

ABSTRACT
In cancer immunotherapy, cytotoxic T or NK cells need to engage cancer cells to initiate the killing. However, in clinical studies and in mouse models, some solid tumors are found with no lymphocytes. It is likely that these tumors will be resistant to all sorts of immunotherapies. Thus, restoring lymphocytic infiltration will be vital to the success of immunotherapies on solid tumors. In order to understand the complex interaction between cancer cells and stromal cells, we propose to establish animal models for studying the tumor microenvironment and to develop and test therapies to restore lymphocytic infiltration of tumors Without lymphocytes infiltrating tumors, all immunotherapies on solid tumors become ineffective.
KEYWORDS: CAR T, cancer biology, chemokine, immunotherapy, interferon gamma, Jak kinases, keratin, melanoma, tumor infiltrating lymphocytes, tumor immunology, tumor microenvironment
Introduction
The percentage of cancer cells within a tumor may vary anywhere from 5% to almost 100%.1 The rest of the cells in a tumor are various immune cells such as lymphocytes, CAFs (cancer associated fibroblasts), TAMs (tumor associated macrophages), MDSCs (myeloid-derived suppressor cells), and endothelium cells of the vasculature and others. As a result of direct interactions between cancer cells and immune cells, pro-inflammatory as well as regulatory cytokines are released, stimulating not only each other, but also affecting other cells such as fibroblasts and macrophages, turning them into CAFs and TAMs. They in turn secrete growth factors and extra-cellular matrices2,3 and as such provide cancer cells with their own milieu, called the tumor microenvironment (TME). TAM-cancer cell interactions take many shapes, from inflammatory, immunomodulatory, angiogenic, to metastatic, depending on the developmental stages of the tumor.4 CAFs, on the other hand, can secrete extracellular matrices (ECMs) that form scaffolds inside the tumor, but under different circumstances, they can activate metalloproteases, cleaving ECMs, and allowing cancer cells to escape the tumor.5 Thus, the TME is determined not only by their association with cancer cells, but also by the dynamical interaction between cancer cells and stromal cells, which includes infiltrating lymphocytes. On the other hand, what gets recruited to the tumor is determined by the TME as well.6 Once lymphocytes enter the tumor, the TME exerts influences on their effectiveness in many ways.7 Upon studying articles on the TME, we have the impression that they do not report the whole picture. First, not all TMEs are alike. Just as there are considerable intra- and inter-tumor heterogeneities of cancer cells, there is a wide spectrum of TMEs. Second, because the formation of a TME is a dynamic process, one can expect the TME of a particular tumor to become altered as it experiences chemotherapies and other changes. Third, to better understand the characteristics of a TME, there needs to be a classification scheme. (Until now, there have been only rudimentary classifications based on (1) the absence or presence of lymphocytes, (2) the microvessel density,8 or (3) those comparing infiltrated T cells, infiltrated B cells, epithelial markers or desmoplasia in stromal components.9) Fourth, there have been few reports of experiments performed to elucidate TMEs, largely because there are so few good mouse models or mouse cancer cell lines.
Because there are many excellent review articles on how cytolytic activities of lymphocytes already in the tumor are moderated by the TME,7,10,11 in this article, we focus on TMEs with very few lymphocytes, how they might have been formed, and how lymphocyte infiltration may be restored. There are various reasons for lymphocytes being blocked from entering a tumor. As examples, when FasL is expressed in the tumor vasculature of some tumor types,12,13 it induces apoptosis in incoming activated T cells in a process called activation induced cell death (AICD). FasL at the tumor vasculature preferentially eliminates CD8+T cells over regulatory T cells (Treg) largely because higher levels of c-FLIP expressed in Treg cells hinder FasL-induced apoptosis. Other tumors express the endothelin B receptor (ETBR) that hinders T cell infiltration into the tumor.14 Yet in other tumors, chemokines required for lymphocyte infiltration are nitrated, making them ineffective.15 These three examples have the same consequence–interfering with lymphocytic infiltration of the tumor. For each case, depending on the kind of immunotherapy, a strategy to restore tumor infiltration of lymphocytes can be devised.7 It must be noted, however, that in the above cases, tumors are expressing chemokines and lymphocytes are recruited to the nearby vasculature, but they fail to enter the tumor. In addition, recent evidence suggests the existence of tumors without the cytokines and chemokines necessary for lymphocyte recruitment to sites on the vasculature adjacent to the tumor. Thus, to restore lymphocytic infiltration into a tumor, it is important to distinguish those tumors that cannot even recruit lymphocytes to the nearby vasculature from those tumors that block entry into the tumor. And for this task, ideas of how TMEs are formed and how they could be modified are needed. In an effort to understand this enormously complex issue, we begin by examining components of multiple TMEs in metastasized melanoma biopsies.
