

Burkholderia multivorans is a member of the Burkholderia cepacia complex, a group of >20 related species of nosocomial pathogens that commonly infect individuals suffering from cystic fibrosis. β-Lactam antibiotics are recommended as therapy for infections due to B.
KEYWORDS: β-lactamases, Burkholderia, β-lactam, AmpC
ABSTRACT
Burkholderia multivorans is a member of the Burkholderia cepacia complex, a group of >20 related species of nosocomial pathogens that commonly infect individuals suffering from cystic fibrosis. β-Lactam antibiotics are recommended as therapy for infections due to B. multivorans, which possesses two β-lactamase genes, blapenA and blaAmpC. PenA is a carbapenemase with a substrate profile similar to that of the Klebsiella pneumoniae carbapenemase (KPC); in addition, expression of PenA is inducible by β-lactams in B. multivorans. Here, we characterize AmpC from B. multivorans ATCC 17616. AmpC possesses only 38 to 46% protein identity with non-Burkholderia AmpC proteins (e.g., PDC-1 and CMY-2). Among 49 clinical isolates of B. multivorans, we identified 27 different AmpC variants. Some variants possessed single amino acid substitutions within critical active-site motifs (Ω loop and R2 loop). Purified AmpC1 demonstrated minimal measurable catalytic activity toward β-lactams (i.e., nitrocefin and cephalothin). Moreover, avibactam was a poor inhibitor of AmpC1 (Ki app > 600 μM), and acyl-enzyme complex formation with AmpC1 was slow, likely due to lack of productive interactions with active-site residues. Interestingly, immunoblotting using a polyclonal anti-AmpC antibody revealed that protein expression of AmpC1 was inducible in B. multivorans ATCC 17616 after growth in subinhibitory concentrations of imipenem (1 μg/ml). AmpC is a unique inducible class C cephalosporinase that may play an ancillary role in B. multivorans compared to PenA, which is the dominant β-lactamase in B. multivorans ATCC 17616.
INTRODUCTION
Burkholderia multivorans is a member of a larger group of pathogens, the Burkholderia cepacia complex (BCC), that includes >20 different species (1, 2). BCC bacteria can cause infections (e.g., pneumonia) in immunocompromised persons or in other individuals, such as those with cystic fibrosis (3–7). Currently, β-lactam antibiotics are recommended as a treatment option for infections due to BCC bacteria (8, 9). However, BCC bacteria are inherently resistant to multiple antibiotics, including β-lactams, in which the mechanistic basis for resistance is correlated with the expression of β-lactamases (10–13).
β-Lactamases are clustered into four-different classes based on structural similarities (classes A, B, C, and D) (14). Class A, C, and D β-lactamases use a serine as the nucleophile, while class B β-lactamases are metalloenzymes, using a Zn2+ ion(s) for catalysis. Analysis of the B. multivorans ATCC 17616 genome revealed the presence of two β-lactamases: PenA, a class A β-lactamase, and AmpC, a class C β-lactamase (Table 1) (15). A LysR-type transcriptional regulator (PenRA) is upstream of blaPenA and is divergently transcribed. Expression of blaPenA is regulated by PenRA through a system analogous to AmpC/AmpR regulatory pathways present in Enterobacteriaceae and Pseudomonas aeruginosa (13, 16).
TABLE 1.
Annotated β-lactamases in the B. multivorans ATCC 17616 genome
| No. | Locus tag; locus; chromosome | Class | Assessment |
|---|---|---|---|
| 1 | BMUL_RS18715; NC_010086; 2 | A | PenA; possesses a signal peptide, SXXK, SDN, and KTG motifs |
| 2 | BMUL_RS20495; NC_010086; 2 | A | Possesses a signal peptide but lacks SXXK, SDN, and KTG motifs |
| 3 | BMUL_RS30175; NC_010087; 3 | C | AmpC1; possesses a signal peptide, SXXK, YSN, and KTG motifs |
PenA was previously characterized and found to possess a very broad hydrolytic profile that includes penicillins, cephalosporins, carbapenems, and the monobactam aztreonam, as well as the β-lactamase inhibitors clavulanic acid, sulbactam, and tazobactam (17). In addition, PenA demonstrated significant sequence heterogeneity within clinical isolates of B. multivorans (18). Here, we analyze the second β-lactamase present in B. multivorans ATCC 17616, AmpC1. Similarly to PenA, AmpC also displays sequence diversity within clinical isolates of B. multivorans. Conversely, AmpC1 from B. multivorans ATCC 17616 possesses a narrow spectrum, and the contribution of AmpC1 to β-lactam resistance appears to be negligible, yet expression of AmpC1 is regulated by β-lactam inducers (e.g., imipenem).
RESULTS AND DISCUSSION
The amino acid sequence of AmpC is diverse compared to those of other AmpC β-lactamases, but the structure is similar.
AmpC possesses the classic motifs (SXXK, YSN, Ω loop, KTG, and R2 loop) found in other AmpC proteins (Fig. 1A). However, AmpC is most similar to other Burkholderia AmpC proteins (75% identity and 80% homology to Burkholderia cenocepacia AmpC [BcAmpC]) (Fig. 1B). Comparisons to prevalent chromosomal (Pseudomonas-derived cephalosporinase 1 [PDC-1], Acinetobacter-derived cephalosporinase 1 [ADC-1], P99 [Enterobacter cloacae AmpC], and Escherichia coli AmpC) and plasmidic (CMY-2 and FOX-4) AmpC proteins reveal lower identity (38 to 46%) and homology (58 to 61%) (Fig. 1B).
FIG 1.

Comparison of AmpC1 from B. multivorans (AmpC1_Bm) with AmpC from B. cenocepacia J2315 (AmpC_Bc) and other prevalent class C β-lactamases (PDC_1 and E. coli AmpC [EcAmpC]). (A) Amino acid sequence alignments using Clustal Ω. Black shading indicates identity, dark-gray shading is similarity, and light-gray and white shading are differences. AmpC motifs (SXXK, YSN, and KTG) are in red. (B) Percent identity and homology between prevalent AmpC proteins and AmpC1.
