

Dried blood spot (DBS) antibiotic assays can facilitate pharmacokinetic (PK) studies in situations where venous blood sampling is logistically and/or ethically challenging. In this study, we aimed to demonstrate the validity of a DBS ceftriaxone assay in a PK study of children with severe illness from Papua New Guinea (PNG), a setting in which health care resources are limited and anemia is common.
KEYWORDS: Papua New Guinea, ceftriaxone, dried blood spots, microsampling, pediatric pharmacology
ABSTRACT
Dried blood spot (DBS) antibiotic assays can facilitate pharmacokinetic (PK) studies in situations where venous blood sampling is logistically and/or ethically challenging. In this study, we aimed to demonstrate the validity of a DBS ceftriaxone assay in a PK study of children with severe illness from Papua New Guinea (PNG), a setting in which health care resources are limited and anemia is common. Using a previously validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay, serial plasma and DBS ceftriaxone concentrations were measured in PNG children aged 5 to 10 years with acute bacterial meningitis or severe pneumonia. The concentration-time data were incorporated into population PK models. Ten children were recruited with an admission hematocrit of 0.22 to 0.52. Raw data demonstrated good correlation between plasma and DBS concentrations (Spearman's rank correlation coefficient [rs] = 0.94 [95% confidence interval, 0.91 to 0.97], P < 0.0001). A marked systematic hematocrit bias was observed, with lower hematocrits resulting in underestimation of DBS-predicted plasma concentration. After adjustment for red cell partitioning and hematocrit bias, a population PK model comparing plasma and DBS-predicted plasma concentrations did not differ in terms of key PK parameters, including clearance, volume of distribution, and residual variability. The performance of the ceftriaxone DBS assay is robust and provides reassurance that this platform can be used as a surrogate for plasma concentrations to provide valid PK and PK/pharmacodynamic studies of severely unwell children hospitalized in a resource-limited setting. It highlights the importance of hematocrit bias in validation studies of DBS assays.
INTRODUCTION
Antimicrobial resistance represents a major threat to global health. One important way of preventing the emergence of antimicrobial resistance and mitigating its consequences is to optimize dosing of antibiotics through pharmacokinetic/pharmacodynamic (PK/PD) studies. A lack of PK/PD data for older antibiotics, such as ceftriaxone, has been identified as a research priority (1). This has particular relevance for neonatal and pediatric patients, especially in resource-limited settings, because studies performed in hospitalized adults from developed countries may not be generalizable due to differences in drug disposition (2, 3), clinical spectrum of disease, and characteristics of the bacterial pathogens (4). Additionally, repeated blood sampling may be unethical in neonates and young children because multiple large-volume venesections could result in adverse hemodynamic effects (5).
The measurement of drug concentrations in dried blood spots (DBS) collected on filter paper represents a promising way of resolving these issues, particularly where there are none of the laboratory facilities required to process, store, and transport blood or plasma samples (6). In a previous study from our group, we developed and validated a DBS ceftriaxone assay using healthy adult Australians. Plasma and DBS concentrations were highly correlated, and the assay limits of quantification (LOQ) and detection (LOD), as well as the thermal stability of the DBS samples, indicated that this approach could be more broadly applied (7). One pertinent clinical situation is severely ill children in the tropics. Our published data suggest that studies using DBS could be performed with sampling blood volumes that were well within accepted limits and without the need for low temperature storage and transportation of samples (7, 8). Children in tropical countries are, however, at high risk of anemia from a variety of genetic, nutritional, and infective causes. This is relevant to DBS PK studies since a “hematocrit bias” is seen as one of the main barriers to the widespread adoption of DBS as a surrogate for plasma measurements in pediatric antibiotic PK studies (6). Hematocrit variability affects blood viscosity and could influence the distribution of blood in the filter paper. Whole blood from patients with low hematocrit may spread more rapidly across the filter paper, resulting in a relatively larger blood spot and underestimation of DBS-predicted plasma concentrations (9).
