

Abstract
Background
ALZ-801 is an oral, small-molecule inhibitor of beta amyloid (Aβ) oligomer formation in clinical development for Alzheimer’s disease (AD). ALZ-801 is a prodrug of tramiprosate with improved pharmacokinetic properties and gastrointestinal tolerability. During clinical studies, we discovered that the primary metabolite of tramiprosate and its prodrug ALZ-801, 3-sulfopropanoic acid (3-SPA), is an endogenous molecule in the human brain and present in the cerebrospinal fluid (CSF) of patients with AD and other neurodegenerative brain diseases.
Objective
The objectives of this research were to (1) identify and confirm the presence of 3-SPA in CSF samples from elderly, drug-naïve patients with memory deficits; (2) quantify the levels of 3-SPA in the CSF of patients with AD from tramiprosate phase III North American (NA) trial; (3) evaluate the in vitro anti-Aβ42 oligomer activity of 3-SPA; and (4) characterize the pharmacokinetics and brain-penetration properties of 3-SPA.
Methods
Lumbar CSF samples from 64 drug-naïve patients with cognitive deficits (Mini-Mental State Examination [MMSE] score range 15–30) and six patients with AD treated with tramiprosate 150 mg twice daily in the phase III trial, at week 78, were analyzed. We used liquid chromatography–tandem mass spectrometry to confirm the structural molecular identity of endogenous 3-SPA with a 3-SPA reference standard and ion-mobility spectrometry–mass spectrometry with molecular dynamics to characterize interactions of 3-SPA with Aβ42 monomers, and the resultant conformational alterations. Rat studies using oral (30 mg/kg) and intravenous (10 mg/kg) doses were conducted to characterize the pharmacokinetic properties and brain penetration of 3-SPA.
Results
We confirmed the presence of 3-SPA in the CSF of drug-naïve patients with cognitive deficits (mean concentration 11.7 ± 4.3 nM). The mean concentration of 3-SPA in patients with AD treated with tramiprosate was 135 ± 51 nM. In vitro studies revealed a multi-ligand interaction of 3-SPA with monomeric Aβ42 that inhibits the aggregation of Aβ42 into small oligomers. Comparisons of the molecular interactions of tramiprosate and 3-SPA with Aβ42 are also presented. Furthermore, in rat preclinical studies, 3-SPA displayed 100% oral bioavailability and 25% brain penetration, indicating that the metabolite is well absorbed and crosses the blood–brain barrier.
Conclusions
We confirmed the endogenous presence of 3-SPA, the major metabolite of tramiprosate, in the CSF of drug-naïve elderly patients with memory deficits due to AD and a variety of other neurodegenerative disorders. The levels of 3-SPA were up to 12.6-fold greater in patients with AD receiving tramiprosate than in drug-naïve patients. In addition, we showed that 3-SPA has potent anti-Aβ oligomer activity, inhibiting aggregation of Aβ42 into small oligomers with efficacy comparable to that of tramiprosate. 3-SPA displays excellent oral availability and brain penetration in rats, suggesting that the higher CSF concentrations of 3-SPA in the human brain after oral administration of ALZ-801 or tramiprosate (and subsequent conversion to 3-SPA) result from the penetration of the metabolite into the central nervous system. These data suggest that 3-SPA is an endogenous agent with potential activity stabilizing the conformational flexibility of Aβ monomers that, in turn, inhibit Aβ misfolding and formation of soluble toxic Aβ oligomers in humans, thereby preventing the initial pathogenic step in the progression of AD. Clinical improvements observed in patients with AD carrying the ε4 allele of the apolipoprotein E gene in tramiprosate phase III studies may in part be explained by the therapeutic effects of excess levels of the metabolite in the brains of these patients. The potential protective role of 3-SPA in AD pathogenesis, as well as its therapeutic role in AD and other neurodegenerative disorders, warrants further investigation.
Key Points
| We describe the discovery and identification of an endogenous Aβ anti-oligomer substance, 3-sulfopropanoic acid (3-SPA), in the human brain. |
| 3-SPA is the primary metabolite of ALZ-801, a prodrug of tramiprosate that is in clinical development for the treatment of Alzheimer’s disease. |
| 3-SPA penetrates the brain and, in a tramiprosate phase III North American (NA) trial, achieved brain concentrations associated with prevention of Aβ42 oligomer formation and clinical outcome benefit in patients with Alzheimer’s disease carrying the ε4 allele of the apolipoprotein E gene. |
Introduction
ALZ-801 is a valine conjugate prodrug of tramiprosate that has been optimized for improved pharmacokinetic properties and gastrointestinal tolerability. Upon oral administration, ALZ-801 is rapidly and fully converted to tramiprosate in plasma and the liver. Tramiprosate, the active agent, readily crosses the blood–brain barrier and reaches a steady-state brain penetration of approximately 40%. Tramiprosate undergoes metabolism into its major metabolite, 3-sulfopropanoic acid (3-SPA) (Fig. 1). Excretion of tramiprosate and the primary metabolite occur via the renal route [1].
Fig. 1.
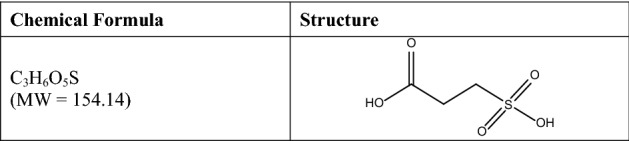
Formula and structure of 3-sulfopropanoic acid. MW molecular weight
During ALZ-801 clinical development studies, we discovered that 3-SPA also occurs as an endogenous agent in the central nervous system (CNS), plasma and urine of drug-naïve humans. Further characterization of the biological activity of the metabolite suggests this substance possesses anti-Aβ oligomer activity that can contribute to the clinical benefit of oral tramiprosate observed in phase III studies in APOE4 (ε4 allele of the apolipoprotein E gene) homozygous and heterozygous Alzheimer’s disease (AD) subjects [2–4].
The extensive clinical and preclinical safety data of oral tramiprosate and ALZ-801, at clinical doses up to the toxicological dose range, show that both the active agent and the metabolite are very well tolerated across rodent and nonrodent species, indicating that the good safety profile is likely because the body is not reacting to these substances as foreign. We also discuss the potential significance of an endogenous “inhibitory” anti-Aβ oligomer substance in the prevention of the pathophysiology of AD and disease progression.