Two homology models of AmpC1 were generated using the B. cenocepacia AmpC crystal structure (Protein Data Bank [PDB] accession no. 5E2G). AmpC1 maintains the expected class C β-lactamase structure; however, the positioning of Q120 in the loop (residues L117 to H126) was nearly 5 Å away from the nucleophilic S64 unlike other AmpC proteins (i.e., CMY-2) (Fig. 2A, purple). The 500-ps molecular dynamics simulation (MDS) of the generated models revealed that the loop region is highly mobile, shifting 10 Å (Fig. 2A, yellow). The second homology model of AmpC1 shows that the loop between residues L117 and H126 was found in a different position (Fig. 2A, magenta). Further refinement of this loop region in AmpC1 was conducted in order to find the most favorable conformation. The loop's energy was optimized using the ab initio loop prediction algorithm LOOPER. The most energetically favorable conformation of the loop was most similar to model 2 (Fig. 2A, cyan). Efforts are under way to determine the crystal structure of AmpC1 to provide additional information on the positioning of the loop.
FIG 2.

(A) Homology models of AmpC1 combined with a loop refinement protocol suggest high flexibility of the L117-to-H126 loop (model 1, purple; model 2, cyan; multiple possible conformations from loop refinement, yellow; best energetically favorable conformation, magenta). The Q120 region has the largest movement (up to 10 Å), possibly changing the shape of the active-site entrance. (B) The amino acids that are replaced in the 27 AmpC variants (Table 2) are highlighted in green. Natural variants of AmpC have amino acid substitutions in two active-site motifs, the Ω loop (blue) (residues D193, R196, and T205) and the R2 loop (orange) (position K290); other active-site regions are the SXXK (with the nucleophilic S64), YSN, and KTG motifs (pink).
AmpC of B. multivorans exhibits considerable sequence heterogeneity.
To determine the sequence diversity of AmpC in B. multivorans, we analyzed the primary amino acid sequence of AmpC proteins from 49 different clinical isolates, and 27 novel AmpC variants were identified (Table 2). The variants had 1 to as many as 13 different amino acid substitutions. The locations of these amino acid substitutions were mapped to the AmpC1 homology model (Fig. 2B). The amino acid substitutions included those in the Ω loop (D193A, R196S, and T205A), as well as the R2 loop (K290T) (Fig. 2B). Amino acid substitutions in the Ω loop and the R2 loop of other AmpC proteins were shown to confer extended-spectrum AmpC phenotypes (e.g., the ability to hydrolyze cefotaxime and ceftazidime) (19).
TABLE 2.
Single amino acid substitutions found in AmpC proteins from 46 clinical isolates of B. multivoransa
| Strain(s) | Amino acid substitution(s), insertion(s), or deletion(s) | No. of differences | Refseq protein accession no. | AmpC designation |
|---|---|---|---|---|
| ATCC 17616, AU21747, AU17545, AU19729, AU10398, AU19518, AU14371, AU28069, AU25543, AU30760, AU20929 | 0 | WP_012218336.1 | AmpC1 | |
| AU19564 | L104P | 1 | WP_105845787.1 | AmpC2 |
| AU13919, AU14328 | T229V, G241A, R309P | 3 | WP_105776316.1 | AmpC3 |
| AU10897 | G98A, T229V, K290T, A363E | 4 | WP_105758864.1 | AmpC4 |
| AU17534 | D76A, T229A, G241A, K290T | 4 | WP_105783420.1 | AmpC5 |
| AU23995 | A129V, R139H, T229A, I244T, A363E | 5 | WP_105810139.1 | AmpC6 |
| AU4507 | A74V, R176Q, T229A, G241A, A363E | 5 | WP_006416937.1 | AmpC7 |
| AU24277 | P18S, T205A, T229A, I244T, A363E | 5 | WP_105835089.1 | AmpC8 |
| AU12481 | A137T, T205A, T229A, I244T, R261Q | 5 | WP_105789398.1 | AmpC9 |
| AU29198 | TL11 M, T229A, D230E, K290T, P304S | 5 | WP_105844745.1 | AmpC10 |
| AU23668 | L104V, A124E, V143 M, T229A, G362D, A363E | 6 | WP_105813500.1 | AmpC11 |
| AU14364 | TL16S, T4A, T229A, G241A, G362D, A363E | 6 | WP_105765510.1 | AmpC12 |
| AU11233 | TL11S, I11F, I33V, E79Q, M184L, T229A, I244T | 7 | WP_105770301.1 | AmpC13 |
| AU26250, AU15954, AU22892 | TL11 M, V89I, R196S, T229V, I244T, K290T, A363E | 7 | WP_105780250.1 | AmpC14 |
| AU15814 | TL16S, T4A, T111A, T229A, G241A, G362D, A363E | 7 | WP_105764661.1 | AmpC15 |
| AU30441 | H7R, I8V, S47A, A141V, T229A, G241A, Q249R, A363D | 8 | WP_105811053.1 | AmpC16 |
| AU30050 | S47A, A141V, D193A, T205A, T229A, G241A, Q249R, A363D | 8 | WP_105793891.1 | AmpC17 |
| AU21015 | I8V, P18S, A165S, D190N, T205A, T229V, I245T, R309P | 8 | WP_107998461.1 | AmpC18 |
| AU14786, AU11772, AU23919 | A17V, S47A, A141V, D193A, T205A, T229A, G241A, Q249R, A363D | 9 | WP_105767194.1 | AmpC19 |
| AU11358 | TL22S, H7R, I8V, S47A, A141V, T205A, T229A, G241A, Q249R, A363D | 10 | WP_105775307.1 | AmpC20 |
| AU22436 | H7R, I8V, S47A, A141V, D193A, T205A, T229A, G241A, Q249R, A363D | 10 | WP_006405341.1 | AmpC21 |
| AU17135, AU10047, AU28442 | H7R, I8V, S47A, A141V, D193A, T229A, G241A, Q249R, P281S, I352 M, A363E | 11 | WP_105752596.1 | AmpC22 |
| AU16734 | H7R, I8V, S47A, A141V, D193A, T229A, G241A, Q249R, P281S, L305R, A363E | 11 | WP_105792096.1 | AmpC23 |
| AU23690, AU11204 | TL22S, H7R, I8V, S47A, A141V, D193A, T229A, G241A, Q249R, P281S, A363E | 11 | WP_105761708.1 | AmpC24 |
| AU19659 | H7R, I8V, S47A, A141V, D193A, T205A, T229A, G241A, Q249R, P281S, L305R, A363E | 12 | WP_105807900.1 | AmpC25 |
| AU30438 | TL22S, H7R, I8V, S47A, A141V, D193A, T205A, T229A, G241A, Q249R, V278I, A363E | 12 | WP_105847138.1 | AmpC26 |
| AU25057 | TL22S, H7R, I8V, S47A, A141V, D193A, T205A, T229A, G241A, Q249R, P281S, A363E | 12 | WP_105838144.1 | AmpC27 |
| AU27706, AU23365 | TL22S, H7R, I8V, I11V, S47A, A141V, D193A, T205A, T229A, G241A, Q249R, A279E, A363E | 13 | WP_069221591.1 | AmpC28 |
| AU18096, AU21596, AU10086 | No class C β-lactamase found |
The AmpC sequences were compared to that of AmpC1 from B. multivorans ATCC 17616.
