

Gene clusters encoding WXG-like proteins and FtsK/SpoIIIE-like P loop ATPases in Firmicutes encode type 7b secretion systems (T7bSS) for the transport of select protein substrates. The Staphylococcus aureus T7bSS assembles in the bacterial membrane and promotes the secretion of WXG-like proteins and effectors. The mechanisms whereby staphylococci extend the T7SS across the bacterial cell wall envelope are not known. Here, we show that staphylococci secrete EssH to cleave their peptidoglycan, thereby enabling T7bSS transport of proteins across the bacterial cell wall envelope.
KEYWORDS: Esx, Staphylococcus aureus, cell wall hydrolase, secretion systems
ABSTRACT
The ESAT-6-like secretion system (ESS) of Staphylococcus aureus is assembled in the bacterial membrane from core components that promote the secretion of WXG-like proteins (EsxA, EsxB, EsxC, and EsxD) and the EssD effector. Genes encoding the ESS secretion machinery components, effector, and WXG-like proteins are located in the ess locus. Here, we identify essH, a heretofore uncharacterized gene of the ess locus, whose product is secreted via an N-terminal signal peptide into the extracellular medium of staphylococcal cultures. EssH exhibits two peptidoglycan hydrolase activities, cleaving the pentaglycine cross bridge and the amide bond of N-acetylmuramyl-l-alanine, thereby separating glycan chains and wall peptides with cleaved cross bridges. Unlike other peptidoglycan hydrolases, EssH does not promote the lysis of staphylococci. EssH residues Cys199 and His254, which are conserved in other CHAP domain enzymes, are required for peptidoglycan hydrolase activity and for S. aureus ESS secretion. These data suggest that EssH and its murein hydrolase activity are required for protein secretion by the ESS pathway.
IMPORTANCE Gene clusters encoding WXG-like proteins and FtsK/SpoIIIE-like P loop ATPases in Firmicutes encode type 7b secretion systems (T7bSS) for the transport of select protein substrates. The Staphylococcus aureus T7bSS assembles in the bacterial membrane and promotes the secretion of WXG-like proteins and effectors. The mechanisms whereby staphylococci extend the T7SS across the bacterial cell wall envelope are not known. Here, we show that staphylococci secrete EssH to cleave their peptidoglycan, thereby enabling T7bSS transport of proteins across the bacterial cell wall envelope.
INTRODUCTION
Bacterial pathogens rely on the secretion of toxins and other effector proteins to implement their virulence strategies during host infection (1). In Gram-negative bacteria, precursors with N-terminal signal peptides are translocated into the periplasmic space, a compartment between the bacterial inner and outer membranes, but not into the extracellular milieu (2). Gram-negative bacteria evolved pathways for the secretion of select proteins across the bacterial envelope (types I, II, and V secretion), the bacterial envelope and the plasma membrane of host cells (type III secretion), or the bacterial envelope and phagosomal membranes for pathogens residing within host cells (type IV secretion) (3). In Gram-positive bacteria, the Sec pathway directs signal peptide-bearing precursors across the bacterial envelope into the extracellular milieu (4). Nonetheless, Actinobacteria and Firmicutes evolved specialized type VIIa and -b secretion systems (T7aSS and T7bSS) to implement specific virulence strategies (5). In mycobacteria, the T7aSS supports the persistent infection of mammalian hosts and a facultative intracellular lifestyle (6–8). T7aSS and T7bSS are defined by two types of genes. All T7SS clusters harbor a gene for the FtsK/SpoIIIE-like P loop ATPase, which fuels the translocation of select protein substrates across the bacterial plasma membrane (9, 10). Another hallmark of the T7SS is the genes coding for WXG-like proteins, small α-helical polypeptides that are secreted and assemble into homo- or heteropolymeric structures (11). WXG-like proteins represent mobile components that extend the T7SS pathway beyond the bacterial membrane and contribute to the secretion of effectors, thereby implementing the specific virulence strategies of various bacterial species (12–14).
Staphylococcus aureus is a commensal of the human nasopharynx and gastrointestinal tract (15–18). Colonization represents the key risk factor for invasive disease, most frequently skin and soft tissue infections but also skeletal, lung, and bloodstream infections, which occur in healthy and immunocompromised individuals (19, 20). When analyzed in a mouse model of bloodstream challenge, S. aureus establishes chronically persistent infections with abscess lesions and lethal outcomes (21). S. aureus mutants with a defective T7bSS, designated ESS (ESAT-6-like secretion system), exhibit diminished persistence, smaller and less abundant abscess lesions, and a reduced bacterial load in host tissues (12, 22). Earlier work characterized five secretion substrates of the S. aureus ESS pathway: EsxA and EsxB with the canonical WXG motif, their pairing partners EsxC and EsxD, and the EssD effector (12, 22–25). For simplicity, EsxA, EsxB, EsxC, and EsxD are referred as WXG-like proteins. A complex formed from EsaA, EssA, and EssB engages WXG-like proteins and the effector for translocation across the bacterial membrane with the help of EssC, the FtsK/SpoIIIE-like P loop ATPase (26). However, the mechanism whereby the ESS pathway secretes protein substrates across the bacterial cell wall has heretofore not been investigated.
The cell wall envelope of S. aureus is comprised of peptidoglycan, a macromolecule with attached wall teichoic acid, capsular polysaccharide, and polypeptides that protects staphylococci from osmotic lysis (27). Peptidoglycan is assembled by the polymerization of its biosynthetic precursor, lipid II [C55-(PO4)2-MurNAc-(l-Ala-d-iGln-(NH2-Gly5)l-Lys-d-Ala-d-Ala)-GlcNAc], thereby generating glycan strands with the repeating disaccharide [→4)-β-MurNAc-(1→4)-β-GlcNAc-(1→]n (28, 29). Wall peptides that are attached to glycan strands are subsequently cross-linked via the cleavage of the amide bond of d-Ala-d-Ala and the generation of a new amide bond between the carboxyl group of d-Ala at position four and the amino group of pentaglycine cross bridges (NH2-Gly5) of neighboring peptidoglycan strands (30, 31). The high degree of peptidoglycan cross-linking and the large (40 to 50 nm) diameter of the staphylococcal cell wall represent a barrier for the secretion of proteins into the extracellular medium (32). To reveal the mechanisms whereby the S. aureus T7bSS translocates its substrates across the cell wall envelope, we studied a heretofore uncharacterized gene, essH, which encodes a murein hydrolase with cysteine, histidine-dependent amidohydrolase/peptidase (CHAP) domain (33).
RESULTS
essH is associated with the staphylococcal ess locus.
Holden and colleagues analyzed ess loci in genome sequences from 153 clinical S. aureus isolates, revealing two conserved and two variable modules (34). Module 1, encompassing secretion machinery genes (esxA-esaA-essA-esaB-essB), and module 4, containing genes that code for two uncharacterized products, are found in the genomes of all isolates; modules 1 and 4 flank the ess locus, whose genes are encrypted in the plus strand of the S. aureus genome (Fig. 1A). Module 2 encompasses essC with conserved 5′- and variable 3′-coding sequences along with other variable genes that encode secretion substrates (esxC, esxB, esxD, and essD in S. aureus USA300 LAC) (Fig. 1A). Module 3 contains genes whose products encompass DUF600 domains (EssI) that associate with the EssD effector in the bacterial cytoplasm and block its nuclease activity (25). Genes encoding membrane proteins with unknown function are also found in module 3. Immediately adjacent to module 1 on the minus strand of the S. aureus genome is an uncharacterized gene (SAUSA300_0277; GenBank accession number ABD20657) that is here designated essH (Fig. 1A). The essH gene is associated with module 1 of ess loci in other Staphylococcus species, including Staphylococcus argenteus BN75, Staphylococcus lugdunensis HKU09-01, and Staphylococcus epidermidis 14.1.R1 (Fig. 1A). Indeed, essH and module 1 represent the most conserved genes of ESS clusters, whereas modules 2, 3, and 4 are more variable (Fig. 1A). essH is absent in strains that lack the ess locus, such as S. epidermidis PM221, which carries an insertion sequence (IS) element replacing essH and modules 1 to 4 (Fig. 1A). The predicted product of essH is 297 amino acids in length, with an N-terminal signal peptide (amino acids 1 to 24), an intervening region without conserved features (amino acids 25 to 124), and a C-terminal CHAP domain (amino acids 125 to 297). The CHAP domain of EssH bears similarity to the MurNAc-l-Ala amidase domain, PRK08581 (NCBI). Both CHAP and PRK08581 belong to the COG3942 superfamily of proteins involved in cell wall biogenesis and the peptidoglycan hydrolase activities of bacteria, archaea, and trypanosomes. In bacteria, CHAP domains are found in murein hydrolases with N-acetylmuramoyl-l-alanine amidase activity and the papain-like fold of the cysteine protease CA peptidase clan CL0125 (35). In S. aureus USA300 LAC, EssH is 1 of 11 proteins with C-terminal CHAP domains (COG3942 superfamily) (Fig. 1B). Two additional proteins, SAUSA300_1923 and SAUSA300_(LytN) (36), harbor CHAP domains (Pfam05257) with reduced sequence conservation and are not shown in the alignment.
FIG 1.

