

Abstract
A set of psychoactive drugs has been analyzed with the use of quantitative structure–activity/property relationships methods. The purpose of this study was to demonstrate both the common and differentiating characteristics of the above-mentioned chemical compounds, physicochemical as well as pharmacological based on the quantum chemical calculations and selected biological activity data and chromatographic retention parameters. During the study, the ab initio model of molecular modeling was performed and PCA, FA, and MLR as the types of chemometric approach. QSAR/QSPR models were proposed based on chosen statistically significant descriptors. The relationship between the structure and biological activity data was able to class and describe the psychoactive properties of the molecules studied. The applied chemometric approaches revealed the influential features of tested structures responsible for their pharmacological activity together with some additional physicochemical properties.
Keywords: Psychoactive drugs, Molecular modeling, Structural analysis, Descriptors, QSAR; QSPR
Introduction
The term psychoactive/psychotropic drug is defined as the collective name of substances whose only or main action is the effect on mental activities. In general, they are agents that act on the central nervous system, but there are also psychotropic agents in medicines belonging to other pharmacological groups. For pharmacological purposes, the division into psycholeptic, psychoanaleptic, and psychodysleptic drugs (hallucinogenic and psychotomimetic) is introduced. For practical purposes, people speak more often about neuroleptics, antidepressants, and anxiolytics. The pharmacological classification is also applied to the chemical classification taking into account the structure of the molecules of the considered drugs (Hendriksen and Groenink 2015; Stahl 2013; Dean 2011; Spiegel 2003).
Psychotropic agents have often been the subject of many works in the field of structure—properties, such as the relationship between the enthalpy of complexation of charge-transfer (CT) complexes with chloranil and their biological activity by a series of neuroleptics (Saucin and van der Vorst 1972), relationships between lipophilicity indices determined by RP-HPLC methods (as well as constants dissociation) and biological activity (Unger and Chiang 1981), pharmacological classification based on retention data in HPLC systems (Nasal et al. 1997) or attempts to predict biological activity for tricyclic neuroleptics and antidepressants based on quantum-chemical calculations of energy values of boundary orbitals e.g. HOMO and LUMO (Cogordan et al. 1999).
The subject of this work was the analysis for data from ab initio calculations and the biological activity data as well as physicochemical parameters presented in the cited papers (Saucin and van der Vorst 1972; Unger and Chiang 1981; Davis and Brody 1966). In addition, chromatographic retention data (Nasal et al. 1997) were also available for some of the compounds considered, which were used as variables dependent on structural parameters.
The aim of this research was to demonstrate both common and differentiating features of the analyzed chemical compounds both in physicochemical and pharmacological terms and confirmed the chemometrics relationship between nonempirical parameters characterizing chemical structure and biological activity.
Materials and methods
Molecules
The following compounds (Fig. 1), considered in the cited papers (Unger and Chiang 1981; Saucin and van der Vorst 1972; Davis and Brody 1966) were selected for the study: acoperon, benperidol, droperidol, haloperidol, spiroperidol, and triperidol from the group of butyrophenone neuroleptics, acetophenazine, chlorpromazine, chlorprothixen, dixyrazine, fluphenazine, levomepromazine, perphenazine, prochlorperazine, promazine, promethazine, thioproperazine, thioridazine, trifluoperazine, and trifluopromazine from the group of tricyclic neuroleptics; and: amitriptiline, clomipramine, desipramine, imipramine, and nortriptiline from the group of tricyclic antidepressants. Data on their structure were available for these compounds.
Figure 1.

Structural formulas of compounds studied
Biological activity and physicochemical parameters data
The study includes literature data on biological activity: D dose (mg/kg) 50% reduction in motor activity of rats expressed as log (1/D) for neuroleptics of butyrophenone derivatives and parts of tricyclic neuroleptics (Saucin and van der Vorst 1972); histamine release as log (1/ED50 × 10−3) for parts of tricyclic neuroleptics and tricyclic antidepressants (Unger and Chiang 1981); ATPase activity inhibition as log [%/(100-%)] at 1 × 10−4 M for parts of tricyclic neuroleptics and imipramine (Davis and Brody 1966). The enthalpy values of −ΔH (kcal/mol) formation of neuroleptic CTs with chloranil (Saucin and van der Vorst 1972) obtained on the basis of spectroscopic measurements of CT complexes, and concern: neuroleptics of the butyrophenone and chlorpromazine derivatives, chlorprothixen, levomepromazine, prochlorperazine, and thioproperazines from the group of tricyclic neuroleptics.
