

Abstract
Hydrophilic drugs are preferred candidates for most routes of drug administration, because of their enhanced solubility and dissolution under aqueous in vivo conditions. However, their hydrophilic nature also leads to decreased permeability across hydrophobic barriers. This is a severe limitation in situations where membrane permeability is the primary factor affecting bioavailability and efficacy of the drug. Highly impermeable cellular membranes or the tight endothelial junctions governing the blood-brain barrier are prime examples of this limitation. In other cases, decreased permeability across mucosal or epithelial membranes may require increased doses, which is an inefficient and potentially dangerous workaround. Covalent conjugation of hydrophilic drugs to hydrophobic moieties like short-chain lipids is a promising strategy for maintaining the critical balance between drug solubility and permeability. This article practically focuses on the production procedure of Lipid drug conjugates (LDCs), various formulation methodologies for preparing LDC nanoparticles with detailed about their in vitro physicochemical characterization at laboratory scale. Moreover, brief overviews on the role of LDCs in novel drug delivery applications as a substrate to various disease therapies are provided.
Graphical Abstract.
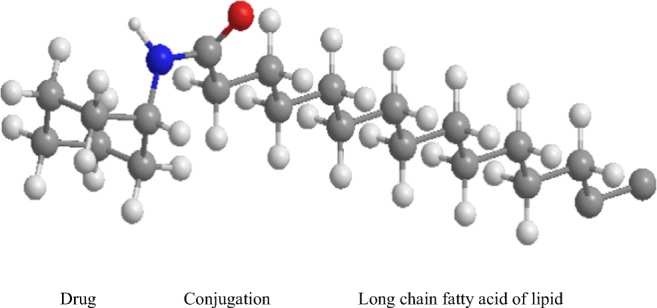
Three dimensional (3-D) schematic representation of LDCs structures.
Keywords: Lipid drug conjugates, Oral administration, Drug delivery, Hydrophilic drug
Introduction
Rapid advances in the field of lipid nanoparticle formulations (LNFs) have caused a revolution to offer superior drug delivery systems for the mitigation of numerous dreadful diseases particularly in the area of pharmaceutical sciences. Such LNFs employed as potential nanocarriers system provides many merits concerning encapsulating hydrophilic drugs, imparting kinetic stability and rigid structural morphology in comparison with other lipid-based vesicular (liposomes, cationic liposomes, leclplex, etc.) and emulsion (lipid emulsions) colloidal nanocarriers systems. LNFs show low cytotoxicity and possess good production scalability also. Development of a wide variety of LNFs viz., solid lipid nanoparticles (SLNs), nanostructured lipid carriers (NLCs), lipid-drug conjugates (LDCs), lipid-polymer hybrid (LPH), etc. for oral drug delivery application has diverted the researcher attention over the years. Apart from these various kinds of LNFs, LDCs attracts increased attention especially in the field of biomedical and pharmaceutical applications. The fundamental vital features of LDCs are the ability of the lipidic matrix to conjugate especially with the water-soluble/hydrophilic drug moieties (Fig. 1) that further offers novel pro-drugs especially in oral drug delivery applications for fulfilling many therapeutic outcomes [1]. Moreover, LDCs technology represents a promising novel approach of controlling and site-specific triggering of the bio-actives of interest. LDCs possess certain advantages like other lipid-based nanocarrier systems, such as [2, 3].
Lipids used for the design of LDCs are biocompatible with the low toxicity profile
Solid at both body and room temperature like SLNs and NLCs.
High capacity to load hydrophilic drugs due to rigid conjugation either by the formation of covalent bond (either by ether or ester formation) or by salt formation with fatty acids
Highly potent hydrophilic drugs with the low therapeutic index can be conjugated easily
LDCs demonstrates high conjugation efficiency with the attached active moieties
It can readily permit through the gastrointestinal tract (GIT).
Possibility the avoidance of GI enzymatic or pH-dependent degradation.
Promotes enhanced oral absorption of the bioactive via lymphatic uptake.
Control/target drug release and its kinetics of conjugated compounds.
Improved stability and bioavailability of many active pharmaceuticals ingredients.
Feasibility of scaling such formulations to bulk industrial level.
Fig. 1.

Three dimensional (3-D) schematic representation of LDCs structures
Such lipid-drug conjugated delivery matrices also possess significant therapeutic potential particularly in targeting especially the hydrophilic drugs at its site of action in severe protozoal infections as well [4].
Necessary production procedure of LDCs at laboratory scale
An insoluble synthetic bulk LDCs are initially either prepared by salt formation conjugated with fatty acids or by covalent linkage to form esters or ethers. Then this is processed with an aqueous surfactant or surfactant mixtures via adopting high shear homogenization techniques to achieve stabilized bulk LDCs formation [4]. The detailed synthesis procedure of bulk LDCs manufacturing is systematically described in section “Initial procedure of synthesizing bulk LDCs”.
Basic ingredients required for the synthesis of LDCs and its NPs
The lipid matrix can be comprised of various fatty acids (e.g., stearic acid, oleic acid, palmitic acid, myristic acid, lauric acid, caprylic acid, squalene etc.), acylglycerols/glycerides (glyceryl monostearate, glyceryl distearate, glyceryl tristearate, glyceryl behenate, glyceryl palmitostearate etc.), waxes (beeswax and cetyl palmitate), various biological membrane derived lipids (phospholipids and sphingomyelins) and mixtures of the same.
Various types of surfactants (including biological membrane lipids) can also be used for the preparation of bulk LDCs. The used surfactants can be lecithin, bile salts (sodium taurocholate, sodium glycolate), sterols (cholesterols), fatty acid ethoxylates, biocompatible non-ionic surfactants (Span −80, Tween 80, poloxamer-188, etc.) and maybe their mixtures which may act as stabilizer [5]. Besides, ligands may be conjugated to this for promoting better tissue or cellular targeting.
Initial procedure of synthesizing bulk LDCs
LDCs can be synthesized in several ways. The hydrophilic drug can be converted to lipophilic prodrug by conjugating its free amino group (if available) with the free carboxylic acid group of fatty acids (Fig. 2) to form an amide linkage [6, 7]. Such reaction can be activated by the addition of some positive catalyst to accelerate the chemical reactions [8]. Similarly, an insoluble synthetic bulk LDCs can also be synthesized by other covalent bonds between lipid and drug moieties via forming either ethers or esters linkage (Fig. 3). It was already observed that drugs which lack carboxyl groups (such as phenytoin) allowing the direct formation of an ester bond with a diglyceride have to be bound to the glycerides by the addition of specific spacers like succinic acid [9]. In other way, researchers were also synthesized LDCs with binary fatty acid salts of various drugs by dissolving the drug and fatty acid salts in different molar ratios in ethanol followed by consequent evaporation of ethanol under reduced pressure at room temperature [10, 11]. After the synthesis of bulk LDCs, it is generally being observed as complete lipophilic with a typical melting point range of approximately 50 to 100 °C reported elsewhere [3].
