, Polymer Chemistry
Graphical Abstract
Highlights
-
•
Dimeric and monomeric zinc heteroscorpionate species were prepared
-
•
Monomeric zinc complex gives irreversible chemical redox and is inactive toward ROP
-
•
Dimeric zinc complex gives reversible chemical redox and is inactive toward ROP
-
•
Oxidation of the zinc benzoxide species occurs at the phosphine moiety
Chemistry; Inorganic Chemistry; Catalysis; Polymer Chemistry
Introduction
Aliphatic polyesters and polycarbonates derived from L-lactide (LA), ɛ-caprolactone (CL), δ-valerolactone (VL), and trimethylene carbonate (TMC) are biodegradable plastics with applications in the biomedical field and food packaging and with other specialty applications (Suriano et al., 2011, Ulery et al., 2011, Tian et al., 2012, Vert, 2005, Nair and Laurencin, 2007, Place et al., 2009, Ruzette and Leibler, 2005, Vroman and Tighzert, 2009, Oerlemans et al., 2010). Utilizing discrete metal complexes for the ring-opening polymerization of these cyclic monomers provides a great degree of control over the microstructure of the resulting polymers. In particular, neutral zinc alkoxide complexes show high activity toward the ring-opening polymerization of cyclic esters and carbonates. Due to the “living” nature of these processes, polymers with high molar mass and low dispersities are typically obtained (Guillaume and Carpentier, 2012, Ajellal et al., 2010, Helou et al., 2010, Helou et al., 2009, Williams et al., 2003, O'Keefe et al., 2001, Dechy-Cabaret et al., 2004, Wu et al., 2006, Platel et al., 2008, Wheaton et al., 2009, Arbaoui and Redshaw, 2010, Huang et al., 2013). In addition, utilizing zinc in polymerizations is advantageous due to the low cost, high abundance, and biocompatibility of the metal, allowing for wide application of the resulting biodegradable plastics.
The prevalent use of zinc in ring-opening polymerizations of cyclic esters and carbonates is matched by the use of hard N/O-based supporting ligands. However, only a few reports use soft donor ligands, such as phosphines, to stabilize the active zinc metal center (Fliedel et al., 2015, D'Auria et al., 2012, Liang et al., 2010). Such interactions can be beneficial due to the hemilabile nature of the zinc-phosphine bond and may result in highly active polymerization systems. The use of redox-active ligands in combination with zinc for ring-opening polymerizations is even less common than the use of soft donor ligands despite the potential benefits (Bakewell et al., 2015). Redox-active ligands provide a means of affecting the reactivity of the metal through changes in the oxidation state of the ligand. For example, in redox-switchable hemilabile ligands, the incorporation of a redox-active group into the ligand framework, in close proximity to a substitutionally labile component, allows to control the strength of the ligand-metal bond, leading to the dissociation of the ligand fragment upon oxidation (Singewald et al., 1995, Sassano and Mirkin, 1995, Higgins and Mirkin, 1995, Slone et al., 1997, Weinberger et al., 2001). In addition, the electronic and steric environment of the transition metal can be controlled without directly affecting its oxidation state (Allgeier and Mirkin, 1998).
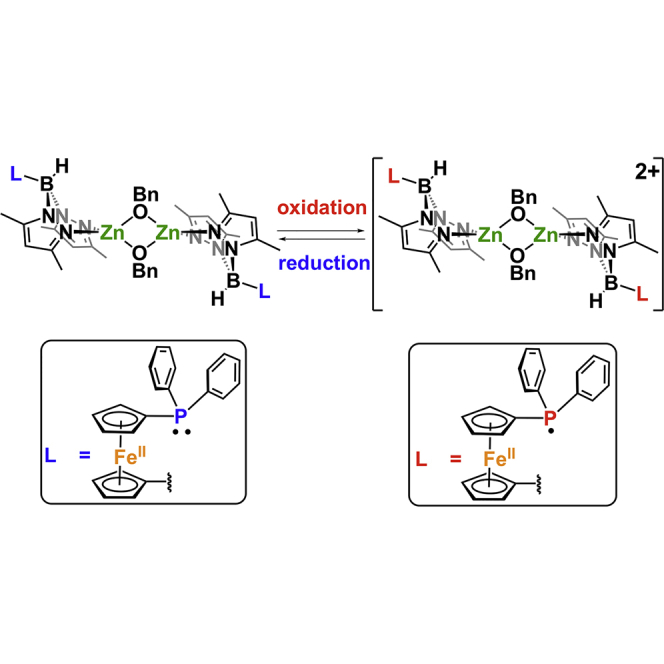
We previously reported the influence of a redox switch on the lability of [fc(PPh2) (BH[(3,5-Me)2pz]2)] ((fcP,B), fc = 1,1′-ferrocenediyl, pz = pyrazole) in a palladium methyl complex in the presence of norbornene (Abubekerov et al., 2016). Upon the oxidation of the ferrocene backbone, the phosphine moiety displayed hemilabile behavior, resulting in norbornene polymerization. We reasoned that using the same ligand to support a more oxophilic metal, such as zinc(II), may yield a beneficial hemilabile interaction between the phosphine and the metal in the reduced state of the supporting ligand. In this case, the oxidation of the ligand could result in the loss of the zinc-phosphine interaction, similar to redox-switchable hemilabile ligands, and in altering the reactivity of a catalyst. Recently, we described the preparation as well as the characterization of [(fcP,B)Zn(μ-OCH2Ph)]2 (Abubekerov et al., 2018). However, in the case of the reduced species, a direct zinc-phosphine interaction was not observed. Despite this finding, and because of our interest in redox-switchable catalytic processes (Abubekerov et al., 2016, Abubekerov et al., 2017, Wang et al., 2014, Wang et al., 2015, Quan and Diaconescu, 2015, Brosmer and Diaconescu, 2015, Abubekerov and Diaconescu, 2015, Upton et al., 2014, Broderick et al., 2011a, Broderick et al., 2011b, Broderick et al., 2011c, Quan et al., 2016, Quan et al., 2017, Lowe et al., 2017, Shepard and Diaconescu, 2016), we set out to investigate the influence of the redox state of fcP,B on the zinc-mediated ring-opening polymerization of cyclic esters and carbonates. In addition, an investigation into the redox and polymerization activity of a monomeric ferrocene-chelating heteroscorpionate zinc complex, (fcP,B)Zn(OPh), is reported.