In light of these considerations, we have performed a second validation study of plasma compared with DBS ceftriaxone concentrations in Papua New Guinean (PNG) children aged between 5 to 10 years who were treated for severe bacterial infections.
RESULTS
Subject characteristics.
Five of the children were male and five were female. The median (range) age, weight, and hematocrit were 8 (5 to 9) years, 20 (13 to 23) kg, and 0.35 (0.22 to 0.52), respectively. Seven children had a clinical diagnosis of acute bacterial meningitis and three had severe pneumonia. One child, for whom there was no body weight recorded, received 1,000 mg ceftriaxone and was assumed to have been 20 kg for purposes of the PK model. There were a total of 73 paired plasma and DBS samples (including predose). Two measured plasma samples were >1,000 mg/liter and were excluded from further analysis, as they were inconsistent with available ceftriaxone PK data and were thought to have arisen from residual ceftriaxone within the intravenous (i.v.) cannula as a result of incomplete flushing. In these samples, the DBS concentrations were also low. Using the remaining raw data without any adjustments, plasma and DBS concentrations were highly correlated (Spearman's rank correlation coefficient [rs] = 0.94 [95% confidence interval, 0.91 to 0.97], P < 0.0001; Fig. 1). The slope (95% confidence interval) of the linear regression line was 0.61 (0.56 to 0.67; Fig. 1).
FIG 1.
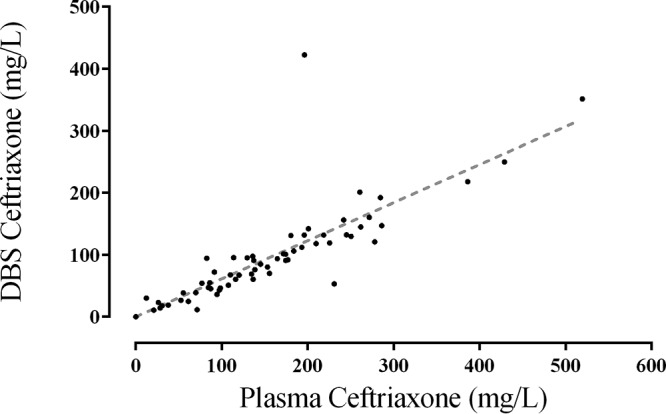
Raw data comparing ceftriaxone concentrations in severely unwell Papua New Guinean children measured from plasma or dried blood spots.
Comparison of plasma and DBS concentrations.
After adjustment for the difference in volume between plasma and whole blood in DBS, there was poor agreement between measured plasma and DBS-derived concentrations, with a mean bias of −12% ± 24% (Fig. 2A). Only 47% of samples were within 20% of each other, lower than the 67% threshold required for incurred sample reanalysis. Accounting for a percentage of the concentration in red cell pellet did not improve the association. Plots of bias versus hematocrit demonstrated a clear correlation (r2 = 0.467). After adjustment for this [through multiplying by 1 + (0.41 − hematocrit) × 1.84], the mean bias was reduced to −2% ± 17% and 75% of the samples were within 20% of each other (Fig. 2C). No further associations were identified as influential.
FIG 2.

Relationship of dried blood spot (DBS) bias in percentage versus hematocrit preadjustment (A) and postadjustment (C), with respective histograms of DBS bias preadjustment (B) and postadjustment (D).
Pharmacokinetic modeling.
After removal of 9 paired predose samples and 6 outliers, there were 56 paired ceftriaxone individual plasma and DBS-derived plasma concentrations available for population PK analysis from 10 pediatric patients. None of the concentrations were below the LOQ. A two-compartment model improved the model fit compared with a single-compartment model, while the addition of a third compartment did not improve the fit. The structural model parameters for were, therefore, clearance (CL), central volume of distribution (VC), intercompartmental clearance (Q), and peripheral volume of distribution (VP), and the interindividual variability (IIV) of CL and VC was able to be estimated. No significant covariate relationships were identified.