Fig. 2.

Schematic synthetic reaction scheme of formation of bulk LDCs via forming an amide bond
Fig. 3.

Schematic synthetic reaction scheme of formation of bulk LDCs via forming an ester bond
Formulation strategies for manufacturing bulk LDCs to LDC-NPs
The procedure mentioned above depicted for the synthesis of bulk LDCs can be further transformed to LDC-NPs via adopting various formulation strategies or methodologies similar to our earlier chapter as we described the design of lipid nanoparticle formulations including SLNs and NLCs [12]. However, LDC-NPs comprised of surfactant stabilized lipid-drug conjugated formulations where the used surfactants diminish the interfacial tension between the dispersed phase (especially water) and the synthesized formulations via enhancing the stabilization of the dispersed particles. Formation of LDC-NPs also causes reduction of particle size and thereby leading to strengthening their mean effective surface area with improved dissolution profile of the conjugated drugs. In Fig. 4, different methods are displayed for the preparation of LDC-NPs. Among which, the maximum number of LDC-NPs manufacturing scheme focused on cold high-pressure homogenization technique and solvent injection method due to their following inherent advantages:
Feasibility of tailoring critical process parameters.
Ease of scale-up.
Uniform particle size distribution and less variable polydispersity index
Improved physical stability of the dispersion from the dispersant.
Avoid temperature induced drug degradation and decomposition.
Fig. 4.
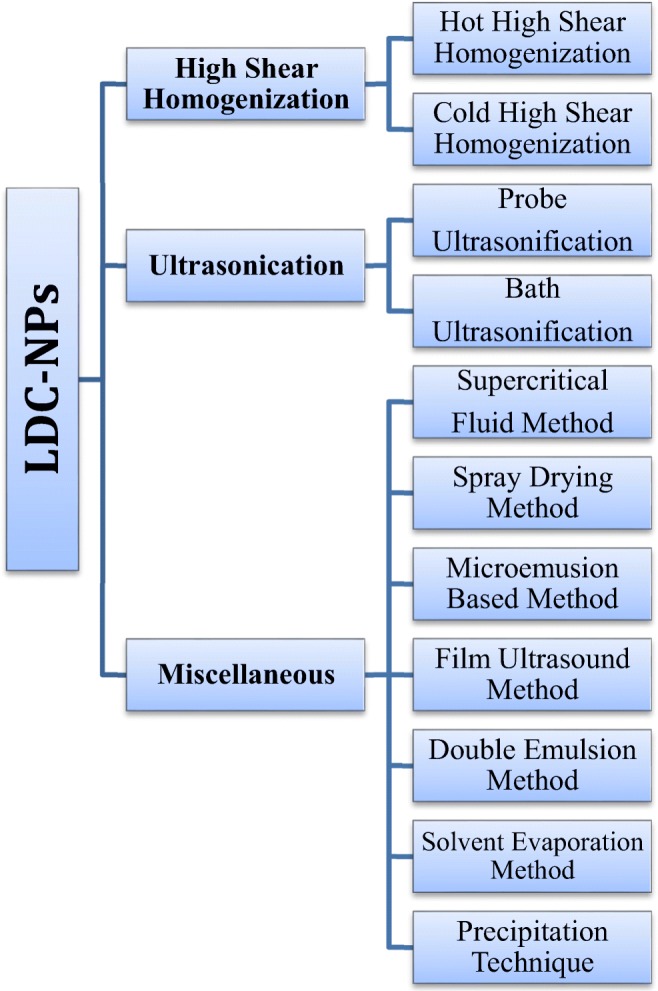
Different methods employed in the preparation of LDC-NPs
Cold high-pressure homogenization technique
It is a straightforward, reliable and suitable homogenization technique for the manufacturing of LDC-NPs from its bulk LDCs. Although this method is entirely different from conventional hot or cold high-pressure homogenization method. Methodically, the synthesized bulk LDC is to be directly dispersed into the cold aqueous systems (kept at refrigerator before dispersion of bulk LDC) containing different surfactant mixture to prevent external partitioning of the conjugated drug into the external aqueous phase. The bulk LDC-surfactant containing aqueous dispersion is to be then further homogenized by high speed/shear homogenizer with a fixed rpm to achieve LDC-NPs with the desired particle size of interest [10]. This method is particularly beneficial for water-loving and temperature-sensitive drugs as it serves as a whole lipidic carrier matrix for a hydrophilic drug for better drug targeting and secondly avoids temperature induced drug decomposition during high shearing. Also, the feasibility of scaling this method for the manufacturing in bulk industrial level and uniform controlling of the desired particle size is also feasible by adopting this method. Thus this method is being currently widely adopted especially in both food and pharmaceutical industry [13].
Solvent injection method
This method is similar to solvent diffusion method. Generally, in this method lipids are to be dissolved in various polar organic solvents like methanol, isopropyl alcohol, acetone, etc. or its different ratio’s solvent mixtures and then to be heated subsequently above the variable melting temperature of the used various lipid matrix. After melting, the melted lipid phase is to be then quickly injected into a surfactant-containing aqueous solution through an injection mode via syringe fitted with a needle, stirred and sonicated to form LDC-NPs. The formed LDC-NPs dispersion is to be then further lyophilized to get fine freeze-dried LDC-NPs [8, 13].
Detailed in vitro physicochemical characterizations
Due to the complex colloidal structure of the delivery system, they’re in vitro characterization possesses is found to be a severe challenge. One can characterize the bulk synthesized LDCs and its NPs as a drug delivery systems in an independent way. Several essential evaluation parameters are associated with the evaluation establishments of LDCs and LDC-NPs. Those following parameters are systematically and separately depicted herein.
Evaluation of synthesized bulk LDC
Percentage yield estimation
The percent yield value of the bulk synthesized LDCs is to be calculated as total amounts of lipid and drug employed during its synthesis by using the following formula [14].
