

Abstract
An explosion of knowledge on the molecular and cellular mechanisms that mediate carcinogenesis has occurred in recent years. Although cancer has existed for over a million years in the human species, effective cures for most cancers that target molecular and cellular pathways have not been achieved. Multiple cellular targets have been examined for preventing or treating cancers including, but not limited to, transcription factors, kinase-mediated cell signaling pathways, and more recently epigenetic targeting of oncogenes and tumor suppressors, and immunomodulation such as chimeric antigen receptor-T cells. Even as the state of knowledge of cancer mechanisms increases, there is considerable room for improvement in preventing and treating cancers. Understanding how a normal cell is transformed into a cancer cell is known but there is considerable tissue and cell type specificity. This has given rise to the field of precision medicine as applied to cancer therapy. Thus, while the development of preventive and treatment regimens has increased, there are certain obstacles that need to be overcome in order to decrease cancer incidence and increase survival of cancer patients. The purpose of this review is to summarize the advances made in cancer biology and how these advances have been used to develop, and hinder, preventive, and therapeutic strategies for cancer.
Keywords: carcinogenesis, mechanisms, targeted chemoprevention, targeted chemotherapy
The field of carcinogenesis has rapidly evolved, in particular since the revolution of molecular biology in the 1970s. The field of carcinogenesis began with the finding that exposure to chemicals is correlated with cancer in chimney sweepers and that painting coal tar on rabbit ears resulted in papillomas (reviewed in Loeb and Harris, 2008). Subsequent studies revealed a link between chemical exposure, metabolism, and damage to deoxyribonucleic acid (DNA) providing somewhat of a more complete understanding of the etiology of carcinogenesis (reviewed in Loeb and Harris, 2008). It is now well accepted that carcinogenesis involves multiple factors that range from exposures to chemicals, mutations in proto-oncogenes and tumor suppressors due to exogenous and endogenous etiologies, and discordant regulation or activities of many critical signaling pathways required for normal cellular homeostasis (Figure 1). The focus of this review is on this range of etiological factors, and how scientists have used this information to develop approaches to help prevent and/or treat cancer in humans. Notably, despite recognition of the causative factors involved in carcinogenesis, progress has been hindered in harnessing the genetic, economic, and environmental factors in the search for preventive and therapeutic strategies. The complexity of this field lends itself to the disparities in our knowledge of cancer etiology, cancer chemoprevention and cancer therapeutics. Thus, the purpose of this review is to address these issues to highlight how increased understanding of cancer cell biology has been used to develop new strategies for preventing and treating this disease, but also hindered progress for cancer chemoprevention and chemotherapy due to the complexities of the signaling involved.
Figure 1.

Ontogeny of events leading to cancer. Chemicals can be metabolized to reactive intermediates that may form an adduct with DNA. In some cases, this adduct can be repaired and no DNA damage results (Upper box on left). In contrast, sometimes the adduct is not repaired causing a mutation in a gene (Upper box, second from left) that may be critical for cancer (proto-oncogene, tumor suppressor). Endogenous metabolism can also cause the generation of reactive intermediates or reactive oxygen species that can bind to DNA and form an adduct or DNA damage, respectively. In some cases, this adduct/damage can be repaired and no alterations in DNA results (Upper box, first from right). In contrast, sometimes the adduct/damage is not repaired causing a mutation in a gene (Upper box, second from right), which may be critical for cancer (proto-oncogene, tumor suppressor). In some case, genes can contain mutations due to inheritance, and this may be in a critical gene that will lead to cancer. Combined, these 2 mechanisms can lead to increased expression of an oncogene and/or decreased expression of a tumor suppressor. Further, there is normal cellular signaling that can be influenced by endogenous or exogenous variables that collectively alter major pathways that regulate proliferation, apoptosis, differentiation, angiogenesis, migration and invasion. With time, these cells can convert to a malignant neoplasm and ultimately cancer.