Association of essH with the Staphylococcus aureus ESS cluster. (A) Genomic organization of ESS clusters of S. aureus USA300 LAC, Staphylococcus argenteus BN75, Staphylococcus lugdunensis HKU09-01, Staphylococcus epidermidis 14.1.R1, and S. epidermidis PM221. Genes in modules 1, 2, 3, and 4 are shown in pink, blue, shades of green, and gray, respectively. Genes flanking the cluster are shown in black; essH is shown in red. DUFs (domains of unknown function) encoded by module 4 genes are listed in the inset. In some strains, module 4 genes are also found upstream of essH. The complete set of modules 1 and 2 genes is only shown for S. aureus USA300 LAC; these genes are conserved in the other species. FocA, formate channel A; HP, hypothetical protein; MarR, multiple antibiotic resistance regulator. (B) CHAP domains of 11 proteins predicted from the genome sequence of S. aureus subsp. aureus USA300_FPR3757 (NCBI RefSeq accession number NC_007793.1) were aligned with Clustal W. The locus tag for essH is SAUSA300_0277. Proteins are aligned with EssH in order of declining sequence conservation. Genes whose products encompass CHAP domains are identified with the last four digits of their locus tag; SAUSA300_0438 and _2579 are listed as Sle1 and LytZ. TraG (pUSA300016) is encoded on a plasmid. Identical, conserved, and similar residues are denoted by asterisks, colons, and periods, respectively, and highlighted in shades of red. Arrows identify the predicted active site residues, Cys199 and His254.
essH is required for S. aureus ESS secretion.
Bacteriophage ϕ85 was used to generate a lysate from strain ΦΝΞ07490, a variant of S. aureus Newman with Bursa aurealis transposon insertion at nucleotide 602 of essH (37). The bacteriophage ϕ85 lysate was used to transduce the ermB marked mutation into S. aureus USA300 LAC* via selection for erythromycin-resistant variants, which were validated by DNA sequencing. When analyzed for growth under T7bSS-inducing conditions, wild-type USA300 LAC* and its essH::ermB variant replicated at a similar rate (Fig. 2A). Cultures of wild-type and essB and essH mutant staphylococci were centrifuged and separated into bacterial cell (pellet) and culture medium (supernatant) fractions. Proteins in each fraction were analyzed by immunoblotting. Antibodies against α-hemolysin (Hla), a secreted protein, and ribosomal protein L6, a cytoplasmic protein, were used as fractionation controls (Fig. 2B and C). Compared to those in wild-type and essB mutant S. aureus, similar amounts of EsxA and EsxC were detected in bacterial cells from essH mutant cultures (Fig. 2B). Unlike the wild-type S. aureus, the essH mutant did not secrete EsxA and EsxC into the culture supernatant (Fig. 2B). This defect in ESS secretion was restored when the essH mutant strain was transformed with pessH, a plasmid for the expression of wild-type essH, but not following transformation with an empty vector control (Fig. 2B). Of note, plasmid-borne expression of essH did not affect staphylococcal growth (Fig. 2A). Plasmid-borne expression of STREPessH, with an in-frame insertion of eight codons (for amino acids WSHPQFEK = STREP tag) after codon 2 of the predicted mature EssH, also did not affect S. aureus growth and restored the secretion of EsxA and EsxC in the essH mutant strain (Fig. 2A and C).
FIG 2.
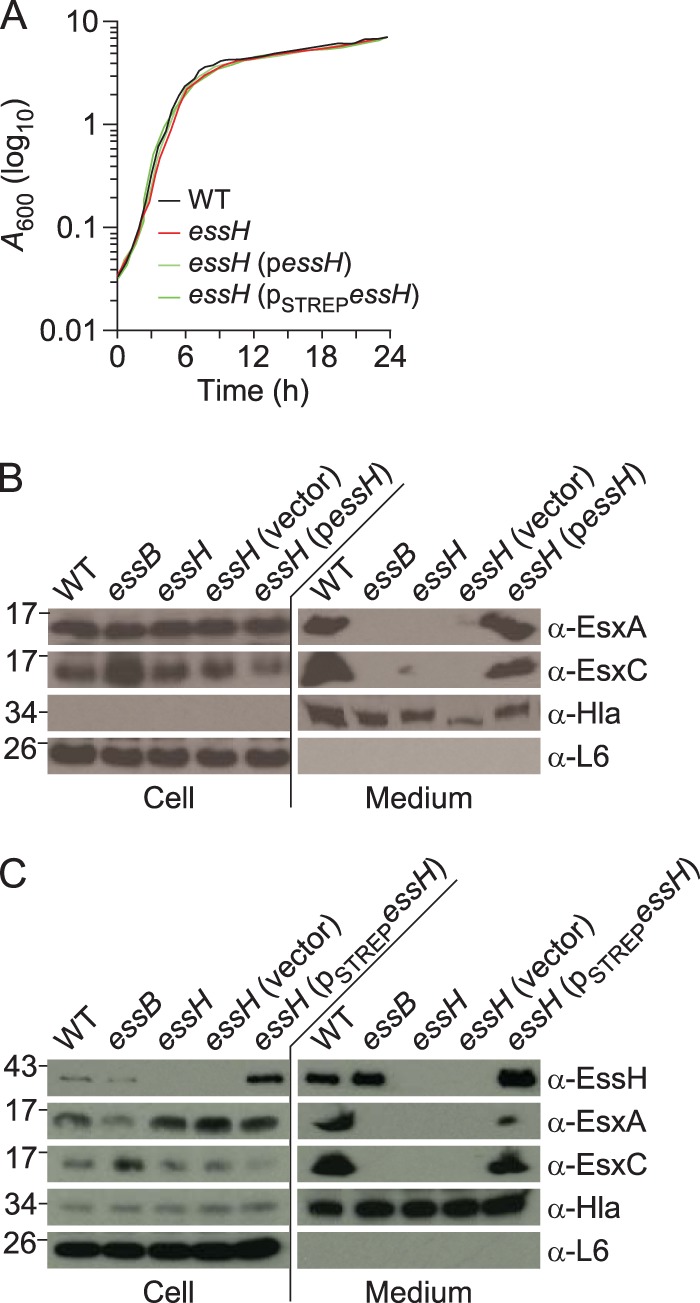
essH is dispensable for S. aureus growth and required for Esx protein secretion. (A) Overnight cultures of bacteria were normalized to an A600 of 5, diluted 1:100 in fresh medium, and grown at 37°C. Growth was monitored as increased absorbance (A600) over 24 h. (B) To assess protein secretion, cultures of S. aureus USA300 LAC* (wild type [WT]) or its isogenic essB, essH, essH(vector), and essH(pessH) variants, were grown to an A600 of 3.0 and centrifuged to separate proteins in the extracellular medium fraction (medium) from staphylococci in the sediment. Bacteria were suspended in PBS and lysed with lysostaphin to release their cellular proteins (cell). Proteins were precipitated with trichloroacetic acid (TCA) and resuspended in buffer such that medium fractions were 25 times more concentrated than cell fractions. Extracts were separated by SDS-PAGE and electrotransferred to PVDF membranes for immunoblot analyses with rabbit polyclonal antibodies specific for EsxA (α-EsxA), EsxC (α-EsxC), α-hemolysin (α-Hla), and ribosomal protein L6 (α-L6). Numbers (in kilodaltons) indicate the migratory positions of molecular weight markers by SDS-PAGE. (C) Cultures of S. aureus USA300 LAC* (WT) or its isogenic essB, essH, essH(vector), and essH(pessHSTREP) variants were grown to an A600 of 3.0, fractionated, and analyzed as described above with the addition of rabbit polyclonal antibodies specific for EssH (α-EssH).
EssH is secreted into the extracellular medium of S. aureus cultures.
The translational product of essH encompasses a predicted 24-residue signal peptide with a type I signal peptidase cleavage site (AQA). To analyze EssH in bacterial extracts, rabbit polyclonal antibodies were raised. DNA corresponding to the coding sequence for the mature portion of STREPEssH was PCR amplified, cloned into pET15b, and expressed in Escherichia coli BL21(DE3). STREPEssH was purified by affinity chromatography on Strep-Tactin–Sepharose and injected into rabbits, and the STREPEssH-specific antibody titer in immune serum (αEssH) was determined via an enzyme-linked immunosorbent assay (ELISA). Immunoblotting experiments with αEssH identified EssH in fractionated cultures from wild-type and essB mutant S. aureus strains: small amounts of EssH were detected within bacterial cells and large amounts in the culture medium (Fig. 2C). EssH could not be detected with αEssH immune serum in fractionated cultures from the essH mutant strain (Fig. 2C). EssH production and secretion into the culture medium were restored following the transformation of essH mutant staphylococci with pSTREPessH but not with the empty vector control (Fig. 2C).
Mutations in essH and essB do not affect Sec-dependent protein secretion.