Lymphocytes infiltrating tumors
By analyzing the data presented by Harlin and colleagues at the University of Chicago16 for the presence of various stromal cells, we could draw the architecture of each tumor, and found that some tumors without infiltrating lymphocytes fall into distinct classes. This group took 44 biopsy samples from metastatic melanoma patients and bulk analyzed their RNA contents using Affimatrix GeneChips.16 For indications that a particular gene expression may be derived from cancer cells, they included in the analysis five melanoma cell lines and three primary melanocytes. The data were put through an unsupervised hierarchical clustering analysis, and genes were aligned along the vertical axis according to the gene cluster analysis, and melanoma samples were aligned along the horizontal axis according to the binary tree generated by the program. From these data (Harlin et al., Cancer Research 2009, Suppl Info 1), we constructed a heatmap (Fig. 1). Clusters of red squares in the lower left corner represent highly expressed genes in the first set of melanoma samples. They are mostly immune cell specific transcripts, suggesting that lymphocytes were infiltrating these tumors. They found that the eighteen Group 1 tumors contained most of the lymphocyte specific transcripts. The rest of the samples were subdivided into two more groups, Group 2 and Group 3 (Fig. 1).
Figure 1.
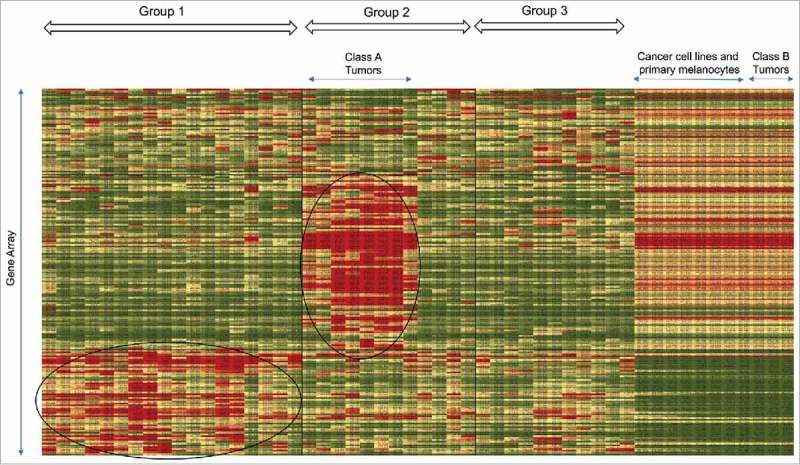
Heatmap generated from unsupervised hierarchical clustering analysis. 44 biopsy samples, 5 cell lines and 3 primary cells were aligned along the horizontal axis while genes were aligned along the vertical axis. The heatmap program generated groups 1–3 and we designated two classes of tumors, Class A and Class B.