Development of effective therapies for the treatment of AD remains an urgent need as no currently available therapies can slow or stop the course of AD. The growing magnitude of the healthcare cost to society is underscored by the number of patients afflicted across geographical regions, with over 5.7 million in the USA [5] and 35 million worldwide [6]. While targeting soluble amyloid aggregates is the only therapeutic approach to date that has shown a disease-modifying effect in patients with AD, no drugs with disease-modification activity, i.e., clinical activity to inhibit or slow the hallmark progressive cognitive and functional decline of AD, have been approved.
Presently, only two classes of drugs are approved, cholinesterase inhibitors and memantine, both symptomatic agents that do not demonstrate efficacy beyond 6 months of treatment in clinical trials. These drug classes, which constitute the current standard of care, target secondary neurotransmitter deficiencies seen in AD, but there is no evidence that they impact the underlying disease pathology.
Emerging anti-amyloid antibodies (e.g., aducanumab and BAN2401) show promise as potential disease-modifying treatments when used at early stages of the disease [7–9]. However, some amyloid immunotherapies have been associated with a dose-dependent risk of amyloid-related imaging abnormalities with edema (ARIA-E), with increased risk reported in APOE4 carriers [7–9]. This presents a major development challenge, since the two highest doses of aducanumab show amyloid clearance and clinical benefit but are associated with an ~ 40% incidence of ARIA-E [9]. A dose titration regimen with aducanumab still shows an ~ 35% incidence of ARIA-E in APOE4 carriers [10]. Although ARIA-E may be asymptomatic or mildly symptomatic in most patients, some patients developed seizures or other serious adverse events. The risk of ARIA-E in patients with AD requires magnetic resonance imaging (MRI) monitoring, which is burdensome in the elderly population and could limit the utility of these drugs in clinical practice.
Soluble low-molecular-weight Aβ42 oligomers are now recognized as key drivers of AD pathogenesis, and increased concentration of Aβ42 oligomers correlates closely with the onset and progression of clinical symptoms [11–13]. Soluble Aβ oligomers have been shown to cause synaptic damage and neuronal death, promote tau phosphorylation, and drive tau pathology [12, 14–19]. Importantly, APOE4/4 AD patients have been shown to have a higher burden of soluble amyloid oligomers [15], which is likely responsible for the earlier onset of disease in this population.
To date, only agents targeting soluble Aβ oligomers or protofibrils such as aducanumab, BAN2401, and ALZ-801/tramiprosate have shown clinical benefits in amyloid-positive patients with AD. Our discovery of an endogenous substance with anti-Aβ oligomer activity that is also a byproduct of the ALZ-801/tramiprosate pathway is suggestive of an important modulatory or inhibitory pathway in the human brain that can protect Aβ monomers from misfolding and aggregation, the hallmark of AD and the key first step in disease pathogenesis. Therefore, for amyloid-positive patients at the early stage of AD, delivery or replenishment of higher levels of an endogenous substance with anti-Aβ oligomer activity that is also safe, as is achieved after ALZ-801 administration, may provide a potential therapeutic and preventive approach.
Objectives
The main objectives of this research were to (1) identify and confirm the presence of 3-SPA in human cerebrospinal fluid (CSF) samples from drug-naïve patients with memory deficits (Mini-Mental State Examination [MMSE] score range: 15–30) using liquid chromatography–tandem mass spectrometry (LC–MS/MS) analyses; (2) quantify the levels of 3-SPA in the CSF of patients with AD from tramiprosate phase III North American (NA) trial; (3) evaluate the in vitro anti-Aβ42 oligomer activity of 3-SPA; and (4) characterize the pharmacokinetics and brain-penetration properties of 3-SPA.
Methods
Identification and Quantitation of 3-Sulfopropanoic Acid (3-SPA) in Human Cerebrospinal Fluid (CSF) by Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
Individual CSF samples were obtained from 64 male and female subjects with cognitive impairment (MMSE range 15–30) caused by a variety of neurogenerative diseases. These patients were referred to the Cognitive Center at the Department of Neurology, Charles University, 2nd Medical Faculty and Motol University Hospital, Prague Czech Republic. The 64 samples were obtained from patients clinically diagnosed with the following conditions: AD dementia (n = 15), mild cognitive impairment (MCI) due to AD (n = 20), Lewy body disease (n = 1), frontotemporal lobar degeneration (n = 17), MCI of other etiology (n = 7), and mixed dementia, vascular disease, or progressive supranuclear palsy (n = 4).
A total of 12 ml CSF was withdrawn via lumbar puncture in the supine position between vertebral body L3 and L5 using an atraumatic needle. The lumbar puncture was conducted between 8 a.m. and 11 a.m. and effectuated immediately after serum sample collection. The CSF was transferred to the CSF laboratory located on the same floor, where it was spun for 5 min at 2000 rpm at room temperature. After centrifugation, the CSF was aliquoted using 0.5 ml tubes and stored immediately at – 80 °C. Only polypropylene tubes were used for CSF withdrawal and storage. The processing time between CSF withdrawal, spinning and freezing was standardized and did not exceed 45 min in total.
The samples were withdrawn from the freezer, shipped on dry ice to Nextcea, Inc. (Woburn, MA, USA), and stored in a freezer set to maintain − 80 °C after receipt. Subjects provided informed consent, in accordance with the Czech Republic ethical guidelines and good clinical practice and the widely recognized consensus protocol for the standardization of CSF collection and biobanking, before the CSF collection and storage were carried out [20, 21].
CSF concentrations of 3-SPA were also quantified in six patients receiving tramiprosate 150 mg twice daily (BID) at week 78 of tramiprosate phase III North American (NA) trial.
Analysis of the CSF sample from drug-naïve patients was performed by Nextcea using LC–MS and LC–MS/MS. A total of 64 human CSF samples were received at Nextcea for analysis. CSF sample analysis of 3-SPA from patients receiving tramiprosate 150 mg BID in tramiprosate phase III North American (NA) trial were determined using a validated LC–MS/MS method at BELLUS Health Inc. (Montreal, Canada).
Derivatization and LC–MS/MS Method
The 3-SPA reference material and human CSF samples were mixed with N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC) and 2,2,2-trifluro ethylamine (TFEA). The samples were vortexed and reacted at room temperature for 30 min. The reactions were centrifuged at 4500 rpm for 5 min. The supernatant was transferred to a new plate for analysis.