AmpC1 possesses a narrow spectrum for β-lactams.
To assess the contribution of AmpC1 to β-lactam resistance, the blaAmpC1 gene was cloned into pBC SK(+) and expressed in E. coli DH10B for susceptibility testing and was cloned into the pGEX-6p-2 plasmid and expressed in E. coli Rosetta-gami 2 DE3 for protein purification and kinetics. In addition, the blaAmpC1 gene was knocked out of B. multivorans ATCC 17616 using allelic replacement followed by subsequent excision via recombination. The blaAmpC1 knockout strain was verified by PCR, and the phenotypic verification is presented in Fig. S1 in the supplemental material. An analytic isoelectric focusing (aIEF) gel overlaid with nitrocefin revealed the absence of AmpC1 activity in the knockout compared to the parental strain.
Susceptibility testing was conducted via disk diffusion and Etest using a panel of select β-lactams (e.g., ampicillin, cephalothin, ceftriaxone, cefotaxime, ceftazidime, and imipenem) (see Fig. S2A in the supplemental material). E. coli producing blaAmpC1 was highly susceptible to all the agents tested, with only minor nonsusceptibility observed for cephalothin, with an intermediate zone of 17 mm (see Fig. S2B in the supplemental material). Subsequently, E. coli bacteria producing blaAmpC1, along with B. multivorans ATCC 17616 ΔblaAmpC1, were subjected to MIC determination via agar dilution. Control strains, including B. multivorans ATCC 17616 and E. coli DH10B pBC SK(+) with blaPenA, as well as an empty vector, were also tested. B. multivorans ATCC 17616 was highly resistant to all of the β-lactams (i.e., ampicillin, cephalothin, and imipenem) tested, and susceptibility to ampicillin was restored with avibactam, as expected from our previous work (Table 3) (20).
TABLE 3.
Susceptibility testing results
| Strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| Ampicillin | Ampicillin-avibactama | Cephalothin | Imipenem | |
| B. multivorans | ||||
| ATCC 17616 | 1,024 | 4 | ≥4,096 | 64 |
| ATCC 17616 ΔblaAmpC1 | 1,024 | 4 | ≥4,096 | 64 |
| E. coli | ||||
| DH10B pBC SK(+) | 2 | 1 | 2 | 1 |
| DH10B pBC SK(+) blaPenA | 4,096 | 4 | 4,096 | 2 |
| DH10B pBC SK(+) blaAmpC1 | 8 | 4 | 32 | 1 |
Ampicillin was varied, and avibactam was held at a constant concentration of 4 μg/ml. CLSI interpretive indices were as follows: Enterobacteriaceae (ampicillin, ≥32 μg/ml, resistant; cephalothin, ≥32 μg/ml, resistant; imipenem, ≥4 μg/ml, resistant; and imipenem, 2 μg/ml, intermediate), nonfermenting Gram-negative bacteria (penicillins, ≥128 μg/ml, resistant; cephalosporins, ≥32 to 64 μg/ml, resistant; and carbapenems, ≥8 to 16 μg/ml, resistant).
The MICs of ampicillin, cephalothin, and imipenem for B. multivorans ATCC 17616 ΔblaAmpC1 were equivalent to those for the parent strain (Table 3). Moreover, E. coli with blaAmpC1 was susceptible to ampicillin (MIC = 8 μg/ml) and imipenem (MIC = 1 μg/ml) but demonstrated resistance to cephalothin (MIC = 32 μg/ml) (Table 3). The observations with AmpC1 are in sharp contrast to those for E. coli producing blaPenA. The MICs for E. coli carrying blaPenA more closely mimicked those of the parent strain, B. multivorans ATCC 17616, with nonsusceptibility to all the agents tested and susceptibility restored with the addition of avibactam (Table 3). The susceptibility testing results suggest that blaPenA contributes significantly to β-lactam resistance in B. multivorans, while AmpC1 plays a minor role.
AmpC proteins typically demonstrate catalytic efficiencies near the diffusion limit (109 M−1 s−1) for select cephalosporins (e.g., cephalothin) (21). Avibactam, a novel β-lactamase inhibitor of the diazabicyclooctane family, inactivates class A, class C, and some class D β-lactamases and, in combination with ceftazidime, restores susceptibility to ceftazidime against B. multivorans (20). Steady-state kinetics were conducted with a panel of β-lactams to evaluate the hydrolytic profile of purified AmpC1, as well as with the β-lactamase inhibitor, avibactam, to assess the ability of avibactam to inactivate AmpC1.
The purified AmpC1 β-lactamase was able to appreciably hydrolyze only cephalothin and nitrocefin; all other β-lactam substrates were not measurably hydrolyzed (see Fig. S2C in the supplemental material). AmpC1 showed high Km values for nitrocefin (176 ± 36 μM) and cephalothin (392 ± 88 μM) (Table 4). However, the kcat value for nitrocefin was considerable, resulting in a catalytic efficiency of 1.4 ± 0.3 μM−1 s−1, which is only ∼2-fold lower than that for PenA (Table 4). The catalytic efficiency for cephalothin was ∼30-fold lower for AmpC1 than for PenA (Table 4). AmpC1 is able to hydrolyze select cephalosporins, but not nearly as well as other AmpC proteins (19). In B. multivorans, PenA is a more efficient β-lactamase than AmpC.