Some S. aureus murein hydrolases are required for Sec-mediated secretion into the extracellular medium (32). To test whether essH mutation causes a general defect in protein secretion into the medium, 1-ml aliquots of staphylococcal cultures were fractioned by centrifugation, separating the proteins in the extracellular medium (medium) from the proteins that sedimented with bacterial cells (Fig. 3). Staphylococci were suspended in sucrose buffer, and the cell wall peptidoglycans were digested with lysostaphin. After centrifugation, the lysostaphin-digested cell walls were removed as supernatant (cell wall) from the protoplast sediment (Fig. 3). Protoplasts were lysed in hypotonic buffer by using a freeze-thaw protocol, and the sample was subjected to ultracentrifugation (100,000 × g), thereby separating the soluble proteins in the supernatant (cytosol) from the proteins that sedimented with staphylococcal membranes (Fig. 3). To detect proteins that are less abundantly secreted, we included a 50× concentrated sample of staphylococcal culture medium in immunoblotting experiments with fractioned USA300 LAC* wild-type and essH and essB mutant cultures. As a control, ribosomal protein L6 was found to be soluble in the cytosol and sedimented with assembled ribosomes (membrane) but was not in the extracellular medium or the cell wall fraction. Mature Hla was found in the extracellular medium and in the membrane fraction. EssE, a component of the T7bSS, was located in the cytosol, whereas EsaA and EssB were located in staphylococcal membranes. Although ESS secretion of EsxC and EssD was abolished in cultures of the essH mutant, neither Sec-mediated secretion of Hla nor production or membrane localization of EsaA and EssB were affected by the essH mutation (Fig. 3). The deletion of the essB gene did not affect Sec-secretion of EssH and Hla but abolished ESS secretion of EsxC and EssD.
FIG 3.
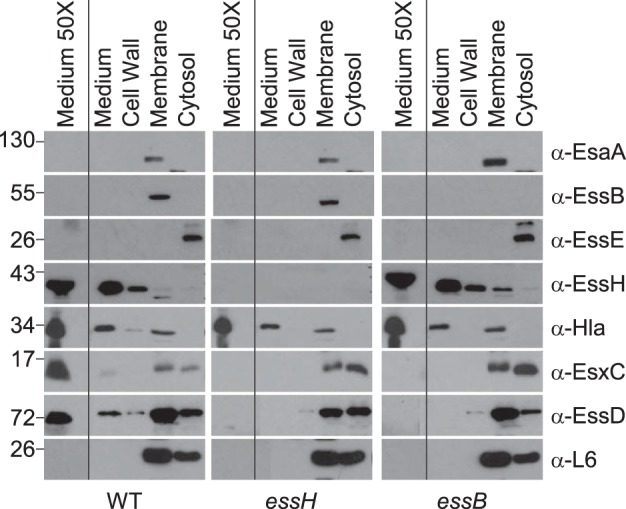
Mutations in essH and essB do not affect Sec protein secretion. Cultures of S. aureus USA300 LAC* WT and essH and essB mutants were grown to an A600 of 3.0 and centrifuged, and proteins in the extracellular medium (medium) were separated from the bacterial sediment. Sediments of 1-ml culture samples were fractionated by suspending staphylococci in sucrose buffer containing lysostaphin. The resulting protoplasts were sedimented by centrifugation. Proteins released by lysostaphin from the bacterial envelope (cell wall) were removed with the supernatant. Protoplasts were lysed and samples ultracentrifuged to sediment membrane proteins (membrane) and separate them from soluble proteins in the supernatant (cytosol). Proteins in all fractions were precipitated with TCA, separated by SDS-PAGE, electro-transferred to PVDF membrane, and analyzed by immunoblotting with rabbit polyclonal antibodies specific for Ess secretion machine components EsaA (α-EsaA), EssB (α-EssB), EssE (α-EssE), murein hydrolase EssH (α-EssH), α-hemolysin (α-Hla), ESS secretion substrates EsxC (α-EsxC), and EssD (α-EssD) as well as ribosomal protein L6 (α-L6). Samples of 50-ml cultures were centrifuged to sediment bacteria, and proteins in the culture medium were precipitated with TCA, washed with acetone, and suspended in sample buffer to generate a concentrated sample of proteins in the culture medium (medium 50×). Numbers on the left indicated kilodaltons.
EssH exhibits peptidoglycan hydrolase activity.
Bacteria from overnight cultures of S. aureus USA300 LAC* were sedimented by centrifugation, washed and suspended in buffer, divided into four aliquots that were treated with 80 nM lysostaphin, 400 nM lysostaphin, and 80 nM STREPEssH or 400 nM STREPEssH, and analyzed for lytic activity by measuring declines in adsorption at 600 nm light (A600) (Fig. 4A). As expected, the addition of 80 nM and 400 nM lysostaphin to staphylococci caused a rapid decline in A600, as the glycyl-glycine endopeptidase binds to and cleaves the peptidoglycans of intact staphylococci (Fig. 4D). In contrast, the treatment with 80 nM or 400 nM STREPEssH did not cause a decline in A600, suggesting that the EssH murein hydrolase is unable to cause bacterial lysis (Fig. 4A). Next, murein sacculi of S. aureus were isolated, extracted with acid, and treated with enzymes to remove secondary cell wall polymers, including teichoic acids, proteins, and polysaccharides. The glycan strands of purified peptidoglycan were then cleaved with mutanolysin, an N-acetylmuramidase, and the sample was split into two equal aliquots. One aliquot was incubated with purified STREPEssH, whereas the other was incubated without enzyme (mock treatment). The samples were analyzed via reversed-phase high-performance liquid chromatography (HPLC). Mock-treated peptidoglycans exhibited the characteristic absorption spectrum of the staphylococcal cell wall: a monomeric wall peptide attached to disaccharide [MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Ala)-GlcNAc] as well as cross-linked dimeric, trimeric, tetrameric, and multimeric species {[MurNAc-(Ala-Gln-(Gly5)Lys-Ala)-GlcNAc]n} (Fig. 4B, black). STREPEssH treatment collapsed the cross-linked peptidoglycan species of mutanolysin-treated peptidoglycans into monomeric or dimeric wall peptides with or without attached mono- or disaccharides (Fig. 4B, red). Matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry experiments identified the m/z of peptidoglycan fragments, which were then interpreted to predict the structures of compounds 1 [MurNAc-Ala-Gln-(Gly)Lys-Ala-Gly4], 2 [Ala-Gln-(Gly5)Lys-Ala-Ala], 3 [MurNAc-(Ala-Gln-(Gly)Lys-Ala-Gly4)-GlcNAc], 4 [MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly4)-GlcNAc], 5 [MurNAc-(Ala-Gln-(Gly)Lys-Ala-Ala)-GlcNAc], 6 [MurNAc-(Ala-Gln-(Gly)Lys-Ala-Ala)-GlcNAc-MurNAc-(Ala-Gln-(Gly)Lys-Ala-Gly)-GlcNAc], 7 [MurNAc-(Ala-Gln-(Gly5)Lys-Ala)-GlcNAc-MurNAc-(Ala-Gln-(Gly)Lys-Ala-Gly4)-GlcNAc], and 8 [MurNAc-Ala-Gln-(MurNAc-Ala-Gln-(Gly)Lys-Ala-Gly5)Lys-Ala-Gly4] (Table 1). These data suggest that STREPEssH cleaves the peptidoglycan of S. aureus at two positions: the amide bond between N-acetylmuramic acid and the l-Ala moiety of the wall peptide and the amide bonds within the pentaglycine cross bridge.
FIG 4.

EssH exhibits amidase and endopeptidase activities. (A) Overnight cultures of S. aureus USA300 LAC* were treated with lysostaphin or STREPEssH, and lysis was measured as a decline in A600 over time. (B) Purified S. aureus peptidoglycans were digested with mutanolysin and split into two samples that were either left untreated (black trace) or were treated with purified STREPEssH (red trace); the carbohydrates in both samples were reduced and analyzed by reversed-phase HPLC. Peaks labeled were desalted and analyzed by MALDI-TOF mass spectrometry; observed m/z are reported in Table 1 alongside their structural interpretation. The asterisk identifies the cross-linked muropeptide dimer of S. aureus (m/z 2,438.59). (C) A sample of purified muropeptide dimer (*) was split into two parts, either left untreated (mock) or treated with STREPEssH (red trace), and analyzed by reversed-phase HPLC. Individual peaks were desalted and analyzed by MALDI-TOF mass spectrometry; observed m/z are reported in Table 2 alongside their structural interpretations. (D) Diagram of cross-linked S. aureus peptidoglycan with arrows identifying the bonds that are cleaved by mutanolysin, lysostaphin, and EssH. mAU, milli-absorbance units; green and blue hexagons, N-acetylglucosamine and N-acetylmuramic acid, respectively.
TABLE 1.
MALDI-TOF mass spectrometry of mutanolysin- and EssHSTREP-treated peptidoglycan fragments
| Compound |
m/z |
Δm/z (calculated − observed) | Predicted structure | |
|---|---|---|---|---|
| Observed | Calculated | |||
| 1 | 1,001.44 | 1,001.12 | 0.32 | MurNAc-Ala-Gln-(Gly)Lys-Ala-Gly4 |
| 2 | 796.75 | 796.94 | 0.19 | Ala-Gln-(Gly5)Lys-Ala-Ala |
| 3 | 1,204.54 | 1,204.33 | 0.21 | MurNAc-(Ala-Gln-(Gly)Lys-Ala-Gly4)-GlcNAca |
| 4 | 1,432.45 | 1,432.6 | 0.15 | MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly4)-GlcNAc |
| 5 | 1,047.47 | 1,048.14 | 0.33 | MurNAc-(Ala-Gln-(Gly)Lys-Ala-Ala)-GlcNAc |
| 6 | 1,980.69 | 1,982.20 | 1.51 | MurNAc-(Ala-Gln-(Gly)Lys-Ala-Ala)-GlcNAc-MurNAc-(Ala-Gln-(Gly)Lys-Ala-Gly)-GlcNAc |
| 7 | 2,368.82 | 2,367.67 | 1.15 | MurNAc-(Ala-Gln-(Gly5)Lys-Ala)-GlcNAc-MurNAc-(Ala-Gln-(Gly)Lys-Ala-Gly4)-GlcNAc |
| 8 | 1,960.67 | 1,961.25 | 0.58 | MurNAc-Ala-Gln-(MurNAc-Ala-Gln-(Gly)Lys-Ala-Gly5)Lys-Ala-Gly4 |
[M+Na]+ sodiated ion.