Chromatographic retention data
Chromatographic data comes from the work of Nasal et al. (1997) and relate to the compounds: chlorpromazine, chlorprothixene, fluphenazine, perphenazine, prochlorperazine, promazine, promethazine, thioridazine, trifluoperazine, trifluopromazine from the group of tricyclic neuroleptics, and: clomipramine, desipramine and imipramine from the group of tricyclic antidepressants. These are logarithm values of the retention coefficients determined on Chiral AGP (log kAGP) fillings and artificial IAM.PC.MG membranes (log kIAM) but also logarithm values of hydrophobicity coefficients determined by the polycratic method on suplex pKb-100 fillings pH 2.5 and 7.4 (log kw2.5Su, log kw7.4Su), Spheri RP-18 pH 2.5 and 7.0 (log kw2.5Sp, log kw7.0Sp), Aluspher RP select B pH 7.3 (log kw7.3Al) and Unisphere-PBD pH 11.7 (log kw11.7Un).
Molecular descriptors
Nonempirical structural indicators—quantum-chemical indicators were calculated during the study. The structure of the tested compounds was studied by molecular modeling using the Gaussian 03W program (Gaussian Inc., Wallingford, CT, USA). The geometry of the molecules was optimized by the Hartree-Fock 6–31G (d, p) method (other designation is 6–31G**) (http://www.gaussian.com/). Among the quantum-chemical indicators were considered: total energy (TE), electronic spatial range (ESE)—the spatial extent of the molecule is defined as the surface covering the volume around the molecule beyond which the electron density is <0.001 eBohr−3, and describes the sensitivity of the molecule to the electric field), the energy of the highest occupied molecular orbitals (E_HOMO), the energy of the lowest unoccupied molecular orbitals (E_LUMO) and the energy difference HOMO and LUMO determined as energy gap (EG). In addition, the following values were used: the largest positive electron charge on atoms (MAX_POS), the largest negative electron charge on atoms (MAX_NEG), the difference between the highest positive and negative charge (ΔQ), total dipole moment (TDM), and isotropic polarization (IPOL). The values of total energy were expressed in atomic energy units a.u. or hartree (1 hartree = 2625.552 kJ·mol−1 or 627.5095 kcal·mol−1 or 27.2116 eV), HOMO energies, LUMO, and energy break were expressed in eV (the above values were converted from a.u. to eV), electronic spatial range in eBohr−3. The values of electron density and electron charges on atoms are in units of elementary charge (e−), dipole moment is expressed in debay (D) and isotropic polarizability in Bohr3 (Bohr = 0.5292·10−10 m = 0.5292 Å). Finally, for the whole group of molecules Dragon 7.0 (Kode Chemoinformatics, Pisa, Italy) software was used to calculate huge set – 5270 of extra descriptors (Todeschini and Consonni 2010; Dragon 7 molecular descriptors 2018 https://chm.kode-solutions.net/products_dragon.php).
Statistical analysis
The data examined the biological activity, physicochemical and retention parameters of compounds were related to their structural indicators using multiparametric regression analysis/multiple regression analysis (MLR) with a stepwise progressive method together with principal component analysis (PCA) and factor analysis (FA) implemented in Statistica 13 (StatSoft, Tulsa, OK, USA) on a personal computer. In PCA, matrix of correlations has been diagonalized and in FA Varimax rotation has been performed.
Results and discussion
The numerical values of all the 10 structural parameters derived from quantum-chemical calculations for all 25 compounds examined are shown in Table 1, the chromatographic retention parameters in Table 2 and finally the biological activity values and physicochemical parameters for selected compounds in Table 3.
Table 1.