TABLE 4.
| Compound | β-Lactamase | Km or Ki app (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|---|
| Cephalothin | PenAc | 71 ± 7 | 221 ± 21 | 3.1 ± 0.3 |
| AmpC1 | 392 ± 88 | 43 ± 5 | 0.11 ± 0.02 | |
| Nitrocefin | PenAc | 142 ± 14 | 460 ± 46 | 3.2 ± 0.3 |
| AmpC1 | 176 ± 36 | 251 ± 25 | 1.4 ± 0.3 | |
| Avibactam | PenAd | 0.5 ± 0.1 | NAe | NA |
| AmpC1 | >600 | NA | NA |
Km and kcat were determined under pseudo-1st-order conditions, and a nonlinear least-square fit of the data to the Henri-Michaelis-Menten equation determined the kinetic parameters. Each experiment was completed in triplicate, and the error values represent the standard errors of the mean.
Ki app was determined using a direct-competition assay under steady-state conditions. Each experiment was completed in triplicate, and the error values represent the standard errors of the mean.
Data were previously published (17).
Data were previously published (20).
NA, not applicable.
Avibactam is a potent inhibitor of PenA (apparent Ki [Ki app] = 500 nM) (Table 4) (20). Conversely, the Ki app endpoint (>600 μM) for AmpC1 could not be determined due to the poor inhibitory activity of avibactam against AmpC1 (Table 4).
Avibactam is able to form an acyl complex with AmpC1.
To determine whether avibactam is able to acylate AmpC1, electrospray ionization-mass spectrometry (ESI-MS) was conducted on AmpC1 incubated at 1:1 and 1:10 AmpC1/avibactam ratios with reactions terminated at 15 min and 24 h. The calculated molecular mass for AmpC1 is 39,748.61 Da, and the theoretical molecular mass for avibactam is 265 Da. The measured masses of apo-AmpC1 were 39,748 ± 5 Da and 39,747 ± 5 Da after 15-min and 24-h incubations (Fig. 3A and B). After a 15-min incubation with avibactam at a 1:1 ratio, only apoenzyme (39,748 Da) was observed; however, at a 1:10 ratio, small amounts of acyl-AmpC1 (40,014 ± 5 Da and 39,933 ± 5 Da) were observed (Fig. 3A and B). The 40,014- ± 5-Da peak corresponds to the addition of the unmodified avibactam (+265 Da) to AmpC1. The 39,933- ± 5-Da peak is also an acyl adduct; however, the mass is decreased by 80 Da, suggestive of the desulfated form of avibactam bound to AmpC1 (22). At 24 h, the acyl complex of AmpC1-avibactam (40,012 ± 5 Da) predominates at both 1:1 and 1:10 ratios (Fig. 3A and B). The desulfated avibactam-acyl complex (39,932 ± 5 Da) is observed in both; in addition, a new minor peak of 39,729 ± 5 Da was observed at the 24-h time point. The 39,729- ± 5-Da peak differs from that of apo-AmpC1 (39,748 Da) by −19 Da, suggestive of loss of a water molecule. Further analysis is under way to determine the significance of this peak. Avibactam is able to acylate AmpC1; however, the kinetic and ESI-MS data suggest that the rate is very low. Conversely, PenA forms an acyl-enzyme complex with avibactam within 15 s of incubation, as detected via ESI-MS (20).
FIG 3.

(A) Mass spectrometry of AmpC1 apoenzyme after 15 min (shaded) and 24 h (nonshaded) of incubation. Shown is mass spectrometry of the AmpC1-avibactam acyl-enzyme complex after 15 min and 24 h of incubation at 1:1 and 1:10 enzyme/avibactam ratios. (B) Summary of mass spectrometry data, including description of peaks formed.
Avibactam forms unfavorable complexes with AmpC1.
Using the homology model of AmpC1 with the more energetically favorable conformation of the L117-to-H126 loop, avibactam was docked into the active site. The active-site pocket of AmpC1 is a deep, narrow groove, resulting in avibactam clashing with some of the side chains during entry into the active site. In the Michaelis-Menten complex of avibactam in AmpC1, the carbonyl of avibactam is positioned in the oxyanion hole (i.e., backbone amides of S64 and S318). However, the carbamate moiety of avibactam is within hydrogen-bonding distance of active-site residues S64, K67, and N152, which decreases the probability of acylation (Fig. 4). The active-site pocket is constraining, preventing productive interactions. The observations of molecular modeling support the high Ki app of avibactam for AmpC1.
FIG 4.
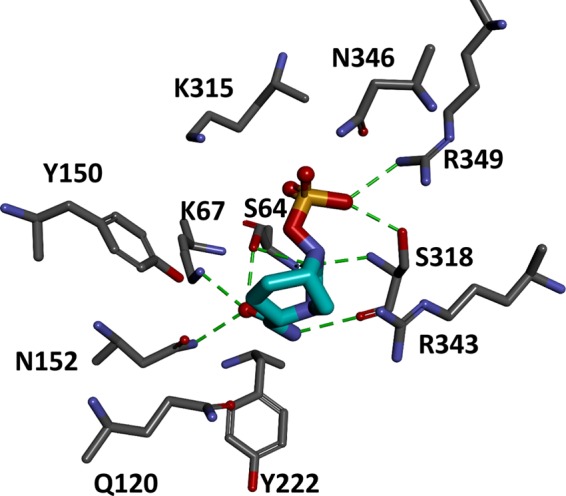
Michaelis-Menten complex of AmpC1 (gray) and avibactam (cyan) revealing nonproductive interactions of avibactam with active-site residues. Potential hydrogen bonding interactions are indicated by dashed green lines.
The morphology of B. multivorans ΔblaAmpC1 is similar to that of wild-type cells. To assess if knocking out blaAmpC1 alters the overall cell shape, B. multivorans ATCC 17616 and the ΔblaAmpC1 strains were stained using BacLight LIVE/DEAD staining and viewed using fluorescence microscopy. The morphology of the ΔblaAmpC1 strain was very similar to that of the wild type; thus, lack of AmpC1 does not affect the cell's shape (see Fig. S1B in the supplemental material).