3-SPA was identified and characterized using LC–MS and LC–MS/MS. Injections were made onto a Thermo Scientific AQUASIL 5 µm, 50 × 2.1 mm column using a Shimadzu autosampler and Ultra Performance Liquid Chromatography pump. Mobile phase A was 0.1% trifluoroacetic acid in water (v/v). Mobile phase B was 0.1% formic acid in 90/10 acetonitrile/water (v/v). The flow rate was 0.35 ml/min. The total running time per sample was 4 min. An API 6500 triple quadrupole mass spectrometer was used for detection. Data were acquired in negative LC–MS and LC–MS/MS modes.
Derivatization, representative chromatograms of 3-SPA in crude native material and human CSF derivatized with EDC and TFEA are shown in Figs. 2, 3, and 4. LC–MS and LC–MS/MS data were acquired using Analyst software (AB Sciex, Foster City, CA, USA). The limit of quantification (LOQ) for the LC–MS/MS method was 0.1 ng/ml, with a dynamic range of 0.1–1000 ng/ml (r = 0.99688 and % coefficient of variation [CV] 5.8% ± 2.0; Alzheon, data on file).
Fig. 2.

Derivatization of 3-sulfopropanoic acid by N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC) and 2,2,2-trifluroethylamine (TFEA) to produce 2‐[(2,2,2‐trifluoroethyl)carbamoyl]ethane‐1‐sulfonic acid
Fig. 3.

Liquid chromatography–tandem mass spectrometry spectra of the authentic 3-sulfopropanoic acid reference standard (derivatized with N ethyl-N′-(3-dimethylaminopropyl) carbodiimide [EDC] and 2,2,2-trifluroethylamine [TFEA])
Fig. 4.

Representative liquid chromatography–tandem mass spectrometry chromatograms for 3-sulfopropanoic acid standard (a) and human cerebrospinal fluid with the identical molecular peak (b); spectra from a single subject with Alzheimer’s disease with a Mini-Mental State Examination score of 20
3-SPA was identified in human CSF by matching the chromatographic retention time and by co-elution of the LC–MS/MS transition ions to the authentic 3-SPA reference standard (synthesized by Paraza Pharma, Montreal, Canada). LC–MS/MS transition ions were selected for monitoring based on 2-[(2,2,2-trifluoroethyl)carbamoyl]ethane-1-sulfonic acid, the product ion spectra of the derivatized 3-SPA reference standard (Fig. 3). Structural matching of 3-SPA in human CSF with an authentic sample as standard was performed by matching the molecular peak of the acid as well as the molecular peaks of the 2-[(2,2,2-trifluoroethyl)-carbamoyl]ethane-1-sulfonic acid derivative, including the MS-MS fragmentation pattern by monitoring two LC–MS/MS transition ions (Fig. 3).
3-SPA Molecular Modeling and Molecular Dynamics Simulations
All molecular modeling was performed using the Schrödinger suite (2015-3; Schrödinger, LLC, New York, NY, USA). Molecular dynamics simulations were run using Desmond [22]. The simulations were run on GeForce GTX Titan Black graphics processing unit (GPU) cards. The Optimized Potential for Liquid Simulations (OPLS 3.0) force field [23] was used to model all interactions, and the SPC model was used for waters.
The 1IYT Aβ42 NMR structure from the Protein Data Bank was used as a starting point for molecular dynamics simulations. This structure is primarily alpha helical and is representative of the peptide in an apolar environment. A 20 Angstrom box of water or a mixed solvent box of 1% 3-SPA in water was added around the peptide using Schrödinger system setup tools. Ions were added to neutralize the charge of the entire system. Simulations were conducted at physiological pH (pH 7.4), equilibrated, and run under constant number, pressure, and temperature (NPT) conditions with periodic boundary conditions [22]. The Nose-Hoover Thermostat and Martina–Tobias–Klein barostat were used to control temperature and pressure, respectively.
Simulations were run in replicates of three for 100 nanoseconds each and the results compiled for analysis. Principal component analysis was performed using ProDy [23] and plotted using custom python scripts.
Ion-Mobility Spectrometry–Mass Spectrometry (IMS–MS)
The conditions used for mass spectrometry, using a Waters Synapt G2-S, were as follows: positive polarity in sensitivity mode, capillary 2.5 kV, nebulizer 2 mbar, source temperature 80 °C, desolvation temperature 60 °C, sample cone setting 35 V, source offset setting 60 V, and mass range 500–4000 m/z. These conditions were maintained throughout the study to ensure consistency of the data and to avoid influencing the detection of oligomers due to preferential ionization conditions.
Samples were directly infused into the mass spectrometer at a flow rate of 10 µl/min using a Protea PM-1000 Syringe Pump and Hamilton 1 ml syringe. Amyloid peptide data were acquired using a Waters Synapt G2-S quadrupole time-of-flight mass spectrometer (Q-TOF MS) with traveling wave ion mobility (Waters Corp., Milford, MA, USA). The data were acquired using the systems sensitivity mode to allow for the detection of the less abundant oligomers. Samples were infused at room temperature. The ion-mobility spectrometry–mass spectrometry (IMS–MS) studies were conducted at Protea, Inc. (Morgantown, WV, USA). The methodology, including the IMS–MS blank control, are detailed in Kocis et al. [2].
Sample Preparation
Recombinant human Aβ42 peptide (1 mg) from BioLegend (99% purity, cat# 843801) was reconstituted in 200 µl of Fisher Optima LC–MS grade water (cat# W6-1) and vortexed vigorously for 2 min to solubilize the peptide, creating a 5 mg/ml solution. Samples were then diluted to a final concentration of 22 pmol/µl and incubated at room temperature for 0, 4, and 24 h. After acquisition of incubated samples was completed, the raw data were analyzed using the Waters MassLynx v2.4 suite with DriftScover v2.7 to visualize drift times for the peptide.
Aβ42 Species Characterization
Aβ42 species characterization using IMS–MS was performed by direct infusion at 22 pmol/µl in water. The peptide was prepared in water to maintain the native state conformation of the peptide and ion-mobility data were acquired to detect and characterize the conformational changes of the native state monomer and any oligomers that may have formed during the incubation.
3-SPA IMS–MS Binding Study
Data for Aβ42 peptide were acquired using a Waters Synapt G2-S Q-TOF MS with traveling wave ion mobility (Waters Corp.). The data were acquired using the systems sensitivity mode to allow for the detection of the less abundant oligomers. Samples were infused at room temperature, as described.
