

Abstract
Purpose of Review
Conventional clinico-pathological features in melanoma patients should be integrated with new molecular diagnostic, predictive, and prognostic factors coming from the expanding genomic profiles. Cutaneous melanoma (CM), even differing in biological behavior according to sun-exposure levels on the skin areas where it arises, is molecularly heterogeneous. The next-generation sequencing (NGS) approaches are providing data on mutation landscapes in driver genes that may account for distinct pathogenetic mechanisms and pathways. The purpose was to group and classify all somatic driver mutations observed in the main NGS-based studies.
Recent Findings
Whole exome and whole genome sequencing approaches have provided data on spectrum and distribution of genetic and genomic alterations as well as allowed to discover new cancer genes underlying CM pathogenesis.
Summary
After evaluating the mutational status in a cohort of 686 CM cases from the most representative NGS studies, three molecular CM subtypes were proposed: BRAFmut, RASmut, and non-BRAFmut/non-RASmut.
Keywords: Cutaneous melanoma, Genetics, Melanoma subtypes, Mutation status, Next-generation sequencing, Single-nucleotide (SNV) and copy number (CNV) variations, UV signature
Introduction
Cutaneous melanoma (CM) is one of the most aggressive malignancies and its incidence is continuously increasing in the Caucasian population [1]. Melanoma is characterized by a molecular heterogeneity, considerably greater than that evidenced by the common histopathological features [2]. The melanoma pathogenesis, referred to as melanomagenesis, is based on the acquisition of sequential alterations in specific genes and pathways controlling metabolic or molecular mechanisms and regulating crucial cell functions [3–6]. During past decades, several tumor suppressor genes and/or oncogenes have been reported to be affected by deleterious mutations or structural alterations [3–6].
The introduction of next-generation sequencing (NGS) strategies is speeding up the efforts to identify the whole pattern of mutations involved in the CM pathogenesis [7]. This emphasizes the need to define all or the vast majority of mutational changes in the different melanoma subtypes in order to further progress into the knowledge of the disease onset and to better match subsets of patients to the most appropriate clinical management. In other words, accurate classifications of the spectra of genetic mutations in melanoma tissues may lead to the development of disease-associated biomarkers that help guide the most appropriate clinical management of the different subsets of patients.
In this review, we focus on main molecular features in CM lesions, as assessed by NGS approaches.
Mutation Status Assessment by NGS-Based Analysis
The NGS assays are used to perform massively parallel sequencing, during which highly redundant fragments of DNA from a single sample are uniformly sequenced [8]. In particular, NGS is a multistep process that typically involves sample acquisition and quality control, DNA isolation and purification, DNA-fragment library preparation, sequencing, and genomic data generation [8]. The most crucial phases in the process are those subsequent to the achievement of the sequence results. Data analysis includes (a) bioinformatics tools for variant identification; (b) variant annotation and prioritization; and (c) interpretation of putative clinical significance. Sequential procedures are used to call somatic sequence variations in tumor tissue samples, after confirming the quality of the sequencing reads [8–10]. Single-nucleotide variations (SNVs), including predicted amino acid changes for non-synonymous mutations and somatic insertion-deletions (indels), are filtered according to the following criteria: (i) coverage of total reads at the variant position; (ii) mutant allele frequency (usually, 10% or more); and (iii) balanced forward and backward reads. Finally, somatic copy number variations (CNVs) are assessed using specific bioinformatics procedures on mapped reads in matched melanoma samples.
Although the NGS methodology may be also used for gene expression studies using RNA substrate, the main application is aimed at identifying mutations and alteration in genes and regulatory elements involved in pathological processes. Prioritizing such genetic variants, by truly distinguishing between “driver” alterations, that are causally related to the cancer development and random “passenger” alterations that have simply accumulated over the tumor growth, represents a major challenge.
NGS Assays in Cutaneous Melanomas: Overview
Recent years have seen an unprecedented growth in the understanding of genetic and genomic changes in melanoma [11••, 12]. Much of the information comes from NGS analyses, including whole exome (WES) and whole genome (WGS) sequencing approaches, and has been instrumental for the discovery of new genes underlying cancer pathogenesis [13•].
For melanoma, the main WES-based study was performed by the Cancer Genome Atlas on 333 melanomas [14••], while the main WGS-based study by Hayward and colleagues on 183 melanomas [15••]. To focus on CM cases only, we evaluated for mutational status a large cohort of such tumor types excluding the few different melanoma subtypes (acral, mucosal, and ocular) from both previous studies and including data from three additional NGS-based studies: a WES analysis on 147 cases [16], a WES analysis on 135 cases [17], and a WGS analysis on 25 cases [18]. Altogether, 686 CM patients were included into the present study (Table 1).
Table 1.
Cutaneous melanoma (CM) cases from the NGS-based studies evaluated for the mutational patterns in the present report
NGS-based findings confirmed the role of more well-known genes (BRAF, NRAS, TP53, CDKN2A, PTEN, MAP 2K1-2, KIT, and RB1) and recently identified genes (NF1, ARID2, PPP6C, RAC1, DDX3X, and IDH1) in melanoma. The scenario of genetic alterations contributing to melanoma gene signature was completed by somatic CNVs, such as gene amplifications in CCND1, CDK4, KIT, MITF, and TERT as well as gene deletions in CDKN2A and PTEN. However, additional candidate cancer genes are likely to be identified since criteria currently applied in NGS analyses for selecting significant mutations do not consent to achieve full statistical power, and driver genes with a mutation frequency ≤ 2% can be missed [19]. Furthermore, different types of mutations are yet to be investigated in detail. In particular, mutations at the 5′-UTR or promoter regions, for their functional impact on gene expression levels, and within the 3′-UTR, for possible interference with the transcript translation activity [20], deserve a more in-depth examination as it is probable that variants at these levels may currently be under-represented. Nonetheless, mutations in other non-coding regions—such as those in transcription factor binding sites [21] or in splicing regions with generation of new transcript isoforms [22]—also deserve a more accurate classification. Finally, the functional role of synonymous mutations as well as of fusion events and all structural changes in melanomagenesis is still to be fully understood.
NGS-Based Mutation Analysis: Results
Considering data from all five studies on CMs, about 75% of genomic sequence variations are represented by C > T substitutions, with another small fraction (< 5%) constituted by CC > TT transitions. These variants are due to the mutagenic effects of ultraviolet (UV) radiation on exposed skin and the entire set of these are indicated as the UV mutation signature [23–24]. For the same UV effects on mutagenesis and interventions of genetic factors [25], the genomic DNA from CM samples has one of the highest mutational burden as compared to that from other cancer types (contributing to the increased responsiveness to immunotherapy [26]). As a confirmation of this, non-cutaneous (i.e., ocular and mucosal) melanomas present a markedly lower mutational load and lack the UV signature [15••, 27]. Overall, the mutation rate in melanomas occurring on chronically sun-exposed skin was found to be approximately five times higher than those on the skin not subject to sun damage, confirming previously reported data (ratio of about 21 mutations/Mb vs. 5 mutations/Mb in the two subgroups, respectively [28•]).
In Table 2, all deleterious gene mutations in our series of 686 melanomas from the above indicated five studies [14••, 15••, 16–18] are reported. Overall, it was confirmed that the most frequent somatic alterations are represented by mutations in BRAF (340; 49.6%) and RAS (202; 29.4%—with NRAS isoform involved in about 94% of RAS-mutated cases). Therefore, about three fourths (76.6%) of CM cases presented a pathogenetic mutation in these two oncogenes. The occurrence of oncogenic RAS mutations is reported in 8/340 (2.4%) cases carrying oncogenic BRAF mutations, confirming that coexistence in the same melanomas of pathogenetic mutations in these two genes is very infrequent. In an additional but still limited fraction of cases (less than 5%), co-occurring BRAF and RAS mutations harbored either a non-activating BRAF variant together with an oncogenic RAS mutation or a RAS variant not recognized as oncogenic together with an activating BRAF mutation. According to the mutation prevalence, the remaining melanoma driver genes could be divided into three groups: one (TP53, NF1, CDKN2A, and ARID2), with mutation frequency between 10 and 20%; the second (PTEN, PPP6C, RAC1, and DDX3X), with mutation frequency between 5 and 9%; and the third one (16 genes), with mutation frequency < 5% (Table 2). Finally, less than one tenth of CMs (45/686; 6.6%) was negative for any genetic alteration.