AmpC1 and PenA expression is induced by imipenem.
Previously, bla gene expression in B. multivorans ATCC 17616 was shown to be inducible by β-lactams (13). However, an E. coli background was used, only blaPenA induction was evaluated, and induction was measured via nitrocefinase activity. Here, the induction of blaAmpC1 and blaPenA expression was assessed via immunoblotting using B. multivorans ATCC 17616 grown in lysogeny broth (LB) in the presence of a β-lactam inducer (i.e., 1 μg/ml imipenem) for different amounts of time (5, 10, 15, 30, 45, 60 75, 90, 105, and 120 min). In the absence of imipenem, PenA protein could not be detected, while a very minor amount of AmpC1 was observed at 75, 90, and 105 min (Fig. 5). The addition of imipenem to the medium resulted in robust expression of AmpC1 and PenA that was time dependent (Fig. 5). The amount of AmpC1 protein produced was greatest between 30 and 90 min and began to decrease by 120 min (Fig. 5). With PenA, at 30 min, the PenA protein level was high, and it remained high up to 120 min (Fig. 5). The presence of 1 μg/ml imipenem in the medium did not alter the growth of B. multivorans during the course of the experiment. Expression of blaAmpC1 and blaPenA is induced by imipenem, supporting a role for these proteins in β-lactam resistance and/or cell wall recycling and metabolism.
FIG 5.

Immunoblotting reveals that protein expression of AmpC1 and PenA is inducible in B. multivorans ATCC 17616 after growth in subinhibitory concentrations of imipenem for different amounts of time. The anti-RecA antibody was used as a loading control.
Conclusions.
In this study, AmpC1 was found to be a weak β-lactamase. Knocking out blaAmpC1 did not impact the susceptibility of B. multivorans; however, blaAmpC1 expression was induced by imipenem. So, what is the true role of AmpC in B. multivorans? Additional investigations are under way to further assess the contribution of AmpC in B. multivorans. The catalytic efficiency of one of the 27 variants of AmpC identified here against β-lactams may be greater than that of AmpC1, thus providing a larger contribution to resistance. Alternatively, AmpC may play some role in peptidoglycan metabolism. For example, AmpH class C low-molecular-weight penicillin-binding proteins (PBPs) characterized in E. coli possess a structure similar to that of AmpC and have bifunctional dd-carboxypeptidase and dd-endopeptidase activities (23, 24). In summary, AmpC from B. multivorans is a unique inducible class C cephalosporinase that may play a subsidiary role compared to PenA, which is the dominant β-lactamase.
MATERIALS AND METHODS
Protein analysis.
AmpC sequences were analyzed by BLASTP and Bl2Seq from the National Center for Biotechnology Information website. Multiple-sequence alignments were generated using CLUSTAL Ω from the European Bioinformatics Institute (EMBL-EBI) and Discovery Studio (25–27).
Whole-genome sequencing (WGS) of B. multivorans.
Genomic DNA was purified from the clinical B. multivorans isolates using a MasterPure Gram-positive DNA purification kit (Epicentre Inc., Madison, WI) as recommended by the manufacturer. The genomes of 49 B. multivorans isolates were sequenced at the J. Craig Venter Institute, Rockville, MD (JCVI) with Illumina NextSeq (two 150-bp reads of sequence data). Paired-end libraries were constructed using Illumina Nextera XT kits. Sequence reads were generated with a target average read depth of ∼100-fold coverage. Sequence reads for each isolate were assembled individually using SPAdes (28) and annotated using the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline (PGAP) (29). Raw DNA sequence reads were submitted to the NCBI Sequence Read Archive (SRA). A blaAmpC gene could not be found in the AU21596, AU18096, and AU10086 genomes.
Bacterial strains and plasmids.
The blaAmpC1 gene was synthesized (Celtek Bioscience, LLC) using the deposited nucleotide sequence from B. multivorans ATCC 17616 and cloned into pET24a(+) using NdeI and XhoI restriction enzymes. The blaAmpC1 gene was subcloned from pET24a(+) to include the ribosomal binding site (RBS) of pET24a(+), using XbaI and XhoI restriction sites; ligated into pBC SK(+); and electroporated in E. coli DH10B for susceptibility testing. The blaAmpC1 gene was also cloned into pGEX-6P-2 (minus nucleotides 1 to 75, encoding the signal peptide) using BamHI and EcoRI restriction sites and transformed into E. coli Rosetta-gami 2 DE3 cells for protein expression. The B. multivorans clinical isolates used in this study were obtained from the Burkholderia cepacia Research Laboratory and Repository (BcRLR) strain collection (20).
The construction of ΔblaAmpC1 was conducted using previously described methods (30). Briefly, nucleotide sequences flanking blaAmpC1 (630 bp upstream and 432 bp downstream) were synthesized as one fragment by Celtek Bioscience, LLC. An NdeI restriction site was added to the pGPI-SceI vector using a Quikchange XL kit via site-directed mutagenesis. The blaAmpC1 flanking sequence was cloned into the pGPI-SceI vector using XbaI and NdeI restriction sites and transformed into E. coli PIR1 cells (Invitrogen). E. coli PIR1 containing pGPI-SceI with the flanking sequences was used as the donor strain, and B. multivorans ATCC 17616 was used as the recipient strain. The “helper strain” was E. coli DH5α(pRK2013). Triparental mating was conducted according to previously published methods (30), and the mixed culture was plated in a single spot on a superoptimal broth (SOB) agar plate containing a nitrocellulose filter and incubated accordingly. The following day, the filter was washed in 1× sterile phosphate-buffered saline (PBS) in order to extract the entire spot of culture and plated onto LB agar with polymyxin B and trimethoprim. The resistant colonies were patched onto phenol red glucose agar plates (5 g peptone, 2.5 g NaCl, 1.8 ml 0.5% phenol red, 7.5 g agar, 50 ml 50% glucose, 450 ml H2O), and those that did not ferment glucose were subjected to PCR to further confirm the first crossover event. The second crossover was performed as described above, with a different donor strain, E. coli DH5α(pDAI-SceI-SacB), using the first-crossover B. multivorans as the recipient and E. coli DH5α(pRK2013) as the helper. Agar plates containing sucrose were used to cure the pDAI-SceI-SacB plasmid as previously described (30). The resulting knockout strain was confirmed by PCR using primers that lie upstream and downstream of blaAmpC; thus, the final PCR product of the blaAmpC knockout was smaller than the wild type. In addition, internal blaAmpC primers were also used to verify the lack of a PCR product with the knockout and the presence of a PCR product with the wild type. All the PCR products were sequenced for verification.