3-SPA (1 mg) was reconstituted in 1 ml of Fisher Optima LC–MS grade water (cat# W6-1) and vortexed vigorously for 2 min until completely dissolved. The sample was then diluted to create 220 and 22,000 pmol/µl solutions to perform a 100-fold and 1000-fold molar excess for the binding experiments with Aβ42 peptide.
Recombinant human Aβ42 peptide (1 mg) was reconstituted in 200 µl of Fisher Optima LC–MS grade water and vortexed vigorously to solubilize to a 5 mg/ml solution. Samples were then diluted to their final concentrations and incubated at room temperature for 0, 4, and 24 h, followed by analysis, as described.
Pharmacokinetics, Oral Absorption and Brain Exposure of 3-SPA in Sprague Dawley Rats
The oral and intravenous pharmacokinetics of 3-SPA were evaluated in male Sprague Dawley (SD) fasted rats at a dose of 30 and 10 mg/kg, respectively (n = 3 per group). The study design and doses used for the pharmacokinetic evaluation of 3-SPA are in the range of standard investigative doses commonly employed to explore and define the single-dose oral and intravenous pharmacokinetic and absorption characteristics of a small molecule in studies in rodents. Animals were housed in a standard facility, with water and food provided ad libitum. 3-SPA was dissolved in saline and administered orally by gavage and intravenously as a bolus. Serial blood samples (approximately 0.2 ml each) were collected from each animal at 0.25, 0.5, 1, 2, 4, 8, and 24 h after dosing into tubes containing K2EDTA and processed for plasma by centrifugation. Plasma samples were stored at – 80 °C until bioanalyses.
A separate group of animals was dosed orally at 30 mg/kg, and terminal brain, CSF, and plasma samples were collected at 1, 2, 6, and 24 h (three animals for each time point) for bioanalyses of 3-SPA in the brain and CSF and to estimate brain penetration relative to plasma concentrations. The in-life study was performed at Agilux Laboratories (Worcester, MA, USA) following quality standards in line with good laboratory practice. Bioanalyses of rat plasma, CSF, and brain were performed using LC–MS/MS at Nextcea. Prior to processing for bioanalysis, the rat brains were perfused to remove pooled blood. Pharmacokinetic data analyses were conducted using Winnonlin Professional v5.0.1 (Pharsight, Mountain View, CA, USA).
Results
Identification and Quantitation of 3-SPA in the CSF of Tramiprosate-Naïve Subjects and Tramiprosate-Treated Patients with Alzheimer’s Disease
The [M-H]− of 3-SPA was detected in human CSF at m/z 234.1 at retention time 1.55 min. The transition ion of the molecular peak of the diacid (234.1/80.9) was selected for quantitation. The lower LOQ (LLOQ) of the LC–MS/MS assay was 0.1 ng/ml for 3-SPA. Table 1 shows the 3-SPA concentrations in human CSF.
Table 1.
Concentrations of 3-sulfopropanoic acid in human cerebrospinal fluid in drug-naïve patients with memory deficits
| Descriptive statistics of patients with memory deficits | All patients | Male | Female |
|---|---|---|---|
| N | 64 | 27 | 37 |
| Age ± SD (years) | 68.6 ± 8.4 | 69.2 ± 8.4 | 68.2 ± 8.3 |
| Clinical diagnosis (n) | |||
| AD | 15 | 4 | 11 |
| MCI due to AD | 20 | 11 | 9 |
| MCI other | 7 | 2 | 5 |
| FTLD | 17 | 9 | 8 |
| Other | 5 | 1 | 4 |
| MMSE mean ± SD | 25.0 ± 3.1 | 25.4 ± 2.4 | 24.6 ± 3.6 |
| Concentration of 3-SPA in CSF (ng/ml [nM]) | |||
| Mean ± SD | 1.8 ± 0.7 (11.8 ± 4.3) | 1.9 ± 0.6 (12.5 ± 4.1) | 1.7 ± 0.7 (11.2 ± 4.4) |
| Median | 1.7 (10.8) | 1.7 (11.1) | 1.6 (10.4) |
| Minimum–maximum | 0.6–4.3 (4.2–27.7) | 0.8–3.8 (5.5–24.7) | 0.6–4.3 (4.2–27.7) |
Data are presented as mean ± standard deviation unless otherwise indicated
AD Alzheimer’s disease, CSF cerebrospinal fluid, FTLD frontotemporal lobular degeneration, MCI mild cognitive impairment, MMSE Mini-Mental State Examination, other includes Lewy body disease, vascular dementia, mixed disease, progressive supranuclear palsy, SD standard deviation, 3-SPA 3-sulfopropanoic acid
The levels of 3-SPA in patients with a variety of cognition-impairing diseases, including AD, ranged from 4.2 to 27.7 nM (Table 1). When related to the CSF concentrations of Aβ42 monomers in patients with AD (0.04–0.1 nM) [24–27], there is an approximately 40- to 700-fold excess of 3-SPA over soluble Aβ42 monomers, which falls within the range at which partial Aβ anti-oligomer aggregation activity may occur in some patients (Table 2). Furthermore, retrospective analyses of the CSF of a subset of patients from tramiprosate phase III North American (NA) trial were also evaluated for the presence of 3-SPA, the primary metabolite of tramiprosate. Table 3 presents descriptive summaries of 3-SPA concentrations in CSF, which were analyzed using a validated method in a different bioanalytical laboratory. The metabolite concentrations were quantified in six patients for whom CSF samples at week 78 were available. The mean CSF concentration of 3-SPA was 144.7 nM (range 112.3–231.8), thus representing a 12.6-fold increase over levels observed in drug-naïve patients.
Table 2.
Comparison of anti-Aβ42 oligomer activity of 3-sulfopropanoic acid vs. tramiprosate at 100:1 and 1000:1 excess ratios of compound:Aβ protein
| Oligomer species | Aβ42 alone | Tramiprosate (100:1) | 3-SPA (100:1) | Tramiprosate (1000:1) | 3-SPA (1000:1) |
|---|---|---|---|---|---|
| Dimer | Y | N | Y | N | N |
| Trimer | Y | Y | Y | N | N |
| Tetramer | Y | Y | Y | N | N |
| Pentamer | Y | Y | Y | N | Y |
| Hexamer | Y | Y | Y | N | N |
| Decamer | Y | N | N | N | N |
N indicates no presence of oligomer species, 3-SPA 3-sulfopropanoic acid, Y indicates the presence of oligomer species
Bold text highlights the results of 3-SPA on inhibition of Aβ42 oligomer sub-species from IMS–MS experiments
Table 3.