Thin layer chromatography (TLC)
TLC is an advantageous technique to confirm the completion of the conjugation process at a different time of intervals. TLC studies have to be carried out for the preliminary confirmation of the formation of desired product after the process of conjugation. For TLC study, optimization of the suitable solvent system is utmost essential to confirm the desired conjugation process. Different ratios of the solvent systems have to be mixed for getting the proper separation of the synthesized, conjugated compound. TLC to be performed at a different time interval of a conjugation process along with the reactants with the process governed by the capillary action on silica gel plates. As time passes by the concentrations of the reactants got depleted, and the new conjugated compound tends to appear at different retention factor (Rf) values. The completion of the reaction can be confirmed by obtaining a precise, sharp spot of the newly synthesized conjugated system where no other spot for the reactants at their respective Rf values should be seen. Rf values of any compound can be quantified by the distance traveled by the solute to the distance traveled by the solvent front and such values further confirmed by keeping them under ultra-violet (UV) lamp (especially for UV sensitive compound) or by using any substance-specific dyes for spot identification.
UV-visible spectroscopy
Majority of the pharmaceutical compound has their specific excitation and emission wavelength; thus there is fixed scanning wavelength (λmax) for each compound, and they were mentioned in several monographs. Completion of the conjugation reaction can also be judged by the help of UV-Visible spectroscopy by the depletion of the absorbance of the reactants after specific time periods. The number of reactants participating in the reaction can be measured by preparing a standard curve of that particular compound using various known concentrations. The time when the absorbance of the reactant/reactants becomes zero will reflect the completion of the reaction [8].
Percent conjugation efficiency (% CEE)
UV-Visible spectroscopy can also determine % CEE by calculating the amount of unconjugated reactants from the initial reactant amount taken for the reactions at a particular wavelength, after confirming that all other components of the reaction mixture and their by-products did not interfere with the analysis of pristine drugs.
Nuclear magnetic resonance (NMR)
1H NMR is a very delicate technique to determine the proton environment of any compound. We can also elucidate the primary structure of the conjugated system by an accurate interpretation of the NMR spectra. NMR is an analytical tool from where we can get the information about the various types of protons present in the conjugate, the number of protons present form the signal strength and also the detailed knowledge of the adjacent proton environment from the splitting of the Spectra’s. For understanding the spectra’s one can perform the NMR spectroscopy for the reactants along with the final conjugated compound. For performing the NMR studies sample preparation is a significant step, and one must choose a particular solvent for solubilizing the reactants/compound. The compounds to be analyzed must be in a pure form, or it can generate unwanted peaks at the spectra which will cause difficulties for interpretation. The compounds are taken in a minute quantity and must be fully solubilized in the deuterated solvents [15, 16]. One can also perform 13C NMR for the interpretation of the carbon environment of the compound.
Fourier transform infra-red (FTIR) spectral analysis
FTIR is an analytical tool where the compound is subjected to generate specific vibrational frequency due to absorption of energy in the infra-red region. The intake of the energy results in producing a vibrational frequency which is definite for every molecule and thus from the vibrational frequency we can get a better idea of the different components present in that molecule. Therefore by accurate interpretation, we can estimate the various molecular components [17, 18]. Thus FTIR analysis will help us to elucidate the conjugation of the molecule and also it will serve as a side study for proper structure elucidation of the conjugate. Samples must be in pure form for the analysis. FTIR data can also reflect the purity of the compound and also one can judge the compatibility amongst the reactants, thus indicating the stability of the conjugated compound.
Mass spectroscopy
Mass spectroscopy is very important and delicate analytical tool, which will help us to elucidate the exact molecular mass of the compound along with possible molecular fragmentation. A complete interpretation of mass spectroscopy can confirm the precise structure of the conjugate. Various ionization techniques can be employed to determine the molecular mass. If the compound is sensitive, we can use matrix-assisted laser desorption/ionization (MALDI) as an analytical tool for elucidating the molecular formula of the conjugates along with its molecular mass.
Characterization of LDC-NPs
After the systemic investigation based on these aforementioned observed evaluation parameters and confirmation of the formation of bulk LDCs, following essential characterizations have to be carried out to establish the formation of LDC-NPs from its bulk LDC further.
Particle size
The size of the particle plays a deciding role for proper GI uptake of the synthesized LDC-NPs and their systemic clearance by the reticuloendothelial system (RES). So it is always important to determine the size of the particle more accurately. It has already been reported that particle size less than 300 nm facilitates intestinal transport of the drug [19]. Dynamic light scattering (DLS) or photon correlation spectroscopy (PCS) method is the more accurate and frequently used techniques for the measurement of particle size and its distribution analysis. DLS measures the intensity fluctuation of the scattered light caused by the Brownian motion of the developed LDC-NPs. However, nanometers to submicron range particle size can easily be measured by this technique [20, 21], which is enough to characterize the particle size of LDC-NPs. Interestingly, DLS or PCS does not measure the particle size directly, instead of that light scattering effects reflects the particle size, as particles are not always perfectly spherical [22]. Therefore, additional techniques, for instance, light microscopy or electron microscopy can provide better contrast and a fast indication of the particle size and shape.
Polydispersity index (PDI)
PDI is very essential to determine the size distribution pattern of the fabricated LDC-NPs. Low PDI value reflects monodisperse particles population from the given LDC-NPs aqueous dispersion. As per many works of literature reports PDI value of less than 0.5 is considered as optimum value and the sample is to be regarded as monodisperse [23, 24]. The determination of such PDI values can be done by dynamic light scattering (DLS) method [25–27].
Zeta potential (ZP)
ZP measures the extent of electrostatic repulsion or attraction between the particles. It is an essential factor to impart stability of any formulation during storage. ZP can be applied to improve the formulation of dispersions, emulsions, and suspensions as ZP can cause dispersion, aggregation or flocculation. The repulsion between similarly charged particle is due to the ZP, i.e., more the ZP value (negative or positive), more the repulsion thus preventing the aggregation of the particles reflecting the stability of the nanoparticle dispersion. Whereas low ZP value leads to the aggregation/flocculation of the particles as the attraction exceeds the repulsion. ZP value of −30 mV is sufficient for stabilizing the conjugated nanodispersion [28]. DLS method can be used for determining the ZP value of the conjugated nanodispersion [25, 27, 29–31].
Electron microscopy analysis
For determination of the shape and surface morphology of the synthesized LDC-NPs, scanning electron microscopy (SEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM) techniques are considered as an essential tool. These techniques may also be used in determining the particle size and its size distribution analysis. Transmission of electrons from the sample surface is the working principle of SEM whereas TEM utilizes electron transport through the sample. SEM and TEM images can also provide an idea of the estimation of particle size and shape. For instance, many studies have been performed and showed spherical shaped nanoparticles [22, 26, 32, 33]. Field emission SEM (FE-SEM) is more sensitive than SEM as it allows detecting samples within nanometer range [34].
