

Toxoplasma gondii, an obligate intracellular parasite replicating in mammalian cells within a parasitophorous vacuole (PV), is an avid scavenger of lipids retrieved from the host cell. Following lipid uptake, this parasite stores excess lipids in lipid droplets (LD).
KEYWORDS: Toxoplasma gondii, lipid droplet, fatty acid uptake and storage, DGAT inhibition, T863 parasiticidal activity
ABSTRACT
Toxoplasma gondii, an obligate intracellular parasite replicating in mammalian cells within a parasitophorous vacuole (PV), is an avid scavenger of lipids retrieved from the host cell. Following lipid uptake, this parasite stores excess lipids in lipid droplets (LD). Here, we examined the lipid storage capacities of Toxoplasma upon supplementation of the culture medium with various fatty acids at physiological concentrations. Supplemental unsaturated fatty acids (oleate [OA], palmitoleate, linoleate) accumulate in large LD and impair parasite replication, whereas saturated fatty acids (palmitate, stearate) neither stimulate LD formation nor impact growth. Examination of parasite growth defects with 0.4 mM OA revealed massive lipid deposits outside LD, indicating enzymatic inadequacies for storing neutral lipids in LD in response to the copious salvage of OA. Toxoplasma exposure to 0.5 mM OA led to irreversible growth arrest and lipid-induced damage, confirming a major disconnect between fatty acid uptake and the parasite's cellular lipid requirements. The importance of neutral lipid synthesis and storage to avoid lipotoxicity was further highlighted by the selective vulnerability of Toxoplasma, both the proliferative and the encysted forms, to subtoxic concentrations of the acyl coenzyme A:diacylglycerol acyltransferase 1 (DGAT1) pharmacological inhibitor T863. T863-treated parasites did not form LD but instead built up large membranous structures within the cytoplasm, which suggests improper channeling and management of the excess lipid. Dual addition of OA and T863 to infected cells intensified the deterioration of the parasite. Overall, our data pinpoint Toxoplasma DGAT as a promising drug target for the treatment of toxoplasmosis that would not incur the risk of toxicity for mammalian cells.
INTRODUCTION
Many immune evasion and replication strategies deployed by protozoan parasites revolve around the manipulation of host cell lipids and lipid-derived metabolites. As an actively dividing organism, the intravacuolar pathogen Toxoplasma gondii is an avid scavenger of lipids, partly due to its inability to synthesize many essential lipids (reviewed in references 1 and 2). This parasite is indeed notorious for exploiting the mammalian host lipidome by suppressing, augmenting, scavenging, remodeling, and metabolizing lipids to successfully multiply within the host cell (1–7). For example, this parasite targets host lipid droplets (LD), multifunctional organelles involved in lipid storage and inflammation regulation, to modulate immune responses (8, 9). In fact, we showed that the number of host LD and the expression of host LD-associated genes peak at the onset of parasite replication in mammalian cells (7). In addition, we demonstrated that Toxoplasma exploits the lipid cargo of host LD for nutritional purposes as it attracts, sequesters, and processes host LD in the parasitophorous vacuole (PV) to access their neutral lipid content. Toxoplasma growth is reduced in mammalian cells depleted of LD (7). When oleic acid (OA) is added to the culture medium, which stimulates mammalian LD biogenesis, Toxoplasma accumulates host LD into the PV and engorges itself uncontrollably with OA (7). Under such conditions, the parasite copes with the vast amounts of OA taken up by storing OA-derived lipids, e.g., acylglycerols and cholesteryl esters (CE), in cytoplasmic LD. These observations reveal that the parasite responds to changes in the neutral lipid environment in the host and that host LD may have an impact on the intracellular development of Toxoplasma.
In addition to host cell LD, Toxoplasma exploits host endolysosomes containing cholesterol originating from plasma low-density lipoprotein (LDL), the main source of cholesterol for the parasite (10). When human LDL is added to the culture medium at a concentration of 1.5 mg/ml (LDL concentrations in human serum, 0.6 to 1.9 mg/ml), the parasite internalizes large amounts of cholesterol, replicates faster, and stores excess cholesterol in LD to avoid the untoward consequences of free cholesterol-induced damage. Under conditions of LDL depletion, the parasite consumes LD-stored cholesterol and slows down its growth (10). Upon supplementation of 0.4 mM OA to the culture medium (OA concentrations in human serum, 0.03 to 3.2 mM) (11), Toxoplasma also activates its enzymatic machinery for OA storage to avoid lipotoxicity. Surprisingly, the parasite also upregulates the expression of a stearoyl coenzyme A (CoA) desaturase homolog (7), involved in the synthesis of OA from stearate, analogously to adipocytes that have evolved to stockpile lipids and preserve nonadipose tissues from damage caused by free lipid accumulation. Between uncontrolled lipid uptake and a limited capacity for lipid storage, Toxoplasma is therefore at risk for cellular dysfunction due to lipid overload.
The T. gondii genome encodes three enzymes essential for storing neutral lipids in cytoplasmic LD: two acyl-CoA:cholesterol acyltransferase (ACAT) enzymes, T. gondii ACAT1 (TgACAT1) and T. gondii ACAT2 (TgACAT2), both involved in producing CE for storage in LD with some fatty acyl-CoA preferences (12–14), and one acyl-CoA:diacylglycerol acyltransferase (DGAT), T. gondii DGAT (TgDGAT), responsible for all triacylglycerol (TAG) synthesis (15). Genetic ablation of TgDGAT is lethal to Toxoplasma, highlighting the importance of TAG synthesis for the parasite. Toxoplasma parasites lacking either ACAT gene display severe growth defects, whereas a double ACAT deletion is not tolerated by the parasite. Pharmacological inhibition of TgACAT enzymes leads to the accumulation of intramembranous free cholesterol, which causes membrane breakdown (13, 14). These observations further underline the essentiality for Toxoplasma of neutral lipid (e.g., fatty acids, TAG, CE) storage, both to survive under conditions of lipid scarcity in the environment and to prevent lipotoxicity due to excessive lipid accumulation.
In this study, we examined the infectivity and intracellular development of Toxoplasma upon supplementation of the culture medium with various fatty acids at physiologically relevant concentrations in human serum. In particular, we addressed the following questions: does the parasite benefit from large amounts of exogenous fatty acids, as observed for other lipids, e.g., cholesterol (10) and sphingolipids (16)? What is the storage capability of Toxoplasma for exogenous fatty acids and acylation of diacylglycerol (DAG) and cholesterol? Is there a threshold in OA concentrations that leads to the saturation of TgDGAT activity for TAG formation? Does Toxoplasma preferentially scavenge and accumulate unsaturated or saturated fatty acids? Compared to mammalian cells, what is the sensitivity of the parasite, both the tachyzoite (proliferative stage) and bradyzoite (semidormant stage forming cysts), toward pharmacological inhibition of TgDGAT?
The aim of our study is to assess whether TgDGAT constitutes a valid drug target, as previously documented for TgACAT enzymes (13, 14). Toxoplasma is an important cause of opportunistic infections in immunocompromised patients (17–19). Current treatment options for toxoplasmosis are limited, poorly tolerated, and inefficient to eliminate cysts in the brain and muscles (19–21). Thus, novel drug targets need to be discovered as the basis for developing more suitable chemotherapeutics. Due to the parasite's gluttony for fatty acids, identifying potential drug targets involved in fatty acid storage pathways may represent a promising approach to control Toxoplasma infections.
RESULTS
OA supplemented to the medium impairs Toxoplasma growth and replication in a concentration-dependent manner.
We previously demonstrated that tachyzoites of Toxoplasma salvage OA (C18:1) proportionally to the concentration of this lipid in the culture medium (7). Upon exposure to 0.4 mM OA (10% fetal bovine serum [FBS] in the culture medium contains approximately 0.01 mM OA), the parasite synthesizes large quantities of CE and acylglycerols, which are then stored in LD. Next, we assessed whether the rapid growth of tachyzoites is impacted by such an avid uptake of OA. We monitored parasite growth by plaque assays in the presence of 0.2 and 0.4 mM OA for 5 days, comprising several cycles of parasite invasion, growth, and egress. Compared to the control condition (no OA added to the medium), long-term exposure of infected monolayers to OA resulted in smaller plaques whose size was proportional to the OA concentrations, with an ∼50% and ∼65% reduction in plaque size being found following incubation with 0.2 mM and 0.4 mM OA, respectively (Fig. 1A).
FIG 1.

Influence of supplemental oleate and palmitate on Toxoplasma replication and development. (A) Toxoplasma development in HFF assessed by plaque assays for 5 days with OA at the indicated concentrations. Data show histograms of the mean area of plaques ± SD from 4 independent experiments. *, P < 0.002, Student's t test. (B) Toxoplasma replication in HFF assessed by tritiated uracil incorporation in the presence of OA at the indicated concentrations at 24 h p.i. Data are percentages ± SD relative to the value for the control (to which no fatty acids were added, the value for which was set as 100%) for biological triplicates with experimental triplicates. *, P < 0.05, Student's t test. (C) Toxoplasma replication assessed by PV size at 24 h p.i. in the presence of OA, which was added to the medium at concentrations ranging from 0 to 0.5 mM. The numbers of parasites per PV in Toxoplasma-infected HFF are displayed as percentages for all PV. Data show the means ± SD for at least 1,000 randomly selected vacuoles from 3 independent experiments. P values were determined by Student's t test. *, P < 0.001; **, P < 0.0001. (D) Transmission EM of intracellular Toxoplasma incubated in the presence of 0.4 mM OA. PV containing 8, 4, 2, or 1 parasite are shown. In addition to LD, osmiophilic membranous structures (red arrowheads) in the residual body (RB), cytoplasm, or PV lumen were detected. hc, host cell; P, parasite. Bars, 250 nm. (E and F) Fluorescence microscopy of HFF infected with RFP-expressing Toxoplasma for 24 h in the presence of 0.5 mM OA, fixed, and stained with BODIPY 493/503. (E) Representative PV for at least 50 infected cells viewed at the same magnification. (F) Many large LD are detected in the parasite cytoplasm, and some are even bulging out from the parasite. (G) Transcriptional profile of TgDGAT upon addition of 0.5 mM OA to the medium for 24 h using real-time qPCR analysis. Data are means of means ± SD from 3 separate assays performed in technical triplicate. *, P < 0.003. (H) Toxoplasma replication in HFF assessed by measurement of tritiated uracil incorporation in the presence of PA at the indicated concentrations at 24 h p.i. Data are percentages ± SD relative to the value for the control (set as 100%) for biological triplicates with experimental triplicates. (I) Fluorescence microscopy of HFF infected with RFP-expressing Toxoplasma for 24 h in the presence of 0.2 or 0.4 mM PA, fixed, and stained with BODIPY 493/503. Representative PV from at least 30 infected cells are shown at the same magnification. Tg, T. gondii.
We next wanted to assess whether the OA-induced growth defect is due to an impairment in parasite replication. By assaying tritiated uracil incorporation in the parasite for 24 h, a significant delay in Toxoplasma replication was observed, corresponding to an ∼20% and ∼35% reduction at 0.1 and 0.4 mM OA, respectively (Fig. 1B). These data were confirmed by enumeration of the parasites per PV after 24 h of incubation with OA at concentrations ranging from 0 to 0.5 mM (nontoxic concentrations for mammalian cells) in 0.1 mM increments (Fig. 1C). The replication defect was proportional to the amounts of OA added to the medium; at 0.3 mM OA, ∼10% of PV contained 16 parasites, whereas ∼40% of PV contained 16 parasites under control conditions. At higher OA concentrations, no large PV were observed, and instead, the majority of PV contained only 4 parasites at 0.4 mM OA and 1 to 2 parasites at 0.5 mM OA.
We conducted electron microscopy (EM) studies to inspect the ultrastructure of intracellular Toxoplasma exposed to 0.4 mM OA (Fig. 1D). Toxoplasma divides by endodyogeny, a process leading to the synchronous formation of two daughter cells within the mother cell. As the daughters emerge, a small structure containing the remnants of the mother, termed the residual body, is evacuated at the basal end of the new progeny and is degraded in the PV lumen when daughter cell formation is complete (22). Confirming our data in Fig. 1C, few PV contained 8 parasites upon incubation with 0.4 mM OA. Within the PV, parasites were also disorganized, in contrast to control parasites, which formed rosette-like structures (see Fig. S1 in the supplemental material). The parasite's cytoplasm was overloaded with large lipid deposits up to 0.4 μm in size, while the residual bodies were abnormally retained and packed with lipid deposits and membranous structures. Additionally, in the smaller PV, the residual body tended to be larger and increasingly filled with osmiophilic deposits. These observations suggest that the machinery for lipid storage in LD is inadequate and potentially saturated, leading to the deleterious accumulation of lipids in the cytoplasm, thereby obstructing the normal endodyogeny process.
To assess the potential oversaturation of parasite LD as lipid reservoirs, parasites were exposed to 0.5 mM OA and stained with the fluorescent dye BODIPY 493/503, which labels LD (Fig. 1E). Single parasites contained numerous, extremely large lipid stores, in contrast to control parasites, which had few, small LD. The oversized LD in the OA-treated parasites were as large as mammalian LD, even protruding from the parasite's cell body and distorting the plasma membrane (Fig. 1F).
We previously showed that at 0.4 mM OA, Toxoplasma upregulates TgDGAT transcription by ∼1.5-fold in an attempt to tolerate the massive uptake of OA (7). However, such an increase in TgDGAT transcripts was likely insufficient, as lipids also accumulated outside LD (Fig. 1D). We next examined to what extent T. gondii could activate its enzymatic machinery for TAG synthesis at concentrations of OA above 0.4 mM. Expression levels of TgDGAT in intracellular parasites exposed to 0.5 mM OA for 24 h were lower than those in parasites for which OA was not added to the medium (Fig. 1G). This reveals the inaptitude of Toxoplasma to adequately respond to OA at 0.5 mM, likely due to a functional deterioration of the parasites puffed up with lipids.
Supplemental palmitate does not impact Toxoplasma replication and lipid stores.
To assess whether the replication defect observed with supplemental OA is specific to this fatty acid, we next incubated Toxoplasma-infected cells with palmitic acid (PA; C16:0), the most common saturated fatty acid in mammalian cells, at concentrations of 0.2 and 0.4 mM (human serum concentrations, 0.3 to 4 mM) (11). In mammalian cells, PA is involved in side chain palmitoylation of proteins to facilitate protein-membrane interactions and subcellular protein trafficking (23). We previously showed that PA is scavenged by Toxoplasma (15); however, PA at 0.2 and 0.4 mM led to neither a growth deficiency nor a growth benefit, as assessed by uracil incorporation (Fig. 1H). Plaque assays in the presence of 0.2 mM PA for 5 days did not reveal any significant differences in plaque sizes under control conditions (82 ± 11 μm3 versus 9 ± 6 μm3 without and with 0.2 mM PA added, respectively), indicating that this fatty acid has no influence on the developmental cycle of T. gondii. Fluorescence microscopy confirmed this finding, showing PV with similar sizes at 24 h postinfection (p.i.) regardless of PA addition to the medium (Fig. 1I). Exposure to 0.4 mM PA did not lead to large LD formation in mammalian cells, indicating that, unlike unsaturated fatty acids (24), saturated fatty acids are not the best substrates for DAG and, thus, do not stimulate LD biogenesis. Similarly, Toxoplasma did not form large LD upon exposure to 0.2 or 0.4 mM PA.