To test the hypothesis that STREPEssH cleaves the peptidoglycan, we purified the mutanolysin-derived monosodiated ion of cross-linked muropeptide with the structure MurNAc-(Ala-Gln-[MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly5)-GlcNAc]Lys-Ala-Ala)-GlcNAc (observed m/z 2,438.59, calculated m/z 2,438.03) (Fig. 4B, asterisk). The muropeptide sample was split into two aliquots. One aliquot was incubated with STREPEssH and the other was incubated without enzyme (mock treatment). The samples were subjected to reversed-phase HPLC, and the absorption peaks were analyzed via MALDI-TOF mass spectrometry (Fig. 4C). STREPEssH cleaved the peptidoglycan substrate with m/z 2,438.59 to generate multiple products that were characterized by MALDI-TOF mass spectrometry to encompass compounds 1 [Ala-Gln-(Gly5)Lys-Ala-Gly], 2 [MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly4)-GlcNAc], 3 [Ala-Gln-[MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly)-GlcNAc]Lys-Ala-Ala], and 4 [MurNAc-(Ala-Gln-(Gly)Lys-Ala-Ala)-GlcNAc] (Table 2). These data demonstrate that STREPEssH indeed cuts the cross-linked disaccharide wall peptide at the amide bond between N-acetylmuramic acid and the l-Ala moiety of the wall peptide (Fig. 4D). Of note, STREPEssH preferentially cuts the pentaglycine cross bridge after the first and the fourth glycyl residues (Table 2 and Fig. 4D).
TABLE 2.
MALDI-TOF mass spectrometry of EssHSTREP-treated peptidoglycan dimers
| Compound |
m/z |
Δm/z (calculated − observed) | Predicted structure | |
|---|---|---|---|---|
| Observed | Calculated | |||
| 1 | 757.60 | 759.93 | 2.33 | Ala-Gln-(Gly5)Lys-Ala-Glyb |
| 2 | 1,432.71 | 1,432.61 | 0.10 | MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly4)-GlcNAca |
| 3 | 1,648.71 | 1,650.93 | 2.22 | Ala-Gln-[MurNAc-(Ala-Gln-(Gly5)Lys-Ala-Gly)-GlcNAc]Lys-Ala-Alab |
| 4 | 1,047.53 | 1,047.14 | 0.39 | MurNAc-(Ala-Gln-(Gly)Lys-Ala-Ala)-GlcNAc |
| 5 | 2,438.59 | 2,438.76 | 0.17 | MurNAc-(Ala-Gln-[MurNac-(Ala-Gln-(Gly5)Lys-Ala-Gly5)-GlcNAc]Lys-Ala-Ala)-GlcNAca |
[M+Na]+ sodiated ion.
[M+H]+ protonated ion.
EssH residues Cys199 and His254 contribute to peptidoglycan hydrolase activity.
An alignment of EssH CHAP domains with the S. aureus USA300 CHAP domain-containing proteins identified the conserved residues Cys199 and His254, which are thought to be involved in catalysis (Fig. 1B). We generated the variant STREPEssHC199A/H254A and purified this protein alongside STREPEssH via Strep-Tactin chromatography (Fig. 5A). Purified murein sacculi of S. aureus were cleaved with mutanolysin, and the samples were split into three equal aliquots. Two aliquots were treated with equal amounts of either STREPEssH or STREPEssHC199A/H254A. As a control, the third aliquot was mock treated. The samples were analyzed via reversed-phase HPLC (Fig. 5B). As expected, mock-treated peptidoglycans exhibited the characteristic absorption spectrum of the mutanolysin-digested cell wall, i.e., a monomeric wall peptide attached to disaccharide and cross-linked multimeric species (Fig. 5B, black), whereas STREPEssH treatment collapsed the cross-linked peptidoglycan species into monomeric or dimeric wall peptides with or without attached mono- or disaccharides (Fig. 5B, red). Importantly, treatment with STREPEssHC199A/H254A had no effect on cross-linked peptidoglycans (Fig. 5B, green). These data suggest that the conserved residues of the CHAP domain, i.e., EssH residues Cys199 and His254, contribute to the peptidoglycan hydrolase activity of this enzyme. The STREPEssHC199A/H254A variant, with substitutions at both conserved residues, cannot cleave the staphylococcal peptidoglycan.
FIG 5.
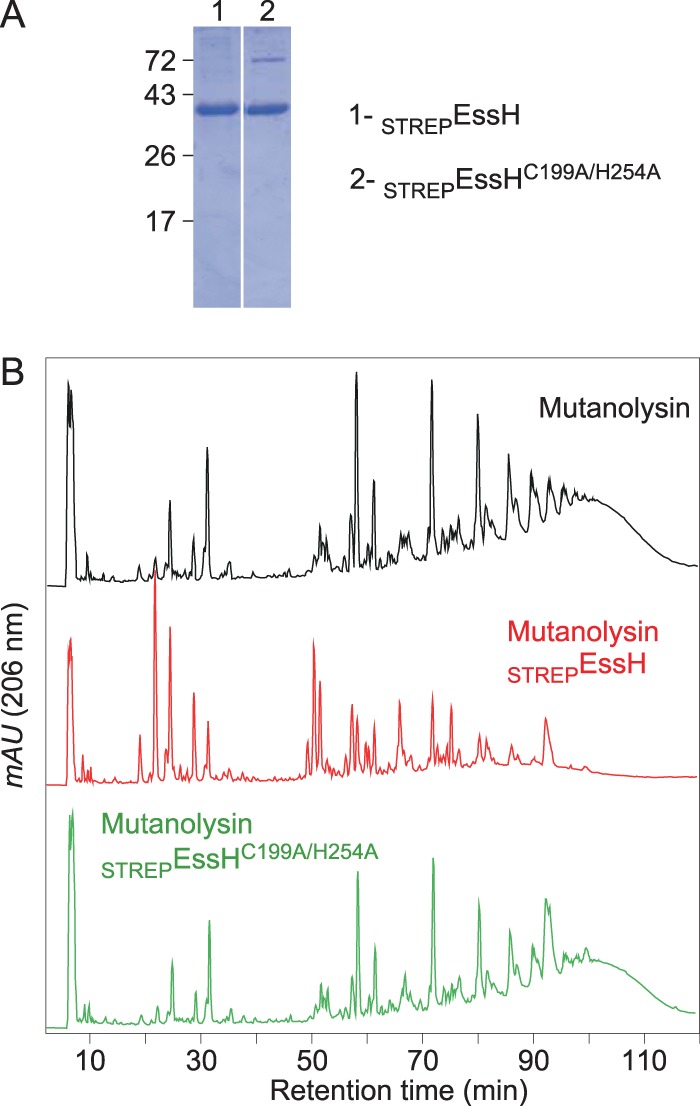
Cys199 and His254 are required for EssH murein hydrolase activity. (A) Coomassie brilliant blue-stained SDS-PAGE gel separating purified STREPEssH (1) and STREPEssHC199A/H254A (2). The other numbers indicate the migratory positions of molecular weight markers (in kilodaltons). (B) Purified S. aureus peptidoglycans were digested with mutanolysin and split into three samples that were either left untreated (black trace) or were treated with STREPEssH (red trace) or STREPEssHC199A/H254A (green trace); the carbohydrates in all samples were reduced and analyzed by reversed-phase HPLC.
EssH peptidoglycan hydrolase activity is required for ESS secretion.
Plasmids pessHC199A, pessHH254A, and pessHC199A/H254A enable the gene expression and synthesis of the EssHC199A, EssHH254A, and EssHC199A/H254A variants, respectively, in the S. aureus essH mutant strain (Fig. 6A). Immunoblotting experiments revealed that S. aureus essH mutants secreted similar amounts of wild-type EssH (encoded by pessH), EssHC199A, EssHH254A, and EssHC199A/H254A (Fig. 6B). As expected, the vector plasmid did not enable the synthesis or secretion of EssH in the S. aureus essH mutant (Fig. 6B). When analyzed for the activity of the ESS pathway in a pairwise comparison, the pessH plasmid restored EsxC secretion from the S. aureus essH mutant, whereas the vector alone, pessHC199A, pessHH254A, and pessHC199A/H254A did not (Fig. 6B). Immunoblot experiments were performed in triplicates to carry out densitometric quantification of immunoreactive signals (Fig. 6C). The amounts of EsxC in cell and medium fractions were calculated relative to the total amount of signal obtained for each strain (the sum of signals in the cell and medium fractions was set at 100%). As a control, the essB mutant was defective in the secretion of EsxC (Fig. 6BC). Further, EssH peptidoglycan hydrolase activity, specifically, the catalytic residues Cys199 and His254 of the CHAP domain, is required for EsxC secretion via the ESS pathway in S. aureus USA300 LAC.