3-sulfopropanoic acid cerebrospinal fluid concentrations (ng/ml) at week 78 in tramiprosate phase III North American (NA) trial
| Descriptive statistics | Tramiprosate 150 mg BID at week 78 |
|---|---|
| N | 6 |
| Mean MMSE ± SD | 22.3 ± 7.9 ng/ml (144.7 ± 51.3 nM) |
| Median 3-SPA concentration | 19.8 ng/ml (128.6 nM) |
| Minimum–maximum 3-SPA concentration | 17.3–35.7 ng/ml (112.3–231.8 nM) |
BID twice daily, SD standard deviation
Anti-Aβ42 Oligomeric Activity of 3-SPA
These results show the effect of modulation of Aß conformational space by 3-SPA leading to the prevention of oligomer formation at the 1000:1 ratio of 3-SPA:Aβ42. The inhibition of Aβ42 oligomerization by 3-SPA was compared with a blank experiment where we identified the full spectrum of soluble oligomeric species from dimer to decamer, as described previously [2]. In addition, we found that 3-SPA:Aβ42 in a 1000:1 ratio yields not only concentration dependency but also time dependency, an anti-Aβ42 oligomeric effect as indicated in Figs. 5 and 6.
Fig. 5.

Ion-mobility spectrometry–mass spectrometry drift time as a function of mass/charge (m/z) after 4-h incubation of Aβ42 with 3-sulfopropanoic acid in ratio 1:1000 of 3-sulfopropanoic acid:Aβ42 with the profile of Aβ42 oligomers
Fig. 6.

Ion-mobility spectrometry–mass spectrometry drift time as a function of mass/charge (m/z) after 24 h of incubation shows the profile of Aβ42 oligomers with 1000-fold excess of 3-sulfopropanoic acid
Table 2 presents a summary of the concentration excess dependency of 3-SPA over Aβ42; 100- versus 1000-fold molar excess of 3-SPA over Aβ42 resulted in a different sub-species profile of inhibition of Aβ42 oligomer formation. The activity is also compared with the same excess dependency of tramiprosate. Near complete prevention of formation of Aβ42 oligomers except for pentamers was shown for 3-SPA. We observed that inhibition of Aβ42 oligomer formation by 3-SPA shows time dependency (Figs. 5, 6). Detection of Aβ42 dimers, trimers, and pentamers under these conditions revealed that 4 h of in vitro incubation was insufficient for complete inhibition of oligomer formation. Taken together, these IMS–MS experiments showed that inhibition of formation of Aβ42 oligomers by 3-SPA is both concentration and time dependent (concentration dependency is indicated in Table 2).
Interestingly, while the functional end results, i.e., inhibition of Aβ42 oligomer formation, are the same for both tramiprosate and its metabolite, 3-SPA, the conformational landscape of the process is not. 3-SPA as a dianion under physiological conditions interacts with cations on amino acid side chains of Aβ42 (Fig. 7). These are protonated amino groups of Lys16, Lys28, and His13,14. At the same time, repulsive forces of 3-SPA dianion and carboxylate groups of Aβ42 are at work. This interplay of ionic interactions contributes to significant conformational changes of Aβ42 monomer species. We also observed that the functional result of 3-SPA is similar to the functional end results found with tramiprosate, i.e., inhibition of Aβ42 oligomer formation [2].
Fig. 7.

Representation of molecular dynamics experiment showing semi-cyclic conformation of Aβ42 in the presence of 1000:1 excess of 3-sulfopropanoic acid
Both IMS–MS data and molecular dynamics (Figs. 3, 4, 5, 6) display multi-ligand binding interactions between 3-SPA and Aβ42 monomers. 3-SPA interacts with Aβ42 via different ionic interactions than does tramiprosate. Interestingly, although employing different ionic binding patterns, under the same conditions, data from IMS–MS and molecular dynamics show qualitatively the same anti-Aβ42 oligomeric result from both compounds. Tramiprosate has shown complete inhibition of Aß42 oligomer formation after 24 h in vitro, whereas 3-SPA has shown the same results except for inhibition of pentamer formation over the same time scale. However, a detailed time course investigation also showed a time-dependent course of oligomer-formation inhibition. After 4 h, 3-SPA inhibits the formation of oligomers, except for dimers, trimers, and pentamers. After sustained 24 h of exposure, the only oligomeric species not inhibited were pentamers of Aβ42. Further investigation of the time-dependent properties of 3-SPA on oligomer formation and inhibitory activity is planned.
These data suggest that the first anti-oligomeric effect of tramiprosate is followed by the second anti-oligomeric effect of tramiprosate metabolite 3-SPA.
Pharmacokinetics and Brain Penetration of Orally Administered 3-SPA in Rats
Figure 8 shows the plasma concentration of a single dose of 3-SPA, dissolved in saline as a clear solution, administered orally and intravenously to rats at a dose level of 30 and 10 mg/kg, respectively. Figure 9 shows the brain, CSF, and corresponding plasma levels after a single oral dose of 3-SPA 30 mg/kg at 1, 2, 6, and 24 h. Mean pharmacokinetic curves were used to calculate pharmacokinetic parameters. Tables 4 and 5 show the resulting pharmacokinetic parameters, oral bioavailability, and brain penetration of 3-SPA in rats.
Fig. 8.
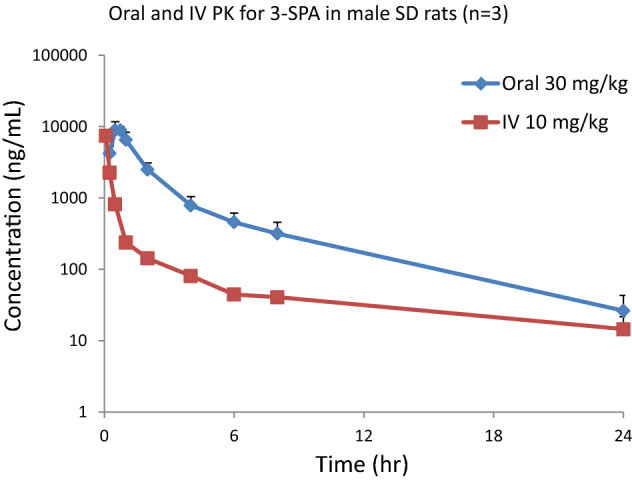
Mean pharmacokinetic curves for single oral and intravenous doses of 3-sulfopropanoic acid in male Sprague Dawley rats (30 and 10 mg/kg). IV intravenous, PK pharmacokinetics, SD Sprague Dawley rats, 3-SPA 3-sulfopropanoic acid
Fig. 9.