In addition to replication defects, Toxoplasma egresses more slowly from host cells upon incubation with 0.4 mM OA.
We wanted to examine whether 0.2 or 0.4 mM OA affects other key steps of Toxoplasma development besides replication. Exit from cells is a crucial step in the propagation of Toxoplasma within a host, and this process is an active event regulated by the parasite (25). To assess the egress ability of T. gondii exposed to supplemental OA, we monitored by live video microscopy the escape of the parasite from host cells over time. The natural egress of Toxoplasma from host cells occurred when the PV size reached 64 to 128 parasites, corresponding to 40 h to 46 h p.i. (not shown). In contrast, when intracellular parasites were exposed to 0.4 mM OA, no egress events were observed at these time points; in fact, even at 76 h p.i., all PV still subsisted in a heavily infected monolayer of fibroblasts (Fig. 2A). Parasites egress was observed only from day 4 onwards (∼96 h p.i.). To investigate the cause of this delay, EM studies were conducted on PV in fibroblasts incubated for 72 h with 0.4 mM OA. Figure 2B panels a to g represent different stages of parasites undergoing egress: intravacuolar parasites formed a tight rosette (Fig. 2B, panel a), which then started to disassemble (Fig. 2B, panels b and c); the luminal content of the PV then became more electron lucent, a sign that the PV membrane was breaking down (Fig. 2B, panel d), allowing parasites to escape the PV but with the parasites remaining trapped in the host cytoplasm, being retained by the host plasma membrane (Fig. 2B, panels e and f); eventually, this membrane ruptured, as illustrated by the electron-lucent host cytoplasm (Fig. 2B, panel g), liberating the parasites into the environment.
FIG 2.

Effect of supplemental OA on Toxoplasma egress from host cells. (A) Live DIC phase microscopy of HFF infected with Toxoplasma for 76 h in the presence of 0.4 mM OA, showing many intact PV, as observed at a low magnification (20×; yellow arrowheads) or at a high magnification (60×; red arrowheads). Bars, 10 μm. An abundance of host LD around the PV is observed. (B) EM of parasites in HFF incubated with 0.4 mM OA for 72 h. (a through g) Different stages of egress: a tightly formed rosette occurs initially (a) but progressively loses its compact structure (b to d), followed by a dwindling of PV membrane (PVM) integrity (e) and, eventually, loss of the PV membrane (f), leading to parasite escape into the host cytoplasm, but the parasite is retained within the host plasma membrane (hPM) with the host nucleus (hn) (g). Bars, 7 μm. (C) Measurement of Toxoplasma egress efficacy following incubation with 0, 0.2, or 0.4 mM OA in the medium. At 24 p.i., egress was chemically induced with A23187, and the time taken to egress (in seconds) was recorded. The ratio of the time taken for treated cells to egress to the time for the control (no OA added) was calculated and plotted as the means of the means ± SD from four independent experiments. The indicated statistically significant differences are between the control and 0.2 mM OA and 0.4 mM OA-treated HFF. *, P < 0.05, Student's t test.
However, the delay in Toxoplasma egress upon OA supplementation may simply be a consequence of slower replication, whereby parasites from small PV are not ready to leave the host cell at 40 h p.i. To examine this possibility, we chemically induced parasite egress using the calcium ionophore A23187 and monitored the time necessary for Toxoplasma to exit from host cells at 24 h p.i. In parasites grown in the absence of OA added to the medium, A23187 induced egress within ∼30 s, regardless of the PV size. In contrast, the time to egress increased by 1.6-fold and 2.2-fold for parasites exposed to 0.2 mM and 0.4 mM OA, respectively. This suggests that the egress defect observed upon exposure to supplemental OA was not due to a smaller PV size but, rather, was due to a decreased efficiency with which OA-overloaded parasites breached the membranes and egressed and/or a failure to respond promptly to the calcium ionophore.
Supplemental unsaturated but not saturated fatty acids lead to LD biogenesis in Toxoplasma.
We next investigated the potential impact of various fatty acids added to the culture at physiological concentrations on LD formation: the saturated fatty acid stearate (SA; C18:0) (11), the polyunsaturated fatty acid linoleate (LA; C18:2), and the monounsaturated palmitoleate (POA; C16:1) (human serum concentrations, 0.1 to 1 mM for SA, 0.2 to 5 mM for LA, and 0.18 to 0.7 mM for PA) (11). In mammalian cells, SA is important for mitochondrial function via stearoylation of proteins (26). LA is involved in the biosynthesis of arachidonic acid, a precursor of some prostaglandins (e.g., prostaglandin E2 [PGE2]) stored in LD (27). Mammalian PGE2 synthesis is induced by Toxoplasma through a protein kinase C-dependent induction of cyclooxygenase-2 (28), resulting in the inhibition of both the Th1-type response and nitric oxide production (8, 9). POA plays a central role in intracellular trafficking and signaling pathways, cell differentiation, and proliferation (29). Red fluorescent protein (RFP)-expressing Toxoplasma was incubated with 0.2 and 0.4 mM SA, LA, or POA for 24 h and analyzed by fluorescence microscopy for PV size and LD using BODIPY 493/503 (Fig. 3). No replication defects occurred after supplementation with SA, as large PV were observed, similar to the findings for control PV without any fatty acid addition. Neither SA-treated parasites nor host fibroblasts contained large LD, indicating that SA, like PA, is a poor substrate for TAG or CE synthesis in Toxoplasma, as reported for mammalian cells (30). Incubation of infected cells with 0.2 mM LA resulted in abnormally small PV with misshapen parasites. Exposure to 0.2 mM LA or POA stimulated the biogenesis of LD in host fibroblasts and Toxoplasma, which exhibited very large LD that accumulated in the cytoplasm.
FIG 3.

Lipid droplet formation in Toxoplasma induced by various fatty acids (FA) and jasmonate. Images were obtained by fluorescence microscopy of uninfected HFF or HFF infected with RFP-expressing Toxoplasma for 24 h in the presence of SA, LA, POA, or JA at the indicated concentrations, fixed, and stained with BODIPY 493/503. Representative PV from at least 25 infected cells are shown at the same magnification.
In parallel, we examined the effect of jasmonic acid (JA) on Toxoplasma PV size and LD. Derived from linolenic acid after oxygenation and cyclization, jasmonates are plant hormones that are responsive to stresses, such as pathogen infections (31). The growth of the apicomplexan parasite Plasmodium is hindered in erythrocytes following incubation with 0.5 mM to 3 mM JA (32). JA also preferentially inhibits the proliferation of cancer cells, leading to the release of cytochrome c, depletion of ATP, and cell death, while it is inactive in nontransformed cells (33). When exposed to 0.2 mM to 2 mM JA, no growth change was observed for Toxoplasma. However, BODIPY 493/503 staining revealed the accumulation of small but abundant LD within the parasite's cytoplasm as well as lipid deposits in the PV, suggestive of dysregulation of lipid homeostatic pathways induced by JA.
Supplemental unsaturated fatty acids impair Toxoplasma replication.
We next quantified our microscopic observations, shown in Fig. 3, of Toxoplasma exposed to supplemental fatty acids by enumerating the parasites per PV at 24 h p.i. (Fig. 4). Incubation of Toxoplasma-infected cells with SA at concentrations up to 0.4 mM had no effect on parasite replication. At 0.05 mM LA, parasites divided at a normal rate, whereas at 0.1 and 0.2 mM, LA had a significant negative impact on replication. To further quantify this growth defect, we generated an algorithm from which the average doubling time under various experimental conditions could be arbitrarily calculated on the basis of the number of parasites per PV (see the equations in Materials and Methods). Corroborating our data in Fig. 1, Toxoplasma incubated with 0.2 mM and 0.4 mM OA replicated, on average, every 8.3 h and 14.6 h, respectively, in comparison to its replication time under control conditions, for which a mean doubling time of 7.5 h was calculated. Parasites exposed to 0.1 mM and 0.2 mM LA had a significantly increased doubling time of 10 h and 193 h, respectively. Exposure of infected cells to 0.2 mM POA slightly decreased Toxoplasma replication, with the calculated doubling time being 12 h. When exposed to 0.2 mM to 2 mM JA, doubling times similar to those of control parasites were calculated.
FIG 4.

Influence of exogenous fatty acids and jasmonate on Toxoplasma replication and parasite lipid storage. (A) Toxoplasma replication in HFF assessed by parasite enumeration per PV at 24 h p.i. in the presence of OA, SA, LA, POA, or JA, added to the medium at the indicated concentrations. Data show the means of the means ± SD for at least 1,000 randomly selected vacuoles from 3 independent experiments. Trypan blue exclusion tests in replicates for evaluating the viability of HFF exposed to these fatty acids for 24 h indicated 98% viable cells under control conditions (no fatty acids added), 97% at 0.2 mM SA, 93% at 0.4 mM SA, 98% at 0.2 mM POA, 97% at 0.05 mM LA, 96% at 0.1 mM LA, 89% at 0.2 mM LA, 99% at 0.2 mM JA, 98% at 1 mM JA, and 97% at 2 mM JA. (B) Predicted doubling times of parasites grown in the presence of the indicated lipids. The values were calculated from the data acquired for panel a using the Predictive Replication Analysis algorithm (see the details in Materials and Methods). *, P < 0.003; **, P < 0.0007; ***, P < 0.00007.
The slow growth of Toxoplasma upon OA supplementation is not due to parasite differentiation into the latent cyst form.
Our results suggest that the replication defect of Toxoplasma in response to OA, POA, and LA could be due to the overaccumulation of lipids in the parasite and the PV, possibly resulting in cellular dysfunctions and impaired metabolism. Alternatively, it remains possible that the reduced replication rates induced by unsaturated fatty acids may result from a parasite stress response, leading to differentiation into a latent cyst form. The conversion of Toxoplasma tachyzoites to bradyzoites (the slow-growing cyst forms) can spontaneously occur in vitro in some cell types (34) but can also be induced in response to environmental stresses, such as changes in temperature and pH, amino acid deprivation, and lower CO2 levels (35, 36). The bradyzoite phenotype is characterized by the incorporation of carbohydrates into the PV membrane, which develops into a thick cyst wall (37). To test the possibility that unsaturated fatty acids act as stressors for the parasite, causing its differentiation into bradyzoites, we incubated infected fibroblasts with OA (as a prototype for unsaturated fatty acids) at 0.2 mM or 0.4 mM for 24 h and monitored the formation of the cyst wall using tetramethyl rhodamine isocyanate (TRITC)-labeled lectin (Fig. S2). No fluorescent staining was observed on the PV membrane of parasites grown in medium supplemented with OA. In comparison, a TRITC-lectin signal was clearly apparent on the PV membrane of parasites cultivated in the absence of CO2 (positive control). As opposed to the Toxoplasma RH strain, the Prugniaud strain is cystogenic; i.e., it is more sensitive to external stressors, leading readily to tachyzoite-to-bradyzoite conversion (36, 38). Under CO2 deprivation conditions, Prugniaud parasites exhibited a very strong fluorescent signal, indicative of cyst wall formation and bradyzoite conversion. When cells were infected with Prugniaud parasites in the presence of 0.2 or 0.4 mM OA and stained with TRITC-lectin, no fluorescence was detected on the PV membrane. These results reveal that the impairment of parasite growth following incubation with supplemental OA is not attributable to a shift in the developmental program of the parasite from tachyzoite to bradyzoite forms.
Upon OA addition, host autophagic organelles gather around the PV but do not appear to be involved in PV degradation.
Mammalian cells exposed to excess OA exhibit increased autophagic activities (39). Excess OA can be utilized to generate autophagosomal membranes for organelle degradation, In addition, lipophagy, i.e., selective autophagy of LD, is triggered in order to regulate lipid metabolism and prevent lipotoxicity. Degradation of the PV via autophagy has been reported for the Toxoplasma cystogenic strains that are susceptible to immune-mediated autophagy induction (40). This process involves the ubiquitination of the PV membrane, the deposition of LC3 around the PV, and the formation of multiple membranes enclosing the PV (41). We therefore investigated whether reduced development of Toxoplasma with supplemental OA could be due to elevated autophagic activities in the host cell, thus representing a harmful environment for the parasite. In our cellular system, we confirmed that supplemental OA induced autophagy in mammalian cells following incubation of fibroblasts expressing green fluorescent protein (GFP)-light chain 3 (LC3) with 0.2 or 0.4 mM OA, as large LC3-containing autophagic structures were detected by microscopy (Fig. S3A). Upon autophagy induction via amino acid depletion, abundant autophagosomes identifiable as large GFP-LC3 puncta were observed in starved cells (positive control), in contrast to the more diffuse fluorescence staining and very small GFP-LC3 puncta seen in cells incubated in normal medium. As in previous studies reporting host autophagosomes gathering around the PV (42, 43), we observed the accumulation of large GFP-LC3 puncta, likely corresponding to autophagic structures, surrounding the PV in infected cells transfected with GFP-LC3. This perivacuolar accumulation of autophagosomal structures was confirmed by an immunofluorescence assay (IFA) using an antibody against LC3; however, the LC3 fluorescent signal was not observed on the PV membrane (Fig. S3B). EM analyses confirmed the proximity of host autophagic structures (some enclosing LD) close to the PV (Fig. S3C). Though accumulating around the PV, host autophagosomal structures fusing with the PV were not seen. These observations make unlikely the destruction of the PV by xenophagy, despite the abundance of autophagosomes in host cells exposed to 0.2 mM OA.
Upon OA supplementation, the host Golgi apparatus undergoes normal fragmentation into ministacks that distribute around the PV.