Atomic force microscopy (AFM)
AFM technique can also be used for the characterization of LNFs [35]. It has already been discussed that electron microscopy can provide a two-dimensional image of the synthesized LDC-NPs whereas AFM is capable of imaging a three-dimensional surface profile of the same tested conjugates. An atomic scaled sharp probe tip is inserted across the sample which provides the structural, mechanical, functional and topographical information of matrix surfaces ranged between nanometer to angstrom region [5, 13]. Depending on the nature of the sample and the serving force the probe can be dragged (contact method) or it can hover just above the sample (non-contact method) [36]. Such microscopy technique can also analyze hydrated and non-hydrated samples containing solvents. Sometimes estimation of particle size can also be done via those electron microscopy techniques [37].
Differential scanning Calorimetry (DSC)
DSC is an essential analytical tool which measures the glass transition temperature as a function of temperature. It reflects the melting and crystallization behavior of the synthesized LDC-NPs. As temperature increases, the crystal lattices breaks down, and data indicates the information about the polymorphism and crystal ordering [38–40]. DSC is also employed to observe physical changes, different lipid/drug modifications possess with various components of test melting point behavior such as a pristine drug, pristine lipid(s), physical mixtures of them, placebo LDC-NPs, etc. along with their thermal enthalpy values.
X-ray powder diffraction (XRD) analysis
XRD is a sophisticated analytical tool which is employed to know the crystalline characteristics of the synthesized LNPs with emphasis on optimized LDC-NPs formulations and other associated test components such as pristine drug, pristine lipid(s), physical mixtures of them, placebo LDC-NPs, etc. The utility of XRD analysis is that this instrument can quickly determine the crystallinity of the tested compounds based on their molecular lattice configuration [41]. Determinations of the crystalline behavior of the samples are crucial as the lipid, and the conjugated drug may undergo some polymorphic transition which may lead to possible undesired physical instability during prolong storage conditions. Lipid crystallinity is also strongly correlated with incorporation or conjugation of the drug and its subsequent rate of release. The pattern of XRD diffractogram reflects the particular molecular arrangement of the lipid matrix, phase transition characteristics, and also predict lipid matrix and drug molecules structure [42, 43].
Fluorescence spectroscopy
Fluorescence spectroscopy can be employed for estimating the lipophilic molecular environment of the synthesized LDC-NPs by tagging with a specific lipophilic fluorescence marker or dye (e.g., Nile red or Coumarin-6). Fluorescent lipophilic dyes those absorption bands differ in size, shape, polarity, and intensity with the surrounding molecular environmental nature [44, 45]. The intensity of the peak will get increased as the particle size gets decreased so by this one can easily compare the particle size of the tested conjugated LDC-NPs samples. The peak intensity also gets increased with increase in lipophilic nature as the lipophilic marker will bind more with the compound.
In vitro drug release study
In vitro drug release from LDC-NPs can be determined by quantifying diffusion of released drug through a dialysis membrane method [46]. To simulate in vivo gastric conditions, the in vitro drug release study is to be performed primarily in simulated intestinal fluid (preferably in the pH range of 6.8 to 7.4) for selective lymphatic uptake of the drug using dialysis sac method with a molecular weight cut-off of 10,000 to 12,000 Da. An accurate quantity of LDC-NPs has to be placed inside the dialysis sac, which is then clipped at both the ends. The whole sac is to be dipped in a suitable volume of dissolution test medium (alkaline pH) with proper stirring at fixed rotation under normal body temperature, i.e., 37 °C (±0.2 °C) for the entire course of the in vitro dissolution study. Aliquots sampling is to be carried out each at pre-set time intervals and immediately replaced by an equal volume of fresh dissolution medium to maintain sink conditions. Further aliquot samples are to be analyzed by suitable analytical instruments and proper conditions as well. The cumulative percent drug released at different intervals of time is to be compared to a standard calibration curve of various concentrations of drug generated by multiple analytical means like UV-visible spectroscopy, fluorescence microscopy, high performance liquid chromatography (HPLC) with suitable detection systems like photo-diode array (PDA), fluorescence spectroscopy, refractive index detection unit etc.
Mechanism of drug release kinetics
The mathematical modeling of drug release kinetics values from LDC-NPs can be determined by applying the obtained cumulative % release value data against different time plots fitted into various established release kinetics model to find the best fit based on regression coefficient value (preferably r2 = 0.999) among the following equations:
Where Qt is the drug release amount at pre-defined time intervals, Q0 is the preliminary drug amount at an initial time (frequently, Q0 = 0), and K0 is the value of zero order release rate constant [47].
First-order rate kinetic equation (log cumulative percentage drug remaining to be released against time):
Where Qt is the drug release amount at pre-defined time intervals, Q0 is the preliminary drug amount at an initial time (frequently, Q0 = 0) and K1 is the value of first order release rate constant.
Where, Qt is the drug release amount at pre-defined time intervals, and K is the value of Higuchi diffusion rate constant [48].
Where, Q0 is the preliminary drug amount at an initial time (frequently, Q0 = 0), Qt is the drug release amount at pre-defined time intervals, Qα is the time taken to complete release of the drug and K is the value of release rate constant.
Stability testing
During prolonged storage period of conditions, the physical stability of LDC-NPs can be estimated by monitoring changes in ZP, mean particle size along with its PDI values, drug content values and by in vitro drug release profiles at different periods of sampling intervals. Determining values at different periods of sampling intervals is critical issues related to the stability of the newly synthesized conjugated compounds. This might govern elevation of particle size, dispersion gelation, and expulsion of drug from lipid crystalline lattice during the period of LDC-NPs storage. The phenomenon of gelation of dispersion occurs because of the lipid network formation and bridges between the spaces of the particles [28].
Statistical analysis
After thorough in vitro physicochemical evaluation, the statistical treatment of the obtained data is much essential to establish the level of significance of the valued acquired data under suitable statistical treatment. Mean, and standard deviation value of each reproducible experimental data are to be used for statistical data analysis.
Role of LDCs in drug delivery applications
A lipophilic prodrug or LDCs is a burgeoning field of nanotechnology that has potential to deliver the drugs in specific target organ of interest. Some updated and exciting application of LDCs in drug delivery field is displayed in Table 1. Taking advantage of lipid metabolic pathways, LDC is designed which could overcome the drug delivery problem and target specificity. The absorption of triglycerides takes place after hydrolysis into fatty acids and monoglycerides by lipase enzyme in the intestinal lumen. Pre-duodenal lipases include lingual and gastric lipases which are stable at acidic pH and hydrolyze the triglycerides at low pH. Hydrolysis of dietary triglycerides is very low by these two common enzymes. Thus, fatty acids and monoglycerides produced after hydrolysis are sufficient for emulsification which prepares the intestinal hydrolysis of lipids. The lipid catabolism in the intestine is mainly performed by phospholipase A2, pancreatic lipase and nonspecific esterase [5, 58].