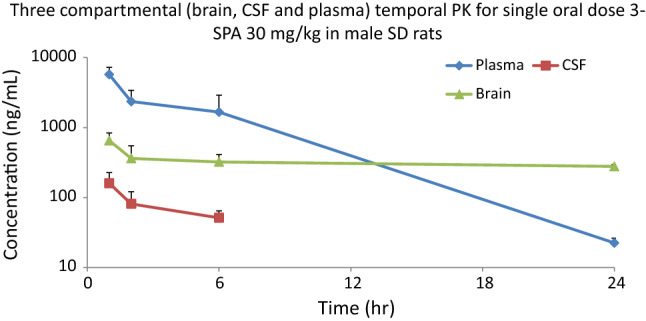
Mean brain, cerebrospinal fluid, and plasma concentration–time course of 3-sulfopropanoic acid after a single oral dose of 30 mg/kg in male Sprague Dawley rats. CSF cerebrospinal fluid, PK pharmacokinetics, SD Sprague Dawley rats, 3-SPA 3-sulfopropanoic acid
Table 4.
Pharmacokinetic parameters of oral and intravenous 3-sulfopropanoic acid in male Sprague Dawley rats (n = 3)
| Parameters | Values |
|---|---|
| Oral pharmacokinetics (30 mg/kg) | |
| t½ | 4.4 h |
| tmax | 0.5 h |
| Cmax | 8943 ng/ml |
| AUC0–t | 18,894 ng/ml × h |
| AUC0–inf | 19,061 ng/ml × h |
| Intravenous pharmacokinetics (10 mg/kg) | |
| t½ | 11.0 h |
| tmax | 0.083 h |
| Cmax | 7484 ng/ml |
| C0 | 13,599 ng/ml |
| AUC0–t | 3407 ng/ml × h |
| AUC0–inf | 3638 ng/ml × h |
| Vz | 43.6 l/kg |
| Cl | 2.7 l/kg/h |
| Vss | 12.9 l/kg |
| Oral bioavailability | ~ 100% |
AUC area under the curve, C0 concentration at time 0, Cl clearance, Cmax maximum observed plasma concentration, t½ half-life, tmax time of maximum observed plasma concentration, Vss apparent volume of distribution at steady state, Vz apparent volume of distribution at terminal phase
Table 5.
Brain penetration of 3-sulfopropanoic acid after oral single-dose administration (30 mg/kg) in fasted male Sprague Dawley rats
| Pharmacokinetic parameters | Plasma | Brain | CSF |
|---|---|---|---|
| t½ (h) | 3.0 | 64.1 | 3.6 |
| Cmax (ng/ml) | 5714.6 | 649.8 | 159.2 |
| AUC0–t (ng/ml × h) | 30001.7 | 7586.5 | 463.6 |
| AUC0–inf (ng/ml × h) | 30099.2 | 33224.4 | 726.2 |
| Brain/plasma AUC ratio | 25.3% | ||
| CSF/brain AUC ratio | 6.1% |
AUC area under the curve, Cmax maximum observed plasma concentration, CSF cerebrospinal fluid, t½ half-life, tmax time of maximum observed plasma concentration
Discussion
This study establishes the existence of an endogenous agent in the brain, 3-SPA, with anti-Aβ oligomer activity. 3-SPA inhibits aggregation of Aβ42 into small oligomers and is also a major metabolite of tramiprosate (3-SPA was previously referred to as NRM5074), which has shown similar anti-Aβ oligomer activity [2] as well as cognitive improvement in a subgroup of APOE4 AD patients in phase III trials [3, 4].
We also describe that, in patients with AD receiving tramiprosate 150 mg BID in a phase III trial, 3-SPA levels in the CSF reached concentrations approximately 12.6-fold greater than those observed in drug-naïve subjects. Importantly, investigative studies of orally administered 3-SPA in rats showed that 3-SPA readily crossed the blood–brain barrier. Therefore, administration of ALZ-801, the prodrug of tramiprosate in development for AD, is also likely to yield high levels of 3-SPA exposure in the brain after oral administration. The discovery of 3-SPA in the human brain, together with its potent anti-Aβ42 oligomer activity, suggests that 3-SPA may contribute to the cognitive benefits of oral tramiprosate in APOE4 homozygous and heterozygous AD patients [3, 4].
Tramiprosate, the active agent of oral prodrug ALZ-801, has previously shown anti-Aβ aggregation and anti-Aβ oligomer effects in preclinical in vitro and in vivo models of AD [28]. 3-SPA displays a concentration-dependent multi-ligand interaction with monomeric Aβ42, which is similar to tramiprosate-binding interactions [2] and differs from the classical 1:1 binding. When concentrations reach a 1000:1 ratio of 3-SPA:Aβ42, the binding interactions stabilize Aβ42 monomer conformation, leading to inhibition of Aβ42 oligomer formation and elongation, as demonstrated by IMS–MS and molecular dynamics. Molecular dynamics analyses also show that 3-SPA binds to Lys16, Lys28, His13, and His14, the key amino acid side chains of Aβ42 that are responsible for amyloid seed formation and neuronal toxicity [14, 29]. Interestingly, the kinetics of oligomer formation inhibition are slower for 3-SPA than for tramiprosate, suggesting an important time-dependency component when the 3-SPA:Aβ42 ratio reaches 1000:1. This finding may have implications for the anti-Aβ oligomer action of 3-SPA, as it implies that sustained excess exposure over time is required for a full effect.
It is further suggested that the 3-SPA levels found in the CSF of drug-naïve subjects may partially inhibit formation of oligomer species. In drug-naïve subjects, the steady state of 3-SPA is likely below the 1000:1 ratio required for complete inhibition of the formation of a full range of oligomeric species. Therefore, the impact of endogenous 3-SPA on Aβ oligomer formation in drug-naïve subjects requires investigation. Furthermore, brain levels of 3-SPA may change over time due to age, disease state, or diurnal variations, opening the possibility that the endogenous anti-oligomer activity of 3-SPA varies considerably over time according to the stage of AD and other neurodegenerative disorders. Consequently, therapeutic replacement or supplementation of the endogenous 3-SPA with oral ALZ-801 may provide an effective and safe approach to achieve sustained 3-SPA levels for maximum anti-Aβ oligomeric activity.