FIG 6.

EssH murein hydrolase activity is required for S. aureus ESS secretion. (A) Diagram of plasmid (vector)-encoded wild-type EssH(pessH) and variants carrying amino acid substitutions C199A, H254A, and C199A/H254A. Blue and red boxes depict the signal peptides and CHAP domains, respectively. Numbers depict lengths of polypeptides in amino acids. (B) Cultures of S. aureus USA300 LAC* (WT) and its essB, essH(vector) (lane 1), essH(pessH) (lane 2), essH(pessHC199A) (lane 3), essH(pessHH254A) (lane 4), and essH(pessHC199A/H254A) (lane 5) variants were grown and fractionated as described in the legend for Fig. 2B. Samples were subjected to immunoblot analyses using rabbit polyclonal antibodies specific for EssH, EsxC, and L6. (C) Densitometry quantification of the abundance of EsxC immune-reactive signal in cell and medium fractions from three independent experiments. Data are analyzed as percentages of total EsxC, i.e., the added densities of protein in cell and medium fractions. Statistical analysis was performed using a two-way ANOVA followed by Tukey's multiple-comparison test.
S. epidermidis essH complements the ESS secretion defect of the S. aureus essH mutant.
S. epidermidis strain 14.1.R1 harbors a chromosomally encoded ess locus, with an essH-like gene located upstream of esxA on the minus strand (Fig. 1A). S. epidermidis essH bears a conserved C-terminal CHAP domain, but its N-terminal domain (amino acids 1 to 142) is not conserved (Fig. 7A). We constructed two new plasmids, namely, pessHSe and pessHSe(SP) (Fig. 7A). Plasmid pessHSe carries the full-length essH from S. epidermidis 14.1.R1, whereas plasmid pessHSe(SP) encodes the S. aureus USA300 LAC essH signal peptide sequence (codons 1 to 24) fused to S. epidermidis 14.1.R1 essH codons 27 to 319 (Fig. 7A). The rationale for this experiment is the observation that the signal peptide of EssHSe is significantly longer than that of S. aureus. When expressed in the S. aureus essH mutant strain, the amino acid differences in the S. epidermidis 14.1.R1 EssH sequence hindered the detection of EssHSe and EssHSe(SP) with immune serum against STREPEssH (recombinant S. aureus EssH) (Fig. 7B). Nevertheless, S. aureus essH(pessHSe) and essH(pessHSe(SP)) strains secreted EsxC at levels similar to those observed with wild-type and essH(pessH) strains, while ESS-dependent secretion was abrogated in both the S. aureus essH(vector) and essB mutant controls (Fig. 7B and C). These results suggest functional conservation of EssH orthologues from S. aureus USA300 LAC and S. epidermidis 14.1.R1 in the staphylococcal ESS pathway.
FIG 7.

Heterologous complementation by S. epidermidis EssH. (A) Diagram of plasmid (vector)-encoded wild-type EssH(pessH), S. epidermidis EssHSe(pessHSe), and EssHSe(SP), a variant of EssHSe secreted via the signal peptide of S. aureus EssH. Blue boxes depict signal peptides, while red and pink boxes depict CHAP domains. Numbers depict lengths of polypeptides in amino acids. (B) Cultures of S. aureus USA300 LAC* (WT) and its essB, essH(vector) (lane 1), essH(pessH) (lane 2), essH(pessHSe) (lane 3), and essH(pessH Se(SP)) (lane 4) variants were grown and fractionated as described in the legend for Fig. 2B. Samples were subjected to immunoblot analyses using rabbit polyclonal antibodies specific for EssH, EsxC, and L6. (C) Quantification of EsxC in cell and medium fractions was performed by quantifying band densities from three independent experiments as described in the legend for Fig. 6C.
SagB peptidoglycan hydrolase activity is not required for ESS secretion.
S. aureus mutants defective for sagB expression exhibit increased glycan chain lengths within their peptidoglycans, which results in altered protein secretion (32). We wondered whether the loss of sagB may also affect the ESS pathway. When analyzed by immunoblotting with specific antibodies and compared to the ESS pathway in wild-type S. aureus USA300 LAC*, the overall levels of EssH and EsxC were slightly diminished (Fig. 8A), likely due to the growth defects associated with the absence of SagB. A quantification of the protein in the supernatant indicated that the secretion of EsxC was unchanged between wild-type, sagB(vector), and sagB(psagB) strains (Fig. 8B). The reduced abundance of EssH and EsxC was reversed upon transformation of the sagB mutant strain with a plasmid enabling the expression of wild-type sagB (psagB) (Fig. 8). Thus, SagB glucosaminidase, which generates the characteristic short-chain glycan strands of S. aureus peptidoglycans, is not required for ESS-mediated secretion.
FIG 8.
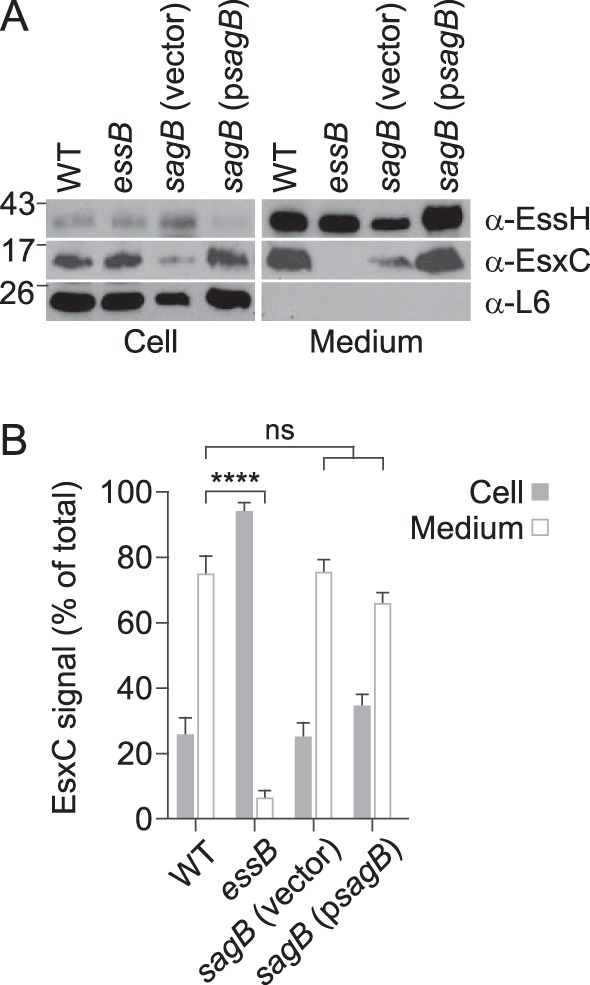
SagB hydrolase is not required for S. aureus ESS secretion. (A) Cultures of S. aureus USA300 (WT), essB, sagB(vector), and sagB(psagB) variants were grown and fractionated as described in the legend for Fig. 2B. Samples were subjected to immunoblot analyses using rabbit polyclonal antibodies specific for EssH, EsxC, and L6. A representative experiment is shown. (B) Quantification of EsxC in cell and medium fractions was performed by quantifying band densities from three independent experiments as described in the legend for Fig. 6C.
DISCUSSION
To date, three S. aureus CHAP domain murein hydrolases have been characterized. Sle1 is secreted into the extracellular medium and binds via its three N-terminal LysM domains to the glycan strands of septal peptidoglycans (38). LytN is secreted via its YSIRK/GXXS signal peptide into the cross wall, i.e., the septal peptidoglycan layer synthesized during staphylococcal cell division (36). Together, Sle1 and LytN cleave the cross wall (septal peptidoglycan) to separate dividing cells, thereby completing the staphylococcal cell cycle (36). ScaH, another CHAP domain hydrolase, is dispensable for the staphylococcal cell cycle (39). In addition to CHAP, ScaH also encompasses an N-acetylglucosaminidase domain whose activity is similar to those of Atl, SagA, and SagB N-acetylglucosaminidases. A S. aureus variant lacking all four glucosaminidases cannot be generated, suggesting that glucosaminidase activity is required for staphylococcal replication (39). Here, we describe the EssH CHAP domain hydrolase of S. aureus. Neither the deletion nor the overexpression of essH affects bacterial replication; however, the deletion of essH abolishes the secretory activity of the S. aureus T7bSS. EssH is synthesized as a precursor and secreted via the canonical Sec pathway. Recombinant EssH exhibits N-acetylmuramoyl-l-alanine amidase and pentaglycyl endopeptidase activities. Specifically, EssH cleaves the amide bond between N-acetylmuramic acid and l-Ala of the wall peptide and the cross bridges of cross-linked wall peptides after the first and fourth glycyl residues. An alignment of EssH with other CHAP domain-containing hydrolases identified the candidate catalytic residues Cys199 and His254, which when mutated to Ala, render the variant EssH inactive both in vitro and in vivo.