Results and Discussion
Synthesis and Characterization of the Oxidized Zinc Benzoxide Complex
We previously described the preparation and investigation of the solution state behavior of [(fcP,B)Zn(μ-OCH2Ph)]2 (Abubekerov et al., 2018). Our studies revealed that this compound retains its dimeric state before and during the ring-opening polymerization of LA and TMC. In addition, we determined that the polymerizations proceeded via a living mechanism, yielding polymers with narrow dispersities and well-controlled molar masses. However, the influence of the redox state of the supporting ligand on the activity of this compound was not investigated.
Electrochemical studies performed on [(fcP,B)Zn(μ-OCH2Ph)]2 show a reversible curve with a redox potential of −0.024 V versus Fc/Fc+ (Figure S33), suggesting that ferrocenium salts may be used as chemical oxidants. On nuclear magnetic resonance (NMR) scale oxidation of the dimeric zinc complex with two equivalents of acetyl ferrocenium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate ([AcFc][BArF]) in C6D6 results in the formation of insoluble red solids and acetyl ferrocene, with only the latter being observed by NMR spectroscopy. Reduction with one equivalent of cobaltocene (Cp2Co) restores the original complex, [(fcP,B)Zn(μ-OCH2Ph)]2, with no apparent decomposition or side products (Equation 1, Figures S1 and S2). On a larger scale, the addition of one equivalent of [AcFc][BArF] to [(fcP,B)Zn(μ-OCH2Ph)]2 led to the isolation of red solids. Attempts to characterize the oxidation product by NMR spectroscopy were unsuccessful; the complex is insoluble in hydrocarbon solvents and rapidly reacts with non-hydrocarbon solvents (tetrahydrofuran [THF], chloroform). The reaction product of THF-d8 with [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 is a paramagnetic complex, which is 31P NMR silent, similar to the previously reported [(fcP,B)PdMe][BArF](Abubekerov et al., 2016). The 11B NMR spectrum (Figure S4) shows a minor shift from δ = −7.2 to −7.8 ppm upon oxidation. The presence of the [BArF] counterion was confirmed by the presence of a singlet at δ = −5.9 and −63.5 ppm in the 11B and 19F NMR spectra (Figures S4 and S5), respectively. Attempts to grow X-ray-quality crystals of the oxidized complex from various neat solvents and solvent combinations were unsuccessful, and only dark red oils were obtained. However, elemental analysis agrees with the formulation [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2.

The lack of a signal in the 31P NMR spectrum of the oxidized complex prompted further investigation into the electronic state of this compound. A determination of the solution-state magnetic susceptibility using the Evans method (Evans, 1959) of [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 was complicated by its lack of solubility in non-polar solvents and its reactivity with polar solvents. The addition of 50–150 equivalents of monomer was necessary to solubilize the catalytically active species in deuterated benzene. In particular, we looked at the effective magnetic moment of in situ-generated [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 in the presence of monomers that could be ring-opened at ambient temperature, such as TMC, VL, and CL. Due to the rapid rate of TMC and VL polymerization, by the time the samples were prepared and taken to the NMR spectrometer (10 min) the polymerization of each monomer was already complete. In the case of TMC, the initial solution state effective magnetic moment was 1.90 μB per dimer, corresponding to a single unpaired electron. Within an hour, the paramagnetic sample decayed to a diamagnetic one (Figure S29). In the presence of VL, the effective initial magnetic moment was determined as 2.60 μB, consistent with two unpaired electrons per dimer, but slowly decreased to 1.34 μB after 2.5 hr (Figure S30). The reaction with CL is the slowest of the three monomers considered and shows the lowest degree of decay of the effective magnetic moment in solution: 3.06 μB after 10 minutes and 1.63 μB after 3.5 hr (Figure S31). These findings suggest that [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 is stable in solution only during the polymerization process and are consistent with the polymerization results described below. Attempts to acquire reproducible superconducting quantum interference device (SQUID) results were unsuccessful due to the sensitive nature of the compound.
Due to the difficulty of characterizing the oxidized complex in solution, we turned to 57Fe Mössbauer spectroscopy on solid samples. At 77 K, with no applied magnetic field, [(fcP,B)Zn(μ-OCH2Ph)]2 shows a doublet with an isomer shift (δ = 0.54(1) mm/s) and quadrupole splitting (ΔEQ = 2.34(1) mm/s) consistent with a low-spin iron(II) complex (Figure 1). Surprisingly, under the same conditions, [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 also displays a doublet with an isomer shift (δ = 0.54(1) mm/s) and quadrupole splitting (ΔEQ = 2.26(1) mm/s) similar to [(fcP,B)Zn(μ-OCH2Ph)]2, and thus also consistent with a low-spin iron(II) complex (Figure 1). Attempts to acquire an X-band electron paramagnetic resonance (EPR) spectrum (in perpendicular mode), both in the solid and solution states, at liquid helium (7 K), liquid nitrogen (92 K), and ambient temperature (293 K), under a variety of different conditions, were unsuccessful. Similar Mössbauer and EPR spectroscopy results were reported for oxidized arylphosphinoferrocenes by Durfey et al. (Durfey et al., 2004) and were attributed to oxidation either of the phosphorus lone pair or of the aryl groups. The presence of an unpaired electron in close proximity of the phosphorus atom would be consistent with the lack of a signal in the 31P NMR spectrum of the oxidized complex. Ligand-based oxidations of ferrocenes, as opposed to iron, were also reported for various phenylphosphinoferrocenes (Verschoor-Kirss et al., 2016) and ferrocenylpyrrole (Verschoor-Kirss et al., 2009) species; however, the inability to observe an EPR signal for these species remains puzzling. Isolated products of chemical oxidations displayed low-spin iron(II) species, as evidenced by 57Fe Mössbauer spectroscopy in the case of a ferrocenylpyrrole complex, and a mix of low-spin iron(II) and low-spin iron(III) species in the case of phenylphosphinoferrocenes. However, the 31P NMR spectra of the oxidized phenylphosphinoferrocenes typically display broadened, but detectible signals.
Figure 1.

Zero-Field 57Fe Mössbauer Spectra of [(fcP,B)Zn(μ-OCH2Ph)]2 and [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2, Recorded as Solid Samples at 77 K
The solid lines are fits with Lorentzian doublets to the experimental spectra using the following isomer shifts δ and quadrupole splittings ΔEQ: δ = 0.54(1) mm/s, ΔEQ = 2.34 mm/s, ΓFWHM = 0.29(1) mm/s (left) and δ = 0.54(1) mm/s, ΔEQ = 2.26(1) mm/s, ΓFWHM = 0.31(1) mm/s (right).