Table 2.
Distribution of genetic and genomic alterations among the 686 CM cases. SNVs, in dark; CNVs, in blue; del, gene deletion; ampl, gene amplification. In red, all cases negative for any genetic/genomic alteration (fully wild-type)


Considering the CNVs whose functional role has been previously demonstrated (see above), inactivation of CDKN2A and PTEN tumor suppressor genes was confirmed to be frequently implicated in melanomagenesis (Table 2). Overall, about one third of CMs (224/686; 32.7%) lack any structural rearrangement in the candidate genes. In Fig. 1, all main genes and related pathways characterized by the NGS-based analyses are represented, evidencing the frequencies of the identified molecular alterations (summing SNVs and CNVs).
Fig. 1.
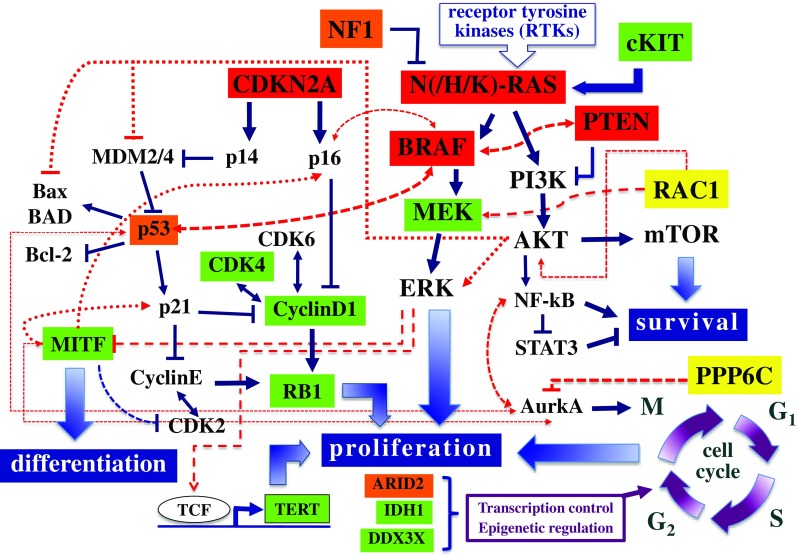
Signal transduction pathways involved in melanomagenesis. Genes are evidenced according to the prevalence of alterations, including SNVs and CNVs, in the series of 686 CM samples: in red, ≥ 20% of cases; in orange, ≥ 10 to < 20%; in yellow, ≥ 5 to < 10%; in green, ≥ 2 to < 5%
The Cancer Genome Atlas classified melanomas in four molecular subtypes: BRAF, RAS, and NF1 mutation carriers, along with the so-called triple wild-type (lack of mutations in all three genes) [14••]. However, NF1 mutations were found—albeit at a lower frequency—in the BRAFmut and RASmut subgroups also, not allowing to appropriately define the subset of NF1mut melanomas as a real independent molecular subtype. This was further confirmed in our series of NGS studies and, therefore, the following three molecular subtypes are here proposed: BRAFmut (Fig. 2), RASmut (Fig. 3), and non-BRAFmut/non-RASmut (Fig. 4). From the evaluation of the mutational status, it appears clearly evident that, even in subtypes characterized by specific main mutations (BRAFmut and RASmut), many additional genes are recurrently mutated (Figs. 2, 3, and 4). For these reasons, there is an increasing need of introducing panel-based testing approaches in order to provide an opportunity for patients to achieve a deeper look at their molecular heterogeneity through an improved clarification of their mutational status [29–32]. Such approaches pave the way for the development of tailored patients’ testing and clinical management.
Fig. 2.
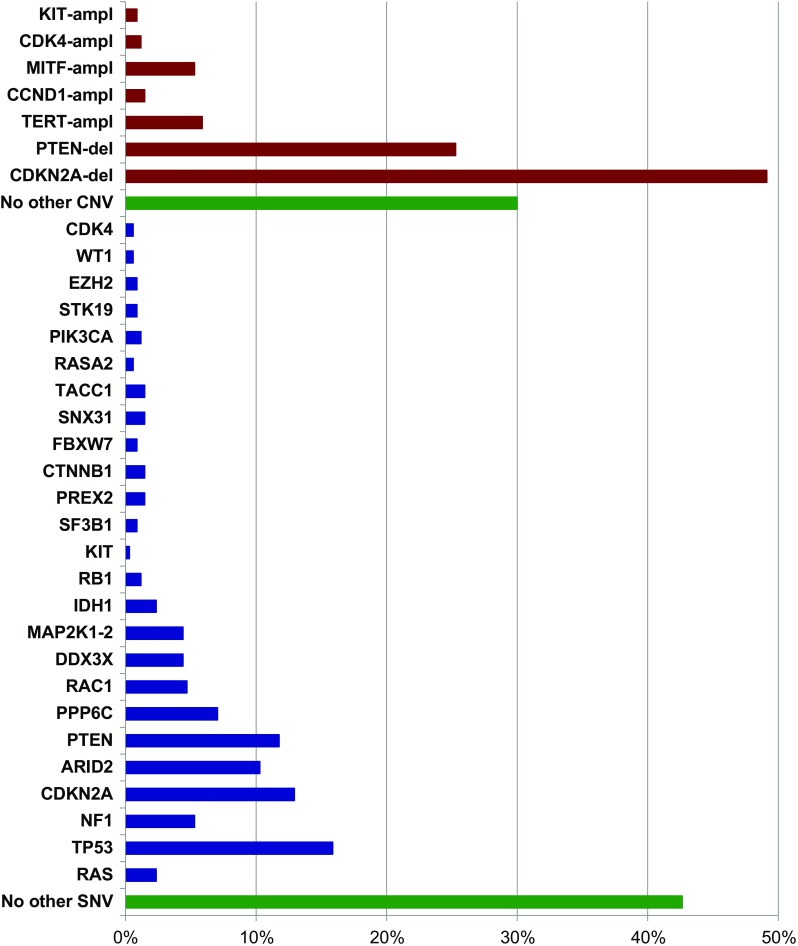
Gene mutation frequencies in the 340 cases with the BRAFmut subtype. In red, copy number variations; in blue, pathogenetic mutations; in green, lack of additional genetic alterations. ampl, amplification; del, deletion
Fig. 3.
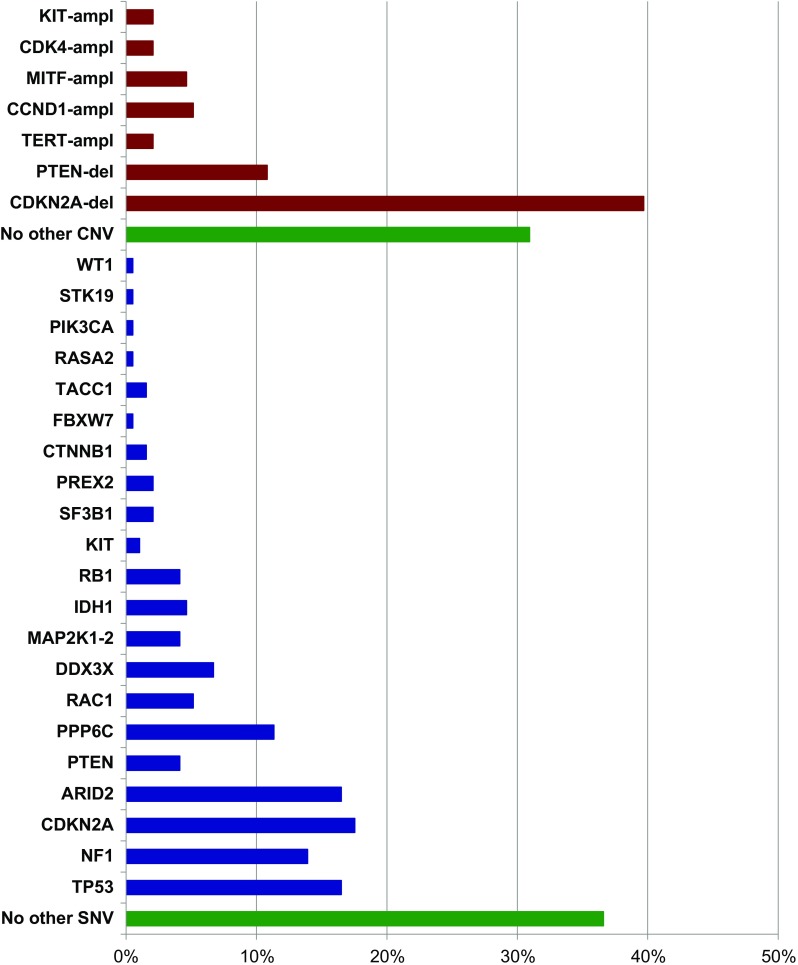
Gene mutation frequencies in the 194 cases with the RASmut subtype. In red, copy number variations; in blue, pathogenetic mutations; in green, lack of additional genetic alterations. ampl, amplification; del, deletion
Fig. 4.