CHEMICAL CARCINOGENESIS: METABOLISM OF CHEMICALS BY XENOBIOTIC METABOLIZING ENZYMES
The discovery of cytochrome P450s (CYPs) in the 1960s (Omura and Sato, 1964a,b) was just the beginning of our understanding that chemicals could be metabolized to reactive intermediates by these enzymes. Electrophilic intermediates from numerous chemicals can form covalent bonds with nucleophilic regions on nucleotides within DNA, in particular proto-oncogenes or tumor suppressors. In the absence of accurate DNA repair of adducts, these chemical modifications can lead to changes in the encoded genes, and this in turn can begin the transition of transforming a normal cell into a cancerous cell. Indeed, a number of DNA “hotspots” have been identified in some genes such as retinoblastoma (RB) or p53 (TP53) that are particularly sensitive to DNA damage by electrophilic intermediates generated by metabolism of chemicals humans are exposed to in the environment (reviewed in Loeb and Harris, 2008). Interestingly, the metabolic pathway that was discovered to bio-activate chemicals and cause cancer, was also determined to be part of a global metabolic network expressed in many species that includes the phase I xenobiotic enzymes, CYPs, and phase II xenobiotic-conjugating enzymes. This is important to note because paradoxically, these pathways also function to detoxify numerous chemicals including drugs. Indeed, it appears that humans and many species have evolved these metabolic pathways to provide mechanisms to detoxify and increase the excretion/elimination of chemicals that could cause cancer/toxicity. This suggests that there is likely a critical balance between bio-activation and detoxification that is influenced by multiple phases I and II enzymes. For example, the Km, Vmax, and turnover number of individual enzymes for their various chemical substrates can all markedly influence the generation and half-life of reactive intermediates, whereas the regulation of expression of xenobiotic enzymes represents another level of control that could alter the relative metabolic activity of an enzyme or enzymes that metabolize chemicals to carcinogens or detoxified compounds.
REGULATION OF XENOBIOTIC METABOLIZING ENZYMES BY TRANSCRIPTION FACTORS
The discovery of nonmembrane bound receptors for hormones and xenobiotics that modulate gene expression advanced our understanding of carcinogenesis and the mechanisms that mediate this disease. For example, the seminal discoveries of the glucocorticoid receptor and the estrogen receptor (ER) (Green et al., 1986; Miesfeld et al., 1984), both transcription factors, provided the foundation for countless experiments testing the hypotheses that endogenous ligands regulated cellular function by regulating expression of genes. This idea was easily expanded into examining how exogenous chemicals might interact with endogenous receptors and disrupt normal physiological function(s). Indeed, this is one of the most fundamental concepts of modern toxicology; that structural similarities between xenobiotics and natural chemicals can cause toxicity/cancer by interfering with normal homeostasis. Thus, as more and more soluble and cell surface receptors were identified and characterized, the possibility that chemicals could cause (or inhibit) cancer through receptor-mediated mechanisms became more plausible. Interestingly, one of the most insightful discoveries in the field of receptor-mediated carcinogenesis was the identification and characterization of the aryl hydrocarbon receptor (AHR) (Burbach et al., 1992). The timing of this discovery was closely related to advances in developmental biology where “knockout” mice were first developed with the use of homologous recombination in embryonic stem cells (Capecchi, 1989). The development of knockout mice was instrumental in deciphering how specific genes/proteins were involved in toxicologic and carcinogenic effects. For example, the first Ahr-null mouse was created in 1995 and provided the first definitive evidence that this soluble receptor mediated the toxicological effects induced by AHR agonists such as tetrachloro-dibenzo-p-dioxin (Fernandez-Salguero et al., 1995, 1996). This set the stage for numerous studies by multiple laboratories examining the role of soluble receptors in toxicity and cancer. For example, while it had been speculated for years that the peroxisome proliferator-activated receptor-α (PPARα) mediated the toxicity and hepatocarcinogenic effects induced by PPARα agonists, the finding that Ppara-null mice were resistant to liver cancer induced by a PPARα agonist (Hays et al., 2005; Peters et al., 1997) provided strong evidence that a receptor-mediated mechanism was required for this effect. Since PPARα agonists do not cause direct DNA damage, they are known as non-genotoxic carcinogens. The critical mutations are likely the