Electronic Structure Calculations
To probe further the electronic structure of [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2, we turned to computational studies by investigating the highest occupied valence states from which the electrons are removed for the model of [(fcP,B)Zn(μ-OCH2Ph)]22+ (see section DFT Calculations, for model description). However, common local and semilocal density functionals cannot address this problem due to the improper description of the highly correlated d orbitals in Fe. The mean field description, i.e., the common exchange-correlation potential in density functional theory (DFT), fails to capture the physics of the localized states due to a self-interaction error (Perdew and Zunger, 1981, Kümmel and Kronik, 2008). This leads to a spurious delocalization of orbitals and an incorrect charge transfer and oxidation energies (Cohen et al., 2008, Kulik, 2015).
We first verified that (semi)local functionals indeed contradict the experimental data. We illustrate this by the density of electronic states obtained with the common generalized gradient approximation (GGA) functional (Perdew et al., 1996) (Figure 2). Projection of the corresponding wave functions onto the atomic orbitals reveals the character of individual states and their mutual hybridization. The top valence region (close to the HOMO at −3.3 eV) is dominated by the localized d orbitals of iron. An Fe d orbital contribution is also found in the lower energy molecular orbitals, but these do not participate energetically in the oxidation process. It is important to note that the phosphorus p orbitals are significantly separated in energy from the top valence region by as much as 0.9 eV. Hence, the GGA results suggest that the Fe atoms are oxidized, in contradiction to the experimental results.
Figure 2.

Total Density of States g(e) for the Top Valence Region
The highest occupied molecular orbital is indicated by a black arrow. Contributions of the atomic-like p and d orbitals of P and Fe are shown by different colors. Note that due to strong hybridization, multiple atomic orbitals contribute to each molecular state.
This conundrum was resolved by mitigating the self-interaction error of the localized states. We resorted to the established approach of correcting the DFT picture by using a site- and orbital-specific potential U derived from the Hubbard model Hamiltonian (Himmetoglu et al., 2014). We estimated the potential for the Fe d states from first principles using linear response theory (Cococcioni and de Gironcoli, 2005) and obtained a U value of 7.7 eV. Such a high value leads to significant changes in the density of states (Figure 2). We observed that the Fe d states are pushed down in energy and significantly hybridize with the valence region below −5 eV. The frontier molecular orbitals are at −4.7 eV with a major contribution from phosphorus p states. Indeed, the highest occupied molecular orbital (HOMO) and HOMO-1 isosurfaces are found mostly around the phosphorus atoms (Figure 3).
Figure 3.

Isosurface for HOMO and HOMO-1 of [(fcP,B)Zn(μ-OCH2Ph)]22+
Yellow and light blue coloring represents the positive and negative real parts of the wave function, respectively. For clarity, the hydrogen atoms were removed from the figure.
The ground state geometry of the molecule is different when optimized for the cation or neutral species. This leads to a slight variation of the U parameter by 0.3 eV, but the character of the top valence state is not affected: for U > 4 eV (i.e., much smaller than the variation due to changes in the geometry), the top valence orbital becomes dominated by phosphorus p states. The GGA + U calculations thus show that Fe remains in its +II oxidation state and the phosphine groups are oxidized.
Polymerization of Cyclic Esters and Carbonates with the Zinc Benzoxide Species
The influence of the ligand oxidation state on the reactivity of the zinc benzoxide complex was examined through the ring-opening polymerization of cyclic esters and carbonates. Due to the difficulty in isolating and manipulating the oxidized complex, it was generated in situ just before use through the addition of two equivalents of [AcFc][BArF] to [(fcP,B)Zn(μ-OCH2Ph)]2. The role of the oxidant as a potential polymerization catalyst was ruled out based on control experiments, which showed no activity under polymerization conditions for LA, TMC, and VL (Figures S14, S15, and S17) and only a minor conversion (<6%) for CL (Figure S16). In all cases, the polymerizations are well controlled with the molar masses of the resulting polymers increasing linearly with conversion, whereas the dispersity values remaining narrow (Tables S1–S6, Figures S47–S52).
Polymerizations of ca. 200 equivalents of LA showed a faster conversion for the reduced than the oxidized compound at various temperatures. At ambient temperature, a 60% conversion was obtained after 24 hr for the reduced species, whereas the oxidized complex reached only 17% conversion. Such a low conversion could be attributed to both the poor solubility of LA and, particularly, to the insolubility of the oxidized complex in benzene at ambient temperature. Performing the polymerizations at 70°C resulted in much shorter reaction times for both complexes (Table 1, entries 1 and 2); complete conversion was observed for the reduced complex and there was 91% conversion for the oxidized complex in 3 hr. The molar masses for the isolated polymers obtained from the oxidized and reduced complexes agree well with the corresponding theoretical molar masses. The dispersity values (Đ) are within the 1.00–1.15 range, suggesting that the polymerization process is well controlled.
Table 1.
Polymerization of Cyclic Esters and Carbonates by [(fcP,B)Zn(μ-OCH2Ph)]2 (“Reduced”) and the In Situ-Generated [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 (“Oxidized”)
| Entry | Compound | Monomer | Time (min) | Conversion (%) | Mn (NMR) | Mn (SEC) | Đ |
|---|---|---|---|---|---|---|---|
| 1 | Reduced | LA | 180 | >99 | 1.40 | 1.38 | 1.14 |
| 2 | Oxidized | LA | 180 | 91 | 1.32 | 1.35 | 1.02 |
| 3 | Reduced | CL | 30 | >99 | 0.90 | 0.89 | 1.09 |
| 4 | Oxidized | CL | 30 | >99 | 1.07 | 1.06 | 1.08 |
| 5 | Reduced | TMC | 90 | 98 | 1.00 | 1.00 | 1.14 |
| 6 | Oxidized | TMC | 15 | >99 | 0.79 | 0.81 | 1.02 |
| 7 | Reduced | VL | 60 | 92 | 1.02 | 1.04 | 1.01 |
| 8 | Oxidized | VL | 20 | 92 | 1.01 | 1.05 | 1.04 |
Conditions: monomer (0.50 mmol), catalyst (0.0025 mmol), oxidant (0.005 mmol), d6-benzene as a solvent (0.5 mL), and hexamethylbenzene (0.025 mmol) as an internal standard. Entries 1–4 were carried out at 70°C and entries 5–8 were performed at ambient temperature; Mn are reported in 104 g/mol; Đ = Mw/Mn.