Group 1 tumors have distinctive attributes. The presence of T cells in Group 1 tumors were confirmed by the presence of TCRα, TCRβ and TCRγ transcripts, and by the presence of transcripts for the T-cell specific adapter proteins Slp76 and Fyb. The presence of immunoglobulin gene transcripts confirmed that B cells also infiltrated Group 1 tumors. Harlin and colleagues showed that lymphocytes were found only in Group 1 melanoma tumors, suggesting that lymphocytes did not infiltrate the rest of the melanoma tumors. Because Group 1 tumor samples included melanoma biopsies from brain, lung, skin and other tissues, the existence of lymphocytes in a tumor seems to be independent of the tumor's location or organ type. On the other hand, macrophages were found in all the tumor samples to varying degrees, but not in the melanoma cancer cell lines and melanocytes. This was because macrophages are found in almost all tissues having been there since the tissues were formed. Monocytes may also be recruited to some tissues by chemokines, and become macrophages. Thus, it is not surprising that macrophages were found in tumors without infiltrating lymphocytes. However, it is difficult to distinguish between macrophages from different origins.17 Harlin and colleagues also showed that the presence of the chemokines CXCL9,10,11 & 13, and CCL 3,4,5,8 & 19 correlated with the presence of lymphocytes in tumor samples. Using an in vitro trans-endothelial cell migration assay and other assays, they showed that chemokines were necessary for lymphocytes to exit the vasculature and enter tumors.16
Tumors without infiltrating lymphocytes
Our main interest is in examining components of the TME in various tumors. Thus, we examine in detail highly expressed transcripts found in the middle of the heatmap (red squares in the vertically stretched oblong circle in the middle of Group 2 tumors). Most of these transcripts are keratinocyte-associated transcripts such as keratins 2A, 10, 14 and 17,18,19 gap junctions,20 aquaporins,21 and desmoplakin.22 These transcripts were also found in cancer cell lines and primary melanocytes (Fig. 2a). Proteins translated from these transcripts play important roles in cell survival in skin tissues.23,24 Furthermore, there are some transcripts such as desmocollins and plakophilins not found in cancer cell lines and primary melanocytes that are important elements in skin tissues for keeping cell-cell contacts such as adherence junctions.25 EGFR and FGFR2 transcripts are also found in these tumors, but not in cancer cell lines and primary melanocytes. Thus, these seem to be tumors in which small cancer cells are tightly packed, but still proliferating with growth factors that might be secreted from CAFs.26
Figure 2.
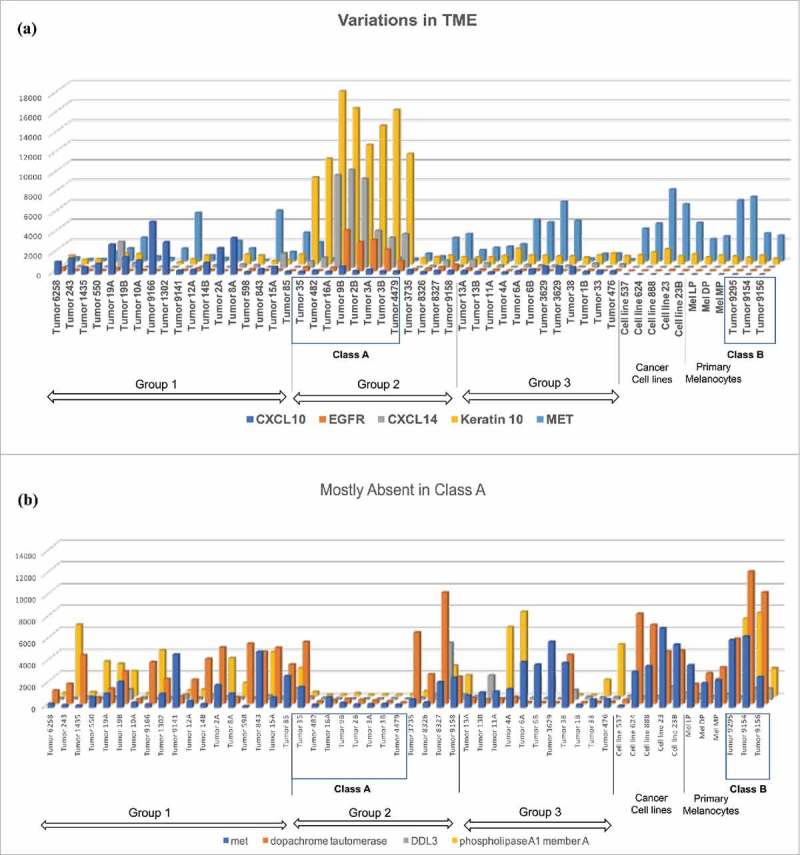
Variation in TME. (a) Most chemokine transcripts were found in Group 1 tumors, except for CXCL14, which was found in Class A tumors. Growth factor receptors such as EGFR and FGFR2 were found in Class A tumors. Keratins 2a,10,14, and 17 were found in Class A tumors. The lower level keratin transcripts were found in cell lines, primary melanocytes and Class B tumors. Proto-oncogene MET transcripts were found in many samples across the groups and classes. (b) Dopachrome tautomerase, known to inhibit macrophage chemotactic response, Drosophila Delta-Like 3 (DDL3), phospholipase A1 Membrane A (PLA1A), and MET were excluded from Class A tumors.