The final model parameter estimates and bootstrap results for both ceftriaxone models are summarized in Table 1. This includes the PK parameters of the structural model and comparisons for each parameter between the plasma and DBS-derived plasma estimates. There were no significant differences for any of the structural or random model parameters. Residual variability was not significantly different. Figure 3 shows goodness-of-fit plots, and Fig. 4 displays prediction-corrected visual predictive checks (pcVPCs) for both the models.
TABLE 1.
Final population pharmacokinetic estimates and bootstrap results for ceftriaxone plasma concentrations and dried blood spot-derived plasma concentration for 10 pediatric patients
| Parameterb | Plasma parameter |
Relative difference in DBS parameter |
P valuea | ||
|---|---|---|---|---|---|
| Estimate | 95% CIc | Estimate | 95% CI | ||
| Objective function value | −143.299 | −260.652 to −77.903 | |||
| Structural model parameters | |||||
| CL (liters/h/70 kg) | 1.05 | 0.54 to 1.67 | 1.01 | 0.9 to 1.17 | 0.856 |
| VC (liters/70 kg) | 5.89 | 4.29 to 16.82 | 1.01 | 0.69 to 1.25 | 0.706 |
| Q (liters/h/70 kg) | 7.38 | 1.07 to 9.29 | 0.829 | 0.63 to 3.49 | 0.726 |
| VP (liters/70 kg) | 10.4 | 5.21 to 16.2 | 1.02 | 0.83 to 1.48 | 0.542 |
| Variable model parameters [shrinkage %] | |||||
| IIV in CL | 68 | 17 to 102 | 1.06 | 0.8 to 1.2 | 0.618 |
| IIV in VC | 80 | 5 to 124 | 0.964 | 0.67 to 1.3 | 0.514 |
| RV | 25 | 12 to 34 | 1.12 | 0.96 to 1.19 | 0.23 |
P value obtained from likelihood ratio testing.
CL (clearance), VC (central volume of distribution), Q (intercompartmental clearance), VP (peripheral volume of distribution), IIV (interindividual variability), and RV (residual variability). IIV presented as .
CI, confidence interval.
FIG 3.

Goodness-of-fit plots for plasma ceftriaxone (A) and plasma ceftriaxone derived from dried blood spots (B).
FIG 4.

(A) Time-concentration profile of ceftriaxone (mg/liter on a log10 scale) for dried blood spot-derived (DBS; open circles) and measured plasma concentrations (closed circles; DBS and plasma data points have been artificially separated to aid comparison). Graphs on the right are the prediction-corrected visual predictive checks (pcVPC) values for dried blood spot-derived (B) and measured plasma (C) concentrations, showing the 10th, 50th, and 90th percentiles with the actual data (solid and dashed lines) within their respective 95% prediction intervals (gray shaded areas).
DISCUSSION
The present data confirm that ceftriaxone can be quantified in DBS as an alternative to measuring plasma concentrations in pediatric PK/PD studies in resource-limited settings, such as PNG. After adjustment for red cell partitioning and hematocrit bias, the population PK parameters did not differ between plasma and DBS-predicted plasma concentrations. Importantly, the residual variability of 25% was similar for each method of ceftriaxone quantification. Although this study was not designed as a formal PK study, the final estimates of CL and VC in these severely unwell PNG children were similar to those in healthy (7) and critically ill adults (10). Accounting for differences in PK methodologies, our data also accord with older studies of ceftriaxone in younger children with meningitis (11).