Contrary to the LA case, the oxidation of the ligand had no observable influence on the rates of polymerization of CL (Table 1, entries 3 and 4). Polymerizations of ca. 200 equivalents of CL at ambient temperature reached completion for both the oxidized and the reduced species in 24 hr. On the other hand, carrying out the polymerizations at 70°C resulted in complete consumption of the monomer within 30 min for both compounds. Similar to LA, the molar masses of the polymers, obtained by size exclusion chromatography (SEC), from both the oxidized and reduced species agree well with the theoretical values. However, the dispersity values (Đ) are slightly narrower and fall between 1.0 and 1.1.
The most pronounced difference between the oxidized and the reduced compounds was observed for the polymerization of TMC (Table 1, entries 5 and 6) and VL (Table 1, entries 7 and 8). The complete conversion of ca. 200 equivalents of TMC was accomplished in under 15 min at ambient temperature utilizing the oxidized species and in up to 90 min for the reduced complex. Similarly, the conversion of ca. 200 equivalents of VL plateaus at 92% for the oxidized species after 20 min at ambient temperature, whereas the same conversion is obtained for the reduced compound after 60 min. Further conversion of VL cannot be obtained with an increased time or elevated temperatures. All polymers show excellent agreement between NMR spectroscopy and SEC molar masses and display narrow dispersity values.
Literature examples of LA and CL polymerization by zinc complexes are rather common (Chisholm et al., 2000, Honrado et al., 2016, Lian et al., 2007, Alonso-Moreno et al., 2008, Schofield et al., 2009, Mou et al., 2014, Garcés et al., 2010), whereas examples of TMC and VL polymerizations are not as prevalent (Vivas et al., 2003, Gowda and Chakraborty, 2010, Helou et al., 2008). Compared with other heteroscorpionate complexes, tris(pyrazolyl)borate complexes, and ligands with similar tripodal frameworks, both the oxidized and the reduced metal complexes display moderate activity for the polymerization of LA and CL (Chisholm et al., 2000, Honrado et al., 2016, Lian et al., 2007, Alonso-Moreno et al., 2008, Schofield et al., 2009, Mou et al., 2014, Garcés et al., 2010). Similarly, the reduced species shows a moderate activity toward the polymerization of TMC and VL, whereas the oxidized compound shows high activity toward the same monomers (Vivas et al., 2003, Gowda and Chakraborty, 2010, Helou et al., 2008).
Synthesis and Characterization of a Zinc Phenoxide Complex
To investigate the possibility of redox-switchable hemilabile ligand behavior we also prepared a monomeric zinc species containing a phosphorus-zinc interaction. The addition of NaOPh to (fcP,B)ZnCl in methylene chloride resulted in the isolation of (fcP,B)Zn(OPh) as orange crystals in 79.8% yield (Equation 2). The solid-state molecular structure of (fcP,B)Zn(OPh) (Figure 4) in crystals of (fcP,B)Zn(OPh)⋅(Et2O) was determined using single-crystal X-ray diffraction. The coordination environment around the zinc center has a distorted tetrahedral geometry with a τ value of 0.88 (Yang et al., 2007). Unlike in the case of [(fcP,B)Zn(μ-OCH2Ph)]2, the phenoxide ligands do not bridge, resulting in a monomeric species. However, attempts to prepare additional monomeric zinc species containing bulky aryloxide or amide groups (e.g., 2,4-di-tert-butylphenoxide, 2,6-dimethylphenoxide, N,N-bis(trimethylsilyl)amide) were not successful.

Figure 4.

Molecular Structure Drawing of (fcP,B)Zn(OPh) from Crystals of (fcP,B)Zn(OPh)•(Et2O) with Thermal Ellipsoids at 50% Probability; Hydrogen Atoms Are Omitted for Clarity
Selected distances (Å) and angles (°): O(1)-Zn(1), 1.9095(15); N(1)-Zn(1), 2.0063(17); N(3)-Zn(1), 1.9921(17); P(1)-Zn(1), 2.3984(5); N(1)-Zn(1)-N(3), 95.55(7); N(1)-Zn(1)-O(1), 115.55(7); N(3)-Zn(1)-O(1), 118.59(7); O(1)-Zn(1)-P(1), 99.79(5); N(1)-Zn(1)-P(1), 120.60(5), P(1)-Zn(1)-N(3), 107.81(5). See also Figure S38.
Electrochemically, (fcP,B)Zn(OPh) shows a reversible redox event with a half-potential of −0.065 V versus Fc/Fc+ (Figure S36). On an NMR scale, the oxidation of (fcP,B)Zn(OPh) with 1.1 equivalents of [AcFc][BArF] in C6D6 results in the formation of insoluble red solids, just like in the case of [(fcP,B)Zn(μ-OCH2Ph)]2. However, the complete oxidation of the starting material does not occur, and a mixture of (fcP,B)Zn(OPh) and AcFc is observed spectroscopically (Figure S22). Similarly, utilizing excess AgBF4 did not result in the complete oxidation of (fcP,B)Zn(OPh), which could still be observed as broad peaks in the 1H NMR spectrum due to the presence of solid silver in the sample (Figure S23).
Polymerization of Cyclic Esters and Carbonates with the Zinc Phenoxide Species
Despite the lack of a robust redox-switchable behavior, we looked into the polymerization activity of (fcP,B)Zn(OPh). Although (fcP,B)Zn(OPh) is a monomeric complex, its propensity to form a dimeric species during the polymerization of cyclic esters and carbonates must be considered. Since [(fcP,B)Zn(μ-OCH2Ph)]2 remains a dimeric species during the ring-opening polymerization of cyclic esters and carbonates, it is worth considering that (fcP,B)Zn(OPh) might dimerize after a single reaction with a monomer. To test this theory, the polymerization of ca. 10 equivalents of LA in the presence of (fcP,B)Zn(OPh) was monitored by NMR spectroscopy (Figure S24). The rate of polymerization is substantially slower compared with that of [(fcP,B)Zn(μ-OCH2Ph)]2, reaching only a 65% conversion of the 10 equivalents after 5 hr at 100°C (Figure S25). However, after the reaction onset, the signals corresponding to (fcP,B)Zn(OPh) do not shift in the 1H NMR spectra (Figure S24); this finding prompted a further look into the polymerization process by diffusion ordered spectroscopy. We previously used this technique to illustrate that our zinc polymerization systems show the same diffusion rate for the catalyst and the polymer (Abubekerov et al., 2018). However, in the present case, the diffusion rate of polylactide (PLA) was substantially different from that of (fcP,B)Zn(OPh), suggesting that (fcP,B)Zn(OPh) does not participate in the polymerization process (Figure S26). Similar results were obtained in the case of TMC. Polytrimethylenecarbonate (PTMC) was formed at a slow rate, but no change was observed for (fcP,B)Zn(OPh) (Figures S27 and S28), suggesting that an impurity or a decomposition product might be responsible for monomer conversion.