Class A tumors have unique gene expression. Eight tumor samples differ from the rest of the melanoma samples by the lack of expression of genes elevated in many melanoma samples, such as phospholipase A1 member A, DDL3 (delta-like 3), and proto-oncogene MET (Fig. 2b). Furthermore, expression of dopachrome tautomerase, a ubiquitously expressed protein that binds to CD74 and regulates macrophage chemotactic responses,27 is also absent in these eight tumors. In short, most of the transcripts found in these tumors were not expressed in other tumors (Fig. 2a), and many of the transcripts found in other tumors were not observed in them (Fig. 2b). We designated these eight tumors, Class A tumors.
It is interesting to note that CXCL14 is the only chemokine with significant presence in Class A tumors (Fig. 2a). There are considerable controversies on the roles of CXCL14.28 It is suspected of playing a pro-tumorigenic role by attracting monocytes into tumors and turning them into tumor-associated macrophages. On the other hand, it is also suggested that CXCL14 may bind to CXCR4 and inhibit its activation.29 If this were the case, because CXCR4 activity is necessary to bring MDSCs to the tumor, a Class A tumor may lack the presence of MDSCs. Because gene expression of tumor cells was performed in bulk instead of analyzing each cell individually, and because there is no single marker for MDSCs, it is difficult to determine the presence of MDSC by these assays, necessitating further studies to clarify the role of CXCL14 in these tumors. To identify CAFs and TAMs also requires multiple markers, and bulk analysis of transcripts from tumors is not suitable. Even with these limitations, Class A tumors might be characterized as tumors filled with small tightly packed cancerous melanocytes, possibly within stratified keratinocytes, and possibly with some CAFs. They contain very few lymphocytes, painting a picture of a “classic” tumor that rejects lymphocyte infiltration.
Class B tumors appear to have a high percentage of cancer cells. The heatmap derived from Harlin's data also showed the existence of three tumors with remarkably similar profiles with cancer cell lines and melanocytes (Fig. 1), suggesting a lack of cellular complexity. Thus, these three tumor biopsies, except for a few macrophages, likely consist mostly of cancer cells. In this regard, Malladi and colleagues at Memorial Sloan Kettering Cancer Center isolated what they call latency competent cells (LCCs) from early stage human, lung and breast cancer cell lines, by injecting human early stage cancer cells into immune-compromised mice that still have NK cells, and isolating those cells that engrafted.30 LCCs show stem-like characteristics and express DKK1, an inhibitor of the WNT/β-catenin pathway, imposing a slow-cycling state with downregulation of ligands for NK cells, thus evading detection by NK cells. Mouse NK cells react with MHC-incompatible cells, virus infected cells, rapidly proliferating cells, cells with activated DNA damage response pathways, and others, much like human NK cells would.31 Thus it seems reasonable to assume that LCCs can escape detection by innate immunity in the human body, and in the absence of an inflammatory reaction caused by immune cells, they may form tumors consisting mostly of cancer cells and very few stromal cells.32 This model explains the existence of tumors with very few stromal cells. We speculate that the three melanoma tumors in Fig. 1 may have been formed by LCCs. We designated these three tumors, Class B tumors.