result of increased oxidative stress that can cause mutations in proto-oncogenes and tumor suppressor genes (Figure 1). Subsequent studies by other laboratories would demonstrate similar requirements for other xenobiotic receptors such as constitutive androstane receptor and pregnane X receptor in mediating the toxicologic/carcinogenic effects of respective agonists (Xie et al., 2000; Yamamoto et al., 2004). Moreover, more complex models involving conditional knockout models, knock-in mouse models, and humanizing mice to express human homologs of specific genes would subsequently provide further insight into the important regulatory roles of soluble receptors in carcinogenesis and species differences induced by chemicals. One of the best examples of this is the demonstration that mice expressing the human PPARα, but not the mouse PPARα, were resistant to hepatocarcinogenic effects induced by PPARα agonists (Morimura et al., 2006; Yang et al., 2008). The mechanism underlying this effect was determined to be due to species differences in the PPARα-dependent regulation of Let-7C micro RNA that in turn regulate an oncogene MYC that in turn increases cell proliferation of hepatic cells with mutant DNA (Shah et al. 2007). This was but one seminal observation that has had major impact on risk assessment as it demonstrated that carcinogenic effects observed in mice may not always be relevant to humans. Additionally, many chemicals that activate xenobiotic receptors that have been shown to mediate their carcinogenic effects also modulate the expression of genes involved in xenobiotic transport and phases I and II xenobiotic metabolism. However, there are many other molecular pathways that can be modulated by activation of soluble receptors. This is important to note because during the same time frame that progress was being made on the molecular pathways mediated by chemical carcinogens that modulated bioactivation and detoxification, many other cellular pathways were also being identified that modulated important signaling including those that regulated cell proliferation, cellular differentiation, apoptosis, and inflammation. Collectively, these observations led to the understanding that chemicals could cause cancer by not only directly causing mutations in critical genes, but also by modulating activity of soluble receptors that in turn regulate expression of xenobiotic metabolizing enzymes, and likely interfering with normal cellular signaling pathways that are central to the fate of cell division and/or cell death.
MOLECULAR AND CELLULAR SIGNALING THAT CAUSE CARCINOGENESIS
Cancer is clearly a complex disease that is mediated by numerous molecular and cellular pathways. The general scheme of events causing cancer is that cells first acquire driver mutations in critical genes that lead to altered expression of proteins with different activities that collectively drive transformation from a normal cell into a cancerous cell. Due to chronic signaling to increase proliferation and/or decrease apoptosis, these cells expand clonally into tumors. As the tumor continues to grow, changes in cellular metabolism occur, which can include increased utilization of different substrates for energy to “feed” the tumor and deprive surrounding normal cells from critical nutrients. New blood vessels can form (angiogenesis) because of the anoxic condition in a tumor that provides for more oxygen to be delivered to the tumor. This is mediated by growth factor/growth factor receptors (eg, vascular endothelial growth factor [VEGF]). As the tumor continues to grow and obtain different mutations, the tumors can become aggressive and begin to migrate and invade cell membranes, ultimately leading to metastasis to other tissues. Once tumors have metastasized, the process can continue and ultimately lead to death.
All of these events are regulated by numerous molecular and cellular mechanisms. Thus, many of the pathways that modulate the processes associated with different cancers are conserved in the sense that there is considerable overlap between different cancers. For example, differential expression/activity of oncogenic proteins or tumor suppressors such MYC or PTEN, respectively, is often observed in many different cell types. Moreover, similar mutations in oncogenic or tumor suppressor genes are often observed in multiple cancers (Kandoth et al., 2013). This is likely related to the changes observed in cellular signaling pathways mediated by RAS, ERK, PDPK1, TP53, and many others as they typically converge on the regulation of cell proliferation, apoptosis, differentiation, angiogenesis, cell migration and invasion, immunological activities, and inflammation. Despite the significant overlap in these signaling pathways, there are clearly unique molecular and cellular signaling signatures for many different cancers.