Analytical isoelectric focusing.
B. multivorans ATCC 17616 and the ΔblaAmpC1 strain were grown in LB to an optical density at 600 nm (OD600) of 0.6. A final concentration of 1 μg/ml of imipenem was added to each culture to induce the expression of β-lactamases, and the cultures were grown for 1 h. Cells were pelleted by centrifugation at 12,000 rpm for 10 min. Crude extracts were prepared and loaded onto a FocusGel 3-10 24S (Serva Electrophoresis GmbH) with a pH gradient from 3 to 10 and electrophoresed using a Multiphor II apparatus. The gels were focused at 4°C at 8 W for 120 min. The detection of β-lactamases was performed using an overlay of 2 mM nitrocefin, a chromogenic β-lactam substrate that turns from orange to red upon hydrolysis by a β-lactamase. Purified PenA or AmpC1 β-lactamases were used as controls.
Fluorescence microscopy.
B. multivorans ATCC 17616 and ΔblaAmpC1 were grown to an OD600 of 0.6 in LB. The cells were pelleted and washed twice in 0.9% NaCl to remove the medium. The cells were resuspended to an OD600 of 1.25 in 1 ml of 0.9% NaCl and stained for 15 min in the dark using LIVE/DEAD BacLight bacterial viability kits (Molecular Probes; L7012) with equal ratios of SYTO9 (3 μl) and propidium iodide (3 μl) stains, according to the manufacturer's instructions. Three microliters of each sample was placed onto a glass slide with a coverslip. Samples were viewed on a Zeiss Axio Imager M1 microscope with a metal halide arc lamp using the green fluorescent protein (GFP) and DSRED filters, and images were obtained at ×400 and ×1,000 (oil immersion) magnification.
Antibiotic susceptibility.
Mueller-Hinton (M-H) agar dilution MICs, according to the Clinical and Laboratory Standards Institute (CLSI) (31, 32), were used to phenotypically characterize strains, as previously described (32).
Protein expression and purification of AmpC1.
E. coli Rosetta 2 DE3 or Origami 2 DE3 cells producing the AmpC1 β-lactamase from the pGEX-6P-2 plasmid were grown, lysed, and processed as previously described (33, 34). The recombinant AmpC1 possesses a cleavable N-terminal glutathione S-transferase (GST) tag, and thus, the crude extracts were run on a GSTrap FF column (GE LifeSciences). The GST tag was cleaved using PreScission protease as previously described (33, 34). The purity of the fractions was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Gels were stained with Coomassie brilliant blue R250, and the protein's mass was verified via ESI-MS. Protein concentrations were determined by measuring absorbance at a wavelength of 280 nm (λ280) and using the protein's extinction coefficient (Δε) (49,850 M−1 cm−1 at 280 nm), which was obtained using the ProtParam tool available through the ExPASy Bioinformatics Resource Portal (31). To maintain full activity, AmpC1 was stored in 10 mM PBS, pH 7.4, with 25% glycerol at −20°C. Three milligrams of purified AmpC1 was sent to New England Peptide for antibody production in rabbits.
Kinetics.
Steady-state kinetic parameters were determined using an Agilent 8453 diode array spectrophotometer, as previously described (32, 35). Enzfitter (Biosoft) was used to fit the data to the Henri-Michaelis-Menten equation and to obtain the kinetic parameters, Vmax and Km, for the β-lactam substrates, nitrocefin and cephalothin, as follows: v = (Vmax × [S])/(Km + [S]), where v indicates velocity and [S] indicates concentration of substrate.
Using a direct-competition assay under steady-state conditions, the Ki app of avibactam was determined for AmpC1 as previously described (32, 35). The data were analyzed according to the following equation to account for the affinity of nitrocefin ([S], ncf) for the lactamase: Ki app (corrected) = Ki app (observed)/(1 + [S]/Km ncf).
ESI-MS.
ESI-MS was conducted on a Waters Synapt G2-Si as previously described (20). Purified AmpC1 was incubated with either a 1:1 or 1:10 enzyme/inhibitor ratio with avibactam. Reactions were terminated by the addition of 0.1% formic acid and acetonitrile.
Molecular modeling.
The AmpC1 homology model was generated using the SWISS-MODEL repository with the B. cenocepacia AmpC crystal structure (PDB accession no. 5E2G) as the template (identity, 298/389 [77%]; similarity, 320/389 [82%]) (36–38). The local quality of the AmpC1 model was estimated using the raw QMEAN scoring function and was found to be 0.7 (39–41). The majority of the residues in AmpC1 had a QMEAN score of 0.8 (1 is maximum for the best model). However, the score for the L117-to-H126 loop was 0.3, indicating that the positioning of the loop was not reliable. The L117-to-H126 loop sits at the entrance to the AmpC1 active site; thus, the model required further refinement. A second homology model generated via the SWISS-MODEL repository had an overall QMEAN score for the protein of 0.73, and the scores for the residues in the L117-to-H126 loop were between 0.6 and 0.8. Around amino acid 300, the QMEAN score was near 0.3; however, the overall quality of the second model, especially for the residues near the active site, was much better. A multistep CHARMm protocol using an ab initio loop prediction algorithm, LOOPER, in BIOVIA Discovery Studio 2017 (DS2017) R2 (Accelrys, Inc. San Diego, CA) was used to generate a set of low-energy conformations for the specified loop region (L117 to H126), and they were scored based on their CHARMm energy. The best-scoring conformation was very similar to that of the second model. Thus, the second AmpC1 model used to construct Michaelis-Menten complexes with avibactam as previously described, using BIOVIA DS2017 molecular-modeling software (32). Intact and acylated avibactams were constructed using Fragment Builder tools and minimized using a Standard Dynamics Cascade protocol of DS2017. The avibactam was automatically docked into the active site of AmpC1 using the CDOCKER module of DS2017 (42).