It is possible that, in patients with AD receiving oral tramiprosate, the active agent of oral prodrug ALZ-801, 3-SPA, contributes to the anti-Aβ oligomer activity of tramiprosate, as 3-SPA readily crosses into the brain and shows potent anti-Aβ oligomer effects. Estimates of mean tramiprosate concentrations in the brain (~ 130 nM) from tramiprosate phase III North American (NA) trial are described in the mechanism of action study [2]. It is likely that the total sustained sum of anti-Aβ42 oligomer agents tramiprosate + 3-SPA available in the brain is responsible for the clinical activity. The projected brain level of the combined tramiprosate + 3-SPA was 375–550 nM, which represents an excess above brain levels of Aβ42 in patients with AD in the range of 5000- to 15,000-fold [1]. ALZ-801, the oral prodrug of tramiprosate with improved pharmacokinetic properties and brain penetration, shows an ~ 30% improvement in brain penetration based on pharmacokinetic/pharmacodynamic (PK/PD) projections using 14C tramiprosate [1]. Increased levels of tramiprosate after ALZ-801 administration reflect the improvement in pharmacokinetics as well as the increase in exposure of the combined products, leading to larger excess concentrations needed to yield full inhibition of Aβ oligomer formation [2]. Therefore, the improved PK/PD profile of ALZ-801 increases the likelihood of a successful clinical outcome in planned efficacy trials. While we did not directly conduct the present experiments at physiological nM concentrations of AB42 because of the sensitivity limitations of the bioanalytical tools available to measure the anti-oligomer activity by IMS–IMS, it is reasonable to project that, based on the law of mass action, the interactions would be similar provided the excess ratio of 3-SPA:AB42 and conditions remain similar. In support of this, the molecular modelling projections support the same interaction when > 1000-fold excess of 3-SPA: A42 is sustained.
Safety data from the combined tramiprosate phase III studies [4] and the safety extension study show a favorable safety profile up to 2.5 years. Furthermore, the brain MRI safety analyses from both phase III trials of tramiprosate revealed no cases of ARIA-E in subjects receiving the active drug. The chronic toxicology studies required for the advancement of ALZ-801 showed that it was well tolerated and exhibited a no observed adverse effect level (NOAEL) in 1-month and 6-month rat studies of 2000 and 1500 mg/kg, respectively. These studies achieved sustained exposure of 3-SPA. The excellent nonclinical safety of 3-SPA is underscored by the fact that, at the 2000 mg/kg of ALZ-801 NOAEL in rats, the sustained levels achieved were 180 millimolar (area under the plasma concentration–time curve [AUC] in h × nM) in the 28-day rat study (Alzheon, data on file). Similarly high levels were also achieved in the 6-month rat toxicology study, where the NOAEL was 1500 mg/kg (Alzheon, data on file). Both the preclinical and clinical safety data for ALZ-801 and oral tramiprosate further underscore the excellent safety and tolerability of the ALZ-801/tramiprosate platform.
The endogenous source of 3-SPA remains unknown, but its molecular structure has moieties suggestive of catabolism of sulphur-containing amino acids such as cysteine. It is thus reasonable to hypothesize that 3-SPA can be produced from cysteine via the cysteine sulfinic acid oxidation and reductive deamination pathway. This potential metabolic pathway is similar to the mammalian biosynthesis of taurine, which starts from cysteine and also proceeds via the cysteine sulfinic acid pathway, via sulfinic acid oxidation and decarboxylation [30]. Consistent with the notion that 3-SPA is produced and eliminated from the body via excretion pathways, the presence of 3-SPA was also observed in urine and plasma in drug-naïve samples (samples obtained from Bioreclamation, NY, USA; Alzheon, data on file). Consistent with this initial observation, an MS-based metabolomics study that examined the urine of healthy volunteer subjects also suggested the presence of 3-SPA in urine, although no bioanalytical structural confirmation was provided [31].
Investigation of the physiological function of 3-SPA and its potential role in aging and the pathogenesis of AD and other neurodegenerative disorders requires additional investigation, and was beyond the scope of this first study to identify, confirm, and characterize 3-SPA. The discovery of an endogenous substance that can prevent Aβ42 oligomer formation suggests the possibility of a protective endogenous anti-Aβ oligomer pathway (“Aβ oligomer brake pathway”) within the human CNS, with the potential to prevent, ameliorate or delay the onset of AD. Such an Aβ oligomer brake pathway could modulate the neurotoxic effects of abnormal Aβ aggregation in the aging human brain. The discovery of an endogenous anti-Aβ oligomeric agent further raises the possibility that processes that govern 3-SPA production and/or serve to regulate the levels of 3-SPA may also play a regulatory or modulatory role in the pathophysiology of Aβ aggregation and elongation of Aβ oligomers that occur in neurodegenerative disorders such as AD. An important additional insight is that 3-SPA, which is present at very high systemic levels in both preclinical and clinical studies, is well tolerated and displays a benign safety profile across species, including humans.
Conclusions
These studies confirm the endogenous presence of 3-SPA, the major metabolite of tramiprosate, in the CSF of drug-naïve elderly patients with memory deficits due to a variety of neurodegenerative disorders. The discovery and demonstration that 3-SPA displays Aβ42 anti-oligomer activity comparable to that of tramiprosate suggests that 3-SPA may contribute to the biological and potential clinical activity of tramiprosate and its prodrug ALZ-801 in patients with AD. The potential protective role of endogenous 3-SPA on normal brain function, and in the pathogenesis of neurodegenerative disorders such as AD, as well as its therapeutic role, warrants further investigation.
Author contributions
JAH wrote the article. All co-authors reviewed and contributed to the article.
Compliance with Ethical Standards
Funding
The studies summarized in this report were supported by Alzheon, Inc. Funding to cover the cost of open access to this article was provided by Alzheon, Inc.
Conflicts of interest
John A. Hey, Petr Kocis, Susan Abushakra, Aidan Power, and Martin Tolar are employees of Alzheon, Inc., and own Alzheon stock options. Jeremy Y. Yu serves as a consultant to Alzheon and owns Alzheon stock options. Jakub Hort and Martin Vyhnálek are independent clinical investigators who collaborated on the study. Dr. Jakub Hort is a member of the Alzheon Scientific Advisory Board and owns Alzheon stock options.