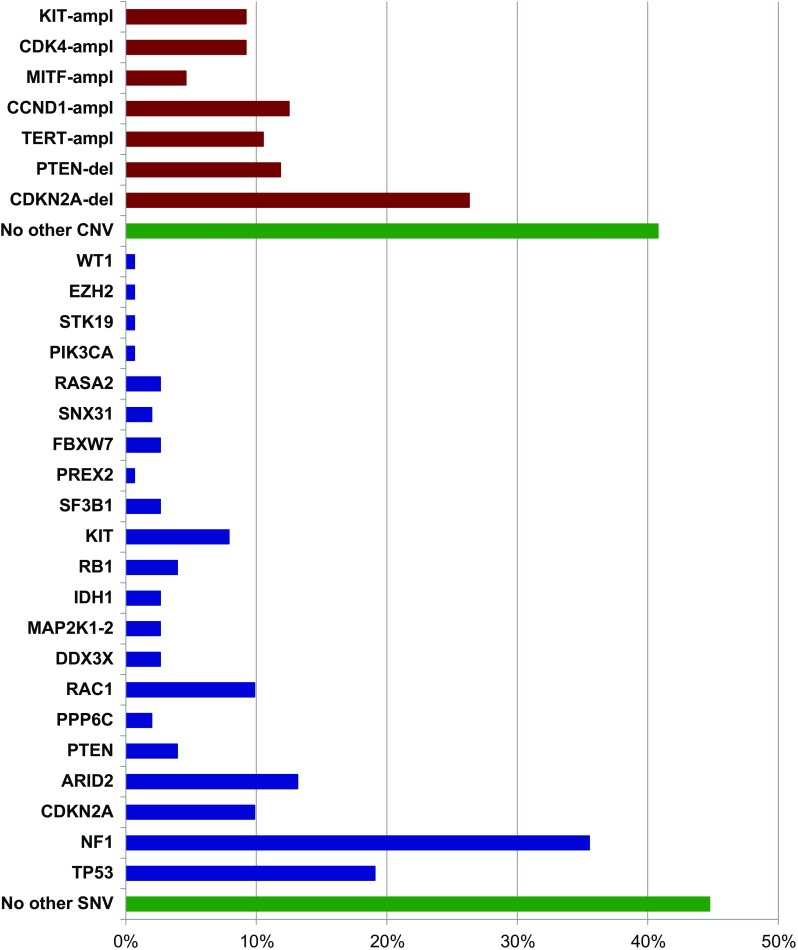
Gene mutation frequencies in the 152 cases with the non-BRAFmut/non-RASmut subtype. In red, copy number variations; in blue, pathogenetic mutations; in green, lack of genetic alterations. ampl, amplification; del, deletion
A description of the genetic alterations identified by NGS-based analyses and characterizing the different molecular subtypes in our series of CM is here provided.
BRAF/MAP 2K1-2 Genes
The RAF kinase family consists of three proteins (ARAF, BRAF, and CRAF), which are all part of the signal transduction cascade named mitogen-activated protein kinase (MAPK) pathway and whose kinase activity is physiologically induced by activation of the upstream RAS protein [33]. Activated BRAF promotes activity of the MEK1-2 kinases, which in turn activate ERK1-2 as final effectors of the MAPK pathway (Fig. 1). In our series, BRAF is mutated in about half (340/686; 49.6%) of cases (Table 2). The most prevalent mutation (about 90% of cases) is represented by a substitution of a valine with glutamic acid at codon 600 (BRAFV600E). Few additional BRAF variants at codon 600 (BRAFV600 mutations) or, less frequently, in other codons within the BRAF kinase region (in particular, BRAFK601E) have been demonstrated to activate the MEK1-2 downstream pathway effectors [3, 11••, 12, 14••]. All these BRAF mutations promote a constitutive stimulation of cell proliferation and tumor growth. Since BRAF is also mutated in common nevi, its oncogenic activation is supposed to be a necessary but not sufficient condition for melanoma development (being considered as an initiation event in melanocyte transformation) [34]. In our CM cohort, BRAFmut carriers were characterized by a higher frequency of PTEN and CDKN2A alterations (including both gene mutations and deletions) in comparison to the RASmut and non-BRAFmut/non-RASmut subtypes (Table 3). As a confirmation of this, it was recently demonstrated that activation of MEK in BRAFmut cells promotes the development, growth, and maintenance of melanoma in vivo when PTEN and/or CDKN2A losses coexist [35].
Table 3.
Distribution of genetic and genomic alterations among the three molecular subtypes of CM cases. SNVs, in dark; CNVs, in blue; del, gene deletion; ampl, gene amplification. In red, negative cases


Finally, BRAF mutations typically occur in younger patients [14••], are less associated with the UV mutation signature, and are more prevalent in melanomas arising on not chronically sun-exposed skin areas [12, 36, 37]. Although the prognostic significance for BRAF mutants remains unclear since controversial results were reported [38–41], the occurrence of BRAF mutations seems to have no impact on the disease-free interval from diagnosis of first-ever melanoma to first distant metastasis, but may worsen the overall survival after disease progression [42•].
MEK1 and MEK2 represent targets downstream of the RAS-RAF-MAPK cascade (Fig. 1). In our series, a small fraction of cases (27/686; 3.9%) carry mutations in the corresponding genes encoding these two kinases (MAP 2K1 and MAP 2K2, respectively). Activating mutations in MAP 2K1-2 represent one of the multiple mechanisms of resistance to BRAF and MEK inhibitors. No difference in the distribution of MAP 2K1-2 mutations among the three CM subtypes was found.
RAS/NF1/RAC1/PREX2 Genes
The three tissue-specific isoforms—HRAS, KRAS, and NRAS—of the RAS gene family encode small GTPase proteins bound to the cytoplasmic membrane. Among them, NRAS is the most commonly mutated gene in CMs (189/686, 27.6%), whereas the involvement of the other two isoforms is minimal (HRAS, 1.5%; KRAS, 1.0%) (Table 2). The oncogenic RAS is able to activate downstream cytoplasmic proteins [11••, 12]: RAF and phosphatidylinositol 3-kinase (PI3K) (Fig. 1). Similarly to BRAF, NRAS mutations occur almost exclusively in a single gene codon (Q61, about 90% of cases); in the remaining 10% of cases, the mutated codon is G12 or G13. Unlike BRAF, NRAS mutations have been described at similar frequencies in melanomas arising in different skin areas [12, 37].
NF1 is a tumor suppressor gene encoding a negative regulator of RAS [43, 44]. NF1 was found mutated in about 14% of CMs from our cohort, partially associated with other mutations in RAS-MAPK pathway genes (such as RASA2, a RASopathy gene activating the RAS-dependent kinase effectors; [45]). Melanomas carrying NF1 mutations present with a higher mutational burden and a markedly evident UV mutation signature [44]. Loss of NF1 function has been related to increased resistance to BRAF and MEK inhibitors [46, 47]. In our series, NF1 mutations have the lowest rate (18/340; 5.3%) in BRAFmut subtype, an intermediate rate (27/194; 13.9%) in RASmut subtype, and the highest prevalence (54/152; 35.5%) in the non-BRAFmut/non-RASmut subtype (Table 3).