Cloning, protein expression, and purification of recA.
The recA gene was amplified from genomic DNA isolated from B. multivorans ATCC 17616. The PCR product obtained was cloned into the pCR-XL-TOPO vector (Invitrogen) and electroporated into E. coli TOP10. The pGEX-6P-2 empty vector (allowing for the addition of a cleavable N-terminal GST tag) and pCR-XL-TOPO recA were digested using BamHI and XhoI restriction enzymes. The digested products, corresponding to pGEX-6P-2 and recA, were ligated overnight at 22°C, and the ligation reaction product was electroporated into E. coli DH10B. The pGEX-6P-2 recA plasmid was verified by DNA sequencing using MCLab. For protein expression, the pGEX-6P-2 recA plasmid was transformed into E. coli Rosetta 2 DE3 cells (Novagen). The RecA protein purification was conducted as described above for AmpC. The protein concentrations were determined by measuring the absorbance at λ280 and using the protein's extinction coefficient (Δε, 14,440 M−1 cm−1 for RecA at λ280). The theoretical mass of RecA (37,765 Da), as well as the extinction coefficient, was obtained using the ProtParam tool on the ExPASy Bioinformatics Resource Portal (31). Three milligrams of purified RecA was sent to New England Peptide for antibody production in rabbits.
Immunoblotting and induction of β-lactamase expression.
To measure β-lactamase induction, immunoblotting was conducted using the polyclonal AmpC1, PenA peptide (18), and RecA antibodies. B. multivorans ATCC 17616 was grown in LB to log phase at an OD600 between 0.6 and 0.7. B. multivorans ATCC 17616 was treated with 1 μg/ml of imipenem to induce expression of blapenA and blaAmpC1, and aliquots of cells were collected after 5, 10, 15, 30, 45, 60, 75, 90, 105, and 120 min of induction. B. multivorans cells not induced with imipenem at the above-mentioned time points were also collected. Subsequently, the cells were pelleted and lysed using stringent periplasmic fractionation to prepare lysates, as previously described (43). These crude extracts were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were blocked in 5% nonfat dry milk in 20 mM Tris-Cl with 150 mM NaCl, pH 7.4 (TBS), for 1 h and probed in 5% nonfat dry milk in TBS with 1 μg/ml of polyclonal AmpC, PenA peptide, or RecA antibodies. The membranes were washed five times for 10 min each time with TBS with 0.05% Tween 20 (TBST), and for protein detection, blots were incubated for 1 h in 1:5,000 dilutions of anti-rabbit secondary horseradish peroxidase (HRP)-conjugated antibodies in 5% nonfat dry milk in TBS. The immunoblots were washed five times for 10 min each time with TBST and developed using an ECL-Plus developing kit (GE Healthcare Life Sciences) or SuperSignal West Femto chemiluminescent substrate (Thermo Scientific) according to the manufacturers' instructions. Fotodyne Luminary/FX was used to capture images.
Accession number(s).
Annotated genomes were deposited in the GenBank WGS repository and can be obtained within BioProject PRJNA434393.
Supplementary Material
ACKNOWLEDGMENTS
The pGPI-SceI, pRK2013, and pDAI-SceI-SacB plasmids were kind gifts from Miguel Valvano of Queen's University Belfast. We thank Brett Hanzlicek for assistance with fluorescence microscopy.
Research reported in this article was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Veterans Affairs Merit Review Program (BX002872 to K.M.P.-W. from the U.S. Department of Veterans Affairs Biomedical Laboratory Research and Development Service). This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under awards U19AI110819 to the JCVI and U19AI109713-SRP to K.M.P.-W. This work was also supported by funding (to J.J.L.) from the Cystic Fibrosis Foundation.
The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States government. The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01140-18.
REFERENCES
- 1.Mahenthiralingam E, Vandamme P. 2005. Taxonomy and pathogenesis of the Burkholderia cepacia complex. Chron Respir Dis 2:209–217. doi: 10.1191/1479972305cd053ra. [DOI] [PubMed] [Google Scholar]
- 2.Vandamme P, Dawyndt P. 2011. Classification and identification of the Burkholderia cepacia complex: past, present and future. Syst Appl Microbiol 34:87–95. doi: 10.1016/j.syapm.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Marson FA, Hortencio TD, Aguiar KC, Ribeiro JD. 2015. Demographic, clinical, and laboratory parameters of cystic fibrosis during the last two decades: a comparative analysis. BMC Pulm Med 15:3. doi: 10.1186/1471-2466-15-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbott IJ, Peleg AY. 2015. Stenotrophomonas, Achromobacter, and nonmelioid Burkholderia species: antimicrobial resistance and therapeutic strategies. Semin Respir Crit Care Med 36:99–110. doi: 10.1055/s-0034-1396929. [DOI] [PubMed] [Google Scholar]
- 5.Hanulik V, Webber MA, Chroma M, Uvizl R, Holy O, Whitehead RN, Baugh S, Matouskova I, Kolar M. 2013. An outbreak of Burkholderia multivorans beyond cystic fibrosis patients. J Hosp Infect 84:248–251. doi: 10.1016/j.jhin.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Chiappini E, Taccetti G, de Martino M. 2014. Bacterial lung infections in cystic fibrosis patients: an update. Pediatr Infect Dis J 33:653–654. doi: 10.1097/INF.0000000000000347. [DOI] [PubMed] [Google Scholar]
- 7.Gautam V, Singhal L, Ray P. 2011. Burkholderia cepacia complex: beyond Pseudomonas and Acinetobacter. Indian J Med Microbiol 29:4–12. doi: 10.4103/0255-0857.76516. [DOI] [PubMed] [Google Scholar]
- 8.Avgeri SG, Matthaiou DK, Dimopoulos G, Grammatikos AP, Falagas ME. 2009. Therapeutic options for Burkholderia cepacia infections beyond co-trimoxazole: a systematic review of the clinical evidence. Int J Antimicrob Agents 33:394–404. doi: 10.1016/j.ijantimicag.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Wuthiekanun V, Peacock SJ. 2006. Management of melioidosis. Expert Rev Anti Infect Ther 4:445–455. doi: 10.1586/14787210.4.3.445. [DOI] [PubMed] [Google Scholar]