In addition to host autophagosomes, Toxoplasma attracts other host organelles that cluster around the PV or that are even associated with the PV membrane, and such a host organelle recruitment is likely to be important for parasite development in mammalian cells (reviewed in reference 44). Previous studies reported on the accumulation of host LD around the PV following the addition of OA to the culture medium (6–8). We next focused on determining if the replication defect of Toxoplasma exposed to excess OA could be attributed to the impaired attraction of host organelles to the PV, e.g., the Golgi apparatus, Rab vesicles, the endoplasmic reticulum (ER), and mitochondria, due to the aggregation of host LD to that area. We monitored the spatial distribution of several host organelles in Toxoplasma-infected cells in the presence of supplemental OA by fluorescence microscopy using mammalian organellar markers or by EM. Under physiological conditions, we illustrated that infection of mammalian cells with Toxoplasma induces dramatic alterations in the morphology and distribution of the host Golgi apparatus (16). Early during infection, the host Golgi apparatus surrounded the PV of Toxoplasma, and later, Golgi apparatus fragments remained around the PV. We investigated, by IFA using an antigiantin antibody, whether this phenomenon occurred in infected cells exposed to 0.2 mM OA. Like under control conditions, we observed a change in the host Golgi apparatus morphology and that Golgi apparatus material encircled the PV (Fig. S4A). The extent of the host Golgi apparatus fragmentation was quantified using MetaScopics quantitative image analysis software (45) at different developmental time points on the basis of the PV size (Fig. S4B). In the presence of 0.2 mM OA, the centroid-to-surface distance of the host Golgi apparatus to small PV (1, 2, and 4 parasites per PV) was significantly larger than that under control conditions, suggesting a slightly delayed accumulation of host Golgi apparatus ministacks around the PV. However, later during infection, the host Golgi apparatus surrounded the PV at levels similar to those seen in control cells, suggesting that perivacuolar LD did not affect the accumulation of Golgi apparatus ministacks at the PV over time.
We next inspected the distribution of host Golgi apparatus fragments relative to that of LD surrounding the PV in HeLa cells expressing GFP-adipose differentiation-related protein (ADRP), a protein located on the LD surface whose ectopic expression enhances LD formation (46). In uninfected cells, both the Golgi apparatus and the majority of LD were located perinuclearly (Fig. S4C). In infected cells, host Golgi apparatus fragments were distributed closer to the PV than host LD, even with 0.2 mM OA treatment. EM studies of infected human foreskin fibroblasts (HFF) in the presence of 0.4 mM OA confirmed the presence of host Golgi apparatus ministacks along the PV membrane (Fig. S4D), as observed under normal conditions (7). Together, these data suggest that host LD do not significantly obstruct the relocation of the host Golgi apparatus at the PV, especially later during infection.
Supplemental OA in the medium does not hinder host Rab vesicle clustering around the PV and internalization into the PV.
In addition to host LD (6–8), some other host organelles are detected within the PV of Toxoplasma (16, 43, 47). For example, we previously demonstrated that Toxoplasma attracts host Golgi apparatus-derived Rab14, Rab30, and Rab43 vesicles and internalizes these sphingolipid-containing vesicles into the PV lumen (16). We wanted to examine the ability of parasites exposed to supplemental OA to reroute host Rab vesicles to the PV. Among Rab GTPases, Rab11a is an evolutionarily conserved protein implicated in regulating vesicular trafficking via the recycling of endosomal compartments and early endosomes to both the trans-Golgi network and the plasma membrane (48). To study the distribution of host Rab11a vesicles, we ectopically expressed a GFP-tagged Rab11a construct in fibroblasts and infected the fibroblasts with RFP-labeled Toxoplasma parasites. Fluorescence imaging of uninfected cells revealed that the GFP-Rab11a signal was primarily perinuclear, whereas upon infection, the distribution of GFP-Rab11a became perivacuolar (Fig. S5). Optical z-sections throughout the PV showed GFP foci within ∼90% of the PV. Under supplemental OA conditions, Rab11a was still detected surrounding the PV and within the PV, suggesting that the presence of host LD did not affect the ability of the parasite to intercept host Rab vesicles.
The accumulation of host LD around the PV does not interfere with the association of the host ER with the PV membrane.
An important feature of the intracellular parasitism of Toxoplasma is the association of the PV membrane with the host ER and mitochondria (reviewed in reference 44). Next, we examined whether the association of these host organelles with the PV is perturbed by the perivacuolar accumulation of LD upon the addition of OA. The subcellular localization of the host ER relative to the PV upon exposure to 0.2 mM OA was examined using antibody against calnexin, and an intense fluorescent staining around the PV similar to that observed in infected cells without OA added was observed (Fig. S6A). We also observed an intense fluorescent perivacuolar staining using an anti-KDEL antibody (not shown). EM analysis of infected cells incubated with 0.4 mM OA revealed host ER elements closely apposed to the PV membrane, with ER-bound ribosomes facing the host cytoplasm (Fig. S6B). Upon supplementation of OA, the host LD were located behind the layer of the host ER attached to the PV membrane, indicating that the parasite could form molecular associations with the host ER independently of the presence of abundant LD encircling the PV.
Upon OA supplementation, the PV membrane associates with host mitochondria.
To test whether the presence of supranumerous host LD hinders the PV-host mitochondrion association, HeLa cells expressing ADRP were infected with Toxoplasma, incubated with 0.2 mM OA, and immunostained with antibody against mitochondrial Tom20. A concentric fluorescent signal similar to that observed under control conditions was detected around the PV (Fig. 5A). Parallel assays conducted on fibroblasts in the presence of 0.2 mM OA confirmed the recruitment of host mitochondria around the PV throughout infection (Fig. 5B). To quantify the recruitment of host mitochondria to the PV under control conditions and in the presence of 0.2 mM OA, the intensity-weighted distance algorithm of MetaScopics (45) was used to measure the fluorescence intensities in an arbitrarily delineated area radiating 7 μm from the PV centroid. No significant difference in the recruitment of this organelle to the PV was observed between these two conditions (Fig. 5C). Interestingly, upon incubation with 0.2 mM or 0.4 mM OA and regardless of the PV size, the Tom20 staining around the PV was thicker than that of mitochondria free in the cytosol (Fig. 5B and D).
FIG 5.

Host mitochondrion recruitment to Toxoplasma PV with supplemented OA. (A) Fluorescence microscopy of uninfected or infected GFP-ADRP-expressing HeLa cells for 24 h without OA (control) or with 0.2 mM OA added to the medium. The cells were stained with anti-Tom20 antibody for host mitochondria (red) or anti-GRA7 antibody (blue). Arrows pinpoint the rim of host mitochondria around PV. Arrowheads pinpoint the PV. (B) Fluorescence microscopy of HFF infected with Toxoplasma for 24 h without OA (control) or with 0.2 mM OA. The HFF were stained with anti-Tom20 (green) or anti-GRA7 (red) antibodies. Blue, DAPI. Arrowheads pinpoint the PV. (C) Quantification of host mitochondrion association with the Toxoplasma PV from the experiments described in the legend to panel B using MetaScopics quantitative image analysis software. Box plots show the average distance, weighted by intensity, of the host mitochondria to the PV boundary, as calculated for host mitochondrial profiles within a 7-μm radius of the PV (data are from >40 infected HFF per condition). There were no statistically significant differences between conditions. (D) Fluorescence microscopy of HFF uninfected (Uninf.) and infected with Toxoplasma for 24 h with 0.4 mM OA added to the medium. The HFF were stained with anti-Tom20 antibody (red) and DAPI (blue). Arrowheads pinpoint the PV.
With supplemental OA, host mitochondria associated with the PV membrane display morphological abnormalities.
The thick fluorescent signal observed specifically on perivacuolar mitochondria was scrutinized in infected cells incubated with 0.2 mM OA for 24 h at the ultrastructural level (Fig. 6). We confirmed that host mitochondria were closely apposed to the PV membrane (Fig. 6A, panel a). Remarkably, most of the host mitochondria associated with the PV appeared abnormally enlarged compared to the mitochondria dispersed in the host cytoplasm (Fig. 6A, panel b). Higher-magnification observations revealed that these large, swollen mitochondria were characterized by a rounded shape, a loss of matrix density, and a distortion of the cristae (Fig. 6B, panel a compared to panel b), which would mirror the intense fluorescent Tom20 signal observed by IFA (Fig. 5B). Some mitochondria associated with the PV exhibited linearization of crista membranes, with abnormal geometrical features (Fig. 6C, panels a and b). Abnormally enlarged mitochondria attached to the PV are likely to be dysfunctional and, as such, may account, at least in part, for the Toxoplasma growth defect noticed with supplemented OA.
FIG 6.

Ultrastructural examination of host mitochondria associated with the PV of Toxoplasma with supplemented OA. (A) EM of a PV of Toxoplasma in HFF exposed for 24 h to 0.2 mM OA, illustrating host mitochondria distributed all around the PV. Host mitochondria associated with the PV show severe cytopathies (arrows in panels a and b), in contrast to host mitochondria in the cytoplasm (double arrows in b). hm, host mitochondrion; hc, host cell; hLD, host LD; P, parasite. Bars, 500 nm. (B) EM of an abnormally swollen host mitochondrion associated with the PV (a), in accordance with IFA observations with Tom20 staining of infected HFF treated for 24 h with 0.2 mM OA (b). Bars, 250 nm. (C) EM of large host mitochondria associated with the PV upon incubation with 0.4 mM OA. The mitochondria exhibit distorted cristae or cristae with angular features (a and b). Bars, 400 nm.
We next wanted to determine if supplementation of saturated fatty acids could alter the morphology of the host mitochondria associated with the PV membrane. Exposure of Toxoplasma to 0.2 mM SA did not result in an enlarged perivacuolar staining of Tom20 compared to exposure to 0.2 mM OA (Fig. S7). Exposure of mammalian cells to excess PA at concentrations up to 0.2 mM for 24 h led to mitochondrial fragmentation, which was associated with increased oxidative stress, mitochondrial depolarization, and a loss of ATP production (49). We confirmed that fibroblasts treated with 0.2 mM PA displayed short mitochondrial profiles stained with anti-Tom20 antibodies, suggestive of mitochondrial fragmentation (see Fig. 10). In infected cells incubated with 0.2 mM PA, we observed fragmented host mitochondria distributed around the PV. Nevertheless, the dysfunctionality of these host organelles with supplemented PA seemed to have no negative consequence for the parasites, whose growth rate was normal in the presence of PA at concentrations up to 0.4 mM (Fig. 1B and C). This suggests that upon OA addition, the host mitochondria attached to the PV that displayed an abnormal morphology are likely functionally impaired, which could be an additional factor causing parasite growth delay.
FIG 10.

MitoTracker staining in T863-treated Toxoplasma. (A and B) Fluorescence microscopy of HFF infected with GFP-expressing Toxoplasma, staining for mitochondria using MitoTracker (MitoT). (A) Infected cells were incubated with T863 at the indicated concentrations for 20 to 22 h, and the cells were compared to cells treated with 10 μM pyrimethamine (PYR) as a negative control (−ve ctl) or the DMSO control. (B) Uninfected cells were treated with 40 μM T863 or the DMSO control strained with MitoTracker. (C) Quantification of the MitoTracker staining of the cells in the experiment whose results are presented in panel A by counting the number of PV containing parasites positively labeled with the mitochondrial probe detecting live cell/metabolically active mitochondria in infected cells. The positive control (+ve ctl) corresponds to conditions without any addition to the culture medium. The percentage of viable PV is graphed. P values were determined by Student's t test. *, P < 0.0095; **, P < 0.0003; ***, P < 0.00003.
Following exposure to 0.5 mM OA, Toxoplasma replication is irreversibly arrested.
We showed that the replication of Toxoplasma is severely impaired upon exposure to 0.5 mM OA (Fig. 1C, E, and F). This prompted us to examine whether Toxoplasma is able to recover from incubation in a medium supplemented with 0.5 mM OA, following transfer to normal culture medium. To assess the competence of a 0.5 mM OA-loaded parasite to resume its growth, we designed pulse-chase experiments in which intracellular parasites were incubated in medium containing 0.5 mM OA, which was then washed away and replaced with control culture medium. Under these conditions, parasite replication and the LD content were analyzed by fluorescence microscopy. In a first set of experiments, the duration of the pulse with 0.5 mM OA was 2 days, and the parasites were allowed to recuperate in normal medium for an additional 2 days (Fig. 7A). However, similar to parasites pulsed with 0.5 mM OA without a chase, all the PV contained a single parasite, revealing the parasite incompetence in recovering after the chase period. Furthermore, these parasites still contained large BODIPY 493/503-labeled LD, in contrast to host cells, indicating that they were unable to degrade or use their LD after the return to control conditions, like mammalian cells.
FIG 7.
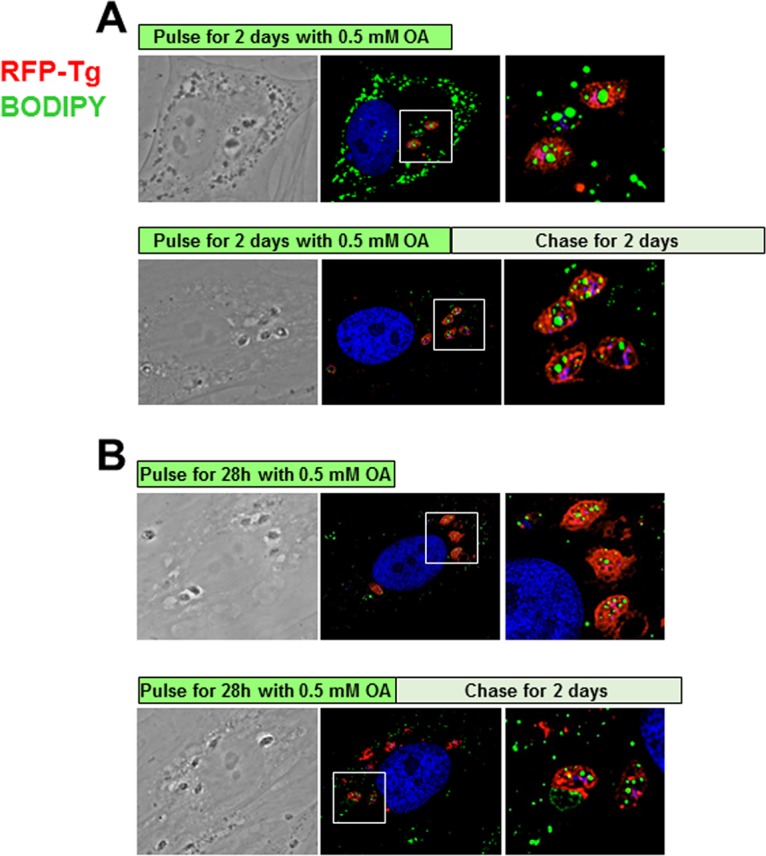
Pulse-chase assays of Toxoplasma exposed to 0.5 mM OA. Fluorescence microscopy of HFF infected with RFP-expressing Toxoplasma for 48 h (A) or 28 h (B) in the presence of 0.5 mM OA, followed by a 48-h chase in control medium or no chase, before fixation and staining with BODIPY 493/503. Representative PV from at least 20 infected cells are shown at the same magnification. The white squares in the middle panels are magnified in the right panels.
