

Abstract
Heat shock proteins are molecular chaperones that are involved in protein folding. In this study, we developed a targeted proteomic method, relying on LC-MS/MS in the parallel-reaction monitoring (PRM) mode, for assessing quantitatively the human heat shock proteome. The method facilitated the coverage of approximately 70% of the human heat shock proteome and displayed much better throughput and sensitivity than the shotgun proteomic approach. We also applied the PRM method for assessing the differential expression of heat shock proteins in three matched primary/metastatic pairs of melanoma cell lines. We were able to quantify ~45 heat shock proteins in each pair of cell lines, and the quantification results revealed that DNAJB4 is down-regulated in the three lines of metastatic melanoma cells relative to the corresponding primary melanoma cells. Interrogation of The Cancer Genome Atlas data showed that lower levels of DNAJB4 expression conferred poorer prognosis in melanoma patients. Moreover, we found that DNAJB4 suppresses the invasion of cultured melanoma cells through diminished expression and activities of matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9). Together, we established, for the first time, a high-throughput targeted proteomics method for profiling quantitatively the human heat shock proteome and discovered DNAJB4 as a suppressor for melanoma metastasis.
Graphical Abstract
Heat shock proteins (HSPs) are molecular chaperones that function in protein folding/unfolding, cell cycle regulation, and protection of cells during stress.1 There are six major HSPs, namely, HSP70, HSP40, HSP60, HSP90, HSP110, and small HSP,2 and some of them were shown to correlate with the progression of multiple types of cancer. For instance, HSP90, a molecular chaperone for protein folding and client protein stabilization,3 is overexpressed in many types of tumors and has been reported to associate with breast cancer progression.4 Thus, targeting HSP90 may inhibit multiple proinvasive pathways5 and enhance cancer immunotherapy.6 On the basis of these discoveries, 13 inhibitors for HSP90 have been developed for clinical evaluation in anticancer therapy.7 Among them, ganetespib has been under clinical trials for the treatment of non-small cell lung cancer (NSCLC)8,9 and breast cancer.10 Likewise, inhibitors for HSP27 and HSP70 have also been developed for clinical studies.9 Hence, comprehensive analysis of heat shock proteins will assist drug discovery and cancer treatment. Current methods for studying heat shock proteins rely on low-throughput Western blot analysis. While mass spectrometry has been widely employed for proteomic analysis,11 no analytical methods have yet been developed for proteome-wide interrogation of heat shock proteins.
Compared to the proteomic analysis in the data-dependent acquisition (DDA) mode, targeted proteomic methods, which involve LC-MS/MS analyses in multiple-reaction monitoring (MRM) or parallel-reaction monitoring (PRM) mode, exhibit much better sensitivity toward peptide detection,12 and thus have become extensively employed in quantitative proteomics studies.13 Owing to the high mass accuracy and resolution provided by the Orbitrap or time-of-flight mass analyzer, PRM provides better accuracy and specificity for quantifying analytes in complex sample matrices;14 hence, it has been widely used in bioanalysis, such as PTM detection15 and metabolite quantification.16 In this work, we developed a PRM-based targeted quantitative proteomic method to interrogate the human heat shock proteome, and we further applied the method for assessing the perturbations of HSPs during melanoma metastasis. Our results revealed many differentially expressed HSPs in paired primary/metastatic melanoma cells, including DNAJB4, which was uniformly down-regulated in the three metastatic lines of melanoma cells relative to the paired primary melanoma cells. We also demonstrated that DNAJB4 suppressed melanoma metastasis by modulating the expression levels and activities of matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9).
EXPERIMENTAL SECTION
Cell Culture.
WM-115 and WM-266–4 cells (ATCC) were cultured in Eagle’s Minimum Essential Medium. IGR-39, IGR-37 (obtained from Prof. Peter H. Duesberg),17 WM-793, and 1205Lu (Wistar Institute) cells were cultured in Dulbecco’s Modified Eagle Medium. All culture media were supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and penicillin (100 IU/mL). The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Approximately 2 × 107 cells were harvested, washed with cold PBS three times, and lysed by incubating on ice for 30 min in CelLytic M (Sigma) cell lysis reagent containing 1% protease inhibitor cocktail. The cell lysates were centrifuged at 9000g at 4 °C for 30 min, and the resulting supernatants were collected. WM-115 and WM-266–4 cells were derived from the primary and metastatic sites of the same melanoma patient.18 IGR-39 and IGR-37 cells were derived from the primary and metastatic sites of another melanoma patient.19 1205Lu cells were initiated from a lung metastasis of WM-793 human melanoma cells after subcutaneous injection into an immune-deficient mouse.20 Cells were cultured in SILAC medium containing [13C6,15N2]-lysine and [13C6]arginine for at least 10 days to promote complete incorporation of isotope-labeled amino acids.21
Plasmid and siRNAs.