Specialized secretion systems assemble in the bacterial envelope to span its membrane and peptidoglycan layers. The estimated pore diameter of peptidoglycan (∼2 nm) represents a physical barrier for the assembly of large macromolecular secretion machines (40), including flagella, type IV pili, and type III (T3SS), type IV (T4SS), and type VI (T6SS) secretion systems (41–43). The genes of peptidoglycan hydrolases supporting the assembly of specialized secretion systems are often clustered with the genes that specify these macromolecular complexes. Most of these enzymes represent lytic transglycosylases that cleave the glycosidic linkages between disaccharide repeats of peptidoglycan (41). For example, the T3SSs of Gram-negative bacteria require the assembly of needle complexes, an element of the secretion machinery that is embedded within the bacterial double membrane envelope and peptidoglycan layer (44). The lytic transglycosidase EtgA forms a 1:1 complex with the EscI inner rod component of the needle complex (45). This interaction triggers murein hydrolase activity and enables the assembly of the T3SS (45, 46). The peptidoglycan layers of Gram-positive bacteria have larger diameters than those of Gram-negative microbes, and in S. aureus, peptidoglycan is subject to remodeling by the SagB glucosaminidase (32). The loss of sagB leads to peptidoglycans with abnormally long glycan strands that impede the travel of proteins across the bacterial cell wall layer following their membrane translocation via the Sec pathway (32). T7bSS in S. aureus is not affected by SagB but requires an active EssH hydrolase. Unlike EtgA, EssH does not display broad lytic activity nor affect glycan strands. Instead, EssH cleaves wall peptides and pentaglycine cross bridges. Interestingly, the transmembrane proteins EsaA and EssB, which have been proposed to form the T7 translocon (26), sediment with the membrane fraction in essH mutant bacteria as occurs in the wild type. Perhaps the T7 translocon fails to extend across the cell wall layer in the essH mutant strain, thereby restricting the travel of the effector proteins.
While the molecular contribution of EssH to T7 secretion remains to be determined, bioinformatics analyses of ess gene clusters clearly reveal a genetic association with essH-like genes. This is exemplified by the S. epidermidis 14.1.R1 clinical isolate (47). Most S. epidermidis isolates lack a T7SS. However, Bruggemann and colleagues noted the presence of an ess gene cluster with aberrant GC content in strain 14.1.R1 (Fig. 1B) (47). The finding that S. epidermidis EssH restores T7 secretion in a S. aureus essH mutant points to a conserved role for this peptidoglycan hydrolase in facilitating ESS-mediated secretion of substrates.
A cell wall hydrolase has not been reported in association with the T7aSS of mycobacterial species. Mycobacterial genomes harbor up to five different T7SS gene clusters, designated ESX-1 through ESX-5 (48). Each T7SS gene cluster encodes core components that assemble into a membrane complex (49); however, the genetic requirements and mechanisms enabling the assembly of these macromolecular complexes in the mycobacterial envelope are not known. Up to 50% of mycobacterial isolates harbor a conjugative plasmid that encompasses components of a T4SS and an intact T7SS gene cluster. A major difference between the plasmid-encoded T7SS and the chromosomal ESX systems is the presence of two genes at the beginning of the plasmid locus that are predicted to encode peptidoglycan hydrolases (50). In conclusion, we propose that the T7bSS of S. aureus is endowed with a conserved essH gene that encodes a hydrolase whose activity is required for the secretion of ESS substrates.
MATERIALS AND METHODS
Media and growth conditions.
S. aureus strains were cultured in tryptic soy broth (TSB) or agar (TSA) at 37°C, unless otherwise stated, and the media were supplemented with 10 μg/ml chloramphenicol for plasmid selection and 0.2% heat-inactivated horse serum (Gibco/Life Technologies) for ESS induction. Bacteriophage ϕ85 transductants of S. aureus USA300 LAC* were selected by supplementing TSA with 200 μg/ml of erythromycin. Escherichia coli was cultured in lysogeny broth (LB) medium or agar at 37°C, supplemented with 100 μg/ml ampicillin for plasmid selection and 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for the production of recombinant proteins.
Bacterial strains and plasmids.
Relevant strains and plasmids are listed in Table 3. S. aureus USA300 LAC* is a variant of the original clone of the epidemic community-acquired methicillin-resistant S. aureus (CA-MRSA USA LAC) strain (51) that has lost plasmid pUSA03 encoding ermC (22). The loss of this plasmid does not alter virulence in mice nor T7S and enables the selection of mutants marked with the ermB allele. S. aureus essH::ermB (referred to as essH in the text) was generated via ϕ85 bacteriophage-mediated transduction of a transposon-disrupted essH allele linked to the erythromycin resistance gene (ermB). Plasmid DNA was passaged through S. aureus RN4220 prior to transformation into USA300 LAC* strains. Plasmid pSEW016 was used for complementation studies. pSEW016 is a derivative of pWWW412, a shuttle vector that carries the promoter and Shine-Dalgarno sequences of the S. aureus hprK gene (52). In pSEW016, the NdeI cloning site of pWWW412 was replaced with SacI.
TABLE 3.
Strains and plasmids used in the study
| Strain, vector, or plasmid | Description | Reference(s) or source |
|---|---|---|
| Strains | ||
| USA300 LAC* | S. aureus USA300 LAC lacking pUSA03 (designated as the wild type [WT]) | 22, 51 |
| RN4220 | S. aureus restriction deficient, cloning intermediate | 56 |
| ΦΝΞ07490 | S. aureus Newman with Bursa aurealis insertion at nucleotide 602 of essH | 37 |
| essH | S. aureus USA300 LAC* essH::ermB | This study |
| essB | S. aureus USA300 LAC* ΔessB | 24 |
| sagB | S. aureus USA300 LAC* ΔsagB::aphA | 32 |
| E. coli DH5α | F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) pho A supE44 λ− thi-1 gyrA96 relA1 | Our collection |
| E. coli BL21 | B F− ompT gal dcm lon hsdSB(rB−mB−) [malB+]K-12(λS) | 57 |
| Vectors and plasmids | ||
| pOS1 | E. coli/S. aureus shuttle vector | 58 |
| pSEW016 | pWWW412 with a modified polylinker herein referred as vector | This study; 52 |
| pET15b | E. coli vector for production of recombinant proteins | Addgene |
| pessH | essH open reading frame cloned into pSEW016 | This study |
| pessHC199A | essHC199A cloned into pSEW016 | This study |
| pessHH254A | essHH254A cloned into pSEW016 | This study |
| pessHC199A/H254A | essHC199A/H254A cloned into pSEW016 | This study |
| pessHSe | S. epidermidis 14.1.R1 essH cloned into pSEW016 | This study |
| pessHSe(SP) | S. aureus USA300 LAC essH nucleotides 1 to 78 fused to S. epidermidis 14.1.R1, essH nucleotides 85 to 960 cloned into pSEW016 | This study |
| pSTREPessH | S. aureus USA300 LAC essH with STREP II tag sequence inserted between nucleotides 72 and 73 and cloned into pSEW016 | This study |
| psagB | sagB cloned into pOS1 | 32 |
| pET15b-STREPessH | essH nucleotides 73 to 894 with N-terminal STREP II tag cloned into pET15b | This study |
| pET15b-STREPessHC199A/H254A | essHC199A/H254A nucleotides 73 to 894 with N-terminal STREP II cloned into pET15b | This study |
Plasmid pessH for complementation studies was constructed by amplifying USA300 genomic DNA by PCR using the primer pairs bearing sequences 5′-CCCCGAGCTCATGAAGAAAACAATTTTACTGACGATGACAACTC-3′ and 5′-CCCCGGATCCTTAATGGATGTAATTATATGATGAAACTTCTGAAGCAGAG-3′. The amplified product was cloned into vector pSEW016, referred to as vector in the text, using SacI and BamHI restriction sites. The replacement of codons 199 and 254 (C199A and H254A, respectively) was achieved by site-directed mutagenesis of pessH using mutation-containing primers 5′-CTTATATACTGCTGGACAAGCAACATGGTATGTCTTTG-3′/5′-CGTTTATCAAAGACATACCATGTTGCTTGTCCAGCAG-3′ and 5′-CAGTAAATGGTCCATTTGGTGCAGTAGCCTACG-3′/5′-CTTTTTCAACGTAGGCTACTGCACCAAATGGACC-3′, respectively. Plasmid pSTREPessH was constructed by amplifying the essH signal peptide sequence by using primers 5′-CCCCGAGCTCATGAAGAAAACAATTTTACTGACGATGACAACTC-3′ and 5′-TTTTTCGAATTGAGGATGTGACCACGTATATGCTTGAGCCGAGTTAGGCG-3′and the mature essH coding sequence with an appended STREP-tag using primers 5′-TGGTCACATCCTCAATTCGAAAAAAATGATAGCAAAACATTAGAAGAAGCAAAGAAAGC-3′ and 5′-CCCCGGATCCTTAATGGATGTAATTATATGATGAAACTTCTGAAGCAGAG-3′. Amplified overlapping DNA fragments were fused by PCR, cloned into pSEW016, and sequenced. For the recombinant production of proteins STREPEssH and STREPEssHC199A/H254A, template DNA was amplified by PCR using the primers 5′-CCCCCCATGGGCTGGTCACATCCTCAATTCGAAAAATATACGAATGATAGCAAAACATTAGAAGAAGCAAAG-3′ and 5′-CCCCGGATCCTTAATGGATGTAATTATATGATGAAACTTCTGAAGCAGAG-3′ and cloned into pET-15b using NcoI and BamHI restriction sites.