NGS analyses revealed pathogenetic mutants in the RAC1 gene, encoding a GTPase able to induce the activity of RAS-dependent pathways and stimulate cell proliferation and migration [16, 45, 48]. Activation of RAC1 may be promoted by mutated PREX2 [18, 48–50]. This gene encodes a PTEN-binding protein and is mutated in 1.5% of CMs from our series (Table 2). In immunodeficient mice, the mutated PREX2 was found to enhance tumorigenesis, suggesting to play an oncogenic role [49].
CDKN2A/RB1/CDK4/CCND1 Genes
The interaction between cyclin D1 (CCND1) and the serine/threonine kinase CDK4/6 is strongly regulating cell proliferation. Once activated, the CDK4/6 protein interacts with the RB1 protein in inducing transcriptional events necessary for cell cycle progression from G1 to S phase [51, 52]. This process is negatively regulated by p16CDKN2A and alterations inactivating this tumor suppressor gene are thought to be related to disease progression: absent in nevi, occurring in primary tumors, and steadily increasing in metastatic melanomas [3, 11••, 12]. The inactivation of p16CDKN2A is due to either genetic (mutations, deletions) or epigenetic (promoter methylation) mechanisms, whereas the amplification of CCND1 is particularly frequent in melanomas negative for BRAF/NRAS mutations, in chronically sun-exposed skin areas [53]. In our series, CDKN2A was found to carry gene mutations in 13.6% of cases (mostly, in RASmut subtype) and gene deletions in 41.4% of cases (mostly, in BRAFmut subtype) (Table 3). The CCND1 gene amplification was found in 5.0% of the cases, with the highest prevalence (12.5%) in the non-BRAFmut/non-RASmut subtype, as compared to BRAFmut (1.5%) or RASmut (5.2%) ones (Table 3).
PTEN/PI3K/AKT/mTOR/KIT Genes
The PI3K-dependent pathway, including the signal transduction cascade with PTEN, AKT, and mTOR proteins, is a strong regulator of melanoma growth and survival [53–55]. In physiological conditions, activated PIK3CA—mostly, consequence of RAS activation—increases the intracellular level of PIP2/PIP3 phosphoinositoles (which are instead reduced by PTEN activity) and the downstream substrates AKT and mTOR are in turn activated (Fig. 1). In our series, constitutive activation of this pathway is due to the acquisition of PTEN alterations (gene mutations and allelic deletions: 7.9 and 18.2%, respectively), with very few cases (0.9%) mutated in PIK3CA gene (Table 3). Oncogenically activated BRAF and silenced PTEN cooperate in melanoma formation and progression, resistance to BRAF/MEK inhibitors, and interference with tumor immune infiltration (suggesting that PI3K-pathway inhibition may represent a strategy in patients receiving targeted therapy in combination with immune checkpoint inhibitors) [56–59].
The mutated or amplified KIT gene, encoding a tyrosine kinase receptor of the cell membrane [53], may activate both MAPK and PI3K-AKT downstream pathways (Fig. 1). In our series of CMs, 2.2% (15/686) of the cases carried a KIT mutation, with a markedly lower prevalence in the BRAFmut and RASmut subtypes (0.3 and 1.0%, respectively), as compared to that (7.9%) found in the non-BRAFmut/non-RASmut subtype (Table 3). Again, coexistence of pathogenetic mutations in BRAF or NRAS or KIT genes is rare, further confirming that oncogenic activations of such genes are mutually exclusive. Analogously, KIT gene amplifications were observed at the low frequency (3.1%); they were more prevalent in non-BRAFmut/non-RASmut subtype (9.2%) than in the other subtypes (BRAFmut, 0.9%, RASmut, 2.1%) (Table 3).
TP53 Gene
Inactivation of the TP53 tumor suppressor gene plays an important role in CM pathogenesis [11••, 12, 36]. Normal intracellular levels of the p53 protein promote the control of cell cycle progression, whereas TP53 upregulation induces mechanisms of protective apoptosis. For its capability in responding to genotoxic damages, p53 levels have been implicated in enhancing or contrasting the UV-driven melanomagenesis [60, 61]. In melanoma, a reduction of the p53 protein levels is commonly reported [3, 11••, 12]. The downregulation of TP53 gene may be due to several mechanisms [62–64]: inactivation of p14CDKN2A causing MDM2 overexpression, or increased stability of MDM2-4 proteins induced by activation of the receptor tyrosine kinase AXL, or TP53 gene silencing through genetic changes (mostly, mutations) or epigenetic deregulations (Fig. 1). As confirmation of previously reported interactions between activated BRAF and silenced TP53 [3, 11••, 12], restoring the intracellular p53 levels may sensitize BRAFmut melanomas to BRAF inhibitors [65].
In our series, pathogenetic mutations of TP53 were present in 16.8% of cases, with a quite similar distribution in the BRAFmut (15.9%), RASmut (16.5%), and non-BRAFmut/non-RASmut (19.1%) subtypes (Table 3).
PPP6C/ARID2/IDH1 Genes
The NGS-based mutation analyses revealed a high frequency of mutations in the genes encoding proteins which regulate the epigenetic mechanisms of gene transcription and expression [66, 67].
Mutations of the PPP6C gene impair the activity of the Aurora A kinase (AurkA), one of the main regulators of the M phase progression into the cell cycle (Fig. 1), causing chromosome instability [68]. Recently, a tight interaction between PPP6C-AurkA and MAPK pathways in controlling proliferative activity and apoptosis in melanoma cells has been demonstrated, contributing to increase the levels of tumor heterogeneity and conferring a resistance signature in BRAF- and NRAS-mutated melanomas [69, 70]. The PPP6C protein deficiency is able to induce cutaneous tumorigenesis in mice treated with the DMBA carcinogen, strongly suggesting a role as tumor suppressor gene [71]. Finally, PP6C mutations were associated with increased autophagy in vitro and in vivo melanoma samples [72]. The PPP6C gene is mutated in 7.1% of CM cells, with similar prevalence in the different stages of tumor progression (6.3% in primary and 7.4% in metastatic melanoma; Table 3).
Among the genes involved in epigenetic regulation, ARID2 encodes subunits of a tumor suppressor complex involved in chromatin remodeling, which has been implicated in pathogenesis of several cancers [36, 73, 74]. Inactivation of genes able to regulate the chromatin remodeling, including ARID2, may enhance the infiltrate of cytotoxic T cells and sensitize melanoma cells to their killing activity [75•]. Furthermore, IDH1 acts as the main source of cytosolic NADPH, protecting cells against reactive oxygen species (ROS) and radiations [76, 77]. Inactivating mutations of these two genes occur in 12.7% (ARID2) and 3.1% (IDH1) of CMs in our series, independently on the BRAF/RAS mutational status (Table 3).
Other Genes in Melanomagenesis
DEAD/H (Asp-Glu-Ala-Asp/His) polypeptide 3,X-linked (DDX3X) is a gene involved in the transcription control by regulating RNA homeostasis [14••, 15••]. DDX3X is involved in the epigenetic regulation of gene expression and its activation induces resistance to kinase inhibitors [78]. The DDX3X mutations were found in 4.7% of CMs, mostly associated with BRAF or RAS mutations (Table 3). Among genes epigenetically modulated by DDX3X, CTNNB1 encodes the beta-catenin protein and is mutated in 1.2% of CMs (Table 3). The CTNNB1 mutations were exclusively observed in the BRAFmut and RASmut subtypes, confirming the functional interaction between beta-catenin signaling and MAPK pathway activity (also through the involvement of recently described p21-activated kinases) [79, 80]. Recent evidence revealed that beta-catenin signaling may be related to a hypo-methylation status, a reduced T cell infiltration, an immune escape condition, and a lower immunogenicity [81, 82].
TERT
The telomerase reverse transcriptase gene (TERT) maintains telomere homeostasis, counteracting the progressive shortening of telomeres during mitoses [83]. Telomerase is present in its active form in stem cells, while it is inactivated in most somatic cells. In majority of cancers, the reactivation of telomerase is crucial for tumor cell survival. Mutations in the TERT gene promoter usually coexist with MAPK pathway activation [84]. TERT was found amplified in 5.8% of CMs from our cohort, with no particular association with the BRAF/NRAS mutational status (Table 3).