In a second set of assays, the pulse duration with 0.5 mM OA was reduced to 28 h followed by 2 days of chase, but no sign of Toxoplasma replication or LD consumption was observed (Fig. 7B). These data suggest that incubation of infected cells with 0.5 mM OA results in irreversible damage to the parasites, which appear to be unable to cope with the massive accumulation of OA that they have ingested, at least for a 28-h time period.
Toxoplasma is highly sensitive to the DGAT1 pharmacological inhibitor T863.
Based on these observations, TAG synthesis seems to be essential for buffering excess internalized fatty acids and preventing lipotoxicity in Toxoplasma. Unlike mammalian cells, T. gondii contains only one DGAT enzyme, which shares 31% identity and 49% similarity with the Homo sapiens DGAT1 (HsDGAT1) and has no sequence homology with Homo sapiens DGAT2 (15). T863 is a selective inhibitor of HsDGAT1, blocking the acyl-CoA binding site of the enzyme, resulting in a blockade of DGAT1-mediated TAG formation (50% inhibitory concentration, ∼120 nM on cultured mammalian cells [50]). To confirm the importance of TAG synthesis and storage in Toxoplasma, we incubated intracellular RFP-expressing Toxoplasma with T863 at concentrations ranging from 1 to 40 μM for 24 h and enumerated the parasites per PV (Fig. 8A). Even at the low concentration of 2.5 μM T863, the PV were, on average, significantly smaller than those in untreated parasites. At 5 μM T863, the replication of the parasites was severely impaired. This replication defect was reflected in the increase in the doubling time, calculated using our algorithm, for parasites treated with T863, with an increase of 2- and 3-fold upon exposure to 10 and 40 μM T863, respectively (Fig. 8B).
FIG 8.

Effect of T863 on Toxoplasma replication, morphology, and lipid body abundance. (A) Toxoplasma replication assessed by PV size at 24 h p.i. in the presence of T863 at the indicated concentrations. The number of parasites per PV in Toxoplasma-infected HFF is displayed as the percentage of PV with that number of parasites among all PV. Data show the means ± SD from at least 20 randomly selected vacuoles from 3 independent experiments. P values were determined by Student's t test. *, P < 0.009; **, P < 0.00001. The results of trypan blue exclusion tests performed in replicates for evaluating the viability of HFF exposed to T863 for 24 h were 97.5% viable cells under the control condition (DMSO) and 97.2% with 80 μM T863. (B) Predicted doubling times of parasites grown in the presence of T863, calculated from the data acquired for panel a using the Predictive Replication Analysis algorithm. (C) Fluorescence microscopy of HFF infected with RFP-expressing Toxoplasma incubated for 24 h with T863 at different concentrations showing differences in PV size and misshapen parasites in T863-treated cells with reduced BODIPY 493/503 signals. Arrowheads pinpoint empty structures. (D) IFA on intracellular Toxoplasma incubated with 80 μM T863 for 24 h and immunostained with anti-GRA7 antibody, confirming that the empty structures were PV. (E) Quantification of Toxoplasma replication in HFF at 24 h p.i., assessed by the use of tritiated uracil incorporation in the presence of 80 μM T863 for 24 h. Data are percentages ± SD relative to the value for the control (DMSO; the value for which was set as 100%) for 4 biological replicates with experimental triplicates. The absolute values were as follows: for experiment 1, 86,951 ± 5,730 cpm for DMSO and 1,604 ± 94 cpm for T863; for experiment 2, 85,902 ± 10,896 cpm for DMSO and 1,745 ± 553 cpm for T863; for experiment 3, 10,836 ± 496 cpm for DMSO and 995 ± 112 cpm for T863. *, P < 0.0001, Student's t test. (F) Fluorescence microscopy of D1D2KO MEF infected with RFP-expressing Toxoplasma incubated for 24 h with 15 μM T863 and stained with BODIPY 493/503 (green).
Fluorescence microscopy of RFP-expressing parasites in fibroblasts exposed to 5 μM T863 for 24 h showed disorganized parasites in the PV, a sign of defects in endodyogeny (Fig. 8C). BODIPY 493/503 LD staining was weaker in parasites treated with 1 μM T863 than in untreated parasites and was almost absent in parasites from 5 μM T863, indicating that this drug inhibits lipid storage in parasite LD. At 10 μM T863, parasite replication was impaired, with PV containing parasites that began to lose their shape definition. At 25 μM T863, some PV contained a large parasitic mass, whereas Toxoplasma parasites treated with 40 μM were barely recognizable in the PV. Upon incubation with 80 μM T863, some PV contained remnants of parasites characterized by a very faint RFP signal, and some vacuole-like structures did not show any fluorescence signals for RFP or DAPI (4′,6-diamidino-2-phenylindole) (Fig. 8C, arrowheads). These empty structures, however, were indeed PV, as IFA using anti-GRA7 antibody showed a fluorescence signal at their periphery, confirming that they had previously contained parasites that had secreted GRA7 prior to disintegrating (Fig. 8D). Monitoring of Toxoplasma replication upon treatment with 80 μM T863 for 24 h showed an ∼95% reduction in the amount of uracil incorporated into the parasites compared to that for the untreated control (Fig. 8E). These observations indicate that T863 has strong parasiticidal activities.
As T863 inhibits mammalian DGAT1 (50), we next wanted to validate TgDGAT1 as a target for T863. Our attempts to purify Toxoplasma treated with 5 μM T863 to analyze the neutral lipid composition by thin-layer chromatography failed due to the fragility of the parasite membranes. We then designed experiments to examine whether T863 reduces neutral lipid storage and, thus, TgDGAT1 activity in intravacuolar Toxoplasma. We infected mouse embryonic fibroblasts (MEF) in which the DGAT1 and DGAT2 genes were knocked out (D1D2KO MEF) (51) with Toxoplasma in the presence of 15 μM T863 for 24 h. In D1D2KO MEF, untreated parasites grew to an average of 4 parasites per PV, and each contained LD. However, T863-treated parasites were dramatically altered in their morphology and contained no LD in their remnant cytoplasm, suggesting that T863 may target TgDGAT1 (Fig. 8F).
In parallel assays, we incubated infected fibroblasts with 15 μM T863 together with 0.2 mM OA for 24 h and stained them with BODIPY 493/503 to determine whether LD could be formed. An absence of the BODIPY signal would suggest that T863 interferes with lipid storage (i.e., DAG acylation), despite the presence of OA. This condition was then compared to one in which parasites were either exposed to 0.2 mM OA in the absence of the drug or incubated with 15 μM T863 without OA addition. The combination of T863 and OA resulted in very small PV each containing a single parasite or, rarely, 2 parasites, in contrast to PV exposed to either T863 or OA alone (Fig. 9A). Measurement of the volume of BODIPY 493/503-positive foci per parasite showed the absence of LD in parasites treated with 15 μM T863 with OA supplementation (Fig. 9B). When 0.5 mM OA was added to the medium with 15 μM T863 for 24 h, the parasites displayed severe growth defects to a similar extent as 80 μM T863-treated parasites (Fig. 8C), suggesting the potentiating effect of OA on treated parasites. These observations indicate that TAG storage is essential for Toxoplasma, especially in the presence of large amounts of exogenous OA.
FIG 9.
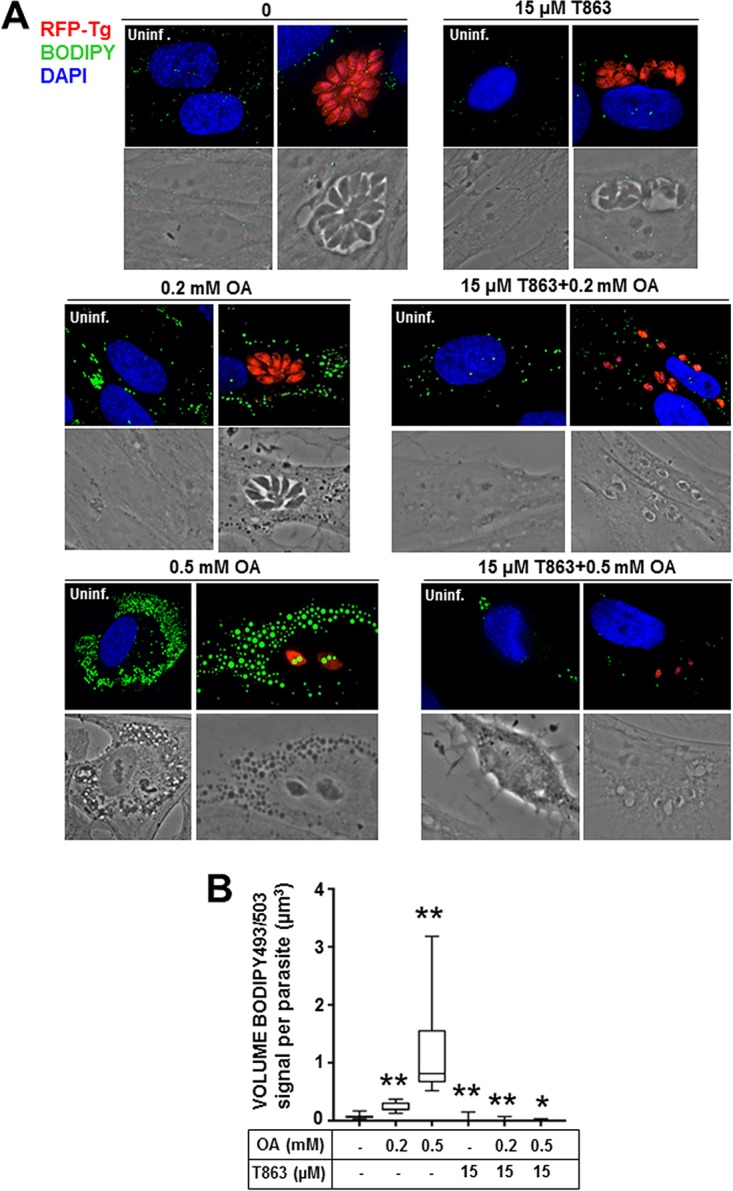
Lipid body abundance in Toxoplasma exposed to T863 and supplemental OA. (A) Fluorescence microscopy of HFF infected with RFP-expressing Toxoplasma incubated with excess OA and/or 15 μM T863 for 24 h and stained with BODIPY 493/503 (green). (B) Quantification of the BODIPY 493/503 signal in Toxoplasma cultivated as described in the legend to panel A. The graph shows the average LD volume, calculated using Volocity software, from BODIPY 493/503 staining. Data (mean volumes ± SD) acquired from three independent experiments. P values were determined by Dunnett's test. *, P < 0.0082; **, P < 0.0001.
Incubation of infected cells with T863 compromises Toxoplasma viability.
To assess the viability of parasites treated with the DGAT1 inhibitor T863, infected cells were exposed to various T863 concentrations (1, 15, or 40 μM) for 20 to 22 h and then incubated with the MitoTracker Red CM-H2Xros. This MitoTracker stains only live, functional mitochondria, as its accumulation in these organelles is dependent on the membrane potential, allowing the discrimination between live and dead cells. The intraparasitic mitochondrial signal significantly waned with increased T863 concentrations, indicative of parasite metabolism damage (Fig. 10A). As a negative control, the antiparasitic drug pyrimethamine, which inhibits dihydrofolate reductase and thus affects the parasite's metabolism (52), was used on infected cells, showing a negligible signal for mitochondria compared to that for the dimethyl sulfoxide (DMSO)-treated control. Infected and uninfected cells did not show any difference in MitoTracker staining at 40 μM T863, confirming the nontoxicity of the drug at this concentration on mammalian cells (Fig. 10B). Quantification of the fluorescence signals for mitochondria within Toxoplasma revealed an ∼15% decline in parasite viability at 1 μM T863 and an ∼70% decline with 7.5 μM the drug. Only 9% and 7% of the parasites were viable at 15 and 40 μM T863, respectively. Control conditions (medium with DMSO or without any addition) showed similar percentages of viable PV, corresponding to 96% and 93%, respectively, while pyrimethamine treatment led to 4% viable Toxoplasma PV. These observations establish that T863 selectively affects Toxoplasma metabolic activities at the lowest concentration of 15 μM, likely consecutively with impaired neutral lipid storage functions.
T863 treatment leads to the massive accumulation of membranous structures in the parasite cytoplasm.
To provide more insights into the cause of the growth arrest and rapid death of Toxoplasma upon T863 treatment, we conducted EM studies on infected cells. At 15 μM T863 for 24 h, the main cytopathy that was consistently observed in all parasites was the buildup of massive membranous tubules, which were packed in the parasite cytoplasm and residual body (Fig. 11A). Under this treatment condition, very few organelles were morphologically recognizable. In an attempt to clarify the origin of the intraparasitic membranous whorls, Toxoplasma parasites were treated with a lower concentration (7.5 μM) of T863. We observed that the ultrastructure of the ER was altered, displaying a swollen appearance, and the distended ER occupied a large volume of the cytoplasm. The nuclear envelope, as an extension of the ER network, was also abnormally dilated. Other organelles, however, were left undisturbed by the DGAT inhibitor (Fig. 11B). No LD were seen in treated parasites. These observations suggest that the parasite ER, wherein TgDGAT is localized (15), could be the prime target of T863, resulting in expansion of this compartment due to the improper acylation of lipids, leading to membrane biogenesis.
FIG 11.

Ultrastructure of Toxoplasma incubated with T863. (A to E) Transmission EM of Toxoplasma-infected HFF incubated without drug or with T863 at 7.5 or 15 μM for 24 h showing cytopathies, such as ER swelling and membranous tubules (yellow arrowheads). No cytopathies were observed in the host cell (hc). a, apicoplast; DG, dense granule; Go, Golgi apparatus; m, mitochondrion; mi, micronemes; n, nucleus; NE, nuclear envelope; RB, residual body. Bars, 300 nm.
Compared to parasites treated with 7.5 μM T863, those exposed to 15 μM the drug exhibited more organellar damage, fragmentation of the cytoplasm, and membrane accumulation, leading to impairment of endodyogeny and daughter formation (Fig. 11C). Some parasites accumulated osmiophilic membranous structures in the PV lumen (Fig. 11D, asterisk) similar to those observed in parasites exposed to 0.4 mM OA (Fig. 1D). In many parasites, the network of tubules initially originating from the ER became more dense and compact in the cytoplasm, reflecting an uncontrolled increase in membrane lipid syntheses (Fig. 11D, arrowheads), and upon rupture of the plasma membrane, these tubules were ultimately discharged loose in the PV lumen (Fig. 11E). No ultrastructural alterations were observed in host cells. These observations highlight the dramatic dysregulation in lipid homeostatic pathways and alterations in parasite membrane organization induced by T863, leading to Toxoplasma death.
Differentiated bradyzoites in culture contain large LD.