Table 1.
Examples of some interesting experiments carried out in respect to the formation and application of LDCs as drug delivery systems
| Lipids used | Conjugation | Drugs used | Significant findings | References |
|---|---|---|---|---|
| Stearic acid and oleic acid | – | Diminazene diaceturate | Targets specific site with prolonged drug release. A possibility of increased uptake of diaminazene by low-density lipoprotein (LDL) receptor at the blood-brain barrier (BBB). It was observed that the formulated LDC nanoparticle avoids the hepatic uptake by plasma protein adsorption pattern. Plasma protein adsorption patterns indicate the avoidance of hepatic uptake with the possibility of increased uptake via the LDL receptors at the BBB. | [4] |
| Stearic acid | Amide group | Cytarabine | The prepared LDC nanoparticles (NPs) showed smooth, spherical particle surface morphology without any aggregations, uniform particle size (136.80 ± 3.24 nm). In vitro, drug release pattern showed biphasic release, i.e., initial fast release almost 15% within one hr. Followed by prolonging release up to 72 h. LDC-NPs were more cytotoxic than Cyt solution at 48 h. LDC-NPs were found to be physically stable concerning size and zeta potential at the refrigerated condition for 90 days. Based on their findings. They concluded that LDC-NPs could be explored for treatment of meningeal leukemia owing to their ability to provide sustained drug release, stability and improved cytotoxicity in a leukemic EL-4 cell line. | [8] |
| Different fatty acids for example stearic acid, oleic acid, myristic acid, lauric acid, caprylic acid and undecanoic acid. | Ester | Phenytoin | Lipid-phenytoin conjugates proved to be suitable substrates for pancreatic lipase in vitro due to the structural resemblance with triglycerides. It also showed significant improvement in the oral BA. | [9] |
| Stearic acid | Amino and hydroxyl group | Decitabine | In vitro release studies showed the initial burst release followed by a sustained release up to 24 h in phosphate buffer saline at pH -7.4 and the data were further studied using kinetic release models which revealed the first-order model as a best-fitting model. Ex vivo gut permeation study showed that there is a nearly fourfold increase in the permeation of the LDC-NPs containing lipid than that for the plain suspension of a drug under the same experimental conditions at the same time. The LDC-NPs of decitabine has higher permeability coefficients which finally lead to the improvement in the oral BA of decitabine. This system also protects the drug from chemical degradation and augmentation in permeability. | [10] |
| Stearic acid and oleic acid | – | Diminazene | LDC-NPs, with 33% (wt/wt) drug loading capacity possess the potential to act as a delivery system for hydrophilic drugs like diminazene diaceturate and that further studies have to demonstrate the usability as a brain delivery system for the treatment of second stage Human African Trypanosomiasis (HAT). Transforming water-soluble hydrophilic drugs into LDC and formation of NPs allows prolonged drug release and targeting to specific sites by intravenous injection. | [11] |
| Phospholipon 100H, Stearic acid and Palmitic acid, | Lipopeptide | Isoniazid | Improved cellular uptake and site-specific delivery of Isoniazid. Lipid monolayer model was used for penetration experiment to examine the membrane affinity of pal-T5-(INH)2 lipopeptide. The penetration lipopeptide is higher into mycolic acid-containing monolayer as compare to phospholipid. | [49] |
| Ricinoleic acid, 12-hydroxy stearic acid | linked to a substrate for a membrane transporter/ receptor via a lipophilic raft | Acyclovir | Biotinylated lipid prodrugs of acyclovir possess enhanced affinity towards sodium-dependent multivitamin transporter (SMVT). These prodrugs appear to be potential candidates for the treatment of oral and ocular herpes virus infections, because of higher expression of SMVT on intestinal and corneal epithelial cells. Their novel prodrug design strategy may help in higher absorption of hydrophilic parent drug. Moreover, this novel prodrug design can result in higher cell permeability of hydrophilic therapeutics such as genes, siRNA, antisense RNA, DNA, oligonucleotides, peptides and proteins. This drug-loaded lipid conjugates also enhanced the BA and site specificity. | [50] |
| Different fatty acids such as Stearic acid, palmitic acid and myristic acid | Amine group of LMWH was conjugated with the carboxylic group of different lipids | Low Molecular Weight Heparin (LMWH) | Significant enhancement of oral bioavailability as different fatty acids showed variable rate of increased bioavailability. The rate order of oral bioavailability was as follows: LMWH-Stearic acid > LMWH-Palmitic acid > LMWH-Myristic acid. Incorporation of these conjugates into SLNs significantly improved the bioavailability of LMWH after oral route administration with insignificant toxicity to different GIT tissue. This strategy holds promise for future applications of oral delivery of LMWH conjugates in the form of SLNs particularly for the treatment of cardiovascular disease. | [51] |
| Squalene | – | Cytarabine | The enhancement of the lipophilic character of cytarabine was obtained by the conjugation to the acyclic isoprenoid chain of squalene, to increase the affinity towards the phospholipid environment of a biological membrane as well as of lipophilic carrier. | [52] |
| C18 fatty acid chain | Ester | Gemcitabine | Showed lower toxicity and promising efficacy in comparison to Gemzar®. This class of compounds with low toxicity may be further explored to circumvent the problems related with Gemzar®. | [53] |
| Stearic acid | – | Diminazene and SIPI, a structural analogon to Diminazene (p,p´-diamidinodiazoaminobenzene | The incubation of nanoparticles in sera of different species results in different adsorption patterns. These differences are difficult to estimate, especially when there is no reference as in the case of mouse plasma. This has to be taken into account when the data is to be correlated with in vivo experiments. | [54] |