MITF
MITF is acting downstream of the BRAF-MEK-ERK signal transduction pathway (Fig. 1). In addition to its involvement in skin pigmentation, MITF plays an important role in proliferation and differentiation of melanocytes [11••, 12]. However, the mechanism of action of MITF is rather complex: low or absent levels of this protein predispose cells to apoptosis; intermediate MITF expression levels promote cell proliferation and survival; high concentration of intracellular MITF proteins induces antiproliferative effects [85]. An association was reported between MITF overexpression, partially regulated by AurkA, and MAPK pathway activation in controlling melanoma cell proliferation and migration [86]. MITF amplification was found in 5.0% of CMs, regardless of the BRAF/RAS mutational status (Table 3).
Mutation Distribution Between Primary and Metastatic Cutaneous Melanomas
Our series of 686 skin melanomas consists of 159 (23.2%) primary tumors and 527 (76.8%) metastatic melanomas. As shown in Table 2, some genetic alterations (BRAF, NF1, ARID2, PTEN, PPP6C, RAC1, DDX3X, MAP 2K1-2, IDH1, RB1, KIT, SF3B1, FBXW7, and PIK3CA mutations; TERT and KIT amplifications) present similar frequencies between primary and metastatic CMs. Alterations may represent initial events promoting cell proliferation and survival, but additional molecular alterations are needed for melanocytic transformation. Prevalence of other genetic alterations (RAS, TP53, CDKN2A, PREX2, and RASA2 mutations; CDKN2A and PTEN deletions, CCND1, MITF, and CDK4 amplifications) is higher in metastatic that in primary melanomas, most likely due to the expansion of cell subpopulations during tumor progression. Finally, a few genetic alterations (CTNNB1, SNX31, TACC1, STK19, EZH2, WT1, and CDK4 mutations) are present in metastatic subsets only, suggesting that these play a role in the advanced phases of the disease.
Overall, the fractions of cases negative for any pathogenetic SNV (14.5 vs. 8.5%) or CNV (54.1 vs. 26.2%) were markedly higher in primary melanomas than in metastatic lesions, respectively.
Conclusions
In addition to the genetic variations, data are emerging that support a role of epigenetic modifications in melanoma pathogenesis [66, 67]. Epigenetic changes having an impact on the expression levels of candidate genes are represented by:
post-translational variations and chromatin remodeling;
methylation of cytosine-guanine dinucleotides (CpG);
gene silencing performed by non-coding RNA.
Small (sncRNAs; < 200 nucleotides) and long (lncRNAs; > 200 nucleotides) non-coding RNAs as well as microRNAs (miRNAs; approximately, 22 nucleotides long) can regulate gene expression through transcription modifications and mRNA stability interfering with translation. As example, a copy number analysis indicated the pathogenetic coexistence of MITF amplification and expression of a specific lncRNA (SAMMSON) in a subset of melanomas. Aberrant DNA methylation [87, 88], up-/downregulation of miRNAs’ activity in controlling key signaling pathways responsible for the melanoma cell growth [89], and occurrence of synonymous mutations with a functional role [90] could further affect melanomagenesis and/or disease behavior.
Considering the entire scenario, the paradigm of melanoma pathogenesis seems to be highly more complicated than expected. A more in-depth knowledge of genes and molecular pathways might help in discriminating melanoma patients for response/resistance to specific treatments (predictive factors) or clinical outcome (prognostic factors). Molecular classification will increasingly be an integral part of the management of patients with cutaneous melanoma.
Conflict of Interest
Giuseppe Palmieri, Maria Colombino, Milena Casula, Antonella Manca, Mario Mandalà, and Antonio Cossu declare they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of the Topical Collection on Melanoma
Contributor Information
Giuseppe Palmieri, Phone: +390792841229, Email: gpalmieri@yahoo.com.
Maria Colombino, Phone: +390792841239, Email: colombinom@yahoo.it.
Milena Casula, Phone: +39 079 2841239, Email: casulam@yahoo.it.
Antonella Manca, Phone: +390792841230, Email: antonella.manca@icb.cnr.it.
Mario Mandalà, Phone: +39 035 267111, Email: mariomandala@tin.it.
Antonio Cossu, Phone: +39 079 229058, Email: cossu@uniss.it.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Sacchetto L, Zanetti R, Comber H, Bouchardy C, Brewster DH, Broganelli P, Chirlaque MD, Coza D, Galceran J, Gavin A, Hackl M, Katalinic A, Larønningen S, Louwman MWJ, Morgan E, Robsahm TE, Sanchez MJ, Tryggvadóttir L, Tumino R, Van Eycken E, Vernon S, Zadnik V, Rosso S. Trends in incidence of thick, thin and in situ melanoma in Europe. Eur J Cancer. 2018;92:108–118. doi: 10.1016/j.ejca.2017.12.024. [DOI] [PubMed] [Google Scholar]
- 2.Grzywa TM, Paskal W, Włodarski PK. Intratumor and intertumor heterogeneity in melanoma. Transl Oncol. 2017;10:956–975. doi: 10.1016/j.tranon.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmieri G, Ombra M, Colombino M, Casula M, Sini M, Manca A, Paliogiannis P, Ascierto PA, Cossu A. Multiple molecular pathways in melanomagenesis: characterization of therapeutic targets. Front Oncol. 2015;5:183. doi: 10.3389/fonc.2015.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paluncic J, Kovacevic Z, Jansson PJ, Kalinowski D, Merlot AM, Huang ML, Lok HC, Sahni S, Lane DJ, Richardson DR. Roads to melanoma: key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim Biophys Acta. 1863;2016:770–784. doi: 10.1016/j.bbamcr.2016.01.025. [DOI] [PubMed] [Google Scholar]
- 5.Lovly CM, Salama AK, Salgia R. Tumor heterogeneity and therapeutic resistance. Am Soc Clin Oncol Educ Book. 2016;35:e585–e593. doi: 10.1200/EDBK_158808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ascierto PA, Agarwala SS, Ciliberto G, Demaria S, Dummer R, Duong CPM, Ferrone S, Formenti SC, Garbe C, Halaban R, Khleif S, Luke JJ, Mir LM, Overwijk WW, Postow M, Puzanov I, Sondel P, Taube JM, Thor Straten P, Stroncek DF, Wargo JA, Zarour H, Thurin M. Future perspectives in melanoma research “melanoma bridge”, Napoli, November 30th-3rd December 2016. J Transl Med. 2017;15:236. doi: 10.1186/s12967-017-1341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meric-Bernstam F, Johnson A, Holla V, Bailey AM, Brusco L, Chen K, et al. A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst. 2015;107 [DOI] [PMC free article] [PubMed]
- 8.Buermans HP, den Dunnen JT. Next generation sequencing technology: advances and applications. Biochim Biophys Acta. 1842;2014:1932–1941. doi: 10.1016/j.bbadis.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 9.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–132. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, Rickaby W, D’Arrigo C, Robson A, Bastian BC. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373:1926–1936. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- 12.Zhang T, Dutton-Regester K, Brown KM, Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016;29:266–283. doi: 10.1111/pcmr.12459. [DOI] [PubMed] [Google Scholar]