Although much is known about lipid metabolism in tachyzoites, the lipid biosynthetic and salvage pathways in the cyst forms are largely unknown. The thick cyst wall is a remnant of the PV membrane that guarantees long-term persistence in the infected host (53). It may then represent a considerable barrier to the diversion and scavenging of host nutrients by bradyzoites, although a study reported that the cyst wall is permeable for molecules up to 10 kDa (54). As a first approach to determine the importance of neutral lipids for chronic infections with T. gondii, we performed EM of differentiated bradyzoites within cultured intracellular cysts (55) to examine whether lipid stores, e.g., LD, are also formed by bradyzoites. Our ultrastructural observations of the ME49 and Prugniaud strains, both closely related to clinical isolates, revealed very large LD in bradyzoites with diameters up to 200 nm, indicative of massive lipid stores in this parasitic stage (Fig. 12A). These LD were closely surrounded by ER tubules, wherein TgACATs and TgDGAT are localized in tachyzoites (13, 15), suggesting that the LD in bradyzoites likely contain CE and TAG. Quantification of the neutral lipid content in differentiated bradyzoites isolated from fibroblasts confirmed that these forms contained significant amounts of free cholesterol, CE, and TAG (Fig. 12B). The presence of LD in bradyzoites was further confirmed by BODIPY 493/503 staining, showing several cytoplasmic fluorescent foci (Fig. 12C). Interestingly, upon the addition of OA to the culture medium, bradyzoites responded similarly to tachyzoites (Fig. 1) by forming more LD (Fig. 12C), suggesting that cyst forms are also capable of accessing exogenous sources of fatty acids and incorporating OA or OA-derived lipids into LD.
FIG 12.

Detection of neutral lipid stores in differentiated bradyzoites in culture. (A) EM of encysted bradyzoites (from the ME49 and Prugniaud [Pru] strains) differentiated for 4 or 7 days showing intracytoplasmic LD surrounded by ER elements. a, amylopectin granule; ap, apicoplast; CW, cyst wall; DG, dense granule; m, mitochondrion; n, nucleus; r, rhoptry; V, Vac compartment. Bars, 600 nm. (B) Quantification of cholesterol, CE, and TAG in encysted ME49 bradyzoites differentiated for 3 days after parasite release from host cells and purification as described previously (7). After washing, parasite lipid concentrations were measured by enzymatic and colorimetric assays. Data are means ± SD from 3 or 4 independent assays. (C) Fluorescence microscopy of encysted ME49 bradyzoites differentiated for 4 days and stained with BODIPY 493/503, TRITC-lectin for the cyst wall, and DAPI. OA at 0.2 or 0.4 mM was added to the medium at day 2 postdifferentiation, showing that the LD number is proportional to the OA concentrations in the medium. Parasite LD are intracytoplasmic, as evidenced by their corresponding images as dark inclusions seen on phase-contrast images (white squares) and by the boxed regions magnified in the orthogonal views (arrows). XY, optical slices taken in the center of the cyst; EF, extended-focus images combining multiple z-slice images.
T863 treatment leads to misshapen cysts and reduced LD in bradyzoites.
Based on the microscopic observations of large LD in bradyzoites, we next wanted to assess the sensitivity of ME49 bradyzoites to T863. In this assay, ME49 tachyzoites were first allowed to develop in fibroblasts overnight and were then incubated in a bradyzoite conversion medium (BCM) in a low-CO2 environment for 3 days to induce cystogenesis. At the end of the first day of bradyzoite conversion, infected cells were incubated with T863 or DMSO for the 2 remaining days. The conversion process was monitored by microscopy for cyst wall formation using TRITC-labeled lectin and for expression of BAG1, the main protein located at the plasma membrane of bradyzoites. The effect of drug treatment was monitored by quantifying the cyst size on the basis of TRITC-lectin labeling using Volocity software. Under control conditions, bradyzoites were BAG1 positive and, on the basis of phase-contrast images and DAPI staining, were densely packed within the cyst, which was delineated by a well-developed cyst wall. The sizes of the cysts were highly variable from one infected cell to another, ranging from very large cysts containing up to 100 parasites to small cysts with ∼5 to 10 parasites, perhaps as a result of asynchronous differentiation and/or second invasion events (Fig. 13A, panels a and b), as demonstrated by wide-ranging values for cyst volumes (Fig. 13B, panel a).
FIG 13.

Effect of T863 on differentiated bradyzoites in culture. (A) (Top) Schema outlining our experimental protocol. (a to f) Fluorescence microscopy of encysted ME49 bradyzoites differentiated for 3 days before staining with TRITC-lectin, DAPI, and BAG1 as a marker of the bradyzoite plasma membrane. At 40 to 42 h postinfection, 80 μM T863 or DMSO (control) was added to the BCM. Following treatment for 2 days, large and small cysts filled with parasites are detected (a and b) and cysts with misshapen parasites are observed under drug treatment (c to f). The images shown have the same magnification. Bars, 17 μm. (B) Quantification of cyst size upon T863 treatment, as described in the legend to panel A. (a) Box plot graphs from 3 independent experiments (40 to 46 cysts per experiment) showing cyst volumes, calculated using Volocity software, where cysts are detected on the basis of TRITC-lectin fluorescence, as described in Materials and Methods. P values were determined by a 2-tailed t test type 3. (b) Graph combining data from the 3 assays whose results are presented in panel a, representing the averaged cyst volume expressed as a percentage of that obtained with the DMSO control (means ± SD; P values were determined by 2-tailed t test type 3).
Upon parasite exposure to 5, 10, or 40 μM T863, no significant differences in cyst sizes were observed between control and treated parasites (not shown), in sharp contrast to the high sensitivity of tachyzoites to this DGAT1 inhibitor at low concentrations (Fig. 8). However, when the concentration of T863 was raised to 80 μM, the drug had severe deleterious effects on bradyzoites, as illustrated in phase-contrast images showing an empty area within the cyst matrix (Fig. 13, panels c to f). This phenotype was further confirmed by sparse DAPI staining, even in large cysts (Fig. 13A, panels c to d), and irregular or weak BAG1 labeling on bradyzoites in smaller cysts (Fig. 13A, panels e and f), indicative of dying parasites. Quantitative size measurement of treated cysts showed smaller cyst volumes (Fig. 13B, panel a), with a significant average reduction of ∼30% compared to the size of control cysts (Fig. 13B, panel b).
To verify the T863 specificity of action on TgDGAT1, we monitored the BODIPY 493/503 staining for LD formation in 80 μM T863-treated bradyzoites. After ME49 tachyzoite infection overnight, we induced bradyzoite conversion as well as LD biogenesis by incubating infected cells in BCM supplemented with 0.2 mM OA under low-CO2 conditions for 24 h prior to treatment with 80 μM T863 for 48 h. Many LD were detected within control parasites in small cysts (not shown) and very large cysts (Fig. 14A, panel a). In contrast, the BODIPY 493/503 staining was very weak in T863-treated cysts, revealing rare and minuscule LD (Fig. 14A, panels b and c). Quantification of the intensity of BODIPY 493/503 fluorescence, normalized to the cyst volume, showed a significantly reduced signal in T863-treated versus control cysts (Fig. 14B, panel a), with an average reduction of ∼50% (Fig. 14B, panel b). These data reveal that bradyzoites are vulnerable to T863, which impedes neutral lipid storage and results in growth defects, although sensitivity occurs at concentrations higher than those observed for tachyzoites.
FIG 14.

Effect of T863 on LD in differentiated bradyzoites in culture. (A) (Top) Schema outlining our experimental protocol. (a to c) Fluorescence microscopy of encysted ME49 bradyzoites before staining with BODIPY 493/503, TRITC-lectin, and DAPI, revealing numerous LD in control cysts (a), while no LD (b) or rare LD (c) were detected in T863-treated parasites. Parasite LD are intracytoplasmic, as evidenced in the orthogonal views (arrows). The images shown have the same magnification. Bars, 17 μm (z-slices) and 19 μm (orthogonal views). (B) Quantification of the BODIPY 493/503 signal in differentiated cysts upon T863 treatment. (a) Box plot graphs from 3 independent experiments (41 to 46 cysts per experiment) showing the sum of BODIPY 493/503 fluorescence per cyst volume corrected for the background as described in Materials and Methods. P values were determined by a 2-tailed t test type 3. (b) Graph combining data from the 3 assays whose results are presented in panel a, representing the averaged sum of the BODIPY 493/503 fluorescence normalized to the cyst volume and corrected for the background (means ± SD; P values were determined by 2-tailed t test type 3).
DISCUSSION
In this study, we examined the growth of Toxoplasma under conditions in which the fatty acid concentrations in the culture medium mimicked the physiological concentrations in human serum. We show that Toxoplasma has a selective avidity for unsaturated fatty acids and document the adverse effects resulting from the accumulation of these lipids within the parasite, including arrest in replication. These observations led us to investigate the importance of proper lipid storage in LD for Toxoplasma. We demonstrate that both the proliferative and encysted forms of the parasite are vulnerable to the DGAT1 inhibitor T863, which interferes with neutral lipid storage. More especially, T863 leads to severe cytopathies associated with abnormal membrane proliferation in the cytoplasm and, eventually, to the death of tachyzoites.
The unsaturated fatty acids OA, LA, and POA are profusely taken up by Toxoplasma and accumulate in gigantic LD, with the consequence of impaired replication. At 0.4 mM OA, the enzymatic activity for fatty acid storage and incorporation into TAG and CE is upregulated (7), but it appears to be insufficient, as illustrated by the massive accumulation of lipids outside LD. This lipid overload is deleterious not only for endodyogeny but also for egress, suggesting a broader weakening of parasite metabolism. At concentrations of 0.5 mM OA, TgDGAT1 expression no longer increases in response to the considerable influx of OA to the parasite, ultimately leading to parasite growth arrest and deterioration. In contrast, the saturated fatty acids PA and SA neither accumulate in LD nor impact the parasite replication rate. These effects of fatty acids on Toxoplasma are in sharp contrast to those of cholesterol and ceramides, both of which boost parasite replication when supplemented to the culture medium (10, 16).
Unexpectedly, this study has revealed that the fatty acids that affect Toxoplasma growth differ from those that impact mammalian cells. In mammalian cells, saturated fatty acids have a strong cytotoxic effect, while unsaturated fatty acids are not toxic, as they even prevent toxicity (56–60). OA is readily incorporated into TAG and induces neither an ER stress response nor peroxisomal H2O2 generation. Conversely, PA excess does not lead to TAG synthesis and induces cell apoptosis, as also reported for SA. Failure of the proper storage of excess lipids in LD causes chronic elevation of circulating fatty acids, which can reach toxic levels within nonadipose tissues due to the deleterious effects of lipid accumulation known as lipotoxicity (61). However, as similarly observed for Toxoplasma, unsaturated fatty acids become toxic for mammalian cells when TAG synthesis is impaired (62). For any cell, the ability to synthesize TAG in response to fatty acid supplementation is critical for protection from lipotoxicity and death. Neutral lipid storage is particularly vital for Toxoplasma, based on its high gluttony for many lipids provided in its environment.
In mammalian cells, cellular fatty acid homeostasis reflects an equilibrium between production processes (e.g., de novo synthetic pathways and hydrolysis of TAG/phospholipids by lipases and import) and utilization processes (e.g., for membrane biosynthesis, energy production through β-oxidation, generation of lipid signaling molecules, posttranslational protein modification, and transcriptional regulation) (63). Fatty acid metabolism in Toxoplasma consists of both synthetic and salvaging pathways (64–66). The parasite can synthesize several major fatty acids through three pathways: cytosolic fatty acid synthetic pathway I (FASI); a unique FASII pathway located in the plastid-like organelle, termed the apicoplast; and the fatty acid elongation system in the ER (66). Pharmacological interference with FASII prevents the completion of parasite cytokinesis; however, supplementation with FASII products, e.g., myristate, PA, or OA, can rescue the cytokinesis defect, which highlights the importance of fatty acid salvage pathways in Toxoplasma (18). However, Toxoplasma fails in controlling the influx of fatty acids when it is provided with large quantities in the culture medium, indicating its ineptness in balancing synthesis, salvage, and the utilization of fatty acids.
Mammalian cells import fatty acids through protein- and non-protein-mediated mechanisms (63). The mechanism of uptake of fatty acids by Toxoplasma is still unknown. The genome of the parasite encodes a variety of acyl-CoA synthases and acyl-CoA binding proteins that might act in the salvage and transport of fatty acids. Among the mammalian fatty acid transporters, the most characterized are the fatty acid binding protein 1/2/3/4 (FABP1/2/3/4), FATP1, and CD63. Toxoplasma contains several genes that exhibit similarity to some of the genes for these proteins, including genes for homologs of mammalian FABP2 (70% identity), FABP3 (70% identity), and FABP4 (84% identity), which may function in fatty acid uptake/membrane translocation.
Toxoplasma exploits many sources of host-derived fatty acids: fatty acids exogenously introduced into the medium, fatty acids synthesized by the host cell, and fatty acids stored in LD by either engulfing host LD into the PV (6, 7) or stimulating lipophagy to liberate fatty acids from TAG (67). The parasite incorporates fatty acids into proteins posttranslationally (68). This is particularly relevant for PA, as palmitoylated proteins are abundant in Toxoplasma and include essential proteins involved in motility (myosin light chain 1, myosin A), cell morphology (PhIL1), and host cell invasion (apical membrane antigen 1 [AMA1]). PA is also a precursor for the de novo synthesis of sphingolipids in T. gondii (69), and various saturated and unsaturated fatty acids are incorporated into phospholipids (70). However, Toxoplasma cannot use fatty acids as sources of ATP, as it lacks homologs encoding adipose triglyceride lipase, which breaks down TAG, fatty acid transporters to the mitochondria (e.g., carnitine palmitoyltransferases), and enzymes for fatty acid oxidation including 3-ketoacyl coenzyme A thiolase, which is a key enzyme in the mitochondrial matrix that generates acetyl-CoA. This uncovers another point of vulnerability for the parasite, as mammalian cells could prevent lipotoxicity by channeling fatty acids to the mitochondrial β-oxidation pathway, which is upregulated with excess fatty acids via the stimulation of the proliferator activated receptor peroxisome proliferator-activated receptor alpha (71). Abounding LD in mammalian cells can also be degraded by lipophagy (72) or expelled to the medium (73). These two processes of LD elimination need to be investigated for Toxoplasma. However, our EM observations on parasites exposed to excess OA did not show any evidence of parasite LD in autophagosomal structures or in the PV lumen. Furthermore, the overload of OA in Toxoplasma leads to irreversible damage, as parasite replication does not resume following transfer to normal medium, suggesting that the parasite is unable to cope with the accumulated LD and return to a normal physiological state.