DFT Calculations
To gain a better understanding of the influence of ligand oxidation on the activity of the catalyst, we turned to DFT. All calculations were carried out with the Gaussian09 program package (Frisch, 2010 [see Supplemental Information for the full reference]) on the Extreme Science and Engineering Discovery Environment (Towns et al., 2014). The methyl groups of the pyrazole substituents were replaced by hydrogen atoms and the phenyl groups of PPh2 were replaced by methyl groups to simplify the calculations (for more details about calculations, see Supplemental Information). Lactide was chosen as the model substrate for the DFT calculations. Although DFT calculations did not describe correctly the electronic structure of [(fcP,B)Zn(μ-OCH2Ph)]22+, the results reported below are still meaningful.
We previously reported a computational study comparing the energies of possible monomeric and dimeric structures of the zinc benzoxide complexes and showed that the dimer [(fcP,B)Zn(μ-OCH2Ph)]2 was more stable (by 3.3 kcal/mol) than the corresponding monomer (fcP,B)Zn(OCH2Ph) (Abubekerov et al., 2018). Calculations conducted at the same level of theory showed that, in the oxidized state, the dimeric species [(fcP,B)Zn(μ-OCH2Ph)]22+ was also greatly favored (20.1 kcal/mol) over the corresponding monomer (Figure 5). This preference was maintained even after the insertion of the first lactide, with the dimeric intermediate being 12.7 kcal/mol lower in energy than the monomeric intermediate (Figure 6). These results suggest that the active species in the reactions involving the oxidized complex is a dimer, similarly to what was previously found for the reduced compound (Abubekerov et al., 2018).
Figure 5.

Optimized Structures for Models of [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2
Figure 6.

Optimized Structures of the Product Obtained after the First Lactide Ring-Opening Event Indicating that the Dimeric Species Is More Energetically Favored than the Monomeric Species
A look at the energy surfaces of the reduced (Abubekerov et al., 2018) and the oxidized complexes during TMC polymerization showed a small difference between the two reactions (Figures S53 and 7, respectively). The highest activation barrier for both reactions corresponds to the ring-opening step (TSII-III), with the activation barrier for the reduced complex being lower by 5.3 kcal/mol. Such a small difference between the two compounds is in agreement with the minor rate differences observed for the ring-opening polymerizations catalyzed by the reduced and oxidized species. These results also agree with the electronic structure calculations described above: since the oxidation of the supporting ligand is centered on the phosphine ligand, it has little impact on the rate of ring-opening polymerization due to the lack of a phosphine-zinc interaction in both the reduced and oxidized states.
Figure 7.

Energy Profile for the Ring-Opening Polymerization of TMC Catalyzed by the Oxidized Form of the Zinc Complex
See also Figure S53.
Conclusions
The zinc complexes supported by a ferrocene-chelating heteroscorpionate ligand showed reversible electron transfer events during cyclic voltammetry experiments. Consequently, their applicability toward redox-switchable catalysis in the ring-opening polymerization of cyclic esters and TMC was investigated. However, [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2 proved difficult to characterize due to its poor solubility and thermally sensitive nature. A combination of NMR and Mössbauer spectroscopies supported the existence of a paramagnetic species, but with the ferrocene iron remaining in a +2 oxidation state. A careful analysis of the electronic structure of the oxidized complex via a computational study concluded that the electron is removed from the phosphorus atom and not iron, as was originally expected.
Different polymerization rates for [(fcP,B)Zn(μ-OCH2Ph)]2 and the corresponding oxidized complex, [(fcP,B)Zn(μ-OCH2Ph)]2[BArF]2, toward the same set of monomers were observed. The differences in the observed reactivity are attributed to the difference in charge distributions between the neutral and the cationic zinc species. More importantly, unlike in other redox-switchable systems reported by us, both oxidation states of the same pre-catalyst reacted with all the substrates investigated. This lack of selectivity is attributed to the absence of a zinc-phosphine interaction in both the reduced and oxidized states of the zinc benzoxide species and was probed via DFT calculations. However, the monomeric zinc phenoxide species, (fcP,B)Zn(OPh), which retains the phosphine-zinc interaction in solution, was found inactive toward the ring-opening polymerization of LA and TMC. Based on the results of this investigation, we are currently developing a monomeric zinc alkoxide species bearing a ferrocene-chelating heteroscorpionate derivative, and are interested in studying its application toward the ring-opening polymerization of cyclic esters and carbonates.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by NSF, Grant 1362999 to P.L.D. and CHE-1048804 for NMR spectroscopy. D.N. acknowledges support by the NSF grant DMR/BSF-1611382. M.E.M. and K.M. acknowledge generous support from the Friedrich-Alexander-University Erlangen-Nürnberg (FAU). Part of the calculations were supported by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575, as the computational project TG-CHE160092. We thank Dr. Jonathan Brosmer for assistance with the elemental analysis experiments.
Author Contributions
M.A. designed and conducted the synthetic and polymerization experiments and wrote the paper, V.V. performed the electronic structure calculations, J.W. performed the DFT calculations, M.E.M. performed the Mössbauer experiments, S.M.Q. performed the polymer characterization, K.M. designed the Mössbauer experiments, D.N. designed the electronic structure calculations, and P.L.D. designed the synthetic and polymerization experiments and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: September 28, 2018
Footnotes
Supplemental Information includes Transparent Methods, 53 figures, 7 tables, and 2 data files and can be found with this article online at https://doi.org/10.1016/j.isci.2018.08.020.
Contributor Information
Karsten Meyer, Email: karsten.meyer@fau.de.
Daniel Neuhauser, Email: dxn@chem.ucla.edu.