Protein purification.
Cultures of E. coli (2 liters) that had been grown to an absorbance at 600 nm (A600) of 2.0 were centrifuged (10,000 × g for 10 min). Sedimented cells were suspended in buffer A (50 mM Tris-HCl [pH 7.5], 150 mM NaCl), and the resulting suspensions were lysed in a French press at 14,000 lb/in2 (Thermo Spectronic, Rochester, NY). Unbroken cells were removed by centrifugation (5,000 × g for 15 min), and the crude lysates subjected to ultracentrifugation (100,000 × g for 1 h at 4°C). Soluble recombinant proteins were subjected via gravity flow to chromatography on Strep-Tactin–Sepharose (IBA) with a packed volume of 1 ml preequilibrated with buffer A. The columns were washed with 20 bed volumes of buffer A and eluted with 4 ml of 2.5 mM desthiobiotin in buffer A. Aliquots of the eluted fractions were mixed with equal volumes of sample buffer and separated on 12% or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. Recombinant proteins were dialyzed against phosphate-buffered saline (PBS) and their concentrations determined with the bicinchoninic acid assay (Pierce).
EssH antiserum.
Purified STREPEssH (100 μg) was emulsified with complete Freund's adjuvant (Difco), and the emulsion was injected subcutaneously into a New Zealand White rabbit purchased from Harlan Sprague Dawley. At 21-day intervals, the animal received two booster immunizations with 100 μg of STREPEssH emulsified with incomplete Freund's adjuvant (Difco). Serum was obtained following the centrifugation of blood samples (10,000 × g for 10 min) and mixed with 0.02% sodium azide for storage.
Fractionation of bacterial cultures and immunoblotting.
To assess protein secretion, S. aureus culture aliquots were centrifuged (10,000 × g for 10 min) to separate proteins in the medium and the cells. The cells from bacterial pellets were washed and lysed with lysostaphin (10 μg/ml for 1 h at 37°C). Proteins in these suspensions (medium or cells) were precipitated by the addition of 10% final concentration trichloroacetic acid, washed in cold acetone, and dried. For the subcellular localization of proteins, pelleted cells were suspended in TSM buffer (50 mM Tris-HCl [pH 7.5], 0.5 M sucrose, 10 mM MgCl2) prior to lysostaphin treatment. The samples were centrifuged at 15,000 × g for 10 min, and the supernatants (cell wall fraction) were transferred to a new tube and protoplasts were lysed by repeated freeze-thawing (3 cycles). The extracts were centrifuged at 100,000 × g for 40 min at 4°C to separate the supernatants containing cytosolic proteins. The pellets containing membrane proteins were suspended in PBS. Proteins in all fractions were precipitated with trichloroacetic acid (TCA), washed with methanol, and dried. All precipitates were solubilized in 100 μl of 0.5 M Tris-HCl (pH 8.0)-4% SDS and heated at 90°C for 10 min. Proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane for immunoblot analysis. The membranes were blocked by incubating in 0.5% milk for 1 h at room temperature. To prevent the binding of primary antibodies to protein A, 0.8 mg of human IgG was added to 10 ml of blocking buffer for 1 h at room temperature prior to the addition of primary polyclonal antibodies (at a dilution of 1:5,000 for T7 proteins or 1:10,000 for L6 and Hla). Membranes were incubated for an additional hour, washed 3 times for 10 min in TBS-T (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween 20), and incubated with 1:10,000 anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technology) for 1 h. The membranes were washed again in TBS-T, and immunoreactive products were revealed by chemiluminescent detection using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). The blots were developed on Amersham Hyperfilm ECL (GE Healthcare Life Sciences).
Biochemical assays.
The general lytic activity of enzymes was assayed using washed cells of S. aureus. Overnight cultures of staphylococci were washed twice with 50 mM Tris-HCl, pH 8.0, and suspended in 650 μl of the same buffer at an absorbance at 600 nm (A600) of ∼1.6. Triplicate sets of 90 μl of cell suspension were aliquoted into 96-well plates. Buffer or enzymes were added to each triplicate set at the indicated concentrations. Changes in A600 were monitored every 5 min at 37°C with agitation in a Synergy HT plate reader (BioTek). The change in absorbance expressed as a percentage of the input for each well was normalized to the negative control and plotted over time.
S. aureus peptidoglycans were prepared in 100 mM phosphate buffer, pH 5.5, as previously described (53), the concentrations were adjusted according to the A600, and they were incubated for 18 h at 37°C with 100 μl of enzyme, either mutanolysin (500 units/ml; Sigma) or STREPEssH (300 μg/ml). After 18 h of incubation with mutanolysin, peptidoglycan samples were boiled for 10 min to quench the reaction, and the samples were centrifuged at 15,000 × g for 15 min. When indicated, soluble material was further incubated with STREPEssH. All samples were neutralized with sodium hydroxide to reach pH 7.0, dried, and reduced via the addition of 250 mM sodium borate and 3 to 5 mg of sodium borohydride. The samples were incubated for 30 min, and the reactions were stopped by the addition of 20% phosphoric acid to reach pH <4.0 as described previously (53). Reduced muropeptides were separated by reversed-phase HPLC on a C18 column (250 mm by 4.6 mm, ODS-Hypersil; Thermo Scientific) as described previously (54). Individual peaks were desalted using reversed-phase ZipTip C18 pipette tips (Millipore) in 50% acetonitrile containing 0.1% trifluoroacetic acid (TFA), and 0.75 μl was cospotted with 0.75 μl of matrix, α-cyano-4-hydroxycinnamic acid, at 10 mg/ml in 50% acetonitrile and 0.1% TFA onto a metal target plate. The samples were subjected to MALDI-TOF mass spectrometry using an Autoflex Speed Bruker MALDI instrument. Ions were detected in reflectron positive mode.
Statistical analyses.
Immunoblot quantification was performed using ImageJ software (National Institutes of Health) (55) and analyzed for statistical significance by two-way analyses of variance (ANOVA) followed by Tukey's multiple-comparison tests with Prism (GraphPad Software). Experiments were repeated three times.
ACKNOWLEDGMENTS
We thank Ryan Jay Ohr for providing reagents and experimental advice and members of the Schneewind and Missiakas laboratory for discussion and comments.
M.B. was supported by a postdoctoral fellowship award from the National Institute of Allergy and Infectious Diseases (NIAID; F32AI140643). The work was supported by grants AI110937 and AI038897 from the NIAID.