Under conditions of added OA, the parasite still recruits host organelles, suggesting that these activities may have occurred prior to the threshold for lipotoxicity being reached. Nevertheless, by attracting host LD and Rab vesicles, Toxoplasma may be even more prone to lipid overload, due to its ability to retrieve their lipid-rich cargo (7, 16, 43). The lack of sensitivity of Toxoplasma toward supplemental PA is surprising, considering that 0.1 mM PA in mammalian cells induces oxidative and ER stress through morphological alterations of mitochondria and ER, leading to apoptosis (49, 74, 75). In the hypothesis that the host mitochondrion and ER association with the PV is important for the intracellular development of the parasite, the questions of why Toxoplasma recruits these dysfunctional organelles and how it survives in PA-damaged cells are thus perplexing.
Upon supplementation of OA, the host mitochondria associated with the PV membrane are morphologically aberrant, while the free cytosolic mitochondria display normal features. Those attached to the PV membrane are enlarged, with a completely disorganized inner membrane topology, evidenced by concentric stacks of inner membrane or honeycomb configurations. In many organisms, the ultrastructure of mitochondria reflects the bioenergetic state of these organelles. This mitochondrial swelling with a distended outer membrane could be a sign of an alteration in the dynamic cycle of mitochondria and the absence of scission. However, it seems unlikely that host mitochondria that are stably anchored to the PV membrane by parasite proteins, e.g., MAF1 (76), could undergo normal cycles of fusion, elongation, and scission. It is known that the molecular composition of the mitochondrial inner membrane could be dramatically modified in the presence of excess OA. Compared to other biological membranes, the mitochondrial inner membrane has an extremely high ratio of protein to lipid mass (75:25), with the cristae being enriched in proteins involved in oxidative phosphorylation, iron/sulfur cluster biogenesis, protein synthesis, and transport of mitochondrial DNA-encoded proteins (77). An attribute of mitochondrial cristae is to minimize the distance between adenine nucleotide translocation and transformation sites to enable the highest possible efficiency for ATP production. Therefore, any changes in the size and shape of cristae may reflect a dysregulation in the rate of ATP production. Abnormal angular cristae could be caused by either an altered protein/lipid composition or an abnormal assembly of proteins, such as ATP synthase, which is involved in the curvature and inner membrane folding (78). Interestingly, the Drosophila ATP6 mutant possesses aberrant mitochondria with a honeycomb appearance, a phenotype that is accompanied by diminished respiration and enhanced production of reactive oxygen species (ROS) (79). The shape of the mitochondrial inner membrane can also be modified by cardiolipins, acidic mitochondrial phospholipids with four fatty acyl side chains (80). These lipids are subjected to a high specificity in fatty acyl remodeling, depending on the cell type, with LA being the predominant lipid of this type. In mammalian mitochondria, the fatty acyl composition of cardiopilins can be influenced by lipids in the environment, e.g., the organism's diet, and any alterations in cardiolipin composition have pathological consequences (81). In the presence of excess OA, LA may be replaced by OA during cardiolipin synthesis in mitochondria, resulting in the abnormal morphological features of these organelles. The physical force of mitochondria attached to the PV membrane may rend these host organelles more susceptible to potential modifications of molecules involved in cellular mitochondrial maintenance and function. We recently demonstrated that Toxoplasma is sensitive to pharmacological inhibition of host CPT-1 and 3-KAT in the fatty acid β-oxidation pathway (7), suggesting that the parasite may take advantage of fatty acids and/or the ATP produced in host mitochondria. However, another study on Toxoplasma-infected fibroblasts showed that blocking of host mitochondrial β-oxidation results in the promotion of parasite growth, presumably due to the increased availability of fatty acids for Toxoplasma (67). These authors suggest a competition between the parasite and host mitochondria for fatty acid acquisition. Though infected cells may limit the access of the parasite to fatty acids by increasing β-oxidation activities, the excessive amounts of unsaturated fatty acids taken up by Toxoplasma are clearly detrimental for its growth, due to its limited storage capacity and inability to oxidize fatty acids to generate ATP.
The abnormal ultrastructure of host mitochondria associated with the PV membrane upon OA supplementation is suggestive of malfunctions in these organelles; thus, perhaps fewer mitochondrial metabolites are available for the parasite. An alternative plausible cause of parasite growth defects in OA-treated cells could be linked to the enhanced production of mitochondrial reactive oxygen species (ROS) within the electron transport chain as a result of increased activities of fatty acid oxidation enzymes (82). It remains to be examined whether host mitochondria associated with the PV membrane are particularly prone to ROS generation, resulting in the destruction of the parasite by oxygen free radicals diffusing inside the PV.
We previously showed that Toxoplasma is vulnerable to disruptions in cholesterol storage pathways: in the absence of TgACAT activity, the parasite accumulates free cholesterol in its membranes, which become fragile, leading to death (13, 14). The effect of the specific DGAT1 inhibitor T863, which competes with oleoyl-CoA to bind DGAT1 and block the DGAT1 function (50), is particularly dramatic for Toxoplasma, as it induces impaired metabolism and growth arrest at very low concentrations. In comparison, mammalian cells can tolerate DGAT1 inhibition or DGAT1 deletion because they can produce TAG through a second enzyme, DGAT2. Strong indirect evidence that T863 targets TgDGAT is supported by (i) the absence of LD in T863-treated parasites by EM observations, confirmed by the loss of the BODIPY 493/503 signal by fluorescence microscopy; (ii) the induction of an abnormal expansion of the ER, where TgDGAT localizes as an initial cytopathy; (iii) the faster parasite death in cells lacking DGAT1 than in wild-type cells at the same T863 concentration; and (iv) the exacerbation of the cidal effect of T863 upon OA supplementation. The higher vulnerability of Toxoplasma cells to T863 than mammalian cells is in accordance with the nonviability of parasites from which TgDGAT, the single gene copy for TAG synthesis, is deleted.
Remarkably, T863-treated parasites build up massive amounts of intracellular membranes, which are stockpiled in the cytoplasm, which likely impedes normal endodyogeny. One plausible explanation could be that, in the face of TgDGAT inhibition, DAG may be diverted to the synthesis of phosphatidylcholine and phosphatidylethanolamine through the Kennedy pathway, with the massive flux of oleoyl acyl-CoA joining newly synthesized phospholipids. To this point, upon T863 treatment, an increase in phosphatidylcholine content has been observed in mammalian cells, but this extra production of phosphatidylcholine has been proven not to be cytotoxic (50). Conversely, mammalian cells overexpressing DGAT1 have decreased acylation and de novo synthesis of phospholipids (83). The amassing of intracellular membranes in the cytoplasm when TAG production is impaired reveals an Achilles' heel of Toxoplasma.
So far, very few studies have been undertaken on the lipid sources and lipid uptake pathways developed by Toxoplasma cyst forms. EM analyses on cyst forms isolated from mouse brains reveal that the cyst wall is 240 nm thick but contains 30-nm circular openings on its surface (54) which allow interchanges with the host cytoplasm and the passage of host macromolecules to the cyst matrix. A recent study reported that T. gondii bradyzoites reorganize the host cytoskeleton and attract host organelles, which distribute around the cyst wall, although to an extent less than that observed for tachyzoites (84). Moreover, exposing converting bradyzoites to 0.2 mM OA leads to LD biogenesis, indicating that the parasite is able to scavenge, deliver to the cyst matrix, and store in LD host-derived fatty acids. Interestingly, LD are particularly large in bradyzoites, which may suggest that cysts rely on lipid stores to survive, as analogously demonstrated for sugar stores, e.g., amylopectin granules that are plentiful in bradyzoites (85). Interfering with the storage of OA-derived lipids impacts bradyzoite development, leading to parasite malformations within the cysts. However, the cyst forms of the ME49 strain are less sensitive to T863 than the tachyzoites of the RH strain. A possible explanation for this reduced sensitivity may be the decreased replication rate of type II (ME49) versus type I (RH) strains and the lower growth rate of bradyzoites, associated with their more quiescent metabolism, than of tachyzoites. A logical inference about these differences in phenotypes would be that bradyzoites require fewer nutrients than tachyzoites to grow and would therefore be less sensitive to nutrient deprivation, yet bradyzoite energy reserves, such as lipid and sugar stores, are likely to be critical for long-term persistence in the host and reactivation into tachyzoites in case of changes in the (immune) status of the host. To this point, it has recently been reported that manipulating the digestion and storage of amylopectin in bradyzoites leads to fewer cysts in mouse brains (86).
Inhibiting TgDGAT1 enzymatic activity to interfere with energy storage in Toxoplasma may be a reasonable therapeutic intervention against toxoplasmosis, including the control of the chronic stage of infection, without incurring a toxicity risk for mammalian cells.
MATERIALS AND METHODS
Reagents and antibodies.
All chemicals were obtained from Sigma (St. Louis, MO) or Thermo Fisher Scientific (Waltham, MA) unless otherwise stated. BODIPY 493/503 (4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene) was purchased from Life Technologies (Carlsbad, CA). T863 {trans-4-[4-(4-amino-7,7-dimethyl-7H-pyrimido[4,5-b][1,4]oxazin-6-yl)phenyl]cyclohexane acetic acid} was obtained from AstraZeneca (Cambridge, UK). Secondary antibodies used for immunofluorescence were conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 350 and were purchased from Invitrogen (Carlsbad, CA). The following primary antibodies were used: rabbit polyclonal anti-GRA7 antibody (47), rabbit monoclonal anti-Tom20 mouse polyclonal (Santa Cruz Biotechnology Inc., Santa Cruz, CA), and mouse monoclonal BAG1 (a generous gift of J.-F. Dubremetz, Université de Montpellier). For OA preparations, sodium oleate was dissolved in H2O at a concentration of 100 mM and then thoroughly mixed by vortexing with 5% fatty acid-free bovine serum albumin (BSA) for a final concentration of 10 mM to ensure BSA-OA complex formation, and the mixture was stored in the dark at 4°C. For PA preparations, sodium palmitate was dissolved in 50% ethanol at a concentration of 100 mM, and OA, SA, POA, LA, and jasmonic acid were prepared similarly to PA but were initially dissolved in 100% ethanol.
Cell lines and culture conditions.
Human foreskin fibroblasts (HFF) and HeLa cells were obtained from the American Type Culture Collection (Manassas, VA). Mouse embryonic fibroblasts (MEF) lacking both DGAT1 and DGAT2 (D1D2KO MEF) were generously provided by Robert Farese (Harvard University, MA) and were grown in Dulbecco's modified Eagle medium (DMEM). All cell lines were grown as monolayers, cultivated in α-minimum essential medium (α-MEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and penicillin-streptomycin (100 units/ml per 100 μg/ml), and maintained at 37°C in 5% CO2 unless specified otherwise.
Parasite cultivation and differentiation.
The tachyzoites from the RH strain (type I lineage) were used throughout this study. In one set of experiments, Toxoplasma parasites (of the RH strain) stably expressing RFP were generously provided by Florence Dzierszinski (McGill University, Montreal, Canada). The cystogenic strains ME49 and Prugniaud (type II lineage; kindly provided by David Roos, University of Pennsylvania) were used to examine lipid stores in differentiated cysts in cultured cells. RH, Prugniaud, and ME49 tachyzoites were propagated in vitro by serial passage in monolayers of HFF (87). To monitor tachyzoite-bradyzoite stage conversion in vitro, coverslips with confluent Toxoplasma-infected HFF were incubated first for 24 h in 5% CO2 (normal culture conditions) and then incubated in bradyzoite conversion medium (BCM), made of RPMI 1640 without NaHCO3 containing 50 mM HEPES (pH 8.2), 3% FBS, and penicillin-streptomycin (100 units/ml per 100 μg/ml), in the absence of CO2 for 3 to 7 days.
Parasite development.
To monitor tachyzoite development, plaque assays were performed using HFF grown until confluence in 6-well plates, infected with 100 Toxoplasma RH parasites per well, and incubated at 37°C for 5 days with excess OA, PA, SA, POA, LA, or JA or in the absence of fatty acids added to the culture medium (control condition). The cells were fixed and stained as described previously (9). The plates were scanned (ScanWizard 5; MicroTek) to calculate the average area of the plaques using Volocity software and graphed in Excel software (Microsoft). Bradyzoite growth was quantified by measurement of cyst volumes. The cysts were identified by staining for the cyst wall using a lectin from Dolichos biflorus conjugated to TRITC (TRITC-lectin). Using Volocity software, the cysts were detected using the Find Objects by Fluorescence Intensity Measurement protocol and the TRITC channel (thresholds were set manually) with a minimum object size of 5 μm, and the volume of the cyst was calculated.
Parasite replication and predictive replication analysis.
Tachyzoite replication was assayed by either uracil incorporation assays or parasite enumeration per PV. For uracil assays, HFF were grown until confluence in 24-well plates prior to infection with 1 × 105 parasites for 20 h at 37°C in α-MEM. Cells were then incubated with 1 μCi of [3H]uracil for 4 h at 37°C, and the samples were processed as described previously (88). To assess the parasite number per PV, coverslips with confluent HFF were infected with RFP-expressing Toxoplasma RH for 30 min, thoroughly washed with phosphate-buffered saline (PBS) to remove extracellular parasites, and incubated with the BSA control or various concentrations of fatty acids for 24 h. Coverslips were fixed with 4% formaldehyde plus 0.02% glutaraldehyde in PBS for 15 min and viewed with a Nikon Eclipse 90i microscope equipped with an oil-immersion Plan Apo 100× objective (numerical aperture [NA], 1.4) and a Hamamatsu GRCA-ER camera (Hamamatsu Photonics, Hamamatsu, Japan). The number of parasites was recorded for at least 1,000 PVs on the coverslip for each condition. The means of the means for each condition are graphed as a percentage of the values for all PVs recorded with standard deviations (SD) using Excel (Microsoft) software.
To further quantify the replicative capacity of Toxoplasma tachyzoites under various conditions, i.e., in the presence of different fatty acids, we created a model wherein the precise growth rate of the parasite could be determined using the data acquired from parasite enumeration per PV.
(i) Weighted average.
A weighted average formula for the average number of parasites per vacuole (PPVavg) was utilized to describe the mean growth of Toxoplasma, calculated on the basis of the number of parasites per vacuole (PPV) at time Texp, which represents the duration of an experiment (in hours). This model also incorporates G, the positive growth coefficient associated with this exponential model, and for Toxoplasma, G is equal to 2, due to its doubling property. If Rmax is the maximum number of replications that occur in an experiment, the number of Toxoplasma parasites per vacuole, Sn, at a given time is then defined by the function f(n) = Gn, where 0 ≤ n ≤ Rmax. Furthermore, in the experiments performed, Rmax did not exceed 4. The number and relative proportions of PPV for each Sn were ascertained by counting the number of parasites per PV under various conditions and categorizing them. If Sp represents the percentage of PPV at each Sn, Sn · Sp = Snw, where Snw is the weighted contribution at stage Sn to PPVavg. The formula for PPVavg can then be expressed by the following:
(ii) Replication period.