| Phospholipid | – | Ibuprofen and Valproic acid | Incubation with phospholipase A2, a phospholipid which has palmitic acid at the SN2 position in glycerol moiety undergoes degradation while the presence of ibuprofen at the SN2 position resists the in vitro hydrolytic degradation of phospholipid. This was due to unnatural S configuration of ibuprofen. | [55] |
| Various lipid-complexed | – | Camptothecin | Camptothecin-lipid conjugate exhibit same antitumor and cytotoxicity properties as that of camptothecin without lipid-complex against multi-drug resistance positive and negative cancer cells. The camptothecin-lipid conjugate achieved the highest concentration at GIT whereas the drug itself alone reached the highest concentration in and pulmonary parenchyma. Besides, it was also reported that in-vivo activity such lipid-drug complex was found to be more potent than pristine drug against intraperitoneal L1210 and P338 leukemia. | [56] |
| Lipidic and glycolipidic amino acids | Lipidic amino acids with peptides and glycolipidic amino acids. | Cephalosporin | Oral absorption of poorly absorbed cephalosporin can be enhanced by conjugation of lipidic and glycolipidic amino acids. Such conjugation enhances the lipophilicity and oral uptake of cephalosporin which intensify the amount of drug in blood and at the site of action. Thus, it was suggested that lipidic amino acid and peptide conjugation of drugs could better alternative for enhanced oral bioavailability of other poorly absorbed drugs. | [57] . |
Conclusion and future perspectives
The collated valuable information reviewed and compiled in this article explored the various lipid-drug conjugates reported for increased oral bioavailability of the drug and site-specific drug targeting. Admixing of lipids with hydrophilic drugs imparted the lipophilicity of the drug in spite of several other benefits. This could lead to enhanced absorption and permeation of drug followed with the highest drug concentration at the site of action. Using the concept of the metabolic fate of fatty acids in our body, drug or molecule employed for linking with fatty acids which resemble the natural triglycerides and undergo same metabolic pathways as natural triglycerides by specific enzymes like phospholipase-A2, pancreatic lipase, and non-specific esterase. The released drug after enzymatic metabolism, it can reach the site of action in maximum concentration as compared to free drug. Thus, this conjugate system might be a potential approach for enhanced oral bioavailability of poorly water-soluble drug in various classes of therapeutic drugs. LDC is a highly versatile platform for poorly soluble drugs and considered as the best approach to get enhanced oral bioavailability of molecules facing the limited oral absorption. Many such drugs are placed in the BCS class II and IV. Creative lipid drug conjugates of these drug molecules in finished formulations may present an improved pharmacokinetic profile in the human body. The approach may be implemented in diverse medicine such as phytomedicine which could be delivered in this new delivery for site-specific and targeted drug delivery. Lipid is naturally digested by the human body and required no much toxicity data to ensure safety issues. Many publications, abstracts, reviews, and patents are available since last decades. This reflected the drawn attention of pharmaceutical scientist involved in research in this domain. There must be a liaison between research academia and pharmaceutical industries to take up this drug delivery approach for successful formulation with improved BA. For comparison, these delivery approaches are considered suitable carrier than other polymeric systems as safer in term of toxicity. It has several distinct advantages such as protection of thermosensitive drug, protection of drug from enzymatic degradation and enhanced oral absorption which result into increased oral absorption. The shelf life stability, sterilization for a parenteral product of lipid-based formulations and large-scale production of lipid drug conjugates are remaining challenges of this carrier system which need to be investigated shortly.
Acknowledgments
The authors are thankful to their respective institution and university for providing access to necessary literature resources and essential library facilities for writing this review article. Recognition also goes to all the authors of papers, books, patents, websites and all other published sources listed in the references that were used to prepare the contents of this review article.
Abbreviations
- LNFs
Lipid Nanoparticle Formulations
- LDCs
Lipid-drug Conjugates
- NPs
Nanoparticles
- GIT
Gastrointestinal Tract
- ZP
Zeta Potential
- PDI
Polydispersity Index
- BA
Bioavailability
- BCS
Biopharmaceutical Classification Systems
Consent for publication
Not applicable.
Declaration of interest
The authors declare that they have no competing interests.
References
- 1.Anthony AA, Mumuni AM, Philip FB. Lipid Nanoparticulate Drug Delivery Systems: A Revolution in Dosage Form Design and Development. Recent Advances in Novel Drug Carrier Systems. Intech Open; 2012. Pp. 107–140.
- 2.Morel S, Terreno E, Ugazio E, Aime S, Gasco MR. NMR relaxometric investigations of lipid nanoparticles (SLN) containining gadolinium (III) complexes. Eur J Pharm Biopharm. 1998;45(2):157–163. doi: 10.1016/S0939-6411(97)00107-0. [DOI] [PubMed] [Google Scholar]
- 3.Muchow M, Maincent P, Müller RH. Lipid nanoparticles with a solid matrix (SLN®, NLC®, LDC®) for oral drug delivery. Drug Dev Ind Pharm. 2008;34(12):1394–1405. doi: 10.1080/03639040802130061. [DOI] [PubMed] [Google Scholar]
- 4.Olbrich C, Gessner A, Kayser O, Mueller RH. Lipid drug conjugate nanoparticles as novel carrier system for the hydrophilic antitrypanosomal drug diminazene aceturate. J Drug Target. 2002;10(5):387–396. doi: 10.1080/1061186021000001832. [DOI] [PubMed] [Google Scholar]
- 5.Das RJ, Baishya K, Pathak K. Recent advancement of lipid drug conjugate as nanoparticulate drug delivery system. Int Res J Pharm. 2013;4(1):73–78. [Google Scholar]
- 6.Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimides for bioconjugation in aqueous media. Bioconjug Chem. 1995;6(1):123–130. doi: 10.1021/bc00031a015. [DOI] [PubMed] [Google Scholar]