Paula L. Diaconescu, Email: pld@chem.ucla.edu.
Data and Software Availability
The cif was deposited with CCDC, #1838242. The optimized coordinates are available as an excel spreadsheet (Data S1) and an xyz file (Data S2) as Supplemental Information.
Supplemental Information
References
- Abubekerov M., Diaconescu P.L. Synthesis and characterization of ferrocene-chelating heteroscorpionate complexes of nickel(II) and zinc(II) Inorg. Chem. 2015;54:1778–1784. doi: 10.1021/ic502691b. [DOI] [PubMed] [Google Scholar]
- Abubekerov M., Khan S.I., Diaconescu P.L. Ferrocene-bis(phosphinimine) nickel(II) and palladium(II) alkyl complexes: influence of the Fe–M (M = Ni and Pd) interaction on redox activity and olefin coordination. Organometallics. 2017;36:4394–4402. [Google Scholar]
- Abubekerov M., Shepard S.M., Diaconescu P.L. Switchable polymerization of norbornene derivatives by a ferrocene-palladium(II) heteroscorpionate complex. Eur. J. Inorg. Chem. 2016;2016:2634–2640. [Google Scholar]
- Abubekerov M., Wei J., Swartz K.R., Xie Z., Pei Q., Diaconescu P.L. Preparation of multiblock copolymers via step-wise addition of l-lactide and trimethylene carbonate. Chem. Sci. 2018;9:2168–2178. doi: 10.1039/c7sc04507g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajellal N., Carpentier J.-F., Guillaume C., Guillaume S.M., Helou M., Poirier V., Sarazin Y., Trifonov A. Metal-catalyzed immortal ring-opening polymerization of lactones, lactides and cyclic carbonates. Dalton Trans. 2010;39:8363–8376. doi: 10.1039/c001226b. [DOI] [PubMed] [Google Scholar]
- Allgeier A.M., Mirkin C.A. Ligand design for electrochemically controlling stoichiometric and catalytic reactivity of transition metals. Angew. Chem. Int. Ed. 1998;37:894–908. doi: 10.1002/(SICI)1521-3773(19980420)37:7<894::AID-ANIE894>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Alonso-Moreno C., Garcés A., Sánchez-Barba L.F., Fajardo M., Fernández-Baeza J., Otero A., Lara-Sánchez A., Antiñolo A., Broomfield L., López-Solera M.I., Rodríguez A.M. Discrete heteroscorpionate lithium and zinc alkyl complexes. synthesis, structural studies, and ROP of cyclic esters. Organometallics. 2008;27:1310–1321. [Google Scholar]
- Arbaoui A., Redshaw C. Metal catalysts for ɛ-caprolactone polymerisation. Polym. Chem. 2010;1:801–826. [Google Scholar]
- Bakewell C., Fateh-Iravani G., Beh D.W., Myers D., Tabthong S., Hormnirun P., White A.J.P., Long N., Williams C.K. Comparing a series of 8-quinolinolato complexes of aluminium, titanium and zinc as initiators for the ring-opening polymerization of rac-lactide. Dalton Trans. 2015;44:12326–12337. doi: 10.1039/c5dt00192g. [DOI] [PubMed] [Google Scholar]
- Broderick E.M., Guo N., Vogel C.S., Xu C., Sutter J., Miller J.T., Meyer K., Mehrkhodavandi P., Diaconescu P.L. Redox control of a ring-opening polymerization catalyst. J. Am. Chem. Soc. 2011;133:9278–9281. doi: 10.1021/ja2036089. [DOI] [PubMed] [Google Scholar]
- Broderick E.M., Guo N., Wu T., Vogel C.S., Xu C., Sutter J., Miller J.T., Meyer K., Cantat T., Diaconescu P.L. Redox control of a polymerization catalyst by changing the oxidation state of the metal center. Chem. Commun. 2011;47:9897–9899. doi: 10.1039/c1cc13117f. [DOI] [PubMed] [Google Scholar]
- Broderick E.M., Thuy-Boun P.S., Guo N., Vogel C.S., Sutter J., Miller J.T., Meyer K., Diaconescu P.L. Synthesis and characterization of cerium and yttrium alkoxide complexes supported by ferrocene-based chelating ligands. Inorg. Chem. 2011;50:2870–2877. doi: 10.1021/ic102076g. [DOI] [PubMed] [Google Scholar]
- Brosmer J.L., Diaconescu P.L. Yttrium-Alkyl complexes supported by a ferrocene-based phosphinimine ligand. Organometallics. 2015;34:2567–2572. [Google Scholar]
- Chisholm M.H., Eilerts N.W., Huffman J.C., Iyer S.S., Pacold M., Phomphrai K. Molecular design of single-site metal alkoxide catalyst precursors for ring-opening polymerization reactions leading to polyoxygenates. 1. Polylactide formation by achiral and chiral Magnesium and Zinc Alkoxides, (η3-L)MOR, where L = Trispyrazolyl- and Trisindazolylborate ligands. J. Am. Chem. Soc. 2000;122:11845–11854. [Google Scholar]
- Cococcioni M., de Gironcoli S. Linear response approach to the calculation of the effective interaction parameters in the $∖mathrm{LDA}+∖mathrm{U}$ method. Phys. Rev. B. 2005;71:035105. [Google Scholar]
- Cohen A.J., Mori-Sánchez P., Yang W. Insights into current limitations of density functional theory. Science. 2008;321:792–794. doi: 10.1126/science.1158722. [DOI] [PubMed] [Google Scholar]
- D'Auria I., Lamberti M., Mazzeo M., Milione S., Roviello G., Pellecchia C. Coordination chemistry and reactivity of zinc complexes supported by a phosphido pincer ligand. Chem. Eur. J. 2012;18:2349–2360. doi: 10.1002/chem.201102414. [DOI] [PubMed] [Google Scholar]
- Dechy-Cabaret O., Martin-Vaca B., Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- Durfey D.A., Kirss R.U., Frommen C., Reiff W.M. Reactions of alkyl- and aryl(ferrocenyl)phosphines with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. Inorg. Chim. Acta. 2004;357:311–315. [Google Scholar]
- Evans D.F. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959:2003–2005. [Google Scholar]