The sequences for siDNAJB4 was 5′-AACCCGGAAUGAGGAGAAGAA-3′.22 The coding sequences of DNAJB4 gene was amplified from a cDNA library from M14 cells by PCR primers 5′-GCTCTAGAGCATTCGAAATGGGGAAA-3′ and 5′-CGGGATCCTCTTCATTCTATGAGGCA-3′. cDNA was subcloned into BamHI- and XbaI-linearized pRK7 vectors. siRNA was transfected using RNAiMAX (Invitrogen) following the manufacturer’s protocol, where non-targeting siRNA (Dharmacon, D-001210–02-20) was used as control. pRK7-DNAJB4 and pRK7-empty vector were transfected into WM-266–4, IGR-37, and 1205Lu cells by using Lipofectamine 2000 (Life Technologies).
LC-PRM Analysis.
To assess the differential expression of heat shock proteins in primary and metastatic melanoma cells, we conducted one forward and one reverse SILAC labeling experiment, where lysates of light-labeled primary melanoma cells and heavy-labeled metastatic melanoma cells were combined at a 1:1 ratio in the forward labeling experiments. The reverse labeling experiments were conducted in the opposite way. All LC-PRM experiments were performed on a Q Exactive Plus quadrupole–Orbitrap mass spectrometer. The mass spectrometer was coupled with an EASY-nLC 1200 system (Thermo Scientific), and the samples were automatically loaded onto a 4 cm trapping column (150 μm i.d.) packed with ReproSil-Pur 120 C18-AQ resin (5 μm in particle size and 120 Å in pore size, Dr. Maisch GmbH HPLC) at 3 μL/min. The trapping column was coupled to a 20 cm fused silica analytical column (PicoTip Emitter, New Objective, 75 μm i.d.) packed with ReproSil-Pur 120 C18-AQ resin (3 μm in particle size and 120 Å in pore size, Dr. Maisch GmbH HPLC). The peptides were then separated using a 140 min linear gradient of 9–38% acetonitrile in 0.1% formic acid and at a flow rate of 300 nL/min. The spray voltage was 1.8 kV. Precursor ions were isolated, at an isolation width of 1.0 m/z, and collisionally activated in the HCD cell with a collision energy of 29 to yield MS/MS.
PRM Data Analysis.
All raw files were processed using Skyline (version 3.5)23 for the generation of extracted-ion chromatograms and peak integration. We imposed a mass accuracy of within 20 ppm for fragment ions during the identification of peptides in the Skyline platform. The targeted peptides were manually checked to ensure that the transitions for multiple fragment ions derived from light and heavy forms of the same peptide exhibit the same elution time in the preselected retention time window. The data were then processed to ensure that the distribution of the relative intensities of multiple transitions associated with the same precursor ion correlates with the theoretical distribution in the MS/MS spectral library entry, which was acquired from shotgun proteomic analysis. The sum of peak areas from all transitions of light or heavy forms of peptides was used for quantification. The Skyline PRM library for heat shock proteins and the raw files for LC-PRM analyses of heat shock proteins for paired melanoma cells were deposited into PeptideAtlas with the identifier number of PASS01177 (http://www.peptideatlas.org/PASS/PASS01177).
TCGA Data Analysis.
OncoLnc24 was employed for Kaplan–Meier survival analysis of melanoma patients using The Cancer Genome Atlas data, where patients were stratified based on the expression levels of DNAJB4 gene being among the top (high group) and bottom (low group) quartiles, respectively. Differences in survival with logrank p-values being less than 0.05 were considered significant.
RESULTS
Development of a High-Throughput PRM Method for the Quantitative Analysis of the Heat Shock Proteome.