The numerical values of 10 structural parameters derived from Gaussian quantum-chemical calculations for all 25 compounds studied
| Compound | TE | ESE | E_HOMO | E_LUMO | EG | MAX_POS | MAX_NEG | ΔQ | TDM | IPOL |
|---|---|---|---|---|---|---|---|---|---|---|
| Aceperon | −1283.84 | 21234.30 | −8.9739 | 2.5415 | 11.5154 | 0.7254 | −0.7283 | 1.4537 | 6.3837 | 242.90 |
| Acetophenazine | −1598.45 | 14981.03 | −7.6523 | 2.3717 | 10.0240 | 0.5402 | −0.7950 | 1.3352 | 1.7719 | 259.79 |
| Amitriptiline | −825.22 | 6515.35 | −8.1413 | 3.5380 | 11.6793 | 0.1801 | −0.5770 | 0.7571 | 1.1089 | 199.06 |
| Benperidol | −1259.67 | 20740.83 | −7.9418 | 2.6286 | 10.5704 | 1.0460 | −0.8566 | 1.9026 | 3.1622 | 228.43 |
| Chlorpromazine | −1620.72 | 8397.08 | −7.7290 | 3.2460 | 10.9750 | 0.3143 | −0.7846 | 1.0989 | 2.7007 | 201.34 |
| Chlorprothixene | −1603.56 | 8999.30 | −7.9483 | 2.6438 | 10.5921 | 0.2668 | −0.5915 | 0.8583 | 2.2333 | 211.05 |
| Clomipramine | −1301.28 | 8666.11 | −8.0945 | 3.5056 | 11.6001 | 0.2790 | −0.7897 | 1.0687 | 2.7070 | 204.08 |
| Desipramine | −803.36 | 6526.89 | −7.8879 | 3.7848 | 11.6727 | 0.2804 | −0.7888 | 1.0692 | 0.9264 | 183.15 |
| Dixyrazine | −1638.63 | 18634.72 | −7.5078 | 3.5674 | 11.0752 | 0.3471 | −0.8013 | 1.1484 | 1.4385 | 269.39 |
| Droperidol | −1258.48 | 19155.09 | −7.9960 | 2.5135 | 10.5095 | 1.0333 | −0.8941 | 1.9274 | 5.3046 | 225.82 |
| Fluphenazine | −1782.30 | 16638.00 | −7.7804 | 2.9826 | 10.7630 | 1.1718 | −0.7955 | 1.9673 | 4.7537 | 246.12 |
| Haloperidol | −1571.75 | 17976.49 | −8.7881 | 2.4400 | 11.2281 | 0.5509 | −0.6697 | 1.2207 | 3.9820 | 220.16 |
| Imipramine | −842.38 | 7331.05 | −7.8795 | 3.7897 | 11.6692 | 0.2800 | −0.7892 | 1.0693 | 0.7071 | 193.13 |
| Levomepromazine | −1314.75 | 8340.75 | −7.3870 | 3.6136 | 11.0006 | 0.4185 | −0.8025 | 1.2209 | 1.5158 | 215.72 |
| Nortriptiline | −786.19 | 6148.49 | −8.0906 | 3.5102 | 11.6008 | 0.2710 | −0.6326 | 0.9036 | 1.6374 | 189.70 |
| Perphenazine | −1905.57 | 14014.45 | −7.6751 | 3.2868 | 10.9619 | 0.3607 | −0.7944 | 1.1551 | 3.8012 | 246.35 |
| Prochlorperazine | −1791.68 | 11122.23 | −7.6588 | 3.2955 | 10.9543 | 0.3613 | −0.7944 | 1.1557 | 2.0514 | 233.31 |
| Promazine | −1161.82 | 7343.41 | −7.1788 | 3.5344 | 10.7132 | 0.3786 | −0.8293 | 1.2078 | 2.5886 | 193.37 |
| Promethazine | −1161.82 | 5419.31 | −7.2528 | 3.6487 | 10.9015 | 0.3699 | −0.8095 | 1.1793 | 2.1065 | 187.65 |
| Spiroperidol | −1298.66 | 20148.70 | −8.6112 | 2.6278 | 11.2390 | 0.7960 | −0.7296 | 1.5256 | 4.3300 | 234.58 |
| Thioproperazine | −2013.05 | 14948.37 | −7.8264 | 2.6357 | 10.4621 | 1.7000 | −0.7947 | 2.4947 | 5.9427 | 270.47 |
| Thioridazine | −1714.32 | 10820.72 | −7.6270 | 3.0993 | 10.7263 | 0.3470 | −0.7869 | 1.1339 | 2.6694 | 247.11 |
| Trifluoperazine | −1668.40 | 12802.53 | −7.7701 | 2.9804 | 10.7505 | 1.1715 | −0.7956 | 1.9672 | 2.5362 | 232.44 |
| Trifluopromazine | −1497.44 | 10070.78 | −7.8596 | 2.9567 | 10.8163 | 1.1740 | −0.7842 | 1.9583 | 3.3624 | 200.41 |
| Triperidol | −1448.48 | 21739.60 | −8.8887 | 2.4065 | 11.2952 | 1.1740 | −0.6691 | 1.8432 | 4.7615 | 219.02 |
Table 2.