Spranger and colleagues examined 197 melanoma patients divided into two groups, one with infiltrating T cells (105 patients) and one without (92 patients). They found a CTNNB1, gain-of-function β-catenin mutation in eight patients, and seven of them had tumors with no T-cell infiltration.33 They suggested that β-catenin induced ATF2 expression, which in turn inhibited CCL4 chemokine expression, a chemokine that facilitates T cell infiltration. Because β-catenin expression is thought to be necessary for the maintenance of stem-like characteristics in cutaneous cancer,34 it will be interesting to test whether Class B tumors express CTNNB1 and still block CCL4 expression, making them resistant to T cell infiltration. Taking into account mutations in β-catenin targeted genes, they argue that increased β-catenin signaling pathways could account for some of the tumors without T cell infiltration, but not nearly all such tumors.
TME diversity
At the cellular level, we can now draw reasonably good pictures of some of the 44 melanoma tumors. (1) Group 1 tumors are infiltrated by lymphocytes. (2) Class A tumors are filled with small tightly packed melanocytes, staining positive with keratin antibodies, and there are very few lymphocytes. (3) Class B tumors are filled mostly with cancer cells and seem to have a limited number of stromal cells. We are not sure what the others look like, but they may look like Group 1 tumors except that they do not have lymphocytes. Thus, we have a very rudimentary phylogenic tree for melanoma tumors. With more data, perhaps we could build a more complex chart much like a Linnaean taxonomy chart which can be interpreted as an evolutionary tree.
We speculate that all the newly metastasized colonies are Class B tumors–small slowly growing stem cell like cancer cells that can evade innate and adaptive immunity. But they do not always stay this way forever. One possible change may come when a solid tumor reaches a certain size and its inner core becomes hypoxic, activating many hypoxia-related programs such as the TGFβ-VEGF based angiogenesis program,35 and interleukin-based infiltration and activation of NK cells into its hypoxic core,36-38 thus initiating many changes to come for Class B tumors, including rapid growth of cancer cells in a well-oxygenated environment. Rapidly growing cancer cells often express NKG2D ligands, attracting and activating NK cells.39,40 Rapid growth of cancer cells inevitably causes a certain number of cells to die. (Even colonies of HeLa and other cancer cells growing in tissue culture experience the death of a small fraction of cells.) Dead or dying cells activate macrophages and dendritic cells, further activating the adaptive arm of the immune system, and release inflammatory cytokines, attracting lymphocytes into the tumor.
There are other possible mechanisms involved in establishing a new cancer colony in a different tissue environment. For example, the fusion of a cancer cell with a nearby healthy cell was observed many times in experimental mouse models.41,42 MDA-MB435, a cancer cell line derived from breast cancer, expresses a number of melanocyte proteins43; it seems possible that a metastasized breast cancer cell had fused with a melanocyte, and thus express melanocyte proteins. These changes could turn Class B tumors into Class A tumors.
Thus, establishing a new colony in a different tissue may involve multiple pathological processes. Faced with this enormously complex problem of deciphering how different TMEs are generated, and how they become resistant to lymphocytic infiltration, we are initiating a project to build an animal model system to study the TME and its development, and TME resistance to lymphocytic infiltration. Before we outline our proposal, however, we briefly describe known mechanisms of lymphocytic infiltration of tumors.
Mechanisms of tumor infiltrating lymphocytes
The well-established model of lymphocyte extravasation is as follows: the flow of lymphocytes slows in vasculatures expressing selectins, then they are halted by integrins such as ICAM-1 and VCAM-1,44 and trans-endothelial migration is triggered by appropriate chemokines.45,46 Pro-inflammatory cytokines and regulatory cytokines trigger necessary components in these pathways.47 For example, expression of all the necessary components for recruiting activated CD8+ T is induced by interferon γ (IFNγ),48 which is released by the innate arm of immunity, reacting to pathogens or in host-vs-graft reactions. The development of immune cells in IFNγ −/− mice is unchanged, and tumor-grafted mice produce CD8+ T cells that can induce cancer-cell killing ex vivo, but in the mice, tumors are not infiltrated by CD8+ T cells and cancer cells remain unaffected.49,50 Thus, IFNγ plays a central role in recruiting CD8+ T cells into tumors. Upon binding to IFNγ, the IFNγ receptor activates JAK kinases and JAK2 expression is associated with tumor-infiltrating lymphocytes.51 Recent findings showed that JAK kinase inhibitors blocked unwanted lymphocyte infiltration in various tissues such as Islets of Langerhans, rheumatoid joints, and bald spots in alopecia, ameliorating the symptoms of corresponding immunological disorders such as insulitis in experimental model mice,52 and rheumatoid arthritis (RA)53 and alopecia54 in patients. In short, INFγ-induced lymphocyte infiltration of tissues is dependent on the IFNγR-JAK2 pathway.