Construction of a PRM Library for Heat Shock Proteins.
The major objective of the present study was to develop a high-throughput analytical method, relying on parallel-reaction monitoring (PRM), for profiling quantitatively the human heat shock proteome. To this end, we first constructed a Skyline PRM library using the retention time, MS, and MS/MS of peptides of heat shock proteins retrieved from shotgun proteomic analyses of the tryptic digestion mixtures of 10 unique cell lines derived from different human tissue origins. These data were obtained from more than 200 LC-MS/MS runs and led to the identification of 11 879 protein groups.
Owing to the high level of similarity in protein sequences from some heat shock proteins, we inspected manually all the identified peptides and incorporated only the unique peptides representing individual heat shock proteins into the library. By including a maximum of five unique peptides for any given heat shock proteins into the library, our current PRM HSP library encompassed 180 unique peptides from 57 distinct human heat shock proteins (Figure 1a, Table S2). This covers approximately 70% of the human heat shock proteome, which contains a total of 84 proteins.25 Among the 57 heat shock proteins in the library, 33, 9, and 5 belong to the HSP40, HSP70, and HSP90 groups, respectively (Figure 1b).
Figure 1.

PRM-based targeted proteomic approach for interrogating the human heat shock proteome. (a) A Venn diagram displaying the numbers of heat shock proteins included in the PRM library and those that could be quantified by the PRM method. (b) A pie chart depicting the protein coverage of different groups of heat shock proteins. (c) Experimental strategy for the PRM-based targeted proteomic approach.
Retention Time Calibration.
To achieve high-throughput detection of HSP peptides, we adopted scheduled PRM analysis, where the mass spectrometer was programmed to acquire the MS/MS of the precursor ions for a limited number of peptides in each 10 min retention time (RT) window. To achieve this, we calculated the normalized RT (iRT) value for each peptide on our target list following a previously published method.26,27 By using 10 tryptic peptides of bovine serum albumin (BSA) as standards, we successfully converted the experimentally determined retention times of the 180 peptides from heat shock proteins into normalized iRT scores (Table S2). The iRT value represents an intrinsic property (i.e., hydrophobicity) of a peptide. Hence, a substantial deviation of measured RT from that projected from the linear plot of RT over iRT is considered a false-positive detection, which is employed as a criterion to validate the results obtained from the PRM assay. The linear RT vs iRT relationship was redefined after every 5–7 LC-PRM runs through the analysis of the tryptic digestion mixture of BSA.
Revelation of Differential Expression of Heat Shock Proteins during Melanoma Metastasis by Scheduled LCPRM Analysis.
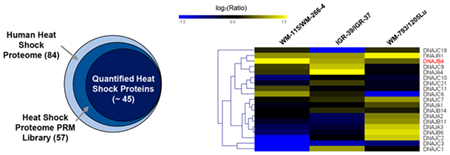
To assess the reprogramming of heat shock proteome during melanoma metastasis, we employed LC-MS/ MS in the PRM mode, together with metabolic labeling using SILAC,21 to examine the differential expression of HSPs in three matched primary/metastatic melanoma cells (i.e., WM-115/WM-266–4, IGR-39/IGR-37, and WM-793/1205Lu. Figure 1c). As shown in Figure 2a, 48 unique HSPs were quantified in WM-115/WM-266–4 paired melanoma cell lines by our PRM-based targeted proteomics method (Figure 2a). All of the quantified peptides for heat shock proteins exhibit an excellent linear fit between the observed retention time and iRT in the library (Figure S1a). Additionally, all 4–6 transitions used for quantification of each peptide from heat shock proteins were eluted at the same retention time with a dot product (dotp) of >0.7 when compared to the same fragment ions found in the MS/MS acquired from shotgun proteomic analysis (Figure S2a),28 suggesting that our method is highly reliable for peptide identification. Furthermore, all the quantified heat shock proteins appeared in both forward and reverse SILAC labeling experiments (Figure 2b). The ratios of quantified peptides obtained from forward and reverse SILAC labeling experiments exhibited an excellent linear fit (Figures 2c and S2b) and displayed a strong correlation (see heat map in Figure S2c), supporting the reproducibility of the analytical method. Moreover, the reproducibility of the method is reflected by the observation that consistent ratios were obtained for different tryptic peptides derived from the same heat shock proteins (Table S3). In this regard, the average relative standard deviations (RSD) among the different quantified peptides from the same heat shock proteins were 11.1% for the data acquired for the WM-115/WM-266–4 paired melanoma cell lines (Table S3).