In our prior study, we demonstrated that red cell partitioning, ceftriaxone protein binding, assay sensitivity, chad positioning, and temperature stability were either noninfluential or could be incorporated as variables in the calculation of DBS-predicted plasma concentrations (7). The key difference between the present study and the initial study in healthy Australian adults was the systematic bias seen with the large variability in hematocrit in the PNG children, consistent with the increased risk of severe anemia in hospitalized children with severe illness (12). In the present group of 10 severely ill PNG children, the hematocrit varied between 0.22 and 0.52. In contrast, there was a tighter hematocrit range in the Australian adults (0.36 to 0.42, with a median of 0.42). In the prior study, we explored the effect of hematocrit values of 0.31 to 0.67 on matrix effect, process efficiency, and absolute recovery (7). However, in this part of the validation process, the entire spot containing a known volume of blood (50 μl) was cut from the filter paper, rather than the standard approach of cutting a single 6-mm chad from a spot with an unknown volume (7). This observation is consistent with recent DBS validation studies demonstrating a clear hematocrit bias (9). Children with the lowest hematocrit had the highest bias, with a consistent underestimation of DBS-predicted plasma concentrations. At a hematocrit of 0.22, bias was approximately −30% to −40%, a value and slope similar to those observed in a study of caffeine concentrations (9). However, a simple linear adjustment for hematocrit removed this bias and enabled accurate estimation of population PK parameters from DBS-predicted plasma concentrations.
Although studies incorporating DBS sampling have many advantages over conventional PK protocols and offer an attractive alternative approach, particularly in very young children from resource-limited settings, we urge a cautious approach. We were able, in the present study, to overcome the ethical challenges of validating this assay in a pediatric population by recruiting older children in the population of interest. Children aged between 5 and 10 years have a much larger blood volume than very young infants and neonates and might therefore be seen as an ideal group for microsampling validation studies in pediatric populations. We believe that similar validation studies are not appropriate in younger children or neonates from similar settings unless low-volume plasma collections are safe, ethical, and feasible.
The present study demonstrates that assessment of hematocrit needs to be incorporated into any PK or PK/PD study where DBS are used, particularly where variability in hematocrit may cause systematic bias. This may present a challenge where hematocrit measurement is not routinely available. One potential alternative to measurement of the hematocrit is to assay potassium directly from the DBS as a surrogate for correction of hematocrit bias (13), but this would require further validation.
This study has some limitations. We aimed to minimize the possibility that residual ceftriaxone in the i.v. cannula may have affected the measured plasma concentrations. Although we applied a standardized method of removal of 3 ml of blood and flushing with normal saline on each occasion, the presence of two samples with physiologically implausible ceftriaxone concentrations of >1,000 mg/liter suggests that care needs to be taken when using this approach for plasma sampling.
Despite emerging scientific interest in microsampling, there is limited industry or regulatory guidance to inform the approaches to validation of DBS bioanalytical assays for use in regulatory studies (6, 14). The FDA draft guideline from 2013 specifies that storage and handling, thermal stability, homogeneity of spotting, hematocrit, carryover, and reproducibility should be considered minimum requirements. However, there is no guidance on the optimal populations, sample size, and reportable outcome requirements for clinical validation studies, sometimes termed bridging studies. Our approach is that, in addition to demonstration of in vitro feasibility and suitability of a microsampling assay, population-specific PK bridging studies should ideally be performed in healthy volunteers (7, 8) and accessible patients with the infection of interest (15). This would confirm that, in addition to adequate correlations between plasma and DBS concentrations, important PK parameters also accord in clinical settings of interest. In demonstrating an important hematocrit bias in PNG children, the present study highlights that extending validation studies to special patient groups should also be considered where possible.
The present detailed approach to clinical validation of a ceftriaxone DBS assay in severely ill PNG children could serve as a template for validation of DBS assays of other antimicrobial agents as part of PK studies and for therapeutic drug monitoring in a variety of health care settings.
MATERIALS AND METHODS
Approvals, patients, and sample collection.