- Fliedel C., Rosa V., Alves F.M., Martins A.M., Aviles T., Dagorne S. P,O-Phosphinophenolate zinc(ii) species: synthesis, structure and use in the ring-opening polymerization (ROP) of lactide, e-caprolactone and trimethylene carbonate. Dalton Trans. 2015;44:12376–12387. doi: 10.1039/c5dt00458f. [DOI] [PubMed] [Google Scholar]
- Frisch M.J. Gaussian Inc; 2010. Gaussian 09 (Revision D.01) [Google Scholar]
- Garcés A., Sánchez-Barba L.F., Alonso-Moreno C., Fajardo M., Fernández-Baeza J., Otero A., Lara-Sánchez A., López-Solera I., Rodríguez A.M. Hybrid scorpionate/cyclopentadienyl magnesium and zinc complexes: synthesis, coordination chemistry, and ring-opening polymerization studies on cyclic esters. Inorg. Chem. 2010;49:2859–2871. doi: 10.1021/ic902399r. [DOI] [PubMed] [Google Scholar]
- Gowda R.R., Chakraborty D. Zinc acetate as a catalyst for the bulk ring opening polymerization of cyclic esters and lactide. J. Mol. Catal. A Chem. 2010;333:167–172. [Google Scholar]
- Guillaume S.M., Carpentier J.-F. Recent advances in metallo/organo-catalyzed immortal ring-opening polymerization of cyclic carbonates. Catal. Sci. Technol. 2012;2:898–906. [Google Scholar]
- Helou M., Miserque O., Brusson J.-M., Carpentier J.-F., Guillaume S.M. Ultraproductive, zinc-mediated, immortal ring-opening polymerization of trimethylene carbonate. Chem. Eur. J. 2008;14:8772–8775. doi: 10.1002/chem.200801416. [DOI] [PubMed] [Google Scholar]
- Helou M., Miserque O., Brusson J.-M., Carpentier J.-F., Guillaume S.M. Highly effective and green catalytic approach toward α,ω-dihydroxy-telechelic poly(trimethylenecarbonate) Macromol. Rapid Commun. 2009;30:2128–2135. doi: 10.1002/marc.200900498. [DOI] [PubMed] [Google Scholar]
- Helou M., Miserque O., Brusson J.-M., Carpentier J.-F., Guillaume S.M. Metal triflates as highly stable and active catalysts for the “immortal” ring-opening polymerization of trimethylene carbonate. ChemCatChem. 2010;2:306–313. [Google Scholar]
- Higgins T.B., Mirkin C.A. Model compounds for polymeric redox-switchable hemilabile ligands. Inorg. Chim. Acta. 1995;240:347–353. [Google Scholar]
- Himmetoglu B., Floris A., De Gironcoli S., Cococcioni M. Hubbard-corrected DFT energy functionals: the LDA+U description of correlated systems. Int. J. Quantum Chem. 2014;114:14–49. [Google Scholar]
- Honrado M., Otero A., Fernández-Baeza J., Sánchez-Barba L.F., Garcés A., Lara-Sánchez A., Rodríguez A.M. Copolymerization of cyclic esters controlled by chiral NNO-scorpionate zinc initiators. Organometallics. 2016;35:189–197. [Google Scholar]
- Huang Y., Wang W., Lin C.-C., Blake M.P., Clark L., Schwarz A.D., Mountford P. Potassium, zinc, and magnesium complexes of a bulky OOO-tridentate bis(phenolate) ligand: synthesis, structures, and studies of cyclic ester polymerisation. Dalton Trans. 2013;42:9313–9324. doi: 10.1039/c3dt50135c. [DOI] [PubMed] [Google Scholar]
- Kulik H.J. Perspective: Treating electron over-delocalization with the DFT+U method. J. Chem. Phys. 2015;142:240901. doi: 10.1063/1.4922693. [DOI] [PubMed] [Google Scholar]
- Kümmel S., Kronik L. Orbital-dependent density functionals: theory and applications. Rev. Mod. Phys. 2008;80:3–60. [Google Scholar]
- Lian B., Thomas C.M., Casagrande O.L., Lehmann C.W., Roisnel T., Carpentier J.-F. Aluminum and zinc complexes based on an amino-bis(pyrazolyl) ligand: synthesis, structures, and use in MMA and lactide polymerization. Inorg. Chem. 2007;46:328–340. doi: 10.1021/ic061749z. [DOI] [PubMed] [Google Scholar]
- Liang L.-C., Lee W.-Y., Tsai T.-L., Hsu Y.-L., Lee T.-Y. Amido phosphine complexes of zinc: synthesis, structure, and catalytic ring-opening polymerization of e-caprolactone. Dalton Trans. 2010;39:8748–8758. doi: 10.1039/c0dt00327a. [DOI] [PubMed] [Google Scholar]
- Lowe M.Y., Shu S., Quan S.M., Diaconescu P.L. Investigation of redox switchable titanium and zirconium catalysts for the ring opening polymerization of cyclic esters and epoxides. Inorg. Chem. Front. 2017;4:1798–1805. [Google Scholar]
- Mou Z., Liu B., Wang M., Xie H., Li P., Li L., Li S., Cui D. Isoselective ring-opening polymerization of rac-lactide initiated by achiral heteroscorpionate zwitterionic zinc complexes. Chem. Commun. 2014;50:11411–11414. doi: 10.1039/c4cc05033a. [DOI] [PubMed] [Google Scholar]
- Nair L.S., Laurencin C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007;32:762–798. [Google Scholar]
- O'Keefe B.J., Hillmyer M.A., Tolman W.B. Polymerization of lactide and related cyclic esters by discrete metal complexes. J. Chem. Soc. Dalton Trans. 2001:2215–2224. [Google Scholar]
- Oerlemans C., Bult W., Bos M., Storm G., Nijsen J.F.W., Hennink W.E. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm. Res. 2010;27:2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J.P., Burke K., Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J.P., Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B. 1981;23:5048–5079. [Google Scholar]
- Place E.S., George J.H., Williams C.K., Stevens M.M. Synthetic polymer scaffolds for tissue engineering. Chem. Soc. Rev. 2009;38:1139–1151. doi: 10.1039/b811392k. [DOI] [PubMed] [Google Scholar]
- Platel R.H., Hodgson L.M., Williams C.K. Biocompatible initiators for lactide polymerization. Polym. Rev. 2008;48:11–63. [Google Scholar]
- Quan S.M., Diaconescu P.L. High activity of an indium alkoxide complex toward ring opening polymerization of cyclic esters. Chem. Commun. 2015;51:9643–9646. doi: 10.1039/c5cc01312g. [DOI] [PubMed] [Google Scholar]