Animal models for studying the TME
Grafting human cancer cells onto immuno-compromised mice have been standard ways to test the growth and metastastic potential of cancer cells. However, in the absence of immune cells, these tumors would not develop proper TMEs in these animals. Thus, grafting cancer cells onto immuno-deficient mice is obviously not the right model to study the TME. Alternatively, tumors in genetically engineered mouse models for a particular cancer type arising from specific mutations (such as PKC mice for studying pancreatic cancer), or tumors generated from mouse cancer cell lines grafted onto wild-type mice could be used. However, both models are unsatisfactory because of the uncertainty as to whether the results could be extended beyond a particular cancer type with a particular set of mutations. A way to test a particular hypothesis on a particular cancer type with a given set of mutations is not readily available. Nevertheless, in this section, first we briefly review the results from those two models. Then, based on recent advances in our understanding of the roles the two separate arms of the immune system (innate and adaptive) play, and the availability of agents that can separate them, we discuss possibilities for making improvements in the existing animal models for studying the TME.
Genetically altered mouse models for studying the TME
One way to study TMEs using animals is to examine tumors of genetically altered model mice that spontaneously generate tumors, such as pancreatic ductal adenocarcinomas (PDAC) formed in transgenic mice expressing KrasG12D and Trp53R172H in the pancreas (KPC mice), or breast cancer formed in mouse mammary tumor virus-driven polyomavirus middle T antigen (MMTV-PyMT) transgenic mice, or prostate cancer formed in prostate-specific PTEN gene-deletion model mice. These tumors in model animals recapitulated various elements of tumor microenvironments found in the clinical settings. For example, TAMs are consistently found in tumors of MMTV-PyMT mice55,56; and CAFs,57 and MDSCs58-60 are consistently found in tumors of PDAC model mice. Furthermore, there are very little CD8+ T cells in the PDACs in PKC mice and breast cancer in MMTV-PyMT mice. Using in vivo and in vitro experiments, the reason for the absence of CD8+ T cells was deciphered; KrasG12D induces granulocyte-macrophage colony-stimulating factor (GM-CSF) production in cancer cells,59 recruiting Gr-1+ CD11b+ myeloid cells into the tumor and developing them into MDSCs.58-60 Consequently CD8+ T cells were excluded from the tumor. CAF released CXCL12 can also inactivate cancer cells57 in recruiting CD8+ T cells to PDAC. Thus, both in pancreatic and breast cancer model mice, GM-CSF secreted from cancer cells recruited myeloid-derived cells and turned them into MDSCs that block lymphocytic infiltration. Whether this is also true in another cancer type with another set of mutations needs to be verified for each cancer type. This constant need for verification is an inevitable consequence of using genetically altered mice to study specific cancer types with specific gene mutations.
There are hundreds of cancer types and thousands of cancer-causing mutations, and we only have a genetically engineered mouse model for very few of them. However, there are over two hundred mouse cancer cell lines and over four thousand human cancer cell lines listed in the ATTC catalogue. Grafting mouse cancer cell lines onto wild-type mice may be a better way to study TMEs. This may prove to be a fruitful endeavor for discovery because only a few mouse cell lines have been grafted onto syngeneic mice.
Grafting mouse cancer cells for studying the TME
Grafting mouse cancer cells onto mice with similar genetic background, can also generate tumors. For example, B16F10 mouse melanoma grafts generated tumors with monocyte-derived MDSCs. In this case, the numeric and functional development of MDSCs was also dependent on GM-CSF,61 and the presence of MDSCs in the tumor blocked T cell infiltration, similar to what we have seen in the pancreatic and breast cancer model mice.