- 13.Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, Heisler LE, Beck TA, Simpson JT, Tonon L, Sertier AS, Patch AM, Jäger N, Ginsbach P, Drews R, Paramasivam N, Kabbe R, Chotewutmontri S, Diessl N, Previti C, Schmidt S, Brors B, Feuerbach L, Heinold M, Gröbner S, Korshunov A, Tarpey PS, Butler AP, Hinton J, Jones D, Menzies A, Raine K, Shepherd R, Stebbings L, Teague JW, Ribeca P, Giner FC, Beltran S, Raineri E, Dabad M, Heath SC, Gut M, Denroche RE, Harding NJ, Yamaguchi TN, Fujimoto A, Nakagawa H, Quesada V, Valdés-Mas R, Nakken S, Vodák D, Bower L, Lynch AG, Anderson CL, Waddell N, Pearson JV, Grimmond SM, Peto M, Spellman P, He M, Kandoth C, Lee S, Zhang J, Létourneau L, Ma S, Seth S, Torrents D, Xi L, Wheeler DA, López-Otín C, Campo E, Campbell PJ, Boutros PC, Puente XS, Gerhard DS, Pfister SM, JD MP, Hudson TJ, Schlesner M, Lichter P, Eils R, Jones DT, Gut IG. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6:10001. doi: 10.1038/ncomms10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, Jakrot V, Kazakoff S, Holmes O, Leonard C, Sabarinathan R, Mularoni L, Wood S, Xu Q, Waddell N, Tembe V, Pupo GM, De Paoli-Iseppi R, Vilain RE, Shang P, LMS L, Dagg RA, Schramm SJ, Pritchard A, Dutton-Regester K, Newell F, Fitzgerald A, Shang CA, Grimmond SM, Pickett HA, Yang JY, Stretch JR, Behren A, Kefford RF, Hersey P, Long GV, Cebon J, Shackleton M, Spillane AJ, RPM S, López-Bigas N, Pearson JV, Thompson JF, Scolyer RA, Mann GJ. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–180. doi: 10.1038/nature22071. [DOI] [PubMed] [Google Scholar]
- 16.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, Zhang H, Zeid R, Ren X, Cibulskis K, Sivachenko AY, Wagle N, Sucker A, Sougnez C, Onofrio R, Ambrogio L, Auclair D, Fennell T, Carter SL, Drier Y, Stojanov P, Singer MA, Voet D, Jing R, Saksena G, Barretina J, Ramos AH, Pugh TJ, Stransky N, Parkin M, Winckler W, Mahan S, Ardlie K, Baldwin J, Wargo J, Schadendorf D, Meyerson M, Gabriel SB, Golub TR, Wagner SN, Lander ES, Getz G, Chin L, Garraway LA. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, Wilmott JS, Gartner JJ, Di Pizio A, Winograd-Katz S, Sindiri S, Rotkopf R, Dutton-Regester K, Johansson P, Pritchard AL, Waddell N, Hill VK, Lin JC, Hevroni Y, Rosenberg SA, Khan J, Ben-Dor S, Niv MY, Ulitsky I, Mann GJ, Scolyer RA, Hayward NK, Samuels Y. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47:1408–1410. doi: 10.1038/ng.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elkon R, Agami R. Characterization of noncoding regulatory DNA in the human genome. Nat Biotechnol. 2017;35:732–746. doi: 10.1038/nbt.3863. [DOI] [PubMed] [Google Scholar]
- 21.Katainen R, Dave K, Pitkänen E, Palin K, Kivioja T, Välimäki N, Gylfe AE, Ristolainen H, Hänninen UA, Cajuso T, Kondelin J, Tanskanen T, Mecklin JP, Järvinen H, Renkonen-Sinisalo L, Lepistö A, Kaasinen E, Kilpivaara O, Tuupanen S, Enge M, Taipale J, Aaltonen LA. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47:818–821. doi: 10.1038/ng.3335. [DOI] [PubMed] [Google Scholar]
- 22.Wiesner T, Lee W, Obenauf AC, Ran L, Murali R, Zhang QF, Wong EW, Hu W, Scott SN, Shah RH, Landa I, Button J, Lailler N, Sboner A, Gao D, Murphy DA, Cao Z, Shukla S, Hollmann TJ, Wang L, Borsu L, Merghoub T, Schwartz GK, Postow MA, Ariyan CE, Fagin JA, Zheng D, Ladanyi M, Busam KJ, Berger MF, Chen Y, Chi P. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature. 2015;526:453–457. doi: 10.1038/nature15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson PA, Pritchard AL, Patch AM, Wilmott JS, Pearson JV, Waddell N, Scolyer RA, Mann GJ, Hayward NK. Mutation load in melanoma is affected by MC1R genotype. Pigment Cell Melanoma Res. 2017;30:255–258. doi: 10.1111/pcmr.12571. [DOI] [PubMed] [Google Scholar]
- 24.Craig S, Earnshaw CH, Virós A. Ultraviolet light and melanoma. J Pathol. 2018;244:578–585. doi: 10.1002/path.5039. [DOI] [PubMed] [Google Scholar]
- 25.Robles-Espinoza CD, Roberts ND, Chen S, Leacy FP, Alexandrov LB, Pornputtapong N, Halaban R, Krauthammer M, Cui R, Timothy Bishop D, Adams DJ. Germline MC1R status influences somatic mutation burden in melanoma. Nat Commun. 2016;7:12064. doi: 10.1038/ncomms12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furney SJ, Turajlic S, Stamp G, Thomas JM, Hayes A, Strauss D, Gavrielides M, Xing W, Gore M, Larkin J, Marais R. The mutational burden of acral melanoma revealed by whole-genome sequencing and comparative analysis. Pigment Cell Melanoma Res. 2014;27:835–838. doi: 10.1111/pcmr.12279. [DOI] [PubMed] [Google Scholar]
- 28.Mar VJ, Wong SQ, Li J, Scolyer RA, McLean C, Papenfuss AT, Tothill RW, Kakavand H, Mann GJ, Thompson JF, Behren A, Cebon JS, Wolfe R, Kelly JW, Dobrovic A, McArthur GA. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res. 2013;19:4589–4598. doi: 10.1158/1078-0432.CCR-13-0398. [DOI] [PubMed] [Google Scholar]
- 29.Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, Malke J, Haydu L, Prieto VG, Tetzlaff M, Ivan D, Wang WL, Torres-Cabala C, Curry J, Roy-Chowdhuri S, Broaddus R, Rashid A, Stewart J, Gershenwald JE, Amaria RN, Patel SP, Papadopoulos NE, Bedikian A, Hwu WJ, Hwu P, Diab A, Woodman SE, Aldape KD, Luthra R, Patel KP, Shaw KR, Mills GB, Mendelsohn J, Meric-Bernstam F, Kim KB, Routbort MJ, Lazar AJ, Davies MA. Beyond BRAF (V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol. 2015;135:508–515. doi: 10.1038/jid.2014.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shitara D, Tell-Martí G, Badenas C, Enokihara MM, Alós L, Larque AB, Michalany N, Puig-Butille JA, Carrera C, Malvehy J, Puig S, Bagatin E. Mutational status of naevus-associated melanomas. Br J Dermatol. 2015;173:671–680. doi: 10.1111/bjd.13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen T, Pajaro-Van de Stadt SH, Yeat NC, Lin JC. Clinical applications of next generation sequencing in cancer: from panels, to exomes, to genomes. Front Genet. 2015;6:215. doi: 10.3389/fgene.2015.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casula M, Colombino M, Manca A, Caracò C, Botti G, Ascierto PA, Lissia A, Cossu A, Palmieri G, Italian Melanoma Intergroup (IMI) Low levels of genetic heterogeneity in matched lymph node metastases from patients with melanoma. J Invest Dermatol. 2016;136:1917–1920. doi: 10.1016/j.jid.2016.05.103. [DOI] [PubMed] [Google Scholar]
- 33.Acosta AM, Kadkol SS. Mitogen-activated protein kinase signaling pathway in cutaneous melanoma: an updated review. Arch Pathol Lab Med. 2016;140:1290–1296. doi: 10.5858/arpa.2015-0475-RS. [DOI] [PubMed] [Google Scholar]
- 34.Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–254. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 35.Yang H, Kircher DA, Kim KH, Grossmann AH, VanBrocklin MW, Holmen SL, Robinson JP. Activated MEK cooperates with Cdkn2a and Pten loss to promote the development and maintenance of melanoma. Oncogene. 2017;36:3842–3851. doi: 10.1038/onc.2016.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding L, Kim M, Kanchi KL, Dees ND, Lu C, Griffith M, Fenstermacher D, Sung H, Miller CA, Goetz B, Wendl MC, Griffith O, Cornelius LA, Linette GP, McMichael JF, Sondak VK, Fields RC, Ley TJ, Mulé JJ, Wilson RK, Weber JS. Clonal architectures and driver mutations in metastatic melanomas. PLoS One. 2014;9:e111153. doi: 10.1371/journal.pone.0111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filipp FV. Precision medicine driven by cancer systems biology. Cancer Metastasis Rev. 2017;36:91–108. doi: 10.1007/s10555-017-9662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbour AP, Tang YH, Armour N, Dutton-Regester K, Krause L, Loffler KA, Lambie D, Burmeister B, Thomas J, Smithers BM, Hayward NK. BRAF mutation status is an independent prognostic factor for resected stage IIIB and IIIC melanoma: implications for melanoma staging and adjuvant therapy. Eur J Cancer. 