Chromatographic parameter values of selected antipsychoctic drugs
| Compound | log kAGP | log kIAM | log kw2.5Su | log kw7.4Su | log kw2.5Sp | log kw7.0Sp | log kw7.3Al | log kw11.7Un |
|---|---|---|---|---|---|---|---|---|
| Chlorpromazine | 2.131 | 1.435 | 1.595 | 4.051 | 1.935 | 2.632 | 3.309 | 4.076 |
| Chlorprothixene | 2.206 | 1.533 | 1.597 | 4.642 | 2.244 | 2.417 | 4.440 | 4.235 |
| Clomipramine | 2.005 | 1.391 | 0.781 | 4.144 | 2.353 | 2.473 | 4.115 | 3.910 |
| Desipramine | 1.595 | 1.031 | 2.134 | 3.020 | 2.015 | 2.341 | 3.171 | 2.888 |
| Fluphenazine | 2.159 | 1.496 | 1.683 | 4.554 | 2.922 | 2.688 | 4.067 | 3.352 |
| Imipramine | 1.670 | 1.097 | 1.391 | 3.535 | 2.082 | 3.158 | 3.133 | 3.020 |
| Perphenazine | 2.283 | 1.393 | 1.635 | 4.305 | 2.997 | 3.092 | 3.256 | 3.070 |
| Prochlorperazine | 2.614 | 1.726 | 1.452 | 4.878 | 1.843 | 2.421 | 4.395 | 3.523 |
| Promazine | 1.890 | 1.165 | 1.556 | 3.492 | 2.338 | 2.808 | 3.794 | 3.294 |
| Promethazine | 1.833 | 1.508 | 1.693 | 4.081 | 1.132 | 3.169 | 3.069 | 3.216 |
| Thioridazine | 2.448 | 1.752 | 2.113 | 4.260 | 2.055 | 2.924 | 3.182 | 4.635 |
| Trifluoperazine | 2.388 | 1.820 | 1.778 | 4.948 | 1.792 | 2.644 | 5.022 | 3.632 |
| Triflupromazine | 1.976 | 1.514 | 1.960 | 4.409 | 2.533 | 2.638 | 3.790 | 4.117 |
Table 3.
Biological activity/physicochemical properties values of selected antipsychotic drugs
| Compound | −ΔH [kcal/mol] | log 1/D | log (1/ED50 × 10−3) | log ATPase activity |
|---|---|---|---|---|
| Aceperon | 0.37 | −1.623 | — | — |
| Acetophenazine | — | — | 2.12 | — |
| Amitriptiline | — | — | 1.85 | — |
| Benperidol | 1.82 | 1.523 | — | — |
| Chlorpromazine | 5.12 | −0.301 | 2.52 | −0.21 |
| Chlorprothixene | 0.19 | 0.022 | 2.57 | — |
| Clomipramine | — | — | 2.22 | — |
| Desipramine | — | — | 1.58 | — |
| Dixyrazine | 4.78 | −0.398 | — | — |
| Droperidol | 0.96 | 1.284 | — | — |
| Fluphenazine | 0.00 | 1.097 | — | — |
| Haloperidol | 5.16 | 1.284 | — | — |
| Imipramine | — | — | 1.79 | −0.99 |
| Levomepromazine | 3.42 | −0.330 | — | — |
| Nortriptiline | — | — | 1.76 | — |
| Perphenazine | — | — | 2.63 | 0.09 |
| Prochlorperazine | 0.00 | 0.137 | 2.75 | 0.87 |
| Promazine | — | — | 2.05 | −1.17 |
| Promethazine | — | — | 1.82 | −0.37 |
| Spiroperidol | 0.69 | 2.000 | — | — |
| Thioproperazine | 0.00 | 0.602 | — | — |
| Thioridazine | — | — | 2.72 | 0.79 |
| Trifluoperazine | — | — | 2.92 | 1.69 |
| Trifluopromazine | — | — | — | 0.27 |
| Triperidol | 5.78 | 1.456 | — | — |