The present study was approved by the PNG Institute for Medical Research Institutional Review Board (14.02), Medical Research Advisory Committee of PNG (14.29), and by the Executive of Port Moresby General Hospital (PMGH). Ten children aged 5 to 10 years old who were admitted to PMGH with clinical signs of severe pneumonia or acute bacterial meningitis and were being treated with ceftriaxone were recruited. Written informed consent was obtained from the parents or guardians of each child. A baseline assessment, including age, weight, and hematocrit was performed and an intravenous (i.v.) cannula was inserted as part of the clinical management plan. A baseline heparinized blood sample (0 h) was taken for drug assay, as well as for routine biochemical and hematologic tests.
Ceftriaxone (Ceftriaxone Sandoz; Sandoz Australia, Pyrmont, NSW, Australia) was administered i.v. at a dose of 50 mg/kg every 12 h as a slow injection over 3 min in accordance with the manufacturer's recommendations and local prescribing guidelines. The i.v. line was then flushed with 10 ml of normal saline. Further 1.5-ml heparinized venous blood samples were drawn at 0.5, 1, 2, 4, 12 (predose), 24 (predose), 30, and 50 h, with prior removal of 3 ml of blood to eliminate dead space effects and flushing with normal saline on each occasion. After centrifugation, plasma and red cell pellets were separated and kept on ice before storage at −80°C. Duplicate mixed capillary DBS on filter paper cards (Whatman 903 Protein Saver cards; GE Healthcare Australia Pty Ltd., Parramatta, NSW, Australia) were collected at each sampling time point from a finger prick. The mixed capillary blood was drawn evenly over the filter paper by capillary action. The DBS cards were then air dried at room temperature for 1 h, placed in an airtight foil envelope with a single desiccant sachet, and transported on ice before storage at −80°C.
Ceftriaxone concentrations from plasma and DBS were measured using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay (7). Consistent with our previous study, the LOQ and LOD were 0.1 mg/liter and 0.05 mg/liter, respectively. For plasma analyses at 1, 5, 10, 50, 100, and 500 mg/liter, the within-run (intraday) coefficients of variation (n = 5) were 7.2%, 5.3%, 8.5%, 6.9%, 2.1%, and 1.5%, and the between-run (interday) coefficients of variation (n = 15) were 9.8%, 8.6%, 8.2%, 7.6%, 6.7%, and 3.2%, respectively. The accuracies of the plasma assay (± standard deviation [SD]; n = 6) at 1, 5, 10, 50, 100, and 500 mg/liter were 93% ± 10%, 104% ± 8%, 93% ± 5%, 100% ± 9%, 103% ± 7%, and 107% ± 2%, respectively. For the DBS analyses at 1, 5, 10, 50, 100, and 500 mg/liter, the within-run coefficients of variation (n = 5) were 3.6%, 2.9%, 2.1%, 7.6%, 8.1%, and 1.8%, and the between-run coefficients of variation (n = 15) were 8.4%, 7.2%, 8.7%, 9.6%, 8.8%, and 3.9%, respectively. The accuracies of the DBS assay (± SD; n = 15) at 1, 5, 10, 50, 100, and 500 mg/liter were 103% ± 9%, 104% ± 8%, 98% ± 6%, 103% ± 10%, 98% ± 9% and 104% ± 4%, respectively.
Data analysis.
Correlations between plasma and DBS ceftriaxone concentrations were assessed using Spearman's rank correlation coefficient (rs) (GraphPad Prism version 6.05; GraphPad Software Inc., La Jolla, CA). To derive plasma concentrations from DBS samples, the relationship between the two was investigated. Although previously quantified for healthy adults, differences in population characteristics (particularly the range of hematocrits) and handling of samples necessitated reexamination of this relationship in the present patient sample. First, outliers were removed using visual inspection. Then DBS samples were adjusted (divided by 1 minus hematocrit) to account for the difference in volume between matrices. The effect of accounting for the small percentage of ceftriaxone detected in the red cell pellet (<10% of the total concentration) was also assessed. To assess the potential effect of various covariates, DBS bias was plotted against hematocrit, measured DBS concentration, time after the first dose, and time after the last dose. Using the FDA definition for incurred sample reanalysis, DBS bias (%) was defined as follows:
These plots of DBS bias were initially inspected visually, and potential trends were assessed as both linear and exponential relationships.