By utilizing the calculated weighted average, PPVavg, and the positive growth coefficient for Toxoplasma, G, we can then calculate the mean number of replications per experiment, Rexp, by using logarithmic properties. The number of replications per experiment can be expressed using the growth formula = PPPavg. With G and PPVavg already being defined, this equation can be solved using the exponent rule of logarithms to remove the exponents from the equation ln() = Rexp · ln(G).
After extracting Rexp from the exponent using the natural log, the equation is Rexp · ln(G) = ln(PPVavg).
By dividing both sides of the equation by ln(G), the number of replications per experiment is expressed as Rexp = ln(PPVavg)/ln(G).
The replication period, Rt, is then subsequently calculated by dividing the total time of the experiment, Texp, by the number of replications per experiment, Rexp, as follows: Rt = Texp/Rexp.
(iii) Growth projection.
With the replication period defined, Rt can be used to project the mean PPV at time T (in hours), PPVn. If PPVi is the initial number of parasites per vacuole, PPVi is equal to 1 since all vacuoles begin with a single parasite. PPVn can then be projected by the following equation: PPVn = PPVi · G(T/Rt).
Parasite egress.
For natural egress assays, HFF were grown until confluence in a 6-well plate and 5 × 106 parasites per well were allowed to invade the cells for 1 h. Noninvading parasites were then washed away and infected cells were incubated for up to 76 h in the presence of 0.2 or 0.4 mM OA. Cells were fixed and observed using an inverted microscope (model TE200; Nikon) equipped with a differential interference contrast (DIC) filter. Images were taken using Spot Advanced software (Diagnostic Instruments, Inc.) and processed using the iMovie program (Apple). For induced egress assays, HFF were grown until confluence in 6-well plates, 5 × 106 parasites per well were allowed to invade the cells for 1 h, and the cells were washed and incubated with 0.2 mM or 0.4 mM OA or without OA for 24 h. Egress was induced by adding the calcium ionophore A23187 (10 μM) (89), and the time taken to egress was recorded by live image capture using an inverted microscope (model TE200; Nikon). The time (in seconds) taken for individual PV to egress from their host cells was recorded using Spot Advanced software and graphed as a ratio of the value under conditions with OA to the value under control conditions (no OA added) in Excel software using the means of means and standard deviations.
Real-time quantitative PCR (qPCR) for transcriptional analyses for TgDGAT.
Confluent HFF were infected with Toxoplasma for 30 min, followed by PBS washes to remove extracellular parasites, and were then incubated with 0.5 mM OA for 24 h. The RNA was extracted and amplified as previously described (7). One hundred nanograms of cDNA, 2.25 μl each of the forward and reverse primers, and 17.5 μl of PowerUp Sybr Green master mix (Applied Biosystems, Foster City, CA) were run on StepOne Plus real-time PCR systems (Applied Biosystems). The data for TgDGAT were normalized to those for the TgGT1 housekeeping gene, and ΔΔCT values were calculated using as the control RNA from cells not exposed to OA in order to assess the relative transcript levels of TgDGAT. The following primers were used: for TgDGAT, forward primer GGA AGT GTG CTA TCC CTT ACA C and reverse primer CTC CCT TAC CAA AGC CGA TAA T, and for TgGT1, forward primer GGC TAT TTT GGC ACC TTT CA and reverse primer AAC GGG AAG ACA AAC CAC AG.
Determination of neutral lipid composition.
Total TAG, cholesteryl ester, and cholesterol contents in cultured bradyzoites isolated from mammalian cells (7) were measured by enzymatic and colorimetric assays using a reagent mixture from Roche Diagnostics (Indianapolis, IN, USA).
Fluorescence microscopy.
Immunofluorescence assays (IFA) on HFF were performed as described previously (90) with a 5-min permeabilization step with 0.3% Triton X-100 in PBS following fixation with 4% formaldehyde (Polysciences, Warrington, PA) plus 0.02% glutaraldehyde in PBS for 15 min. Detection was performed with anti-GRA7 (1:500), anti-Tom20 (1:100), or anti-BAG1 (1:200) diluted in PBS containing 3% BSA, followed by Alexa Fluor-conjugated IgG diluted 1:2,000 in PBS containing 3% BSA. BODIPY 493/503 (a 1:100 dilution in PBS) or TRITC-lectin (a 1:250 dilution in PBS containing 3% BSA) labeling was performed after the secondary incubation step for 30 min, followed by three 5-min PBS washes prior to staining with DAPI (4′,6-diamidino-2-phenylindole). Coverslips were mounted using ProLong Diamond antifade mountant (Life Technologies) to minimize bleaching during microscopy. Cells were viewed with a Nikon Eclipse 90i microscope equipped with an oil-immersion Plan Apo 100× objective (NA, 1.4) and a Hamamatsu GRCA-ER camera (Hamamatsu Photonics, Hamamatsu, Japan). Optical z-sections with 0.2-μm spacing were acquired using Volocity software (PerkinElmer, Waltham, MA). The images were deconvolved using an iterative restoration algorithm, and the registry was corrected using Volocity software. Volocity software and Photoshop software (Adobe) were used to adjust the brightness levels and to crop and resize the images obtained. To quantify the host organelle-PV interaction, we used the quantitative image analysis software MetaScopics, using the Intensity by Distance algorithm or the Centroid to Surface Distance algorithm as previously described (45). The quantification of the BODIPY 493/503 signal within Toxoplasma (tachyzoites) was performed using Volocity software: the Crop tool was used to delineate the PV, and the Find Object tool was used to highlight the BODIPY staining. This was followed by the calculation of the sum of the BODIPY volume by the program and graphing of the results. The BODIPY 493/503 signal in Toxoplasma cysts was measured by first using the Find Objects by Fluorescence Intensity measurement protocol to highlight the cyst, as described above. Then, the sum of the BODIPY 493/503 fluorescence intensity within the cyst was measured. To normalize to the size of the cyst, the sum intensity value was divided by the cyst volume. Finally, to subtract the background fluorescence, the sum of the BODIPY 493/503 fluorescence intensity was measured for a region of interest (ROI) of known size placed in an area of background fluorescence. This value was divided by the volume of the ROI and then subtracted from the value associated with the cyst for each image. The level of detection of the MitoTracker signal within the parasite was done using Volocity software, where the Colocalization tool was used to detect areas where the green fluorescence from the parasite overlapped the red fluorescence of the mitochondria. If no colocalization was observed, the parasites were deemed not viable.
Electron microscopy.
For transmission EM, HFF infected with tachyzoites (RH strain) or bradyzoites (ME49 and Prugniaud strains) were fixed in 2.5% glutaraldehyde (EM grade; Electron Microscopy Sciences, Hatfield, PA) in 0.1 M sodium cacodylate buffer (pH 7.4) for 1 h at room temperature and processed as described previously (91), before examination with a Philips CM120 electron microscope (Eindhoven, the Netherlands) under 80 kV.
Statistical methods.
Data are displayed in box plots using Kaleidagraph software (Synergy Software) or graphed in Excel software (Microsoft). Means and standard deviations were calculated from three independent experiments using Excel software (Microsoft). P values were calculated using Student's t test in Excel software.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully thank the members of the I. Coppens laboratory for helpful discussions during the course of this work and the technical staff from the Johns Hopkins Microscopy Facility. We acknowledge the generous providers of plasmids and cell lines.
This study was supported by NIH grant AI060767 and AI138714 to I.C.
The funder agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00347-18.
REFERENCES
- 1.Coppens I. 2013. Targeting lipid biosynthesis and salvage in apicomplexan parasites for improved chemotherapies. Nat Rev Microbiol 11:823–835. doi: 10.1038/nrmicro3139. [DOI] [PubMed] [Google Scholar]
- 2.Ramakrishnan S, Serricchio M, Striepen B, Bütikofer P. 2013. Lipid synthesis in protozoan parasites: a comparison between kinetoplastids and apicomplexans. Prog Lipid Res 52:488–512. doi: 10.1016/j.plipres.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramakrishnan S, Docampo MD, MacRae JI, Ralton JE, Rupasinghe T, McConville MJ, Striepen B. 2015. The intracellular parasite Toxoplasma gondii depends on the synthesis of long-chain and very long-chain unsaturated fatty acids not supplied by the host cell. Mol Microbiol 97:64–76. doi: 10.1111/mmi.13010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naemat A, Elsheikha HM, Al-Sandaqchi A, Kong K, Ghita A, Notingher I. 2015. Analysis of interaction between the apicomplexan protozoan Toxoplasma gondii and host cells using label-free Raman spectroscopy. Analyst 140:756–764. doi: 10.1039/C4AN01810A. [DOI] [PubMed] [Google Scholar]
- 5.Ihara F, Nishikawa Y. 2014. Starvation of low-density lipoprotein-derived cholesterol induces bradyzoite conversion in Toxoplasma gondii. Parasit Vectors 7:248. doi: 10.1186/1756-3305-7-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X, Binns D, Reese ML. 2017. The coccidian parasites Toxoplasma and Neospora dysregulate mammalian lipid droplet biogenesis. J Biol Chem 292:11009–11020. doi: 10.1074/jbc.M116.768176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nolan SJ, Romano JD, Coppens I. 2017. Host lipid droplets: an important source of lipids salvaged by the intracellular parasite Toxoplasma gondii. PLoS Pathog 13:e1006362. doi: 10.1371/journal.ppat.1006362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mota LA, Roberto Neto J, Monteiro VG, Lobato CS, Oliveira MA, da Cunha M, D'Ávila H, Seabra SH, Bozza PT, DaMatta RA. 2014. Culture of mouse peritoneal macrophages with mouse serum induces lipid bodies that associate with the parasitophorous vacuole and decrease their microbicidal capacity against Toxoplasma gondii. Mem Inst Oswaldo Cruz 109:767–774. doi: 10.1590/0074-0276140119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomes AF, Magalhães KG, Rodrigues RM, de Carvalho L, Molinaro R, Bozza PT, Barbosa HS. 2014. Toxoplasma gondii-skeletal muscle cells interaction increases lipid droplet biogenesis and positively modulates the production of IL-12, IFN-γ and PGE2. Parasit Vectors 7:47. doi: 10.1186/1756-3305-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coppens I, Sinai AP, Joiner KA. 2000. Toxoplasma gondii exploits host low-density lipoprotein receptor-mediated endocytosis for cholesterol acquisition. J Cell Biol 149:167–180. doi: 10.1083/jcb.149.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdelmagid SA, Clarke SE, Nielsen DE, Badawi A, El-Sohemy A, Mutch DM, Ma DW. 2015. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS One 10:e0116195. doi: 10.1371/journal.pone.0116195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonda S, Ting LM, Novak S, Kim K, Maher JJ, Farese RV Jr, Ernst JD. 2001. Cholesterol esterification by host and parasite is essential for optimal proliferation of Toxoplasma gondii. J Biol Chem 276:34434–34440. doi: 10.1074/jbc.M105025200. [DOI] [PubMed] [Google Scholar]
- 13.Nishikawa N, Quittnat F, Stedman TT, Voelker DR, Choi JY, Zahn M, Yang M, Joiner KA, Coppens I. 2005. Cholesteryl ester synthesis and lipid body biogenesis in Toxoplasma are controlled by host lipids: characterization of two novel isoforms of ACAT. Cell Microbiol 7:849–867. doi: 10.1111/j.1462-5822.2005.00518.x. [DOI] [PubMed] [Google Scholar]
- 14.Lige B, Sampels V, Coppens I. 2013. Characterization of a second sterol-esterifying enzyme in Toxoplasma highlights the importance of cholesterol storage pathways for the parasite. Mol Microbiol 87:951–967. doi: 10.1111/mmi.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quittnat F, Nishikawa Y, Stedman TT, Voelker DR, Choi JY, Zahn MM, Murphy RC, Barkley RM, Pypaert M, Joiner KA, Coppens I. 2004. On the biogenesis of lipid bodies in ancient eukaryotes: synthesis of triacylglycerols by a Toxoplasma DGAT1-related enzyme. Mol Biochem Parasitol 138:107–122. doi: 10.1016/j.molbiopara.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Romano JD, Sonda S, Bergbower E, Smith ME, Coppens I. 2013. Toxoplasma gondii salvages sphingolipids from the host Golgi through the rerouting of selected Rab vesicles to the parasitophorous vacuole. Mol Biol Cell 24:1974–1995. doi: 10.1091/mbc.e12-11-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luft BF, Remington JS. 1992. Toxoplasma encephalitis in AIDS. Clin Infect Dis 15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 18.Nissapatorn V, Lee C, Quek KF, Leong CL, Mahmud R, Abdullah KA. 2004. Toxoplasmosis in HIV/AIDS patients: a current situation. Jpn J Infect Dis 57:160–165. [PubMed] [Google Scholar]
- 19.Tenter AM, Heckeroth AR, Weiss M. 2000. Toxoplasma gondii: from animals to humans. Int J Parasitol 30:1217–1258. doi: 10.1016/S0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furrer H, Opravil M, Bernasconi E, Telenti A, Egger M. 2000. Stopping primary prophylaxis in HIV-1-infected patients at high risk of Toxoplasma encephalitis. Swiss HIV Cohort Study. Lancet 355:2217–2218. [DOI] [PubMed] [Google Scholar]
- 21.Dannemann BR, Israelski DM, Remington JS. 1988. Treatment of toxoplasmic encephalitis with intravenous clindamycin. Arch Intern Med 148:2477–2482. [PubMed] [Google Scholar]
- 22.Anderson-White BR, Beck JR, Chen CT, Meissner M, Bradley PJ, Gubbels MJ. 2012. Cytoskeleton assembly in Toxoplasma gondii cell division. Int Rev Cell Mol Biol 298:1–31. doi: 10.1016/B978-0-12-394309-5.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legrand P, Rioux V. 2010. The complex and important cellular and metabolic functions of saturated fatty acids. Lipids 45:941–946. doi: 10.1007/s11745-010-3444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yen CLE, Stone SJ, Koliwad S, Harris C, Farese RV. 2008. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49:2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blackman MJ, Carruthers VB. 2013. Recent insights into apicomplexan parasite egress provide new views to a kill. Curr Opin Microbiol 16:459–464. doi: 10.1016/j.mib.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, Miller AK, Vidal-Puig A, Teleman AA. 2015. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 525:124–128. doi: 10.1038/nature14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weller PF, Monahan-Earley RA, Dvorak HF, Dvorak AM. 1991. Cytoplasmic lipid bodies of human eosinophils. Subcellular isolation and analysis of arachidonate incorporation. Am J Pathol 138:141–148. [PMC free article] [PubMed] [Google Scholar]