First of all, the analysis of PCA was performed for the geometry-optimized structures. At the outset, a PCA analysis was carried out of only non-empirical data in order to check whether and to what extent they could be useful in order to classify the compounds in question. The following results were obtained: PC1 is about 50.2%, PC2 is equal to 22.6% and PC3 ~10.9%. PC1 has the greatest impact on the maximum positive charge on atoms (MAX_POS), the difference between the highest positive and negative charge on atoms (ΔQ) and isotropic polarization (IPOL). It is interesting that electrons are considered polar parameters, while bulk parameters (TE, ESE) have an approximately smaller impact on PC1. On the other hand, PC2 influences mostly the largest negative electron charge on atoms (MAX_NEG) and the energy of the highest occupied molecular orbital (E_HOMO). PC3 has the greatest impact on IPOL and again ΔQ but also appears MAX_NEG and MAX_POS with smaller impact Fig. 1.
Factor analysis (FA) performed with the same calculation conditions coincides completely with the results for the molecules obtained by PCA method (FA1 ~50.2% and FA2 ~22.6%).
Figure 2 represents scatter plots of the scores according to performed PCA and FA. Analyzing PCA 3D scatter plots of the score we can distinguish three type of clusters; the first one includes: acepron, benperidol, droperidol, haloperidol, spiroperidol, triperidol, and also fluphenazine, trifluopromazine, trifluoperazine, thioproperazine; the second cluster containes: amitriptiline, clomipramine, desipramine, imipramine, nortriptiline also chlorpromazine, levomepromazine, promazine, promethazine; the third cluster includes: acetophenazine, chlorprothixene, dixyrazine, perphenazine, prochlorperazine, and thioridazine. We can observe that the first cluster mostly involves neuroleptics derived from butyrophenone and other very potent neuroleptics, the second one mostly belongs to tricyclic antidepressants (dibenzoazepine and dibenzoheptadiene derivatives), and the third one mostly to tricyclic neuroleptics.
Figure 2.
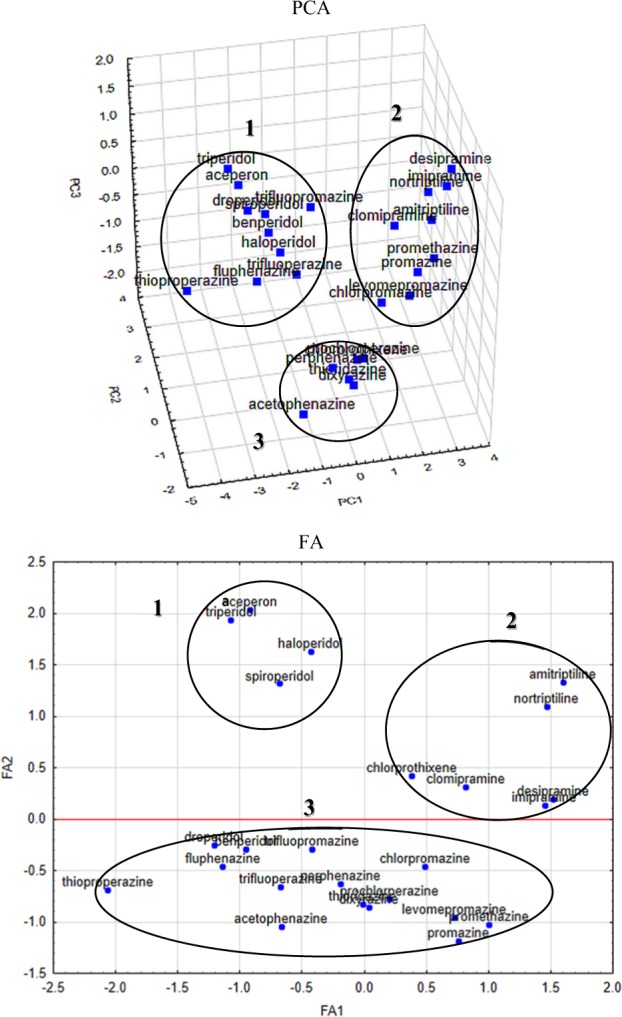
Three-dimensional scatter plots of the scores derived from the quantum-chemical calculations of structural parameters of the first three components obtained using PCA (PC1 50.21%, PC2 22.63%, PC3 10.89%) together with two-dimensional plot of scores the first two factors obtained by FA (FA1 50.21%, FA2 22.63%)
On the other hand, analyzing FA 2D scatter plots of scores we can also distinguish three types of clusters: the first one includes: acepron, droperidol, haloperidol, and spiroperidol; the second: amitriptiline, clomipramine, desipramine, imipramine nortriptiline, and chlorprothixene and finally the third one includes all the others studied psychoactive drugs.