Pharmacokinetic modeling.
Loge plasma concentration-time data sets for ceftriaxone in plasma and the derived ceftriaxone plasma concentrations from DBS samples were analyzed simultaneously by nonlinear mixed effects modeling using NONMEM v 7.2.0 (ICON Development Solutions, Ellicott City, MD) with a Visual FORTRAN 10.0 compiler (Intel). The first-order conditional estimation (FOCE) with interaction estimation method was used. During the model-building process, the minimum value of the objective function (OFV), goodness-of-fit plots (including conditional weighted residuals [CWRES]), and predictive checks were used. Significance using a chi-square distribution was set at P < 0.01 for the difference in OFV between nested models. Allometric scaling was employed a priori, with volume terms multiplied by (wt/70)1.0 and clearance terms by (wt/70)0.75 (16). Residual variability (RV) equivalent to a proportional model on the normal scale was used for the log-transformed data. Base models were parameterized using VC (central volume of distribution), CL (clearance), VP, and Q (peripheral volumes of distribution[s] and their respective intercompartmental clearance[s]). Both data sets were modeled simultaneously, with an additional variable for each parameter to estimate the difference between the two data sets.
One-, two-, and three-compartment models (ADVAN 1, 3, and 11) with bolus dosing were tested. Once the structure of the models was established, interindividual variability (IIV) and correlations between IIV terms were estimated, where supported by the data. To enable comparison of the plasma and DBS data, an additional parameter was included to enable estimation of the differences in each fixed and random parameter for the DBS model.
Finally, relationships between model parameters with gender, age, and hematocrit were assessed through inspection of scatterplots and boxplots and were subsequently evaluated within NONMEM. A stepwise forward inclusion and backward elimination method was used with a significance of P < 0.05 required for inclusion of a covariate relationship and P < 0.01 to retain a covariate relationship.
Model evaluation.
A bootstrap using Perl-speaks-NONMEM (PsN) with 1,000 samples was performed, and the parameters derived were summarized as median and 2.5th, and 97.5th percentiles (95% empirical confidence intervals [CI]) to facilitate evaluation of final model parameter estimates. In addition, prediction-corrected visual predictive checks (pcVPCs) were performed with 1,000 data sets simulated from the final models using PsN. The observed 10th, 50th, and 90th percentiles were plotted with their respective simulated 95% CIs. Numerical predictive checks (NPCs) were performed to complement the pcVPCs in assessing the predictive performance of the model. Given that both models had the same structure, it was possible to quantify these differences statistically using a chi-square distribution with 2 degrees of freedom to compare the difference between the resultant OFVs.
ACKNOWLEDGMENTS
This study was funded through NHMRC project grant (1047105). T.M.E.D. is supported by an NHMRC Practitioner Fellowship (1058260). B.R.M. was supported by an NHMRC Early Career Fellowship (1036951).