One of the earliest recognition of a nuclear receptor as a prognostic factor for breast cancer was that expression of the ER was increased in woman with breast cancer as compared with controls (Knight et al., 1977). This suggested that altered ER-dependent signaling may be critical for breast cancer etiology due to transcriptional regulation of genes that promote tumorigenesis. Oncogenic proteins are another classic example of proteins that promote cell proliferation, inhibit apoptosis or prevent differentiation of cancer cells. Oncogenes are derived from proto-oncogenes that encode “normal” proteins that in turn regulate cell proliferation, apoptosis or differentiation. However, when these genes attain specific mutations, they can become oncogenic and change their normal role from maintaining homeostasis to promoting tumorigenic signaling. There are many classes of oncogenes including: (1) receptor tyrosine kinases (eg, epidermal growth factor receptor [EGFR], VEGFR, or HER2/neu) that regulate growth and differentiation; (2) cytoplasmic tyrosine kinases (eg, SRC or the ABL gene) that regulate cell proliferation, differentiation, migration, and survival; (3) cytoplasmic serine/threonine kinases (eg, RAF or cyclin-dependent kinases [CDKs]) that regulate cell cycle control, proliferation, differentiation, apoptosis, and survival; (4) membrane linked GTPases (eg, RAS) that regulate cell proliferation; and (5) transcription factors (eg, MYC) that indirectly regulate cell proliferation. Although there are clear examples that chronic inflammation can promote carcinogenic signaling including proliferation, cell survival and migration (Coussens and Werb, 2002), more recent evidence has also demonstrated that enhancing immune function in cancer patients can actually modulate the immune cells to target the tumor and actually stop tumor growth by increasing an immune response against the cancerous tumor (Gong et al., 2018; Grigor et al., 2017). The latter approach is gaining significant traction in the field and represents complex signaling pathways that have been used to develop highly effective therapeutic treatment strategies for cancer patients.
Given the amount of knowledge on the molecular and cellular signaling that drives cancer, the question arises whether tailored approaches should be developed for inhibiting or enhancing a specific activity to prevent and treat cancer? Both the similarities and disparities in cancer etiologies hamper the development of strategies for cancer prevention and treatment. Nevertheless, there are clear examples of successful strategies that have been developed that markedly improved the lifespan of cancer patients in some instances.
TARGETING MOLECULAR AND CELLULAR SIGNALING TO INHIBIT CARCINOGENESIS
Given the knowledge gained in the past 20–30 years, it is not surprising that significant progress has been made in targeting some molecular and cellular pathways to help prevent or treat different cancers (Figure 2). For example, as noted above, the expression of ER was positively correlated with breast cancer in women. This led to the development of antiestrogenic compounds in an attempt to develop drugs that help prevent the progression and inhibit this disease. Tamoxifen is an ER antagonist that has proven to be fairly effective at increasing the lifespan of breast cancer patients that express ER in their tumors (Early Breast Cancer Trialists’ Collaborative et al., 2011; Jaiyesimi et al., 1995). The mechanism by which tamoxifen is thought to function is by preventing ER-dependent regulation of genes that promote cancer cells from proliferating. This is an excellent example of an adjuvant chemotherapeutic drug that has remained effective for many years based on the original finding that expression of a soluble receptor was correlated with breast cancer progression. Thus, by preventing the soluble receptor from transcriptionally regulating target genes, breast cancer can be markedly inhibited and the lifespan of a breast cancer patient can be markedly increased.
Figure 2.