Figure 2.

Performances of PRM-based targeted proteomic approach for interrogating the perturbations in expression of heat shock proteins during melanoma metastasis. (a) Differential expression of heat shock proteins in WM-115/WM-266–4 paired melanoma cells. (b) A Venn diagram displaying the overlap between quantified heat shock proteins from the forward and reverse SILAC labelings of WM-115/WM-266–4 paired melanoma cells. (c) Correlation between the ratios obtained from forward and reverse SILAC labeling experiments. (d) A heat map showing the differences in expression of heat shock proteins in three pairs of primary/metastatic melanoma cell lines. Genes were clustered according to Euclidean distance. The data in (a) and (d) represent the mean of the results obtained from one forward and one reverse SILAC labeling, and Table S3 lists the ratios obtained from individual measurements.
We also analyzed the SILAC samples from paired WM-115/WM-266–4 melanoma cells by using LC-MS/MS in the DDA mode. After prefractionation using a strong cation-exchange (SCX) column,29 20 fractions were subjected to LC-MS/MS analysis in the DDA mode. The results from this analysis only led to the quantification of 36 heat shock proteins. Meanwhile, our scheduled PRM method allowed for the quantification of 48 heat shock proteins in two LC-PRM runs without prefractionation, demonstrating the superior sensitivity and throughput of the PRM method (Figure S1b).
A total of 43 and 44 unique heat shock proteins were quantified in IGR-39/IGR-37 and WM-793/1205Lu paired melanoma cells, respectively. The reliability and reproducibility of the quantification results were similar to those obtained from the WM-115/WM-266–4 paired melanoma cells (Figure S1c–f and Table S3). Our quantification results showed that 7, 10, and 20 heat shock proteins were up-regulated, and 18, 8, and 5 were down-regulated in primary (WM-115, IGR-39, and WM-793 cells, respectively) relative to the corresponding metastatic (WM-266–4, IGR-37, and 1205Lu) melanoma cells (Figures 2a, S3, 2d, and Table S3). We also assessed the differential expression of DNAJB4, DNAJC3, and DNAJB1 (HSP40) in paired WM-115/WM-266–4 melanoma cells by using Western blot analysis. The ratios obtained from Western blot are in keeping with those obtained from PRM analyses (Figures 3a and S4), demonstrating that the PRM method is capable of profiling accurately the differential expression of heat shock proteins. In the meantime, we found that HSPB1 (HSP27), which was previously shown to suppress the invasive ability and the activities of secreted matrix metalloproteinases (MMPs) in A375 malignant melanoma cells,30 is up-regulated in the two primary melanoma cells, WM-115 and IGR-39 (Figure 2d).
Figure 3.

DNAJB4 is down-regulated in metastatic melanoma cells. (a) Western blot for the validation of the expression levels of DNAJB4 in the three pairs of melanoma cells. 115, 266–4, 39, 37, 793, and 1205 denote WM-115, WM-266–4, IGR-39, IGR-37, WM-793, and 1205Lu cells, respectively. (b) PRM traces for the quantification of DNAJB4 protein in three pairs of melanoma cells from forward and reverse SILAC labeling experiments. (c) Quantitative comparison of ratios of DNAJB4 obtained from PRM and Western blot analysis (n = 3). (d) Kaplan–Meier survival analysis showing that higher levels of expression of the DNAJB4 gene confers better prognosis of melanoma patients.
Down-Regulation of DNAJB4 in Metastatic Melanoma Cells and Its Modulation of the Invasive Capabilities of Cultured Melanoma Cells.