- 10.Cheung TK, Ho PL, Woo PC, Yuen KY, Chau PY. 2002. Cloning and expression of class A β-lactamase gene blaA(BPS) in Burkholderia pseudomallei. Antimicrob Agents Chemother 46:1132–1135. doi: 10.1128/AAC.46.4.1132-1135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godfrey AJ, Wong S, Dance DA, Chaowagul W, Bryan LE. 1991. Pseudomonas pseudomallei resistance to β-lactam antibiotics due to alterations in the chromosomally encoded β-lactamase. Antimicrob Agents Chemother 35:1635–1640. doi: 10.1128/AAC.35.8.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tribuddharat C, Moore RA, Baker P, Woods DE. 2003. Burkholderia pseudomallei class A β-lactamase mutations that confer selective resistance against ceftazidime or clavulanic acid inhibition. Antimicrob Agents Chemother 47:2082–2087. doi: 10.1128/AAC.47.7.2082-2087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trepanier S, Prince A, Huletsky A. 1997. Characterization of the penA and penR genes of Burkholderia cepacia 249 which encode the chromosomal class A penicillinase and its LysR-type transcriptional regulator. Antimicrob Agents Chemother 41:2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob Agents Chemother 54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poirel L, Rodriguez-Martinez JM, Plesiat P, Nordmann P. 2009. Naturally occurring class A β-lactamases from the Burkholderia cepacia complex. Antimicrob Agents Chemother 53:876–882. doi: 10.1128/AAC.00946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhar S, Kumari H, Balasubramanian D, Mathee K. 2018. Cell-wall recycling and synthesis in Escherichia coli and Pseudomonas aeruginosa—their role in the development of resistance. J Med Microbiol 67:1–21. doi: 10.1099/jmm.0.000636. [DOI] [PubMed] [Google Scholar]
- 17.Papp-Wallace KM, Taracila MA, Gatta JA, Ohuchi N, Bonomo RA, Nukaga M. 2013. Insights into β-lactamases from Burkholderia species, two phylogenetically related yet distinct resistance determinants. J Biol Chem 288:19090–19102. doi: 10.1074/jbc.M113.458315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becka SA, Zeiser ET, Marshall SH, Gatta JA, Nguyen K, Singh I, Sutton G, Greco C, Fouts DE, LiPuma JJ, Papp-Wallace KM. 18 June 2018. Sequence heterogeneity of the PenA carbapenemase in clinical isolates of Burkholderia multivorans. Diagn Microbiol Infect Dis. doi: 10.1016/j.diagmicrobio.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacoby GA. 2009. AmpC β-lactamases. Clin Microbiol Rev 22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papp-Wallace KM, Becka SA, Zeiser ET, Ohuchi N, Mojica MF, Gatta JA, Falleni M, Tosi D, Borghi E, Winkler ML, Wilson BM, LiPuma JJ, Nukaga M, Bonomo RA. 2017. Overcoming an extremely drug resistant (XDR) pathogen: avibactam restores susceptibility to ceftazidime for Burkholderia cepacia complex isolates from cystic fibrosis patients. ACS Infect Dis 3:502–511. doi: 10.1021/acsinfecdis.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulychev A, Mobashery S. 1999. Class C β-lactamases operate at the diffusion limit for turnover of their preferred cephalosporin substrates. Antimicrob Agents Chemother 43:1743–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Leiza SM, de Pedro MA, Ayala JA. 2011. AmpH, a bifunctional DD-endopeptidase and DD-carboxypeptidase of Escherichia coli. J Bacteriol 193:6887–6894. doi: 10.1128/JB.05764-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Ω. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R. 2015. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43:W580–W584. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. 2013. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aubert DF, Hamad MA, Valvano MA. 2014. A markerless deletion method for genetic manipulation of Burkholderia cenocepacia and other multidrug-resistant Gram-negative bacteria. Methods Mol Biol 1197:311–327. doi: 10.1007/978-1-4939-1261-2_18. [DOI] [PubMed] [Google Scholar]
- 31.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. The proteomics protocols handbook. Humana Press, New York, NY. [Google Scholar]
- 32.Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA. 2010. Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob Agents Chemother 54:890–897. doi: 10.1128/AAC.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papp-Wallace KM, Becka SA, Taracila MA, Zeiser ET, Gatta JA, LiPuma JJ, Bonomo RA. 2017. Exploring the role of the Ω-loop in the evolution of ceftazidime resistance in the PenA β-lactamase from Burkholderia multivorans, an important cystic fibrosis pathogen. Antimicrob Agents Chemother 61:e01941-16. doi: 10.1128/AAC.01941-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C, Caselli E, Prati F, van den Akker F, Bonomo RA. 2013. Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J Med Chem 56:1084–1097. doi: 10.1021/jm301490d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papp-Wallace KM, Taracila M, Wallace CJ, Hujer KM, Bethel CR, Hornick JM, Bonomo RA. 2010. Elucidating the role of Trp105 in the KPC-2 β-lactamase. Protein Sci 19:1714–1727. doi: 10.1002/pro.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bienert S, Waterhouse A, de Beer TA, Tauriello G, Studer G, Bordoli L, Schwede T. 2017. The SWISS-MODEL repository; new features and functionality. Nucleic Acids Res 45:D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guex N, Peitsch MC, Schwede T. 2009. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PDBViewer: a historical perspective. Electrophoresis 30(Suppl 1):S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 39.Benkert P, Tosatto SC, Schomburg D. 2008. QMEAN: a comprehensive scoring function for model quality assessment. Proteins 71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 40.Benkert P, Biasini M, Schwede T. 2011. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T. 2017. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep 7:10480. doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu G, Robertson DH, Brooks CL, Vieth M. 2003. Detailed analysis of grid-based molecular docking: a case study of CDOCKER—a CHARMm-based MD docking algorithm. J Comput Chem 24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- 43.Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, Bonomo RA. 2012. Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: exploring the roles of Arg220 and Glu276. Antimicrob Agents Chemother 56:4428–4438. doi: 10.1128/AAC.05769-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.