Statistically significant Gaussian parameters characterizing biological activity are presented in the following equations:
| 1 |
R = 0.9150, R2 = 0,8372, F = 61.7126, s = 0.1813, p < 0.0001, n = 14, k0 = 0
| 2 |
R = 0.7896, R2 = 0.6235, F = 11.5949, s = 0.6559, p = 0.0114, n = 9, k0 = 0
Similarly, statistically significant Gaussian parameters characterizing chromatographic retention values are presented below:
| 3 |
R = 0.9608, R2 = 0.9231, F = 60.0945, s = 0.3036, p < 0.0001, n = 13, k0 = 0
| 4 |
R = 0.7713, R2 = 0.5949, F = 16.1514, s = 0.6648, p = 0.0020, n = 13, k0 = 0
| 5 |
R = 0.8364, R2 = 0.6996, F = 25.6215, s = 0.5724, p = 0.0004, n = 13, k0 = 0
| 6 |
R = 0.6378, R2 = 0.4068, F = 7.5449, s = 0.8044, p = 0.0190, n = 13, k0 = 0
| 7 |
R = 0.5936, R2 = 0.3524, F = 5.9859, s = 0.8405, p = 0.0324, n = 13, k0 = 0
| 8 |
R = 0.6671, R2 = 0.4450, F = 8.8226, s = 0.0127, p = 0.7780, n = 13, k0 = 0
Obtained results in most with TE and E_LUMO (TDM and ESE in individual cases) may indicate the specific nature of interactions between the drug molecule and the receptor.
In the final step, progressive stepwise multiple regression analysis was performed but with the use of a huge set of additional descriptors obtained by the professional software for the analyzed compounds with owned biological activity and chromatographic retention parameter values. Model equations for statistically significant descriptors are presented on below set of derived equations.
Biological activity/physicochemical properties values described by the Dragon software are as follows:
| 9 |
R = 0.9537, R2 = 0.9095, F = 30.1882, s = 0.3472, p < 0.0001, n = 13, k0 = 0
| 10 |
R = 0.9858, R2 = 0.9718, F = 103.6188, s = 0.1937, p < 0.0001, n = 13, k0 = 0
| 11 |
R = 0.9873, R2 = 0.9748, F = 128.4855, s = 0.1813, p < 0.0001, n = 14, k0 = 0
| 12 |
R = 0.9891, R2 = 0.9783, F = 135.5797, s = 0.1699, p < 0.0001, n = 9, k0 = 0
Analogous chromatographic retention parameters described by the Dragon software are presented below:
| 13 |
R = 0.9804, R2 = 0.9612, F = 74.4953, s = 0.2272, p < 0.0001, n = 13, k0 = 0
| 14 |
R = 0.9845, R2 = 0.9692, F = 94.6584, s = 0.2024, p < 0.0001, n = 13, k0 = 0
| 15 |
R = 0.9506, R2 = 0.9036, F = 46.8890, s = 0.3400, p < 0.0001, n = 13, k0 = 0
| 16 |
R = 0.9532, R2 = 0.9086, F = 29.8380, s = 0.3490, p < 0.0001, n = 13, k0 = 0
| 17 |
R = 0.9718, R2 = 0.9444, F = 50.8709, s = 0.2725, p < 0.0001, n = 13, k0 = 0
| 18 |
R = 0.9873, R2 = 0.9748, F = 115.9598, s = 0.1834, p < 0.0001, n = 13, k0 = 0
The full list of molecular descriptors obtained from Dragon software with their designations is presented in Table 4.