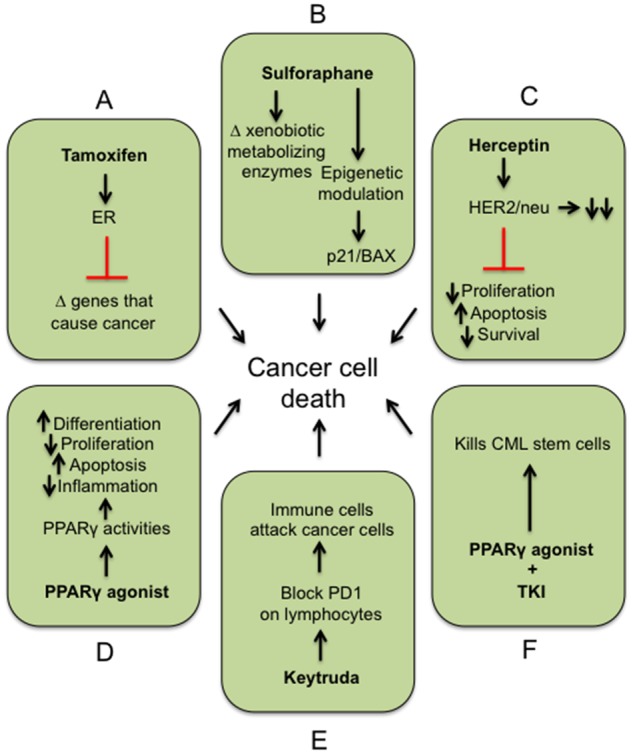
Examples of targeted approaches for preventing/treating cancers. A, Tamoxifen and ER antagonist prevent ER from transcribing genes that cause cancer. B, Sulforaphane alters expression of xenobiotic metabolizing enzymes and epigenetically modified genes such as p21 and Bcl-2-associated X protein. C, Herceptin binds to the HER2 receptor leading to degradation and prevention of the HER2 from mediating cellular events that cause cancer. D, Ligand activation of PPARγ causes pleiotropic effects including inducing differentiation and apoptosis, and inhibiting proliferation and inflammation. E, Keytruda binds to cancer cells expressing programed cell death protein 1 (PD-1), which triggers immune cells to attack the cancer cells. F, Combining a PPARγ agonist with a TKI can collectively target chronic myeloid leukemia stem cells. Combined, all of these pathways when modulated with the described drug(s) can cause cell death of the cancer cell(s).
Chemopreventive agents have also been developed based on preclinical and clinical data showing that specific compounds modulate key molecular and cellular mechanisms that promote tumorigenesis. A good example of a natural product that has shown promise in both preclinical and clinical trials is sulforaphane. Sulforaphane is an isothiocyanate found in cruciferous vegetables such as broccoli. It is a classic example of a natural product that can target multiple molecular cellular pathways that modulate cancer. For example, sulforaphane can help detoxify chemical carcinogens by inducing glutathione-S-transferases and inhibiting expression of CYPs (Amjad et al., 2015). These effects could help prevent initial mutagenic effects of chemicals required for cancer initiation. However, sulforaphane also has been shown to epigenetically modify expression of genes allowing for enhanced expression of proteins such as p21 or Bcl-2-associated X protein, which inhibits cell cycle progression and induction of apoptosis, respectively (Amjad et al., 2015). Thus, sulforaphane exhibits pleiotropic effects in cancer models and in clinical trials that indicate this natural product can effectively inhibit molecular and cellular signaling pathways that promote diseases such as bladder or prostate cancers and supports the establishment of clinical trials to examine the efficacy of sulforaphane in cancer patients (Amjad et al., 2015; Leone et al., 2017; Tortorella et al., 2015). This also illustrates the suitability of using compounds that target more than a single molecular or cellular pathway to prevent or treat cancer because they likely explain the enhanced efficacy observed as compared with other candidate molecules.
HER2 is a membrane bound receptor with intracellular tyrosine kinase activity that is often overexpressed in a number of cancers in particular breast cancer (Coussens et al., 1985; Mitri et al., 2012). Herceptin (also known as trastuzumab) is a monoclonal antibody-based drug that specifically targets HER2/neu and causes down-regulation of this receptor. This prevents the intracellular activities of HER2/neu including mitogen-activated protein kinase, phosphoinositide 3-kinase/protein kinase B, protein kinase C and signal transducer, and activator of transcription 5. In doing so, this drug effectively blocks multiple pathways by binding to the HER2/neu receptor and blocks downstream signaling that causes inhibition of cell cycle progression, induction of apoptosis, and inhibition of cell survival that collectively stop the tumor from growing. Interestingly, this drug was initially not effective for all breast cancer patients, but subsequent studies revealed that it was the breast cancer patients that exhibited overexpression of HER2/neu who responded. This and observations from other studies led to the development of “personalized” medicine based on genomics. In other words, one required a specific genotype (in this case overexpression of the HER2/neu gene in their cancer), and if this was present, then the drug Herceptin was effective. This paradigm has become increasingly more common and illustrates how drugs can be used more effectively based on the genetic makeup of the tumor.