- 7.Pignatello R, Spampinato G, Sorrenti V, Di Giacomo C, Vicari L, McGuire JJ, et al. Lipophilic methotrexate conjugates with antitumor activity. Eur J Pharm Sci. 2000;10(3):237–245. doi: 10.1016/S0928-0987(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 8.Sharma P, Dube B, Sawant K. Synthesis of Cytarabine lipid drug conjugate for treatment of meningeal leukemia: development, characterization and In vitro cell line studies. J Biomed Nanotechnol. 2012;8(6):928–937. doi: 10.1166/jbn.2012.1464. [DOI] [PubMed] [Google Scholar]
- 9.Scriba GK. Phenytoin-lipid conjugates as potential prodrugs of phenytoin. Arch Pharm. 1993;326(8):477–481. doi: 10.1002/ardp.19933260810. [DOI] [PubMed] [Google Scholar]
- 10.Neupane YR, Sabir MD, Ahmad N, Ali M, Kohli K. Lipid drug conjugate nanoparticle as a novel lipid nanocarrier for the oral delivery of decitabine: Ex-vivo gut permeation studies. Nanotechnology. 2013;24(41):1–11. doi: 10.1088/0957-4484/24/41/415102. [DOI] [PubMed] [Google Scholar]
- 11.Olbrich C, Gessner A, Schröder W, Kayser O, Müller RH. Lipid drug conjugate nanoparticles of the hydrophilic drug diminazene-cytotoxicity testing and mouse serum adsorption. J Control Release. 2004;96(3):425–435. doi: 10.1016/j.jconrel.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee S., Pillai J. Nanoarchitectonics for Smart Delivery and Drug Targeting. 2016. Lipid Nanoparticle Formulations for Enhanced Antituberculosis Therapy; pp. 285–314. [Google Scholar]
- 13.Das S, Chaudhury A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech. 2011;12(1):62–76. doi: 10.1208/s12249-010-9563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee S, Chattopadhyay P, Ghosh A, Goyary D, Karmakar S, Veer V. Influence of process variables on essential oil microcapsule properties by carbohydrate polymer-protein blends. Carbohydr Polym. 2013;93(2):691–697. doi: 10.1016/j.carbpol.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 15.Wen B, Sun Y, Xu Y, Sun J, Liu X, Wang Y, et al. Pharmacokinetic characteristics of the cytarabine prodrug, ilecytarabine, after intravenous and oral administration to rats. Asian J Pharm Sci. 2008;3:200. [Google Scholar]
- 16.Knothe G, Kenar JA. Determination of the fatty acid profile by 1H-NMR spectroscopy. Eur J Lipid Sci Technol. 2004;106:88–96. doi: 10.1002/ejlt.200300880. [DOI] [Google Scholar]
- 17.Ferreira L, Vidal MM, Gil MH. Evaluation of poly(2- hydroxyethyl methacrylate) gels as drug delivery systems at different pH value. Int J Pharm. 2000;194(2):169–180. doi: 10.1016/S0378-5173(99)00375-0. [DOI] [PubMed] [Google Scholar]
- 18.Ren S, Yang S, Zhao Y, Yu T, Xiao X. Preparation and characterization of an ultrahydrophobic surface based on a stearic acid self-assembled monolayer over polyethyleneimine thin films. Surf Sci. 2003;546(2–3):64–74. doi: 10.1016/j.susc.2003.09.018. [DOI] [Google Scholar]
- 19.Charman WN, Stella VJ, editors. Lymphatic transport of drugs. Boca Raton: CRC Press; 1992. [Google Scholar]
- 20.Müller RH, Runge SA, Ravelli V, Thünemann AF, Mehnert W, Souto EB. Cyclosporine-loaded solid lipid nanoparticles (SLN®):drug-lipid physicochemical interactions and characterization of drug incorporation. Eur J Pharm Biopharm. 2008;68(3):535–544. doi: 10.1016/j.ejpb.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Müller RH, Runge S, Ravelli V, Mehnert W, Thünemann AF, Souto EB. Oral bioavailability of cyclosporine: solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int J Pharm. 2006;317(1):82–89. doi: 10.1016/j.ijpharm.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Liu C, Weiwei Z, Zhang N. Enhanced gastrointestinal absorption of N-3-O-toluyl-fluorouracil by cationic solid lipid nanoparticles. J Nanopart Res. 2010;12(3):975–984. doi: 10.1007/s11051-009-9648-4. [DOI] [Google Scholar]
- 23.Zhang J, Fan Y, Smith E. Experimental design for the optimization of lipid nanoparticles. J Pharm Sci. 2009;98(5):1813–1819. doi: 10.1002/jps.21549. [DOI] [PubMed] [Google Scholar]
- 24.Anton N, Benoit J-P, Saulnier P. Design and production of nanoparticles formulated from nano-emulsion templates-a review. J Control Release. 2008;128(3):185–199. doi: 10.1016/j.jconrel.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Xie S, et al. Formulation, characterization, and pharmacokinetics of praziquantel- loaded hydrogenated castor oil solid lipid nanoparticles. Nanomedicine London. 2010;5(5):693–701. doi: 10.2217/nnm.10.42. [DOI] [PubMed] [Google Scholar]
- 26.Sanjula B, Shah FM, Javed A, Alka A. Effect of poloxamer 188 on lymphatic uptake of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J Drug Target. 2009;17(3):249–256. doi: 10.1080/10611860902718672. [DOI] [PubMed] [Google Scholar]
- 27.Paliwal R, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5(2):184–191. doi: 10.1016/j.nano.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN) dispersions. Int J Pharm. 1998;168(2):221–229. doi: 10.1016/S0378-5173(98)00092-1. [DOI] [Google Scholar]
- 29.Radomska-Soukharev A. Stability of lipid excipients in solid lipid nanoparticles. Adv Drug Deliv Rev. 2007;59(6):411–418. doi: 10.1016/j.addr.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee B, Santra K, Pattnaik G, Ghosh S. Preparation, characterization and in vitro evaluation of sustained release protein-loaded nanoparticles based on biodegradable polymers. Int J Nanomedicine. 2008;3(4):487–496. doi: 10.2147/IJN.S3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai MJ, Huang YB, Wu PC, Fu YS, Kao YR, Fang JY, et al. Oral apomorphine delivery from solid lipid nanoparticles with different monostearate emulsifiers: pharmacokinetic and behavioral evaluations. J Pharm Sci. 2011;100(2):547–557. doi: 10.1002/jps.22285. [DOI] [PubMed] [Google Scholar]
- 32.Varshosaz J, Minayian M, Moazen E. Enhancement of oral bioavailability of pentoxifylline by solid lipid nanoparticles. J Liposome Res. 2010;20(2):115–123. doi: 10.3109/08982100903161456. [DOI] [PubMed] [Google Scholar]
- 33.Kakkar V, Singh S, Singla D, Kaur IP. Exploring solid lipid nanoparticles to enhance the oral bioavailability of curcumin. Mol Nutr Food Res. 2010;55(3):495–503. doi: 10.1002/mnfr.201000310. [DOI] [PubMed] [Google Scholar]