As noted above, our PRM method has revealed the differential expression of a known suppressor for melanoma metastasis, i.e., HSPB1. We next asked whether any other differentially expressed heat shock proteins may act as drivers or suppressors for melanoma metastasis. In this vein, we found that DNAJB4 was down-regulated in all three metastatic melanoma cells relative to the corresponding primary melanoma cells, which we validated by Western blot analysis (Figure 3a–c). In addition, Kaplan–Meier survival analysis of The Cancer Genome Atlas (TCGA) data24 showed poorer prognosis for those melanoma patients with lower levels of mRNA expression of the DNAJB4 gene (Figure 3d), indicating that DNAJB4 may suppress melanoma metastasis. Along this line, DNAJB4 was previously shown to be a suppressor for lung cancer metastasis.31
To explore the potential roles of DNAJB4 in melanoma metastasis, we next examined how the migratory and invasive abilities of WM-115 and WM-266–4 cells are modulated by the expression level of DNAJB4. Our results showed that, after knocking down the expression of DNAJB4 using siRNA (Figure S5), the invasive ability of WM-115 cells increased significantly (Figures 4a and S6a). Reciprocally, ectopic overexpression of DNAJB4 led to diminished invasive ability of WM-266–4 cells (Figures 4a and S6b). However, no significant alterations in the migratory abilities were observed for WM-115 or WM-266–4 cells upon genetic manipulation of the expression level of DNAJB4 (Figures 4b and S6a,b). Likewise, we found that ectopic overexpression of DNAJB4 led to significant diminutions in invasive abilities of the two other metastatic melanoma lines (i.e., IGR-37 and 1205Lu cells, Figures 4a and S7), though no apparent alteration in migratory abilities was observed (Figures 4b and S7). In addition, siRNA-mediated knockdown of DNAJB4 in the two other lines of primary melanoma cells (i.e., IGR-39 and WM-793) elicited marked elevations in migration and invasion abilities (Figures 4a,b, S5, and S7). Cumulatively, the above results suggested that DNAJB4 suppresses melanoma metastasis in cultured cells.
Figure 4.

DNAJB4 modulates the invasive capacities of melanoma cells. (a, b) Quantification data showing the effects of expression levels of DNAJB4 on the invasive (a) and migratory (b) abilities of melanoma cells. The data represent the mean and standard deviation of results obtained from three parallel experiments. O.E. represents ectopic overexpression. (c) Representative images showing the effect of siRNA-mediated knockdown of DNAJB4 on the migration and invasion of WM-793 primary melanoma cells.
Suppression of Melanoma Cell Invasion by DNAJB4 via Regulation of Matrix Metalloproteinases (MMPs).
Having demonstrated the role of DNAJB4 in suppressing the invasive capabilities of melanoma cells, we next explored the mechanisms through which the invasive capacities of melanoma cells are regulated by DNAJB4. Matrix metalloproteinases (MMPs) are cancer-associated, secreted, zinc-dependent endopeptidases that assume important roles in degrading extracellular matrix (ECM) proteins and in promoting cancer metastasis.32 Moreover, metastatic cancer cells often display augmented levels of MMPs relative to primary tumor cells. Among the human MMPs, MMP-2 (gelatinase A) and MMP-9 (gelatinase B) are two major proteases responsible for remodeling the ECM environment and facilitating cancer metastasis.33
To examine the potential involvements of MMP-2 and MMP-9 in DNAJB4-mediated alterations in the invasive capabilities of melanoma cells, we assessed, by using a gelatin zymography assay, how the activities of secreted MMPs in melanoma cells are affected by the expression level of DNAJB4. Our results demonstrated that the activities of secreted MMP-2 and MMP-9 decreased (Figures 5 and S8) in the three lines of metastatic melanoma cells (WM-266–4, IGR-37, and 1205Lu) with ectopic overexpression of DNAJB4. On the other hand, siRNA-mediated knockdown of DNAJB4 led to marked elevations in the activities of MMP-2 and MMP-9 in the three lines of primary melanoma cells (WM-115, IGR-39, and WM-793. Figures 5 and S8), suggest that DNAJB4 inhibits the activity of secreted MMPs.
Figure 5.

DNAJB4 modulates the enzymatic activities of MMP-2 and MMP-9. (a) Representative gelatin zymography assay result showing the changes in activities of secreted MMP-2 and MMP-9 in WM-793 cells upon treatment with control, non-targeting siRNA, or siRNA targeting DNAJB4. (b) Quantification results showing the modulation of activities of secreted MMP-2 and MMP-9 in primary melanoma cells upon siRNA-induced knockdown of DNAJB4 (left) or in metastatic melanoma cells upon ectopic overexpression of DNAJB4 (right). The data represent the mean and standard deviation of results obtained from three parallel experiments and were normalized to the results obtained for primary cells treated with control non-targeting siRNA (left) or metastatic melanoma cells treated with control empty vector (right).