Transcription factors such as nuclear receptors can regulate drug metabolizing enzymes and many other cell signaling pathways. The PPARγ is a good example of a receptor that has been targeted for the prevention and treatment of cancers. Although this receptor was initially identified and characterized as being required for adipocyte differentiation (Tontonoz et al., 1994), subsequent studies demonstrated that activation with specific ligands could prevent tumorigenesis in preclinical models (reviewed in Peters et al., 2012). For example, ligand activation of PPARγ can promote terminal differentiation, inhibit cell proliferation, angiogenesis, migration and invasion, metastasis, and inflammation (Frohlich and Wahl, 2015; Glass and Saijo, 2010; Mueller et al., 1998; Ninomiya et al., 2014; Wan et al., 2011). The mechanisms that mediate these effects include PPARγ-dependent modulation of proteins including CDKs, CYCLINs, MYC, PTEN, and others that regulate apoptosis and inflammation (reviewed in Frohlich and Wahl, 2015). This is a good example of how activating a nodal nuclear receptor can be used to prevent and treat cancers through mechanisms that target multiple cellular signaling molecules.
Modulation of the immune system has been targeted for years to prevent and treat different cancers. For many years, the thought was that chronic inflammation promotes tumorigenesis, and thus, antiinflammatory agents such as nonsteroidal antiinflammatory drugs were used with mixed success for cancer prevention (Coussens and Werb, 2002). However, recent discoveries have led to a paradigm shift and now there is solid evidence that promoting immune cell activities can have profound effects on specific cancers. For example, pembrolizumab (Keytruda) is a drug that is used to effectively treat subsets of cancer patients with inoperable melanoma or nonsmall cell lung cancer (Pardoll, 2012). In order for this drug to function, the cancer cells must express programed cell death protein 1 ligand (PD-1L) and not have mutations in the EGFR or anaplastic lymphoma kinase genes. Keytruda blocks PD1 on lymphocytes, which allows immune cells to target and destroy cancer cells. This is accomplished because PD1 typically prevents the immune system from attacking the body’s own cells. There is clear evidence that targeting the immune system and inflammation is a complex prospect for cancers and that there may be opposing mechanisms that can effectively be targeted for preventing and treating cancers.
As cancer prevention and therapies continue to evolve, targeting more than one cellular or molecular target may provide even more powerful approaches to increase the efficacy of treatment for cancer patients. For example, tyrosine kinase inhibitors (TKIs) have been used to treat chronic myeloid leukemia (CML), but a complete molecular response is not typically observed. However, treatment with TKI and the PPARγ ligand pioglitazone specifically targeted CML stem cells and was shown to potentially prevent recurrence of this disease (Glodkowska-Mrowka et al., 2016; Prost et al., 2015). This is but one example of how combining agents can more effectively target molecular and cellular pathways for preventing and treating cancers and is currently the focus of many studies. The general thought is that if some inhibition can be obtained with one target, then trying to target another pathway will markedly improve the clinical outcome for cancer patients. Much hope remains that combinatorial approaches like this will markedly improve strategies to prevent and treat cancer.
FUTURE DIRECTIONS
The future of cancer research holds much promise, but there is likely considerable time that will be required before cures for all cancers will be obtained. From the early days of discovering how chemicals can cause cancer in animal models, to more recent times when there are new prevention and treatment strategies that have markedly extended the lifespan of cancer patients, these advances would not be possible without the continuum of research that underscores these achievements. There remains a need to identify molecular and cellular pathways that are changed before, during and after cells convert from normal to cancer cells. It would be naïve to suggest that all molecular and cellular pathways that cause cancer are clearly understood. And still, given the difference in longevity in cancer patients observed in 2018 as compared with that observed in 1950, considerable advances have clearly been made. Although part of this improvement can be attributed to better and more effective screening strategies (eg, mammography, colonoscopy, screening for biomarkers, etc.), the advances made in elucidating the molecular and cellular pathways that drive carcinogenesis have also been major contributors to the development of new drugs and strategies that contribute to the improved prognosis for cancer patients. However, significant gaps in knowledge remain. Learning how to customize the targeting of pathways based on genomic, proteomic, epigenetic, metabolomic, and microbiome analyses will likely yield major advances, especially when coupled with higher throughput approaches to analyze these data with new bioinformatic techniques. It is likely that combinatorial approaches targeting multiple molecular and cellular pathways is going to be the most effective way to prevent or treat cancers; much like the treatment for human immunodeficiency virus. One major question that has evaded cancer researchers to date is why and how does genotype influence efficacy of cancer prevention/treatment? Whether there will ever be a day when cancer patients will be able to choose treatments based on genotype/phenotype, and effectively stop this disease that affects so many people in the world remains to be seen but is clearly within our grasp. Thus, while it is possible to suggest that our current state of knowledge of cancer biology has been a friend to many cancer patients, .there are many cancer patients that likely view the gaps in our knowledge as a foe
FUNDING
This work was supported in part by the National Institutes of Health (CA124533 and CA140369 to J.M.P.) and (ES022186, ES028288, and ES026684 to A.D.P.).