- 28.Peng BW, Lin JY, Zhang T. 2008. Toxoplasma gondii induces prostaglandin E2 synthesis in macrophages via signal pathways for calcium-dependent arachidonic acid production and PKC-dependent induction of cyclooxygenase-2. Parasitol Res 102:1043–1050. doi: 10.1007/s00436-007-0873-4. [DOI] [PubMed] [Google Scholar]
- 29.De Fabiani E. 2011. The true story of palmitoleic acid: between myth and reality. Eur J Lipid Sci Technol 113:809–811. doi: 10.1002/ejlt.201100187. [DOI] [Google Scholar]
- 30.Sampath H, Ntambi JM. 2005. The fate and intermediary metabolism of stearic acid. Lipids 40:1187–1191. doi: 10.1007/s11745-005-1484-z. [DOI] [PubMed] [Google Scholar]
- 31.Ahmad P, Rasool S, Gul A, Sheikh SA, Akram NA, Ashraf M, Kazi AM, Gucel S. 2016. Jasmonates: multifunctional roles in stress tolerance. Front Plant Sci 7:813. doi: 10.3389/fpls.2016.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gold D, Pankova-Kholmyansky I, Fingrut O, Flescher E. 2003. The antiparasitic actions of plant jasmonates. J Parasitol 89:1242–1244. doi: 10.1645/GE-94R. [DOI] [PubMed] [Google Scholar]
- 33.Raviv Z, Cohen S, Reischer-Pelech D. 2013. The anti-cancer activities of jasmonates. Cancer Chemother Pharmacol 71:275–285. doi: 10.1007/s00280-012-2039-z. [DOI] [PubMed] [Google Scholar]
- 34.Ferreira-da-Silva MF, Takács AC, Barbosa HS, Gross U, Lüder CG. 2009. Primary skeletal muscle cells trigger spontaneous Toxoplasma gondii tachyzoite-to-bradyzoite conversion at higher rates than fibroblasts. Int J Med Microbiol 299:381–388. doi: 10.1016/j.ijmm.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Dzierszinski F, Nishi M, Ouko L, Roos DS. 2004. Dynamics of Toxoplasma gondii differentiation. Eukaryot Cell 3:992–1003. doi: 10.1128/EC.3.4.992-1003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skariah S, McIntyre MK, Mordue DG. 2010. Toxoplasma gondii: determinants of tachyzoite to bradyzoite conversion. Parasitol Res 107:253–260. doi: 10.1007/s00436-010-1899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boothroyd JC, Black M, Bonnefoy S, Hehl A, Knoll LJ, Manger ID, Ortega-Barria E, Tomavo S. 1997. Genetic and biochemical analysis of development in Toxoplasma gondii. Philos Trans R Soc Lond B Biol Sci 352:1347–1354. doi: 10.1098/rstb.1997.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asgari Q, Keshavarz H, Shojaee S, Motazedian MH, Mohebali M, Miri R, Mehrabani D, Rezaeian M. 2013. In vitro and in vivo potential of RH strain of Toxoplasma gondii (type I) in tissue cyst forming. Iran J Parasitol 8:367–375. [PMC free article] [PubMed] [Google Scholar]
- 39.Singh R, Kaushik Y, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. 2009. Autophagy regulates lipid metabolism. Nature 458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selleck EM, Orchard RC, Lassen KG, Beatty WL, Xavier RJ, Levine B, Virgin HW, Sibley LD. 2015. A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-γ-activated human cells. mBio 6:e01157-. doi: 10.1128/mBio.01157-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coppens I. 2017. How Toxoplasma and malaria parasites defy first, then exploit host autophagic and endocytic pathways for growth. Curr Opin Microbiol 40:32–39. doi: 10.1016/j.mib.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Weiss LM, Orlofsky A. 2010. Coordinate control of host centrosome position, organelle distribution, and migratory response by Toxoplasma gondii via host mTORC2. J Biol Chem 285:15611–15618. doi: 10.1074/jbc.M109.095778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romano JD, Nolan SJ, Porter C, Ehrenman K, Hartman EJ, Hsia RC, Coppens I. 2017. The parasite Toxoplasma sequesters diverse Rab host vesicles within an intravacuolar network. J Cell Biol 216:4235–4254. doi: 10.1083/jcb.201701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romano JD, Coppens I. 2013. Host organelle hijackers: a similar modus operandi for Toxoplasma gondii and Chlamydia trachomatis: co-infection model as a tool to investigate pathogenesis. Pathog Dis 69:72–86. doi: 10.1111/2049-632X.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nolan SJ, Romano JD, Luechtefeld T, Coppens I. 2015. Neospora caninum recruits host cell structures to its parasitophorous vacuole and salvages lipids from organelles. Eukaryot Cell 14:454–473. doi: 10.1128/EC.00262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miura S, Gan JW, Brzostowski J, Parisi MJ, Schultz CJ, Londos C, Oliver B, Kimmel AR. 2002. Functional conservation for lipid storage droplet association among Perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J Biol Chem 277:32253–32257. doi: 10.1074/jbc.M204410200. [DOI] [PubMed] [Google Scholar]
- 47.Coppens I, Dunn JD, Romano JD, Pypaert M, Zhang H, Boothroyd JC, Joiner KA. 2006. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125:261–274. doi: 10.1016/j.cell.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 48.Wilcke M, Johannes L, Galli T, Mayau V, Goud B, Salamero J. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J Cell Biol 151:1207–1220. doi: 10.1083/jcb.151.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jheng HF, Tsai PJ, Guo S-M, Kuo LH, Chang CS, Su IJ, Chang CR, Tsai YS. 2012. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32:309–319. doi: 10.1128/MCB.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao J, Zhou Y, Peng H, Huang X, Stahler S, Suri V, Qadri A, Gareski T, Jones J, Hahm S, Perreault M, McKew J, Shi M, Xu X, Tobin JF, Gimeno RE. 2011. Targeting acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J Biol Chem 286:41838–41851. doi: 10.1074/jbc.M111.245456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, Han X, Brownell N, Gross RW, Zechner R, Farese RV Jr. 2011. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J Lipid Res 52:657–667. doi: 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Derouin F, Chastang C. 1989. In vitro effects of folate inhibitors on Toxoplasma gondii. Antimicrob Agents Chemother 33:1753–1759. doi: 10.1128/AAC.33.10.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferguson DJ, Hutchison WM. 1987. An ultrastructural study of the early development and tissue cyst formation of Toxoplasma gondii in the brains of mice. J Parasitol Res 73:483–491. doi: 10.1007/BF00535321. [DOI] [PubMed] [Google Scholar]
- 54.Lemgruber L, Lupetti P, Martins-Duarte ES, De Souza W, Vommaro RC. 2011. The organization of the wall filaments and characterization of the matrix structures of Toxoplasma gondii cyst form. Cell Microbiol 13:1920–1932. doi: 10.1111/j.1462-5822.2011.01681.x. [DOI] [PubMed] [Google Scholar]
- 55.Di Cristina M, Dou Z, Lunghi M, Kannan G, Huynh MH, McGovern OL, Schultz TL, Schultz AJ, Miller AJ, Hayes BM, van der Linden W, Emiliani C, Bogyo M, Besteiro S, Coppens I, Carruthers VB. 2017. Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nat Microbiol 2:17096. doi: 10.1038/nmicrobiol.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gehrmann W, Würdemann W, Plötz T, Jörns A, Lenzen S, Elsner M. 2015. Antagonism between saturated and unsaturated fatty acids in ROS mediated lipotoxicity in rat insulin-producing cells. Cell Physiol Biochem 36:852–865. doi: 10.1159/000430261. [DOI] [PubMed] [Google Scholar]
- 57.Plötz T, Hartmann M, Lenzen S, Elsner M. 2016. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr Metab 13:16–27. doi: 10.1186/s12986-016-0076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aardema H, Vos PL, Lolicato F, Roelen BA, Knijn HM, Vaandrager AB, Helms JB, Gadella BM. 2011. Oleic acid prevents detrimental effects of saturated fatty acids on bovine oocyte developmental competence. Biol Reprod 85:62–69. doi: 10.1095/biolreprod.110.088815. [DOI] [PubMed] [Google Scholar]
- 59.Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. 2011. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther 339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mantzaris MD, Tsianos EV, Galaris D. 2011. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J 278:519–530. doi: 10.1111/j.1742-4658.2010.07972.x. [DOI] [PubMed] [Google Scholar]
- 61.Schaffer JE. 2003. Lipotoxicity: when tissues overeat. Curr Opin Lipidol 14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 62.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE. 2003. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaffer JE. 2002. Fatty acid transport: the roads taken. Am J Physiol Endocrinol Metab 282:E239–E246. doi: 10.1152/ajpendo.00462.2001. [DOI] [PubMed] [Google Scholar]
- 64.Mazumdar J, Striepen B. 2007. Make it or take it: fatty acid metabolism of apicomplexan parasites. Eukaryot Cell 6:1727–1735. doi: 10.1128/EC.00255-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramakrishnan S, Docampo MD, MacRae JI, Pujol FM, Brooks CF, van Dooren GG, Hiltunen K, Kastaniotis AJ, McConville MJ, Striepen B. 2012. Apicoplast and endoplasmic reticulum cooperate in fatty acid biosynthesis in apicomplexan parasite Toxoplasma gondii. J Biol Chem 287:4957–4971. doi: 10.1074/jbc.M111.310144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martins-Duarte ÉS, Carias M, Vommaro R, Surolia N, de Souza W. 2016. Apicoplast fatty acid synthesis is essential for pellicle formation at the end of cytokinesis in Toxoplasma gondii. J Cell Sci 129:3320–3331. doi: 10.1242/jcs.185223. [DOI] [PubMed] [Google Scholar]
- 67.Pernas L, Bean C, Boothroyd JC, Scorrano L. 2018. Mitochondria restrict growth of the intracellular parasite Toxoplasma gondii by limiting its uptake of fatty acids. Cell Metab 27:886–897. doi: 10.1016/j.cmet.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 68.Foe IT, Child MA, Majmudar JD, Krishnamurthy S, van der Linden WA, Ward GE, Martin BR, Bogyo M. 2015. Global analysis of palmitoylated proteins in Toxoplasma gondii. Cell Host Microbe 18:501–511. doi: 10.1016/j.chom.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sonda S, Sala G, Ghidoni R, Hemphill A, Pieters J. 2005. Inhibitory effect of aureobasidin A on Toxoplasma gondii. Antimicrob Agents Chemother 49:1794–1801. doi: 10.1128/AAC.49.5.1794-1801.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welti R, Mui E, Sparks A, Wernimont S, Isaac G, Kirisits M, Roth M, Roberts CW, Botté C, Maréchal E, McLeod R. 2007. Lipidomic analysis of Toxoplasma gondii reveals unusual polar lipids. Biochemistry 46:13882–13890. doi: 10.1021/bi7011993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. 2002. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest 109:121–130. doi: 10.1172/JCI0214080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V. 2014. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol 24:609–620. doi: 10.1016/j.cub.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lam T, Harmancey R, Vasquez H, Gilbert B, Patel N, Hariharan V, Lee A, Covey M, Taegtmeyer H. 2016. Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Discov 2:16061. doi: 10.1038/cddiscovery.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brookheart RT, Michel CI, Schaffer JE. 2009. As a matter of fat. Cell Metab 10:9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. 2006. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 76.Pernas L, Adomako-Ankomah Y, Shastri AJ, Ewald SE, Treeck M, Boyle JP, Boothroyd JC. 2014. Toxoplasma effector MAF1 mediates recruitment of host mitochondria and impacts the host response. PLoS Biol 12:e1001845. doi: 10.1371/journal.pbio.1001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vogel F, Bornhövd C, Neupert W, Reichert AS. 2006. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol 175:237–247. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zick M, Rabl R, Reichert AS. 2009. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta 1793:5–19. doi: 10.1016/j.bbamcr.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 79.Celotto AM, Frank AC, McGrath SW, Fergestad T, Van Voorhies WA, Buttle KF, Mannella CA, Palladino MJ. 2006. Mitochondrial encephalomyopathy in Drosophila. J Neurosci 26:810–820. doi: 10.1523/JNEUROSCI.4162-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ikon N, Ryan RO. 2017. Cardiolipin and mitochondrial cristae organization. Biochim Biophys Acta 1859:1156–1163. doi: 10.1016/j.bbamem.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bradley RM, Stark KD, Duncan RE. 2016. Influence of tissue, diet, and enzymatic remodeling on cardiolipin fatty acyl profile. Mol Nutr Food Res 60:1804–1818. doi: 10.1002/mnfr.201500966. [DOI] [PubMed] [Google Scholar]
- 82.Schönfeld P, Wojtczak L. 2008. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med 45:231–241. doi: 10.1016/j.freeradbiomed.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 83.Bagnato C, Igal RA. 2003. Overexpression of diacylglycerol acyltransferase-1 reduces phospholipid synthesis, proliferation, and invasiveness in simian virus 40-transformed human lung fibroblasts. J Biol Chem 278:52203–52211. doi: 10.1074/jbc.M305760200. [DOI] [PubMed] [Google Scholar]
- 84.Paredes-Santos TC, Martins-Duarte ES, de Souza W, Attias M, Vommaro RC. 2018. Toxoplasma gondii reorganizes the host cell architecture during spontaneous cyst formation in vitro. Parasitology 145:1027–1038. [DOI] [PubMed] [Google Scholar]
- 85.Dubey JP, Lindsay DS, Speer CA. 1998. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev 11:267–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sugi T, Tu V, Ma Y, Tomita T, Weiss LM. 2017. Toxoplasma gondii requires glycogen phosphorylase for balancing amylopectin storage and for efficient production of brain cysts. mBio 8:e01289-17. doi: 10.1128/mBio.01289-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roos DS, Donald RG, Morrissette NS, Moulton AL. 1994. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol 45:27–63. doi: 10.1016/S0091-679X(08)61845-2. [DOI] [PubMed] [Google Scholar]
- 88.Striepen B, Soldati-Favre D. 2013. Genetic manipulation of Toxoplasma gondii, p 408–409. In Weiss LM, Kim K (ed), Toxoplasma gondii: the model apicomplexan—perspectives and methods. Academic Press, London, United Kingdom. [Google Scholar]
- 89.Endo T, Sethi KK, Piekarski G. 1982. Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp Parasitol 53:179–188. doi: 10.1016/0014-4894(82)90059-5. [DOI] [PubMed] [Google Scholar]
- 90.Joiner KA, Fuhrman SA, Mietinnen H, Kasper LL, Mellman I. 1990. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor transfected fibroblasts. Science 249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 91.Fölsch H, Pypaert M, Schu P, Mellman I. 2001. Distribution and function of AP-1 clathrin adaptor complexes in polarized epithelial cells. J Cell Biol 152:595–606. doi: 10.1083/jcb.152.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.