We also assessed, by employing qRT-PCR, how the mRNA expression levels of MMP2 and MMP9 genes in melanoma cells are regulated by the expression levels of DNAJB4. Our results showed that ectopic overexpression of DNAJB4 in the three lines of metastatic melanoma cells (WM-266–4, IGR-37, and 1205Lu) suppressed the expression of MMP2 and MMP9 genes (Figure S9). siRNA-mediated knockdown of DNAJB4 in the primary melanoma cells (WM-115, IGR-39, and WM-793), however, induced heightened mRNA expression levels of MMP2 and MMP9 (Figure S9). Together, DNAJB4 may suppress melanoma metastasis by inhibiting the transcription of genes encoding MMP-2 and MMP-9 and by diminishing the activities of secreted MMPs.
DISCUSSION
In this study, we developed, for the first time, a PRM-based targeted quantitative proteomic method for the comprehensive analysis of the heat shock proteome in cultured human cells. Our PRM library contained 57 heat shock proteins, which encompassed approximately 70% of the human heat shock proteome. We showed that the method exhibited higher throughput and superior sensitivity than the shotgun proteomic method.
We also applied this method to investigate the reprogramming of the heat shock proteome during melanoma metastasis by analyzing the differential expression of heat shock proteins in three matched pairs of primary/metastatic melanoma cell lines, WM-115/WM-266–4, IGR-39/IGR-37, and WM-793/1205Lu. HSPB1 (HSP27), a previously reported suppressor for melanoma metastasis,30 was found to be down-regulated in two metastatic melanoma cells based on PRM analysis. Moreover, DNAJB4, whose elevated expression confers better survival in melanoma patients, was found to be consistently down-regulated in all three lines of metastatic melanoma cells. We also demonstrated that DNAJB4 suppressed melanoma cell invasion by inhibiting the expression and activity of MMP-2 and MMP-9.
DNAJB4 was found to interact with AP-2α to repress the expression of AP-2α target genes in lung cancer cells,34 and MMP234 and MMP935 genes could be transcriptionally regulated by AP-2α. Our RT-qPCR result showed that the mRNA levels of MMP2 and MMP9 genes could be modulated by the expression levels of DNAJB4 in all three pairs of melanoma cells. Thus, the suppressed expression of the MMP2 and MMP9 genes by DNAJB4 could be attributed to the binding between DNAJB4 and AP-2α in melanoma cells.
Secretion of MMP-9 was shown to be regulated by SRC tyrosine kinase,36 which is known to promote the metastatic transformations of different types of cancers,37 including melanoma.38 On the grounds of our observation that diminished expression of DNAJB4 could elicit increased secretion of MMP-9 and the previous finding that DNAJB4 could act as an endogenous inhibitor for SRC,31 we reason that the increased secretion of MMP-9 in WM-793 cells may arise from the elevated activity of SRC induced by siRNA-mediated knockdown of DNAJB4.
Aside from DNAJB4, several other heat shock proteins were found to be differentially expressed in primary/metastatic melanoma cells. These proteins could potentially play critical roles in melanoma metastasis. For instance, DNAJC3, which is commonly up-regulated in the three lines of metastatic melanoma cells, is known to inhibit the phosphorylation of eIF2α.39 The phosphorylation of eIF2α has been demonstrated to be associated with cancer progression;40 therefore, DNAJC3 could also be a potential driver for melanoma metastasis. Melanoma is one of the most common cancers in the United States, and metastasis contributes to the mortality of the majority of melanoma patients.41 Our targeted proteomics method led to the discovery of novel potential promoters or suppressors of melanoma metastasis, which provided important new knowledge for understanding the etiology of melanoma progression.
Taken together, we developed, for the first time, a high-throughput and robust PRM-based targeted proteomic method for the quantitative analysis of the human heat shock proteome. It can be envisaged that the method can be generally applicable for assessing how cells respond to extracellular stimuli (e.g., upon exposure to environmental toxicants or heat shock protein inhibitors) by altering the expression of heat shock proteins.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA210072). The authors would like to thank Prof. Peter H. Duesberg for providing some of the cell lines used in this study.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b00986.