REFERENCES
- 1.Mouton JW, Ambrose PG, Canton R, Drusano GL, Harbarth S, MacGowan A, Theuretzbacher U, Turnidge J. 2011. Conserving antibiotics for the future: new ways to use old and new drugs from a pharmacokinetic and pharmacodynamic perspective. Drug Resist Updat 14:107–117. doi: 10.1016/j.drup.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Ayalew K, Nambiar S, Yasinskaya Y, Jantausch BA. 2003. Carbapenems in pediatrics. Ther Drug Monit 25:593–599. doi: 10.1097/00007691-200310000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Suyagh M, Collier PS, Millership JS, Iheagwaram G, Millar M, Halliday HL, McElnay JC. 2011. Metronidazole population pharmacokinetics in preterm neonates using dried blood-spot sampling. Pediatrics 127:e367–e374. doi: 10.1542/peds.2010-0807. [DOI] [PubMed] [Google Scholar]
- 4.Elliott SR, Beeson JG. 2008. Estimating the burden of global mortality in children aged <5 years by pathogen-specific causes. Clin Infect Dis 46:1794–1795. doi: 10.1086/588049. [DOI] [PubMed] [Google Scholar]
- 5.Howie SR. 2011. Blood sample volumes in child health research: review of safe limits. Bull World Health Organ 89:46–53. doi: 10.2471/BLT.10.080010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parker SL, Dorofaeff T, Lipman J, Ballot DE, Bandini RM, Wallis SC, Roberts JA. 2016. Is there a role for microsampling in antibiotic pharmacokinetic studies? Expert Opin Drug Metab Toxicol 12:601–614. doi: 10.1080/17425255.2016.1178238. [DOI] [PubMed] [Google Scholar]
- 7.Page-Sharp M, Nunn T, Salman S, Moore BR, Batty KT, Davis TM, Manning L. 2016. Validation and application of a dried blood spot ceftriaxone assay. Antimicrob Agents Chemother 60:14–23. doi: 10.1128/AAC.01740-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knippenberg B, Page-Sharp M, Salman S, Clark B, Dyer J, Batty KT, Davis TM, Manning L. 2016. Validation and application of a dried blood spot assay for biofilm-active antibiotics commonly used for treatment of prosthetic implant infections. Antimicrob Agents Chemother 60:4940–4955. doi: 10.1128/AAC.00756-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Kesel PM, Lambert WE, Stove CP. 2015. Does volumetric absorptive microsampling eliminate the hematocrit bias for caffeine and paraxanthine in dried blood samples? A comparative study. Anal Chim Acta 881:65–73. doi: 10.1016/j.aca.2015.04.056. [DOI] [PubMed] [Google Scholar]
- 10.Garot D, Respaud R, Lanotte P, Simon N, Mercier E, Ehrmann S, Perrotin D, Dequin PF, Le Guellec C. 2011. Population pharmacokinetics of ceftriaxone in critically ill septic patients: a reappraisal. Br J Clin Pharmacol 72:758–767. doi: 10.1111/j.1365-2125.2011.04005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steele RW, Eyre LB, Bradsher RW, Weinfeld RE, Patel IH, Spicehandler J. 1983. Pharmacokinetics of ceftriaxone in pediatric patients with meningitis. Antimicrob Agents Chemother 23:191–194. doi: 10.1128/AAC.23.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manning L, Laman M, Rosanas-Urgell A, Michon P, Aipit S, Bona C, Siba P, Mueller I, Davis TM. 2012. Severe anemia in Papua New Guinean children from a malaria-endemic area: a case-control etiologic study. PLoS Negl Trop Dis 6:e1972. doi: 10.1371/journal.pntd.0001972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Kesel PM, Capiau S, Stove VV, Lambert WE, Stove CP. 2014. Potassium-based algorithm allows correction for the hematocrit bias in quantitative analysis of caffeine and its major metabolite in dried blood spots. Anal Bioanal Chem 406:6749–6755. doi: 10.1007/s00216-014-8114-z. [DOI] [PubMed] [Google Scholar]
- 14.Kothare PA, Bateman KP, Dockendorf M, Stone J, Xu Y, Woolf E, Shipley LA. 2016. An integrated strategy for implementation of dried blood spots in clinical development programs. AAPS J 18:519–527. doi: 10.1208/s12248-015-9860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Page-Sharp M, Coward J, Moore BR, Salman S, Marshall L, Davis TME, Batty KT, Manning L. 2017. Penicillin dried blood spot assay for use in patients receiving intramuscular benzathine penicillin G and other penicillin preparations to prevent rheumatic fever. Antimicrob Agents Chemother 61:e00252-17. doi: 10.1128/AAC.00252-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]