2014;50:2668–2676. doi: 10.1016/j.ejca.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 39.Carlino MS, Haydu LE, Kakavand H, Menzies AM, Hamilton AL, Yu B, Ng CC, Cooper WA, Thompson JF, Kefford RF, O'Toole SA, Scolyer RA, Long GV. Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer. 2014;111:292–299. doi: 10.1038/bjc.2014.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutkowski P, Gos A, Jurkowska M, Switaj T, Dziewirski W, Zdzienicki M, Ptaszyński K, Michej W, Tysarowski A, Siedlecki JA. Molecular alterations in clinical stage III cutaneous melanoma: correlation with clinicopathological features and patient outcome. Oncol Lett. 2014;8:47–54. doi: 10.3892/ol.2014.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas NE, Edmiston SN, Alexander A, Groben PA, Parrish E, Kricker A, Armstrong BK, Anton-Culver H, Gruber SB, From L, Busam KJ, Hao H, Orlow I, Kanetsky PA, Luo L, Reiner AS, Paine S, Frank JS, Bramson JI, Marrett LD, Gallagher RP, Zanetti R, Rosso S, Dwyer T, Cust AE, Ollila DW, Begg CB, Berwick M, Conway K, GEM Study Group Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015;1:359–368. doi: 10.1001/jamaoncol.2015.0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 43.Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. 2017;11:13. doi: 10.1186/s40246-017-0109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cirenajwis H, Lauss M, Ekedahl H, Törngren T, Kvist A, Saal LH, Olsson H, Staaf J, Carneiro A, Ingvar C, Harbst K, Hayward NK, Jönsson G. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol. 2017;11:438–451. doi: 10.1002/1878-0261.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, Serin M, Bosenberg M, Ariyan S, Narayan D, Sznol M, Kluger HM, Mane S, Schlessinger J, Lifton RP, Halaban R. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47:996–1002. doi: 10.1038/ng.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, Merghoub T, Ribas A, Chapman PB, Yaeger R, Taylor BS, Schultz N, Berger MF, Rosen N, Solit DB. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:2340–2350. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ranzani M, Alifrangis C, Perna D, Dutton-Regester K, Pritchard A, Wong K, Rashid M, Robles-Espinoza CD, Hayward NK, McDermott U, Garnett M, Adams DJ. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2015;28:117–119. doi: 10.1111/pcmr.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kunz M. Oncogenes in melanoma: an update. Eur J Cell Biol. 2014;93:1–10. doi: 10.1016/j.ejcb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Lissanu Deribe Y, Shi Y, Rai K, Nezi L, Amin SB, Wu CC, Akdemir KC, Mahdavi M, Peng Q, Chang QE, Hornigold K, Arold ST, Welch HC, Garraway LA, Chin L. Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc Natl Acad Sci U S A. 2016;113:E1296–E1305. doi: 10.1073/pnas.1513801113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lissanu DY. Mechanistic insights into the role of truncating PREX2 mutations in melanoma. Mol Cell Oncol. 2016;3:e1160174. doi: 10.1080/23723556.2016.1160174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, Do H, Mitchell C, Cullinane C, Liu W, Fox SB, Dutton-Regester K, Hayward NK, Jene N, Dobrovic A, Pearson RB, Christensen JG, Randolph S, McArthur GA, Sheppard KE. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27:590–600. doi: 10.1111/pcmr.12228. [DOI] [PubMed] [Google Scholar]
- 52.Yadav Vipin, Chen Shih-Hsun, Yue Yong Gong, Buchanan Sean, Beckmann Richard P., Peng Sheng-Bin. Co-targeting BRAF and cyclin dependent kinases 4/6 for BRAF mutant cancers. Pharmacology & Therapeutics. 2015;149:139–149. doi: 10.1016/j.pharmthera.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 53.Lee B, Sandhu S, McArthur G. Cell cycle control as a promising target in melanoma. Curr Opin Oncol. 2015;27:141–150. doi: 10.1097/CCO.0000000000000159. [DOI] [PubMed] [Google Scholar]
- 54.Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, Davies MA, Grossmann AH, VanBrocklin MW, McMahon M, Holmen SL. AKT1 activation promotes development of melanoma metastases. Cell Rep. 2015;13:898–905. doi: 10.1016/j.celrep.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, Wheler JJ, Kurzrock R. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377–387. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, Comin-Anduix B, Ribas A. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20:3446–3457. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silva JM, Bulman C, McMahon M. BRAFV600E cooperates with PI3K signaling, independent of AKT, to regulate melanoma cell proliferation. Mol Cancer Res. 2014;12(3):447–463. doi: 10.1158/1541-7786.MCR-13-0224-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, Pineda C, Geukes Foppen MH, Scolyer R, Song JY, Verbrugge I, Hoeller C, Dummer R, Haanen JB, Long GV, Blank CU. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology. 2016;5:e1238557. doi: 10.1080/2162402X.2016.1238557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zuo Q, Liu J, Huang L, Qin Y, Hawley T, Seo C, Merlino G, Yu Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene. 2018;37(24):3275–3289. doi: 10.1038/s41388-018-0205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi YS, Fisher DE. UV and melanoma: the TP53 link. Cell Res. 2014;24(10):1157–1158. doi: 10.1038/cr.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viros A, Sanchez-Laorden B, Pedersen M, Furney SJ, Rae J, Hogan K, Ejiama S, Girotti MR, Cook M, Dhomen N, Marais R. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature. 2014;511:478–482. doi: 10.1038/nature13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu M, Miller P, Lu X. Restoring the tumour suppressive function of p53 as a parallel strategy in melanoma therapy. FEBS Lett. 2014;588:2616–2621. doi: 10.1016/j.febslet.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 63.Lankenau MA, Patel R, Liyanarachchi S, Maharry SE, Hoag KW, Duggan M, Walker CJ, Markowitz J, Carson WE, 3rd, Eisfeld AK, de la Chapelle A. MicroRNA-3151 inactivates TP53 in BRAF-mutated human malignancies. Proc Natl Acad Sci U S A. 2015;112:E6744–E6751. doi: 10.1073/pnas.1520390112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Polo A, Luo Z, Gerarduzzi C, Chen X, Little JB, Yuan ZM. AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex. J Mol Cell Biol. 2017;9:154–165. doi: 10.1093/jmcb/mjw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krayem M, Journe F, Wiedig M, Morandini R, Najem A, Salès F, van Kempen LC, Sibille C, Awada A, Marine JC, Ghanem G. p53 reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K) BRAF melanoma to vemurafenib. Eur J Cancer. 2016;55:98–110. doi: 10.1016/j.ejca.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 66.Sarkar D, Leung EY, Baguley BC, Finlay GJ, Askarian-Amiri ME. Epigenetic regulation in human melanoma: past and future. Epigenetics. 2015;10:103–121. doi: 10.1080/15592294.2014.1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JJ, Sholl LM, Lindeman NI, Granter SR, Laga AC, Shivdasani P, Chin G, Luke JJ, Ott PA, Hodi FS, Mihm MC, Jr, Lin JY, Werchniak AE, Haynes HA, Bailey N, Liu R, Murphy GF, Lian CG. Targeted next-generation sequencing reveals high frequency of mutations in epigenetic regulators across treatment-naïve patient melanomas. Clin Epigenetics. 2015;7:59. doi: 10.1186/s13148-015-0091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gold HL, Wengrod J, de Miera EV, Wang D, Fleming N, Sikkema L, Kirchhoff T, Hochman T, Goldberg JD, Osman I, Gardner LB. PP6C hotspot mutations in melanoma display sensitivity to Aurora kinase inhibition. Mol Cancer Res. 2014;12:433–439. doi: 10.1158/1541-7786.MCR-13-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hilton JF, Shapiro GI. Aurora kinase inhibition as an anticancer strategy. J Clin Oncol. 2014;32:57–59. doi: 10.1200/JCO.2013.50.7988. [DOI] [PubMed] [Google Scholar]