Detailed experimental conditions for Western blot, real time quantitative PCR, migration and invasion assay, and gelatin zymography assay; images and quantification data from Western blot, gelatin zymography assay, and migration/invasion assay; bar graphs showing the LCPRM data (PDF)
Table S2: Proteins and peptides included in the Skyline human heat shock protein library (XLSX)
Table S3: Expression levels of heat shock proteins in WM-115/WM-266–4, IGR-39/IGR-37, and WM-793/ 1205Lu pairs of melanoma cells obtained from PRM method (XLSX)
REFERENCES
- (1). Feder ME; Hofmann GE Annu. Rev. Physiol 1999, 61, 243–282. [DOI] [PubMed] [Google Scholar]
- (2). Wu J; Liu T; Rios Z; Mei Q; Lin X; Cao S Trends Pharmacol. Sci 2017, 38, 226–256. [DOI] [PubMed] [Google Scholar]
- (3). Trepel J; Mollapour M; Giaccone G; Neckers L Nat. Rev. Cancer 2010, 10, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4). Pick E; Kluger Y; Giltnane JM; Moeder C; Camp RL; Rimm DL; Kluger HM Cancer Res 2007, 67, 2932. [DOI] [PubMed] [Google Scholar]
- (5). Koga F; Kihara K; Neckers LEN Anticancer Res 2009, 29, 797–807. [PubMed] [Google Scholar]
- (6). Mbofung RM; McKenzie JA; Malu S; Zhang M; Peng W; Liu C; Kuiatse I; Tieu T; Williams L; Devi S; et al. Nat. Commun 2017, 8, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7). Sidera K; Patsavoudi E Recent Pat. Anti-Cancer Drug Discovery 2014, 9, 1–20. [PubMed] [Google Scholar]
- (8). Ramalingam S; Goss G; Rosell R; Schmid-Bindert G; Zaric B; Andric Z; Bondarenko I; Komov D; Ceric T; Khuri F; Samarzija M; Felip E; Ciuleanu T; Hirsh V; Wehler T; Spicer J; Salgia R; Shapiro G; Sheldon E; Teofilovici F; Vukovic V; Fennell D Ann. Oncol 2015, 26, 1741–1748. [DOI] [PubMed] [Google Scholar]
- (9). Hendriks LEL; Dingemans A-MC Expert Opin. Invest. Drugs 2017, 26, 541–550. [DOI] [PubMed] [Google Scholar]
- (10). Jhaveri K; Chandarlapaty S; Lake D; Gilewski T; Robson M; Goldfarb S; Drullinsky P; Sugarman S; Wasserheit-Leiblich C; Fasano J; Moynahan ME; D’Andrea G; Lim K; Reddington L; Haque S; Patil S; Bauman L; Vukovic V; El-Hariry I; Hudis C; Modi S Clin. Breast Cancer 2014, 14, 154–160. [DOI] [PubMed] [Google Scholar]
- (11). Skultety L; Hernychova L; Toman R; Hubalek M; Slaba K; Zechovska J; Stofanikova V; Lenco J; Stulik J; Macela A Ann. N. Y. Acad. Sci 2005, 1063, 115–122. [DOI] [PubMed] [Google Scholar]
- (12). Lange V; Picotti P; Domon B; Aebersold R Mol. Syst. Biol 2008, 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). Marx V Nat. Methods 2013, 10, 19–22. [DOI] [PubMed] [Google Scholar]
- (14). Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Mol. Cell. Proteomics 2012, 11, 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15). Bilan V; Selevsek N; Kistemaker HAV; Abplanalp J; Feurer R; Filippov DV; Hottiger MO Mol. Cell. Proteomics 2017, 16, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Zhou J; Liu H; Liu Y; Liu J; Zhao X; Yin Y Anal. Chem 2016, 88, 4478–4486. [DOI] [PubMed] [Google Scholar]
- (17). Bloomfield M; Duesberg P Mol. Cytogenet 2016, 9, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Westermark B; Johnsson A; Paulsson Y; Betsholtz C; Heldin CH; Herlyn M; Rodeck U; Koprowski H Proc. Natl. Acad. Sci. U. S. A 1986, 83, 7197–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Martin J-M; Luis J; Marvaldi J; Pichon J; Pic P Eur. J. Biochem 1989, 180, 435–439. [DOI] [PubMed] [Google Scholar]