Table 4.
List of statistically significant molecular descriptors characterizing biological activity/physicochemical properties/chromatographic retention obtained from Dragon software (Eqs. 9–18)
| Descriptor | Full name | Block |
|---|---|---|
| Biological activity/physicochemical properties | ||
| CATS2D_09_DA | CATS (Chemically Advanced Template Search) 2D Donor–Acceptor at lag 09 | CATS2D |
| RDF095v | Radial Distribution Function - 095 / weighted by van der Waals volume | RDF descriptors |
| H4m | H autocorrelation of lag 4/weighted by mass | GETAWAY (GEometry, Topology, and Atom-Weights AssemblY) descriptors |
| H-053 | H attached to C0(sp3) with 2X attached to next C | Atom-centered fragments |
| PJI3 | 3D Petitjean shape index | Geometrical descriptors |
| AVS_B(e) | Average vertex sum from Burden matrix weighted by Sanderson electronegativity | 2D matrix-based descriptors |
| DISPm | Displacement value/weighted by mass | Geometrical descriptors |
| Mor08u | Signal 08/unweighted | 3D-MoRSE (3D-Molecule Representation of Structures based on Electron diffraction) descriptors |
| MATS6s | Moran autocorrelation of lag 6 weighted by I-state | 2D autocorrelations |
| Eig04_AEA(bo) | eigenvalue n. 4 from augmented edge adjacency mat. weighted by bond order | Edge adjacency indices |
| GATS5s | Geary autocorrelation of lag 5 weighted by I-state | 2D autocorrelations |
| Retention parameters | ||
| H1v | H autocorrelation of lag 1/weighted by van der Waals volume | GETAWAY descriptors |
| DISPi | Displacement value/weighted by ionization potential | Geometrical descriptors |
| H2u | H autocorrelation of lag 2/unweighted | GETAWAY descriptors |
| X1A | Average connectivity index of order 1 | Connectivity indices |
| GATS5m | Geary autocorrelation of lag 5 weighted by mass | 2D autocorrelations |
| Mor06p | Signal 06/weighted by polarizability | 3D-MoRSE descriptors |
| R2v | R autocorrelation of lag 2/weighted by van der Waals volume | GETAWAY descriptors |
| CMC-50 | Ghose–Viswanadhan–Wendoloski CMC drug-like index at 50% | Drug-like indices |
| R1e+ | R maximal autocorrelation of lag 1/weighted by Sanderson electronegativity | GETAWAY descriptors |
| Mor09i | Signal 09/weighted by ionization potentia | 3D-MoRSE descriptors |
| B08[C-N] | Presence/absence of C-N at topological distance 8 | 2D Atom Pairs |
| H6m | H autocorrelation of lag 6/weighted by mass | GETAWAY descriptors |
| ATS6s | Broto–Moreau autocorrelation of lag 6 (log function) weighted by I-state | 2D autocorrelations |
| H4i | H autocorrelation of lag 4/weighted by ionization potential | GETAWAY descriptors |
| ALOGP2 | Squared Ghose–Crippen octanol-water partition coeff. (logP^2) | Molecular properties |
| HATS1v | Leverage-weighted autocorrelation of lag 1/weighted by van der Waals volume | GETAWAY descriptors |
| R8v | R autocorrelation of lag 8/weighted by van der Waals volume | GETAWAY descriptors |
Performed predictions with the use of professional software and the wide range of molecular descriptors provide more detail information about the studied molecules.
The obtained statistically significant molecular descriptors belong to different classes but we can distinguish some of the common one’s classes between them. In our opinion it is difficult to indicate the most important equation between the proposed because the datasets are relatively small, therefore we wanted to focus rather on the same block of descriptors appearing in the presented equations.