Grafting B16F10 melanoma onto CD5-deficient mice cause tumors to grow much more slowly compared to the same cancer cells grafted onto wild-type mice.62 Even though CD5 is a negative regulator of TCR signaling in thymocytes and mature T lymphocytes,63 CD5- deficient mice are healthy and their hematopoietic development seems largely intact.64 Initially, B16F10 melanoma-derived tumors in CD5-deficient mice were infiltrated with all sorts of T cells, and they exhibited a more activated phenotype (up to 2 weeks post-injection), compared to the tumors injected in the wild-type mice. However, in the subsequent weeks, there was an increase in activation induced cell death (AICD) of CD8+ T cells by FasL released from cancer cells, and tumors grew larger at the later stage (week 2–3 of post-injection), and had fewer infiltrating T lymphocytes.
Lastly, mouse fibrosarcoma cancer cells (CSA1M) isolated from Rous sarcoma virus infected mice, can be grafted subcutaneously onto wild-type mice. However, unlike the above three examples, the tumors generated from the grafts were infiltrated with lymphocytes, at least initially. However, 8–10 weeks post-infection, the levels of IFNγ declined precipitously, as did the percentages for CD4+ and CD8+ T infiltrating lymphocytes, while marked increase in IL-10 and IL-4 were observed, and Mac-1+ Gr-1+ myeloid suppressor cells (MSCs) accumulated in the late stage tumors.65 In short, the rate of decline in tumor infiltrating CD8+ T cells was slower in CSA1M-derived fibrasarcoma compared to B16F10-derived melenoma, and associated molecules involved were similar but not exactly the same. Thus, both with genetically engineered mouse models and murine syngeneic tumor models, tumors infiltrated with lymphocytes recruited myeloid cells and turned them into MDSCs so that eventually they became devoid of lymphocytes, in similar manners. Further, the absence of CD5 prolonged the duration of lymphocyte infiltrated TMEs.
Injecting the same cancer cells onto mice with different genetic background can also yield very different results. For example, when CSA1M cells were grafted onto IFNγ−/− mice, there was no lymphocytic infiltration at the begining.49 We note that hematopoietic development in IFNγ−/− mice was largely intact,66 and CSA1M-reacting CD8+ cells were generated ex vivo. When these CSA1M-reacting CD8+ cells were injected into CSA1M bearing mice, it seemed that they could not infiltrate the tumor at the earliest stage.49 In fact, lymphocytes do not infiltrate CSA1M tumor in IFNγ−/− mice because inflammatory signals are not transduced, and CD8+ T cells are not even recruited to the vasculature near the tumor, the same reason that JAK kinase inhibitors blocked lymphocytic infiltration to the Islets of Langerhans in type I diabetes mice, and to the rheumatoid joints in RA patients. Had the researchers searched for MDSCs in CSA1M tumors generated in IFNγ−/− mice, we speculate that they would not have found one. In other words, CSA1M tumors in IFNγ−/− mice did not become resistant to lymphocytic infiltration, they are so from the beginning.
Based on the current available data, we present a model for solid tumor development. Tumors transition from a lymphocyte null tumor (such as Class B tumors in melanoma biopsies) – Category I (Cat I), to a lymphocyte-infiltrated tumor – Category II (Cat II), and then to a MDSC containing tumor devoid of lymphocytes – Category III (Cat III). To progress from Cat I to Cat II, inflammatory IFNγ signaling is required. To transition from Cat II to Cat III, cytokines that recruit myeloid cells (either Gr-1+ CD11b+ cells or Mac-1+ Gr-1+ cells) into the tumor, and turn them into MDSCs, such as GM-CSF is needed. The absence of CD5 could fill tumors with all sorts of activated CD8+ T cells, and prolong development of tumors at Cat II, but not halt it. The clinically important issue is to find ways to revert Cat III tumors to Cat II tumors.