- 34.Sahana B, Santra K, Basu S, Mukherjee B. Development of biodegradable polymer-based tamoxifen citrate-loaded nanoparticles and effect of some manufacturing process parameters on them: a physicochemical and in vitro evaluation. Int J Nanomedicine. 2010;5(7):621–630. doi: 10.2147/IJN.S9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shahgaldian P, Da Silva E, Coleman AW, Rather B, Zaworotko MJ. Para-acyl-calixarene based solid lipid nanoparticles (SLNs): a detailed study of preparation and stability parameters. Int J Pharm. 2003;253(1–2):23–38. doi: 10.1016/S0378-5173(02)00639-7. [DOI] [PubMed] [Google Scholar]
- 36.Schwarz C, Freitas C, Mehnert CW, Muller RH. Sterilization and physical stability of drug-free and etomidate- loaded solid lipid nanoparticles. Proc Int Symp Control Release Bioact Mater. 1995;22:766–767. [Google Scholar]
- 37.Zur Muhlen A, et al. Atomic force microscopy studies of solid lipid nanoparticles. Pharm Res. 1996;13(9):1411–1416. doi: 10.1023/A:1016042504830. [DOI] [PubMed] [Google Scholar]
- 38.Jenning V, Thunemann A, Gohla S. Characterisation of a novel solid lipid nanoparticle carrier system based on binary mixtures of liquid and solid lipids. Int J Pharm. 2000;199(2):167–177. doi: 10.1016/S0378-5173(00)00378-1. [DOI] [PubMed] [Google Scholar]
- 39.Sari A, Akcay M, Soylak M, Onal A. Polymer-stearic acid blends as form-stable phase change material for thermal energy storage. J Sci Ind Res. 2005;64:991–996. [Google Scholar]
- 40.Dicko A, et al. Biophysical characterization of a liposomal formulation of cytarabine and daunorubicin. Int J Pharm. 2010;391(1–2):248–259. doi: 10.1016/j.ijpharm.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Souto EB, Mehnert W, Muller RH. Polymorphic behavior of Comprito l888 ATO as bulk lipid and as SLN and NLC. J Microencapsul. 2006;23(4):417–433. doi: 10.1080/02652040600612439. [DOI] [PubMed] [Google Scholar]
- 42.Bunjes H, Steiniger F, Richter W. Visualizing the structure of triglyceride nanoparticles in different crystal modifications. Langmuir. 2007;23(7):4005–4011. doi: 10.1021/la062904p. [DOI] [PubMed] [Google Scholar]
- 43.Estella-Hermoso de Mendoza A, Rayo M, Mollinedo M, Blanco-Prieto MJ. Lipid nanoparticles for alkyl lysophospholipid edel fosine encapsulation: development and in vitro characterization. Eur J Pharm Biopharm. 2008;68(2):207–213. doi: 10.1016/j.ejpb.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 44.Huang ZR, Hua SC, Yang YL, Fang JY. Development and evaluation of lipid nanoparticles for camptothecin delivery: a comparison of solid lipid nanoparticles, nanostructured lipid carriers, and lipid emulsion. Acta Pharmacol Sin. 2008;29(9):1094–1102. doi: 10.1111/j.1745-7254.2008.00829.x. [DOI] [PubMed] [Google Scholar]
- 45.Banerjee S, Roy S, Nath Bhaumik K, Kshetrapal P, Pillai J. Comparative study of oral lipid nanoparticle formulations (LNFs) for chemical stabilization of antitubercular drugs: physicochemical and cellular evaluation. Artif Cells Nanomed Biotechnol. 2018;26:1–19. doi: 10.1080/21691401.2018.1431648. [DOI] [PubMed] [Google Scholar]
- 46.Stela G, Esther I. Conjugates for cancer therapy and diagnosis, patent application number: 20110275590; Publication date: 11, October (2011).
- 47.Wyatt DA. Taking poorly water-soluble compounds through discovery. In: Recent advances in the formulations and development of poorly soluble drugs. Bulletin Technique Gattefosse. 1999:31–39.
- 48.Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50:874–875. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 49.Penzesa CB, Schnoller D, Horvati K, Kiss E. Membrane affinity of antituberculosis drug conjugate using lipid monolayer containing mycolic acid. Colloids Surf A Physicochem Eng Asp. 2012;413(5):142–148. doi: 10.1016/j.colsurfa.2012.02.013. [DOI] [Google Scholar]
- 50.Vadlapudia AD, Vadlapatla RK, Kwatra D, Earla R, Samanta SK. Targeted lipid-based drug conjugates: a novel strategy for drug delivery. Int J Pharm. 2012;434(1–2):315–324. doi: 10.1016/j.ijpharm.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paliwal R, Shivani RP, Govind PA, Suresh PV. Biomimetic solid lipid nanoparticles for oral bioavailability enhancement of low molecular weight heparin and its lipid conjugates: In vitro and in-vivo evaluation. Mol Pharm. 2011;8(4):1314–1321. doi: 10.1021/mp200109m. [DOI] [PubMed] [Google Scholar]
- 52.Sarpietro MG, Ottimo S, Giuffrida MC, Rocco F, Ceruti M, Castelli F. Synthesis of n-squalenoyl cytarabine and evaluation of its affinity with phospholipid bilayers and monolayers. Int J Pharm. 2011;406(1–2):69–77. doi: 10.1016/j.ijpharm.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 53.Ali SM, Khan AR, Ahmad MU, Chen P, Sheikh S, Ahmad I. Synthesis and biological evaluation of gemcitabine-lipid conjugate. Bioorg Med Chem Lett. 2005;15(10):2571–2574. doi: 10.1016/j.bmcl.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 54.Gessner A, Olbrich C, Schroder W, Kayser O, Muller RH. The role of plasma proteins in brain targeting: species-dependent protein adsorption patterns on brain-specific lipid drug conjugate (LDC) nanoparticles. Int J Pharm. 2001;214(1–2):87–91. doi: 10.1016/S0378-5173(00)00639-6. [DOI] [PubMed] [Google Scholar]
- 55.Kurz M, Scriba GK. Drug-phospholipid conjugates as potential prodrugs: synthesis, characterization, and degradation pancreatic phospholipase A2. Chem Phys Lipids. 2000;107(2):143–157. doi: 10.1016/S0009-3084(00)00167-5. [DOI] [PubMed] [Google Scholar]
- 56.Sugarman SM, Zou Y, Wasan K, Poirot K, Kumi R, Reddy S, et al. Lipid-complexed camptothecin: formulation and initial biodistribution and antitumor activity studies. Cancer Chemother Pharmacol. 1996;37(6):531–538. doi: 10.1007/s002800050425. [DOI] [PubMed] [Google Scholar]
- 57.Toth I, Hughes RA, Dekany G, Hillery AM, Ward P. Synthesis and oral uptake studies of lipidic and glyco-lipidic conjugates of β- lactam antibiotics. Liebigs Annalen Der Chemie. 1994;1994(7):685–688. doi: 10.1002/jlac.199419940709. [DOI] [Google Scholar]
- 58.Lambert DM. Rationale and applications of lipids as prodrug carriers. Eur J Pharm Sci. 2000;11(2):S15–S27. doi: 10.1016/S0928-0987(00)00161-5. [DOI] [PubMed] [Google Scholar]