- Quan S.M., Wang X., Zhang R., Diaconescu P.L. Redox switchable copolymerization of cyclic esters and epoxides by a zirconium complex. Macromolecules. 2016;49:6768–6778. [Google Scholar]
- Quan S.M., Wei J., Diaconescu P.L. Mechanistic studies of redox-switchable copolymerization of lactide and cyclohexene oxide by a zirconium complex. Organometallics. 2017;36:4451–4457. [Google Scholar]
- Ruzette A.-V., Leibler L. Block copolymers in tomorrow's plastics. Nat. Mater. 2005;4:19–31. doi: 10.1038/nmat1295. [DOI] [PubMed] [Google Scholar]
- Sassano C.A., Mirkin C.A. Degenerate exchange reactions: a novel and general way to determine the thermodynamic perturbations on transition metal complexes that result from ligand oxidation. J. Am. Chem. Soc. 1995;117:11379–11380. [Google Scholar]
- Schofield A.D., Barros M.L., Cushion M.G., Schwarz A.D., Mountford P. Sodium, magnesium and zinc complexes of mono(phenolate) heteroscorpionate ligands. Dalton Trans. 2009:85–96. doi: 10.1039/b813116c. [DOI] [PubMed] [Google Scholar]
- Shepard S.M., Diaconescu P.L. Redox-switchable hydroelementation of a cobalt complex supported by a ferrocene-based ligand. Organometallics. 2016;35:2446–2453. [Google Scholar]
- Singewald E.T., Mirkin C.A., Stern C.L. A redox-switchable hemilabile ligand: electrochemical control of the coordination environment of a RhI complex. Angew. Chem. Int. Ed. 1995;34:1624–1627. [Google Scholar]
- Slone C.S., Mirkin C.A., Yap G.P.A., Guzei I.A., Rheingold A.L. Oxidation-state-dependent reactivity and catalytic properties of a Rh(I) complex formed from a redox-switchable hemilabile ligand. J. Am. Chem. Soc. 1997;119:10743–10753. [Google Scholar]
- Suriano F., Coulembier O., Hedrick J.L., Dubois P. Functionalized cyclic carbonates: from synthesis and metal-free catalyzed ring-opening polymerization to applications. Polym. Chem. 2011;2:528–533. [Google Scholar]
- Tian H., Tang Z., Zhuang X., Chen X., Jing X. Biodegradable synthetic polymers: preparation, functionalization and biomedical application. Prog. Polym. Sci. 2012;37:237–280. [Google Scholar]
- Towns J., Cockerill T., Dahan M., Foster I., Gaither K., Grimshaw A., Hazlewood V., Lathrop S., Lifka D., Peterson G.D. XSEDE: accelerating scientific discovery. Comput. Sci. Eng. 2014;16:62–74. [Google Scholar]
- Ulery B.D., Nair L.S., Laurencin C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. B Polym. Phys. 2011;49:832–864. doi: 10.1002/polb.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton B.M., Gipson R.M., Duhovic S., Lydon B.R., Matsumoto N.M., Maynard H.D., Diaconescu P.L. Synthesis of ferrocene-functionalized monomers for biodegradable polymer formation. Inorg. Chem. Front. 2014;1:271–277. [Google Scholar]
- Verschoor-Kirss M., Kreisz J., Feighery W., Reiff W.M., Frommen C.M., Kirss R.U. Synthesis and chemical oxidation of 3-ferrocenylpyrrole and ferrocenyl-substituted triazoles: iron versus ligand based oxidation. J. Organomet. Chem. 2009;694:3262–3269. [Google Scholar]
- Verschoor-Kirss M.J., Hendricks O., Verschoor C.M., Conry R., Kirss R.U. Chemical oxidation of ferrocenyl(phenyl)phosphines and ferrocenyl(phenyl)phosphine chalcogenides. Inorg. Chim. Acta. 2016;450:30–38. [Google Scholar]
- Vert M. Aliphatic polyesters: great degradable polymers that cannot do everything. Biomacromolecules. 2005;6:538–546. doi: 10.1021/bm0494702. [DOI] [PubMed] [Google Scholar]
- Vivas M., Mejías N., Contreras J. Ring-opening polymerization of lactones initiated by diphenylzinc–coinitiator systems. Polym. Int. 2003;52:1005–1009. [Google Scholar]
- Vroman I., Tighzert L. Biodegradable polymers. Materials. 2009;2:307. [Google Scholar]
- Wang X., Brosmer J.L., Thevenon A., Diaconescu P.L. Highly active yttrium catalysts for the ring-opening polymerization of ɛ-caprolactone and δ-valerolactone. Organometallics. 2015;34:4700–4706. [Google Scholar]
- Wang X., Thevenon A., Brosmer J.L., Yu I., Khan S.I., Mehrkhodavandi P., Diaconescu P.L. Redox control of group 4 metal ring-opening polymerization activity toward l-lactide and ɛ-caprolactone. J. Am. Chem. Soc. 2014;136:11264–11267. doi: 10.1021/ja505883u. [DOI] [PubMed] [Google Scholar]
- Weinberger D.A., Higgins T.B., Mirkin C.A., Stern C.L., Liable-Sands L.M., Rheingold A.L. Terthienyl and poly-terthienyl ligands as redox-switchable hemilabile ligands for oxidation-state-dependent molecular uptake and release. J. Am. Chem. Soc. 2001;123:2503–2516. doi: 10.1021/ja0030008. [DOI] [PubMed] [Google Scholar]
- Wheaton C.A., Hayes P.G., Ireland B.J. Complexes of Mg, Ca and Zn as homogeneous catalysts for lactide polymerization. Dalton Trans. 2009:4832–4846. doi: 10.1039/b819107g. [DOI] [PubMed] [Google Scholar]
- Williams C.K., Breyfogle L.E., Choi S.K., Nam W., Young V.G., Hillmyer M.A., Tolman W.B. A highly active zinc catalyst for the controlled polymerization of lactide. J. Am. Chem. Soc. 2003;125:11350–11359. doi: 10.1021/ja0359512. [DOI] [PubMed] [Google Scholar]
- Wu J., Yu T.-L., Chen C.-T., Lin C.-C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006;250:602–626. [Google Scholar]
- Yang L., Powell D.R., Houser R.P. Structural variation in copper(i) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, [small tau]4. Dalton Trans. 2007:955–964. doi: 10.1039/b617136b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.