In the case of biological activity/physicochemical properties of analyzed psychoactive drugs, the most often appeared geometrical descriptors together with GETAWAY and atom fragments descriptors followed by the 2D autocorrelations and 3D-MoRSE descriptors. Experimental equations confirmed the very important role of geometric and topologic properties of the molecules together with electronic ones. Furthermore, interesting information we received also analyzing the retention parameters values on different chromatographic columns and in different chromatographic conditions. There was a confirmation in eight of the experimental cases particularly important role of GETAWAY descriptors and also molecular properties descriptors (e.g. ALOGP2, characterizing octanol-water partition coefficient) followed by appeared again 2D autocorrelations and 3D-MoRSE descriptors, so we can observe that similar group of descriptors play a dominant role in pharmacological role and physicochemical properties of examined psychoactive structures.
Conclusion
Based on the above discussion, the results can be drawn as follows. The largest influence on the values of both biological activity/physicochemical properties and chromatographic retention parameters among the 10 quantum-chemical parameters considered (Gaussian software) are most often the total energy (TE) and frontier orbital energy LUMO (E_LUMO). On the other hand, we can distinguish GETAWAY (GEometry, Topology, and Atom-Weights AssemblY) descriptors next to 3D-MoRSE (3D-Molecule Representation of Structures based on Electron diffraction) descriptors from Dragon software, which leads to the assumption that a functional dependency exists. These parameters seem to be particularly important for psychoactive activity and properties of analyzed structures, which is related to the hypotheses regarding the mechanism of action of compounds with this type of elements of structure and mostly their pharmacological classification. It seems advisable also to continue research in the extended database of molecules and their experimental values of pharmacological/physicochemical properties.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Cogordan JA, Mayoral M, Angeles E, Toscano RA, Martinez R. Neuroleptic and antidepressant tricyclic compounds: theoretical study for predicting their biological activity by semiempirical, density functional, and Hartree–Fock methods. Int J Quant Chem. 1999;71:415–432. doi: 10.1002/(SICI)1097-461X(1999)71:5<415::AID-QUA3>3.0.CO;2-0. [DOI] [Google Scholar]
- Davis PW, Brody TM. Inhibition of Na+K+-activated adenosine triphosphatase activity in rat brain by substituted phenothiazines. Biochem Pharmacol. 1966;15:703–710. doi: 10.1016/0006-2952(66)90004-9. [DOI] [PubMed] [Google Scholar]
- Dean CE. Psychopharmacology: a house divided. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1–10. doi: 10.1016/j.pnpbp.2010.08.028. [DOI] [PubMed] [Google Scholar]
- Dragon 7 molecular descriptors (2018). https://chm.kode-solutions.net/products_dragon.php. Accessed 12 March 2018
- Hendriksen H, Groenink L. Back to the future of psychopharmacology: a perspective on animal models in drug discovery. Eur J Pharmacol. 2015;759:30–41. doi: 10.1016/j.ejphar.2015.03.020. [DOI] [PubMed] [Google Scholar]
- Official Gaussian Website (2018). http://www.gaussian.com/. Accessed 12 March 2018
- Nasal A, Buciński A, Bober L, Kaliszan R. Prediction of pharmacological classification by means of chromatographic parameters processed by principal component analysis. Int J Pharm. 1997;159:43–55. doi: 10.1016/S0378-5173(97)00267-6. [DOI] [Google Scholar]
- Saucin M, van der Vorst A. On the formation of charge transfer complexes between neuroleptic drugs and chloranil. Biochem Pharmacol. 1972;21:2673–2680. doi: 10.1016/0006-2952(72)90016-0. [DOI] [PubMed] [Google Scholar]
- Spiegel R. Psychopharmacology: an introduction. 4th edn. Hoboken: Wiley-Blackwell; 2003. [Google Scholar]
- Stahl SM. Stahl’s essential psychopharmacology neuroscientific basis and practical applications. 4th edn. Cambridge: Cambridge University Press; 2013. [Google Scholar]
- Todeschini R, Consonni V (2010) Molecular descriptors for chemoinformatics: volume i: alphabetical listing /volume ii: appendices, references, vol 41. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
- Unger SH, Chiang GH. Octanol-physiological buffer distribution coefficients of lipophilic amines by reversed-phase high-performance liquid chromatography and their correlation with biological activity. J Med Chem. 1981;24:262–270. doi: 10.1021/jm00135a006. [DOI] [PubMed] [Google Scholar]