REFERENCES
- Amjad A. I., Parikh R. A., Appleman L. J., Hahm E. R., Singh K., Singh S. V. (2015). Broccoli-derived sulforaphane and chemoprevention of prostate cancer: From bench to bedside. Curr. Pharmacol. Rep. 1, 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach K. M., Poland A., Bradfield C. A. (1992). Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. U. S. A. 89, 8185–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi M. R. (1989). Altering the genome by homologous recombination. Science 244, 1288–1292. [DOI] [PubMed] [Google Scholar]
- Coussens L., Yang-Feng T. L., Liao Y. C., Chen E., Gray A., McGrath J., Seeburg P. H., Libermann T. A., Schlessinger J., Francke U., et al. (1985). Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 230, 1132–1139. [DOI] [PubMed] [Google Scholar]
- Coussens L. M., Werb Z. (2002). Inflammation and cancer. Nature 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists’ Collaborative G., Davies C., Godwin J., Gray R., Clarke M., Cutter D., Darby S., McGale P., Pan H. C., Taylor C., et al. (2011). Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 378, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Pineau T., Hilbert D. M., McPhail T., Lee S. S., Kimura S., Nebert D. W., Rudikoff S., Ward J. M., Gonzalez F. J. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P. M., Hilbert D. M., Rudikoff S., Ward J. M., Gonzalez F. J. (1996). Aryl-hydrocarbon receptor-deficient mice are resistant to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179. [DOI] [PubMed] [Google Scholar]
- Frohlich E., Wahl R. (2015). Chemotherapy and chemoprevention by thiazolidinediones. Biomed. Res. Int. 2015, 845340.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C. K., Saijo K. (2010). Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 10, 365–376. [DOI] [PubMed] [Google Scholar]
- Glodkowska-Mrowka E., Manda-Handzlik A., Stelmaszczyk-Emmel A., Seferynska I., Stoklosa T., Przybylski J., Mrowka P. (2016). PPARγ ligands increase antileukemic activity of second- and third-generation tyrosine kinase inhibitors in chronic myeloid leukemia cells. Blood Cancer J. 6, e377.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Chehrazi-Raffle A., Reddi S., Salgia R. (2018). Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 6, 8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S., Walter P., Kumar V., Krust A., Bornert J. M., Argos P., Chambon P. (1986). Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 320, 134–139. [DOI] [PubMed] [Google Scholar]
- Grigor E. J. M., Fergusson D. A., Haggar F., Kekre N., Atkins H., Shorr R., Holt R. A., Hutton B., Ramsay T., Seftel M., et al. (2017). Efficacy and safety of chimeric antigen receptor T-cell (CAR-T) therapy in patients with haematological and solid malignancies: Protocol for a systematic review and meta-analysis. BMJ Open 7, e019321.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays T., Rusyn I., Burns A. M., Kennett M. J., Ward J. M., Gonzalez F. J., Peters J. M. (2004). Role of peroxisome proliferator-activated receptor-α (PPARα) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis 26, 219–227. [DOI] [PubMed] [Google Scholar]
- Jaiyesimi I. A., Buzdar A. U., Decker D. A., Hortobagyi G. N. (1995). Use of tamoxifen for breast cancer: Twenty-eight years later. J. Clin. Oncol. 13, 513–529. [DOI] [PubMed] [Google Scholar]
- Kandoth C., McLellan M. D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J. F., Wyczalkowski M. A., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight W. A., Livingston R. B., Gregory E. J., McGuire W. L. (1977). Estrogen receptor as an independent prognostic factor for early recurrence in breast cancer. Cancer Res. 37, 4669–4671. [PubMed] [Google Scholar]