Ethical standards
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Informed Consent
Drug naïve subjects provided informed consent in accordance with the Czech Republic ethical guidelines.
References
- 1.Hey JA, Yu Jeremy Y, Versavel M, Abushakra A, Kocis P, Power A, Kaplan PL, Amedio J, Tolar M. Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer’s disease. Clin Pharmacokinet. 2018;57:315–333. doi: 10.1007/s40262-017-0608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kocis P, Tolar M, Yu J, Sinko W, Ray S, Blennow K, Fillit H, Hey JA. Elucidating the Abeta42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: integrating molecular analytical methods. Pharmacokinetic and clinical data. CNS Drugs. 2017;31:495–509. doi: 10.1007/s40263-017-0434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abushakra S, Porsteinsson A, Scheltens P, Sadowsky C, Vellas B, Cummings J, Gauthier S, Hey JA, Power A, Wang P, Shen L, Tolar M. Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer’s disease suggest disease modification potential. J Prev Alzheimers Dis. 2017;4:149–156. doi: 10.14283/jpad.2017.26. [DOI] [PubMed] [Google Scholar]
- 4.Abushakra S, Porsteinsson A, Vellas B, Cummings J, Gauthier S, Hey JA, Power A, Hendrix S, Wang P, Shen L, Sampalis J, Tolar M. Clinical benefits of tramiprosate in Alzheimer’s disease are associated with higher number of APOE4 alleles: the ‘‘APOE4 gene-dose effect’’. J Prev Alz Dis. 2016;3:219–228. doi: 10.14283/jpad.2016.115. [DOI] [PubMed] [Google Scholar]
- 5.Alzheimer’s disease facts and figures. Alzheimer’s Dement 2018;14(3):367–429.
- 6.Word Alzheimer Report. London: Alzheimer’s Disease International (ADI); 2016.
- 7.Lasser R, Ostrowitz S, Scheltens P, et al. Efficacy and safety of Gantenerumab in prodromal AD: results from Scarlet road—a global, multicenter trial. Alzheimer’s association international conference (AAIC); 2015 (abstract ID 5963).
- 8.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of Bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 10.Viglietta V, O’Gorman J, Williams L, Chen T, Enayetallah A, Chiao P, Hock C, Nitsch RM, Haeberlein AB, Sandrock A. Aducanumab titration dosing regimen: 12-month interim analysis from prime, a randomized double blind, placebo controlled phase Ib study in patients with prodromal or mild Alzheimer’s disease. J Prev Alzheimers Dis. 2016;3(suppl 1):378. [Google Scholar]
- 11.Caselli RJ, Dueck AC, Osborne D, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med. 2009;361:255–263. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL. Amyolid beta oligomerization in Alzheimer’s dementia vs. high pathology controls. Ann Neurol. 2013;73(1):104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viola KL, Klein WL. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129:183–206. doi: 10.1007/s00401-015-1386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Usui K, Hulleman JD, Paulsson JF, Siegel SJ, Powers ET, Kelly JW. Site-specific modification of Alzheimer’s peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. PNAS. 2009;106:18563–18568. doi: 10.1073/pnas.0804758106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto T, Serrano-Pozo A, Hori Y, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J Neurosci. 2012;32:15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono K, Yamada M. Low-n oligomers as therapeutic targets of Alzheimer’s disease. J Neurochem. 2011;117:19–28. doi: 10.1111/j.1471-4159.2011.07187.x. [DOI] [PubMed] [Google Scholar]
- 17.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from A 1–42 are potent central nervous system neurotoxins. PNAS. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin M, Selkoe DJ. Systematic time analysis of time-dependent neural effects of soluble amyloid oligomers in culture and in vivo: prevention by scyllo-inositol. Neurobiol Dis. 2015;82:152–163. doi: 10.1016/j.nbd.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanderstichele H, Bibl M, Engelborghs S, et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: a consensus paper from the Alzheimer’s biomarkers standardization initiative. Alzheimers Dement. 2012;8(1):65–73. doi: 10.1016/j.jalz.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Hort J, Glosova L, Vyhnalek M, Bojar M, Skoda D, Hladikova M. The liquor tau protein and beta amyloid in Alzheimer’s disease. Cesk Slov Neurol N. 2007;70(1):30–36. [Google Scholar]
- 22.Shivakumar D, Harder E, Damm W, Friesner RA, Sherman W. Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J Chem Theory Comput. 2012;8:2553–2558. doi: 10.1021/ct300203w. [DOI] [PubMed] [Google Scholar]
- 23.Bakan A, Meireles LM, Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pannee J, Portelius E, Minthon L, Gobom J, Andreasson U, Zetterberg H, et al. Reference measurement procedure for CSF Abeta1-42 and the CSF Abeta1-42 /Abeta1-40 ratio—a crossvalidation study against amyloid PET. J Neurochem. 2016;139:651–658. doi: 10.1111/jnc.13838. [DOI] [PubMed] [Google Scholar]
- 26.Herukka S-K, Rummukainen J, Ihalainen J, Und Zu Fraunberg von M, Koivisto AM, Nerg O, et al. Amyloid-beta and tau dynamics in human brain interstitial fluid in patients with suspected normal pressure hydrocephalus. J Alzheimers Dis. 2015;46:261–269. doi: 10.3233/JAD-142862. [DOI] [PubMed] [Google Scholar]
- 27.Lue L-F, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/S0002-9440(10)65184-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, et al. Targeting soluble Aβ peptide with tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28:537–547. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Sinha S, Lopes DHJ, Bitan G. A key role for lysine residues in amyloid β-protein folding, assembly, and toxicity. ACS Chem Neurosci. 2012;3:473–481. doi: 10.1021/cn3000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehninger AL. Lehninger principles of biochemistry. 4. New York: W.H Freeman; 2005. [Google Scholar]
- 31.Zhang Tong, Creek Darren J, Barrett Michael P, Blackburn Gavin, Watson David G. Evaluation of coupling reversed phase. Aqueous normal phase, and hydrophilic interaction liquid chromatography with Orbitrap mass spectrometry for metabolomic studies of human urine analytical chemistry. Anal Chem. 2012;84:1994–2001. doi: 10.1021/ac2030738. [DOI] [PubMed] [Google Scholar]