- (20). Simon H-G; Risse B; Jost M; Oppenheimer S; Kari C; Rodeck U Cancer Res 1996, 56, 3112. [PubMed] [Google Scholar]
- (21). Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (22). Tsai M-F; Wang C-C; Chang G-C; Chen C-Y; Chen H-Y; Cheng C-L; Yang Y-P; Wu C-Y; Shih F-Y; Liu C-C; Lin H-P; Jou Y-S; Lin S-C; Lin C-W; Chen WJ; Chan W-K; Chen JJW; Yang P-CJ Natl. Cancer Inst 2006, 98, 825–838. [DOI] [PubMed] [Google Scholar]
- (23). MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24). Anaya J PeerJ. Comput. Sci 2016, 2, e67. [Google Scholar]
- (25). Kampinga HH; Hageman J; Vos MJ; Kubota H; Tanguay RM; Bruford EA; Cheetham ME; Chen B; Hightower LE Cell Stress Chaperones 2009, 14, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Proteomics 2012, 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27). Miao W; Xiao Y; Guo L; Jiang X; Huang M; Wang Y Anal. Chem 2016, 88, 9773–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28). de Graaf EL; Altelaar AF; van Breukelen B; Mohammed S; Heck AJ J. Proteome Res 2011, 10, 4334–4341. [DOI] [PubMed] [Google Scholar]
- (29). Marchetti N; Fairchild JN; Guiochon G Anal. Chem 2008, 80, 2756–2767. [DOI] [PubMed] [Google Scholar]
- (30). Aldrian S; Trautinger F; Fröhlich I; Berger W; Micksche M; Kindas-Mügge I Cell Stress Chaperones 2002, 7, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Chen CH; Chang WH; Su KY; Ku WH; Chang GC; Hong QS; Hsiao YJ; Chen HC; Chen HY; Wu R; Yang PC; Chen JJ; Yu SL Oncogene 2016, 35, 5674–5685. [DOI] [PubMed] [Google Scholar]
- (32). Bauvois B Biochim. Biophys. Acta 2012, 1825, 29–36. [DOI] [PubMed] [Google Scholar]
- (33). Roomi MW; Monterrey JC; Kalinovsky T; Rath M; Niedzwiecki A Oncol. Rep 2009, 21, 1323–1333. [DOI] [PubMed] [Google Scholar]
- (34). Chang T-P; Yu S-L; Lin S-Y; Hsiao Y-J; Chang G-C; Yang P-C; Chen JJ W. Cancer Res 2010, 70, 1656–1667. [DOI] [PubMed] [Google Scholar]
- (35). Schwartz B; Melnikova VO; Tellez C; Mourad-Zeidan A; Blehm K; Zhao YJ; McCarty M; Adam L; Bar-Eli M Oncogene 2007, 26, 4049–4058. [DOI] [PubMed] [Google Scholar]
- (36). Cortes-Reynosa P; Robledo T; Macias-Silva M; Wu SV; Salazar EP Matrix Biol 2008, 27, 220–231. [DOI] [PubMed] [Google Scholar]
- (37). Homsi J; Cubitt C; Daud A Expert Opin. Ther. Targets 2007, 11, 91–100. [DOI] [PubMed] [Google Scholar]
- (38). Hanna SC; Krishnan B; Bailey ST; Moschos SJ; Kuan PF; Shimamura T; Osborne LD; Siegel MB; Duncan LM;O’Brien ET 3rd; Superfine R; Miller CR; Simon MC; Wong KK; Kim WY J. Clin. Invest 2013, 123, 2078–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39). Gao D; Bambang IF; Putti TC; Lee YK; Richardson DR; Zhang D Lab. Invest 2012, 92, 200. [DOI] [PubMed] [Google Scholar]
- (40). Podszywalow-Bartnicka P; Cmoch A; Wolczyk M; Bugajski L; Tkaczyk M; Dadlez M; Nieborowska-Skorska M; Koromilas AE; Skorski T; Piwocka K Oncotarget 2016, 7, 79706–79721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). Zbytek B; Carlson JA; Granese J; Ross J; Mihm MC Jr.; Slominski A Expert Rev. Dermatol 2008, 3, 569–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.