- Leone A., Diorio G., Sexton W., Schell M., Alexandrow M., Fahey J. W., Kumar N. B. (2017). Sulforaphane for the chemoprevention of bladder cancer: Molecular mechanism targeted approach. Oncotarget 8, 35412–35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L. A., Harris C. C. (2008). Advances in chemical carcinogenesis: A historical review and prospective. Cancer Res. 68, 6863–6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesfeld R., Okret S., Wikstrom A. C., Wrange O., Gustafsson J. A., Yamamoto K. R. (1984). Characterization of a steroid hormone receptor gene and mRNA in wild-type and mutant cells. Nature 312, 779–781. [DOI] [PubMed] [Google Scholar]
- Mitri Z., Constantine T., O'Regan R. (2012). The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemother. Res. Pract. 2012, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura K., Cheung C., Ward J. M., Reddy J. K., Gonzalez F. J. (2006). Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor α to Wy-14, 643-induced liver tumorigenesis. Carcinogenesis 27, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller E., Sarraf P., Tontonoz P., Evans R. M., Martin K. J., Zhang M., Fletcher C., Singer S., Spiegelman B. M. (1998). Terminal differentiation of human breast cancer through PPARγ. Mol. Cell 1, 465–470. [DOI] [PubMed] [Google Scholar]
- Ninomiya I., Yamazaki K., Oyama K., Hayashi H., Tajima H., Kitagawa H., Fushida S., Fujimura T., Ohta T. (2014). Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol. Lett. 8, 2709–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T., Sato R. (1964). The carbon monoxide-binding pigment of liver microsomes. I. evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378. [PubMed] [Google Scholar]
- Omura T., Sato R. (1964b). The Carbon Monoxide-Binding Pigment of Liver Microsomes. Ii. Solubilization, Purification, and Properties. J. Biol. Chem. 239, 2379–2385. [PubMed] [Google Scholar]
- Pardoll D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. M., Cattley R. C., Gonzalez F. J. (1997). Role of PPARα in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14, 643. Carcinogenesis 18, 2029–2033. [DOI] [PubMed] [Google Scholar]
- Peters J. M., Shah Y. M., Gonzalez F. J. (2012). The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 12, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prost S., Relouzat F., Spentchian M., Ouzegdouh Y., Saliba J., Massonnet G., Beressi J. P., Verhoeyen E., Raggueneau V., Maneglier B., et al. (2015). Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 525, 380–383. [DOI] [PubMed] [Google Scholar]
- Shah Y. M., Morimura K., Yang Q., Tanabe T., Takagi M., Gonzalez F. J. (2007). Peroxisome proliferator-activated receptor α regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol. Cell. Biol. 27, 4238–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P., Hu E., Spiegelman B. M. (1994). Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79, 1147–1156. [DOI] [PubMed] [Google Scholar]
- Tortorella S. M., Royce S. G., Licciardi P. V., Karagiannis T. C. (2015). Dietary sulforaphane in cancer chemoprevention: The role of epigenetic regulation and HDAC inhibition. Antioxid. Redox. Signal. 22, 1382–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Z., Shi W., Shao B., Shi J., Shen A., Ma Y., Chen J., Lan Q. (2011). Peroxisome proliferator-activated receptor γ agonist pioglitazone inhibits β-catenin-mediated glioma cell growth and invasion. Mol. Cell. Biochem. 349, 1–10. [DOI] [PubMed] [Google Scholar]
- Xie W., Barwick J. L., Downes M., Blumberg B., Simon C. M., Nelson M. C., Neuschwander-Tetri B. A., Brunt E. M., Guzelian P. S., Evans R. M. (2000). Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 406, 435–439. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Moore R., Goldsworthy T. L., Negishi M., Maronpot R. R. (2004). The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 64, 7197–7200. [DOI] [PubMed] [Google Scholar]
- Yang Q., Nagano T., Shah Y., Cheung C., Ito S., Gonzalez F. J. (2008). The PPAR α-humanized mouse: A model to investigate species differences in liver toxicity mediated by PPAR α. Toxicol. Sci. 101, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]