- 70.Puig-Butille JA, Vinyals A, Ferreres JR, Aguilera P, Cabré E, Tell-Martí G, Marcoval J, Mateo F, Palomero L, Badenas C, Piulats JM, Malvehy J, Pujana MA, Puig S, Fabra À. AURKA overexpression is driven by FOXM1 and MAPK/ERK activation in melanoma cells harboring BRAF or NRAS mutations: impact on melanoma prognosis and therapy. J Investig Dermatol. 2017;137:1297–1310. doi: 10.1016/j.jid.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 71.Hayashi K, Momoi Y, Tanuma N, Kishimoto A, Ogoh H, Kato H, Suzuki M, Sakamoto Y, Inoue Y, Nomura M, Kiyonari H, Sakayori M, Fukamachi K, Kakugawa Y, Yamashita Y, Ito S, Sato I, Suzuki A, Nishio M, Suganuma M, Watanabe T, Shima H. Abrogation of protein phosphatase 6 promotes skin carcinogenesis induced by DMBA. Oncogene. 2015;34:4647–4655. doi: 10.1038/onc.2014.398. [DOI] [PubMed] [Google Scholar]
- 72.Wengrod J, Wang D, Weiss S, Zhong H, Osman I, Gardner LB. Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci Signal. 2015;8:ra27. doi: 10.1126/scisignal.aaa0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cajuso T, Hänninen UA, Kondelin J, Gylfe AE, Tanskanen T, Katainen R, Pitkänen E, Ristolainen H, Kaasinen E, Taipale M, Taipale J, Böhm J, Renkonen-Sinisalo L, Mecklin JP, Järvinen H, Tuupanen S, Kilpivaara O, Vahteristo P. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int J Cancer. 2014;135:611–623. doi: 10.1002/ijc.28705. [DOI] [PubMed] [Google Scholar]
- 74.Duan Y, Tian L, Gao Q, Liang L, Zhang W, Yang Y, Zheng Y, Pan E, Li S, Tang N. Chromatin remodeling gene ARID2 targets cyclin D1 and cyclin E1 to suppress hepatoma cell progression. Oncotarget. 2016;7:45863–45875. doi: 10.18632/oncotarget.10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, Long HW, Yuan GC, Doench J, Brown M, Liu XS, Wucherpfennig KW. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359(6377):770–775. doi: 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caino MC, Altieri DC. Molecular pathways: mitochondrial reprogramming in tumor progression and therapy. Clin Cancer Res. 2016;22:540–545. doi: 10.1158/1078-0432.CCR-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kunz M, Hölzel M. The impact of melanoma genetics on treatment response and resistance in clinical and experimental studies. Cancer Metastasis Rev. 2017;36:53–75. doi: 10.1007/s10555-017-9657-1. [DOI] [PubMed] [Google Scholar]
- 78.Nozaki K, Kagamu H, Shoji S, Igarashi N, Ohtsubo A, Okajima M, Miura S, Watanabe S, Yoshizawa H, Narita I. DDX3X induces primary EGFR-TKI resistance based on intratumor heterogeneity in lung cancer cells harboring EGFR-activating mutations. PLoS One. 2014;9:e111019. doi: 10.1371/journal.pone.0111019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yeh I, Lang UE, Durieux E, Tee MK, Jorapur A, Shain AH, Haddad V, Pissaloux D, Chen X, Cerroni L, Judson RL, LeBoit PE, McCalmont TH, Bastian BC, de la Fouchardière A. Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat Commun. 2017;8:644. doi: 10.1038/s41467-017-00758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu H, Liu S, Zhang G, Wu B, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, Zhang W, Chen G, Cheng C, Zeng J, Wu LW, Zhang J, Liu X, Xu W, Krepler C, Sproesser K, Xiao M, Miao B, Liu J, Song CD, Liu JY, Karakousis GC, Schuchter LM, Lu Y, Mills G, Cong Y, Chernoff J, Guo J, Boland GM, Sullivan RJ, Wei Z, Field J, Amaravadi RK, Flaherty KT, Herlyn M, Xu X, Guo W. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature. 2017;550:133–136. doi: 10.1038/nature24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Massi D, Romano E, Rulli E, Merelli B, Nassini R, De Logu F, Bieche I, Baroni G, Cattaneo L, Xue G, Mandalà M. Baseline β-catenin, programmed death-ligand 1 expression and tumour-infiltrating lymphocytes predict response and poor prognosis in BRAF inhibitor-treated melanoma patients. Eur J Cancer. 2017;78:70–81. doi: 10.1016/j.ejca.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 82.Nsengimana Jérémie, Laye Jon, Filia Anastasia, O’Shea Sally, Muralidhar Sathya, Poźniak Joanna, Droop Alastair, Chan May, Walker Christy, Parkinson Louise, Gascoyne Joanne, Mell Tracey, Polso Minttu, Jewell Rosalyn, Randerson-Moor Juliette, Cook Graham P., Bishop D. Timothy, Newton-Bishop Julia. β-Catenin–mediated immune evasion pathway frequently operates in primary cutaneous melanomas. Journal of Clinical Investigation. 2018;128(5):2048–2063. doi: 10.1172/JCI95351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heidenreich B, Kumar R. TERT promoter mutations in telomere biology. Mutat Res. 2017;771:15–31. doi: 10.1016/j.mrrev.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 84.Bai X, Kong Y, Chi Z, Sheng X, Cui C, Wang X, Mao L, Tang B, Li S, Lian B, Yan X, Zhou L, Dai J, Guo J, Si L. MAPK pathway and TERT promoter gene mutation pattern and its prognostic value in melanoma patients: a retrospective study of 2,793 cases. Clin Cancer Res. 2017;23:6120–6127. doi: 10.1158/1078-0432.CCR-17-0980. [DOI] [PubMed] [Google Scholar]
- 85.Seberg HE, Van Otterloo E, Cornell RA. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res. 2017;30:454–466. doi: 10.1111/pcmr.12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pathria G, Garg B, Borgdorff V, Garg K, Wagner C, Superti-Furga G, Wagner SN. Overcoming MITF-conferred drug resistance through dual AURKA/MAPK targeting in human melanoma cells. Cell Death Dis. 2016;7:e2135. doi: 10.1038/cddis.2015.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wouters J, Vizoso M, Martinez-Cardus A, Carmona FJ, Govaere O, Laguna T, Joseph J, Dynoodt P, Aura C, Foth M, Cloots R, van den Hurk K, Balint B, Murphy IG, McDermott EW, Sheahan K, Jirström K, Nodin B, Mallya-Udupi G, van den Oord JJ, Gallagher WM, Esteller M. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017;15:101. doi: 10.1186/s12916-017-0851-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Micevic G, Theodosakis N, Bosenberg M. Aberrant DNA methylation in melanoma: biomarker and therapeutic opportunities. Clin Epigenetics. 2017;9:34. doi: 10.1186/s13148-017-0332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fattore L, Costantini S, Malpicci D, Ruggiero CF, Ascierto PA, Croce CM, Mancini R, Ciliberto G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget. 2017;8:22262–22278. doi: 10.18632/oncotarget.14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gartner JJ, Parker SC, Prickett TD, Dutton-Regester K, Stitzel ML, Lin JC, Davis S, Simhadri VL, Jha S, Katagiri N, Gotea V, Teer JK, Wei X, Morken MA, Bhanot UK, NISC Comparative Sequencing Program. Chen G, Elnitski LL, Davies MA, Gershenwald JE, Carter H, Karchin R, Robinson W, Robinson S, Rosenberg SA, Collins FS, Parmigiani G, Komar AA, Kimchi-Sarfaty C, Hayward NK, Margulies EH, Samuels Y. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc Natl Acad Sci U S A. 2013;110:13481–13486. doi: 10.1073/pnas.1304227110. [DOI] [PMC free article] [PubMed] [Google Scholar]