REFERENCES
- 1.Green ER, Mecsas J. 2016. Bacterial secretion systems: an overview. Microbiol Spectr 4:VMBF-0012-2015. doi: 10.1128/microbiolspec.VMBF-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kudva R, Denks K, Kuhn P, Vogt A, Muller M, Koch HG. 2013. Protein translocation across the inner membrane of Gram-negative bacteria: the Sec and Tat dependent protein transport pathways. Res Microbiol 164:505–534. doi: 10.1016/j.resmic.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Costa TR, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, Waksman G. 2015. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol 13:343–359. doi: 10.1038/nrmicro3456. [DOI] [PubMed] [Google Scholar]
- 4.Schneewind O, Missiakas D. 2014. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim Biophys Acta 1843:1687–1697. doi: 10.1016/j.bbamcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Unnikrishnan M, Constantinidou C, Palmer T, Pallen MJ. 2017. The enigmatic Esx proteins: looking beyond mycobacteria. Trends Microbiol 25:192–204. doi: 10.1016/j.tim.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. 2007. Type VII secretion—mycobacteria show the way. Nat Rev Microbiol 5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 7.Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. 1996. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol 178:1274–1282. doi: 10.1128/jb.178.5.1274-1282.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C. 2005. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol 174:3570–3579. doi: 10.4049/jimmunol.174.6.3570. [DOI] [PubMed] [Google Scholar]
- 9.Zoltner M, Ng WM, Money JJ, Fyfe PK, Kneuper H, Palmer T, Hunter WN. 2016. EssC: domain structures inform on the elusive translocation channel in the type VII secretion system. Biochem J 473:1941–1952. doi: 10.1042/BCJ20160257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg OS, Dovala D, Li X, Connolly L, Bendebury A, Finer-Moore J, Holton J, Cheng Y, Stroud RM, Cox JS. 2015. Substrates control multimerization and activation of the multi-domain ATPase motor of type VII secretion. Cell 161:501–512. doi: 10.1016/j.cell.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallen MJ. 2002. The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol 10:209–212. doi: 10.1016/S0966-842X(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 12.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci U S A 102:1169–1174. doi: 10.1073/pnas.0405620102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sysoeva TA, Zepeda-Rivera MA, Huppert LA, Burton BM. 2014. Dimer recognition and secretion by the ESX secretion system in Bacillus subtilis. Proc Natl Acad Sci U S A 111:7653–7658. doi: 10.1073/pnas.1322200111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garufi G, Butler E, Missiakas D. 2008. ESAT-6-like protein secretion in Bacillus anthracis. J Bacteriol 190:7004–7011. doi: 10.1128/JB.00458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kluytmans J, van Belkum A, Verbrugh H. 1997. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10:505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Belkum A, Melles DC, Nouwen J, van Leeuwen WB, van Wamel W, Vos MC, Wertheim HF, Verbrugh HA. 2009. Co-evolutionary aspects of human colonisation and infection by Staphylococcus aureus. Infect Genet Evol 9:32–47. doi: 10.1016/j.meegid.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 17.von Eiff C, Becker K, Machka K, Stammer H, Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study group. N Engl J Med 344:11–16. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- 18.Bhalla A, Aron DC, Donskey CJ. 2007. Staphylococcus aureus intestinal colonization is associated with increased frequency of S. aureus on skin of hospitalized patients. BMC Infect Dis 7:105. doi: 10.1186/1471-2334-7-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pallin DJ, Egan DJ, Pelletier AJ, Espinola JA, Hooper DC, Camargo CA Jr. 2008. Increased U.S. emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann Emerg Med 51:291–298. doi: 10.1016/j.annemergmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HK, Missiakas D, Schneewind O. 2014. Mouse models for infectious diseases caused by Staphylococcus aureus. J Immunol Methods 410:88–99. doi: 10.1016/j.jim.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burts ML, DeDent AC, Missiakas DM. 2008. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol Microbiol 69:736–746. doi: 10.1111/j.1365-2958.2008.06324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson M, Aly KA, Chen YH, Missiakas D. 2013. Secretion of atypical protein substrates by the ESAT-6 secretion system of Staphylococcus aureus. Mol Microbiol 90:734–743. doi: 10.1111/mmi.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson M, Chen YH, Butler EK, Missiakas DM. 2011. EsaD, a secretion factor for the Ess pathway in Staphylococcus aureus. J Bacteriol 193:1583–1589. doi: 10.1128/JB.01096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohr RJ, Anderson M, Shi M, Schneewind O, Missiakas D. 2017. EssD, a nuclease effector of the Staphylococcus aureus ESS pathway. J Bacteriol 199:e00528-16. doi: 10.1128/JB.00528-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aly KA, Anderson M, Ohr RJ, Missiakas D. 2017. Isolation of a membrane protein complex for type VII secretion in Staphylococcus aureus. J Bacteriol 199:e00482-17. doi: 10.1128/JB.00482-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarre WW, Schneewind O. 1999. Surface proteins of Gram-positive bacteria and the mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev 63:174–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatterjee AN, Park JT. 1964. Biosynthesis of cell wall mucopeptide by a particulate fraction from Staphylococcus aureus. Proc Natl Acad Sci U S A 51:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strominger JL, Izaki K, Matsuhashi M, Tipper DJ. 1967. Peptidoglycan transpeptidase and d-alanine carboxypeptidase: penicillin-sensitive enzymatic reactions. Fed Proc 26:9–22. [PubMed] [Google Scholar]
- 30.Tipper DJ, Strominger JL. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc Natl Acad Sci U S A 54:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tipper DJ, Strominger JL. 1968. Biosynthesis of the peptidoglycan of bacterial cell walls. XII. Inhibition of cross-linking by penicillins and cephalosporins: studies in Staphylococcus aureus in vivo. J Biol Chem 243:3169–3179. [PubMed] [Google Scholar]
- 32.Chan YG, Frankel MB, Missiakas D, Schneewind O. 2016. SagB glucosaminidase is a determinant of Staphylococcus aureus glycan chain length, antibiotic susceptibility, and protein secretion. J Bacteriol 198:1123–1136. doi: 10.1128/JB.00983-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman A, Rawlings ND. 2003. The CHAP domain: a large family of amidases including GSP amidase and peptidoglycan hydrolases. Trends Biochem Sci 28:234–237. doi: 10.1016/S0968-0004(03)00061-6. [DOI] [PubMed] [Google Scholar]
- 34.Warne B, Harkins CP, Harris SR, Vatsiou A, Stanley-Wall N, Parkhill J, Peacock SJ, Palmer T, Holden MT. 2016. The Ess/type VII secretion system of Staphylococcus aureus shows unexpected genetic diversity. BMC Genomics 17:222. doi: 10.1186/s12864-016-2426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanz-Gaitero M, Keary R, Garcia-Doval C, Coffey A, van Raaij MJ. 2014. Crystal structure of the lytic CHAP(K) domain of the endolysin LysK from Staphylococcus aureus bacteriophage K. Virol J 11:133. doi: 10.1186/1743-422X-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frankel MB, Hendrickx AP, Missiakas DM, Schneewind O. 2011. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J Biol Chem 286:32593–32605. doi: 10.1074/jbc.M111.258863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. 2004. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A 101:12312–12317. doi: 10.1073/pnas.0404728101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, Hayashi I, Yamagishi J, Komatsuzawa H, Sugai M. 2005. Identification and molecular characterization of an N-acetylmuramyl-l-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol Microbiol 58:1087–1101. doi: 10.1111/j.1365-2958.2005.04881.x. [DOI] [PubMed] [Google Scholar]
- 39.Wheeler R, Turner RD, Bailey RG, Salamaga B, Mesnage S, Mohamad SA, Hayhurst EJ, Horsburgh M, Hobbs JK, Foster SJ. 2015. Bacterial cell enlargement requires control of cell wall stiffness mediated by peptidoglycan hydrolases. mBio 6:e00660. doi: 10.1128/mBio.00660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demchick P, Koch AL. 1996. The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J Bacteriol 178:768–773. doi: 10.1128/jb.178.3.768-773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koraimann G. 2003. Lytic transglycosylases in macromolecular transport systems of Gram-negative bacteria. Cell Mol Life Sci 60:2371–2388. doi: 10.1007/s00018-003-3056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheurwater EM, Burrows LL. 2011. Maintaining network security: how macromolecular structures cross the peptidoglycan layer. FEMS Microbiol Lett 318:1–9. doi: 10.1111/j.1574-6968.2011.02228.x. [DOI] [PubMed] [Google Scholar]
- 43.Weber BS, Hennon SW, Wright MS, Scott NE, de Berardinis V, Foster LJ, Ayala JA, Adams MD, Feldman MF. 2016. Genetic dissection of the type VI secretion system in Acinetobacter and identification of a novel peptidoglycan hydrolase, TagX, required for its biogenesis. mBio 7:e01253-16. doi: 10.1128/mBio.01253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubori T, Matsushima Y, Nakamura D, Uralil J, Lara-Tejero M, Sukhan A, Galan JE, Aizawa S-I. 1998. Supermolecular structure of the Salmonella typhimurium type III protein secretion system. Science 280:602–605. doi: 10.1126/science.280.5363.602. [DOI] [PubMed] [Google Scholar]
- 45.Burkinshaw BJ, Deng W, Lameignere E, Wasney GA, Zhu H, Worrall LJ, Finlay BB, Strynadka NC. 2015. Structural analysis of a specialized type III secretion system peptidoglycan-cleaving enzyme. J Biol Chem 290:10406–10417. doi: 10.1074/jbc.M115.639013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.García-Gómez E, Espinosa N, de la Mora J, Dreyfus G, Gonzalez-Pedrajo B. 2011. The muramidase EtgA from enteropathogenic Escherichia coli is required for efficient type III secretion. Microbiology 157:1145–1160. doi: 10.1099/mic.0.045617-0. [DOI] [PubMed] [Google Scholar]
- 47.Christensen GJ, Scholz CF, Enghild J, Rohde H, Kilian M, Thurmer A, Brzuszkiewicz E, Lomholt HB, Bruggemann H. 2016. Antagonism between Staphylococcus epidermidis and Propionibacterium acnes and its genomic basis. BMC Genomics 17:152. doi: 10.1186/s12864-016-2489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Houben EN, Korotkov KV, Bitter W. 2014. Take five - type VII secretion systems of mycobacteria. Biochim Biophys Acta 1843:1707–1716. doi: 10.1016/j.bbamcr.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 49.Beckham KS, Ciccarelli L, Bunduc CM, Mertens HD, Ummels R, Lugmayr W, Mayr J, Rettel M, Savitski MM, Svergun DI, Bitter W, Wilmanns M, Marlovits TC, Parret AH, Houben EN. 2017. Structure of the mycobacterial ESX-5 type VII secretion system membrane complex by single-particle analysis. Nat Microbiol 2:17047. doi: 10.1038/nmicrobiol.2017.47. [DOI] [PubMed] [Google Scholar]
- 50.Ummels R, Abdallah AM, Kuiper V, Aâjoud A, Sparrius M, Naeem R, Spaink HP, van Soolingen D, Pain A, Bitter W. 2014. Identification of a novel conjugative plasmid in mycobacteria that requires both type IV and type VII secretion. mBio 5:e01744-14. doi: 10.1128/mBio.01744-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 52.Bubeck Wardenburg J, Williams WA, Missiakas D. 2006. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Nat Acad Sci U S A 103:13831–13836. doi: 10.1073/pnas.0603072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Jonge BL, Chang YS, Gage D, Tomasz A. 1992. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J Biol Chem 267:11248–11254. [PubMed] [Google Scholar]
- 54.Glauner B, Holtje J-V, Schwarz U. 1988. The composition of the murein of Escherichia coli. J Biol Chem 263:10088–10095. [PubMed] [Google Scholar]
- 55.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 57.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 58.Schneewind O, Mihaylova-Petkov D, Model P. 1993. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J 12:4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]