Disrupting SEMA4D
Semaphorin 4D (SEMA4D) is a ubiquitously expressed protein that plays a role in axon-guidance,67 osteoclasis68 and modulation of the TME.69 Recently Evans and colleagues reported that SEMA4D was at the invasive edges of growing tumors, and disrupting SEMA4D with an antibody treatment caused increased infiltration of lymphocytes into the tumor, causing in some cases, complete tumor regression.70 These experiments were performed by grafting the mouse cancer cell lines Colon26 and Tubo.A5 onto wild-type mice. The authors believed that dendritic cells and TAMs were the source of SEMA4D at the invasive edge,69 and because SEMA4D dimers hinder cell migration in in vitro experiments,71 they argue that SEMA4D proteins hinder lymphocyte infiltration in these animals. Because SEMA4D deficient mice seem to have a largely intact immune system72 with only minor defects in organs due to cell migration defects,73 had they grafted the same mouse cells lines to SEMA4D deficient mice, it is reasonable to expect that either a tumor would not have formed, or at least, tumors would have been infiltrated with lymphocytes. Unfortunately, these experiments were not performed. Thus, much like FasL, SEMA4D released from within the tumor creates a barrier for lymphocyte infiltration. Our one concern is that unlike the well documented existence of FasL at tumor vasculature in clinical samples,13 SEMA4D at the tumor border regions had not been previously reported. Instead, SEMA4D staining seems to be distributed throughout the tumor (see for example, Human Protein Atlas Immunostaining data). Thus, we are not certain how common this phenomenon is.
It is known that some cancer cells exist in defined protective environments. For example, Yan-Gao Man reported that some usually benign breast cancer cells were encapsulated in myoepithelium-derived smooth muscle actin (SMA) coated capsules.74 A typical capsule contained several thousand cancer cells. SMA-coated capsules could potentially expel cytokines and chemokines secreted from cancer cells, trapping them inside, thus silencing signals from within the tumor. On the other hand, because most lymphocytes express metalloproteases, and SMA fibers are known to be re-organized by metalloproteases,75 a SMA coated capsule may not be a definite hindrance to lymphocytic infiltration once lymphocytes found their way to the capsule. There is no clinical data showing lymphocytes inside SMA-coated tumor capsules. However, Yan-Gao Man suggested that these tumors were usually not inflammatory until tumor cell budding starts from the capsule. This budding was often observed just before tumor invasion.74 Structures such as SMA-coated capsules or desmoplasia around tumors must at the least affect the rate of lymphocytic infiltration.9 Thus, SMA-coated tumor capsules may represent a very early stage of tumor development. Just when cancer cells escape their original site, they are surrounded by epithelial cells and form a capsule and find a niche in a new tissue. We note that these SMA-coated tumor capsules have not been observed in genetically engineered mouse models, nor in syngeneic cancer cell transfer models. However, by bulk analyzing RNA from SMA-coated clinical tumor capsules, assembling their components in ex vivo experiments and injecting them into mouse bodies, perhaps SMA-coated tumor capsules in mice could be recreated, which may inform how tumor cell budding from the SMA-coated capsule is initiated.
In order to find the extent of TME varieties, more bulk analysis of RNA from clinical tumor samples could show detailed components of each TME, and the in-depth analysis could reveal new classes of TMEs and new charts of various TMEs. Based on the new charts, models of how a tumor could develop would be built. However, in order to test the developmental models of tumors, better animal models for studying TMEs are needed. Briefly described below is our proposal for establishing new animal models for studying TMEs.
Grafting human cancer cells onto genetically altered mice for studying TME
First, it is possible to create tumors without infiltrating lymphocytes in all the above mouse models of cancer, whether they are genetically engineered mice or mouse cancer cells grafted onto syngeneic mice, by pre-treating mice with IFNγ neutralizing agents. When these agents are withdrawn, it is expected that lymphocytes will infiltrate tumors. In the presence of GM-CSF, MDSCs would accumulate, eventually blocking lymphocyte entry, and return the tumor to a state devoid of lymphocytes, allowing time to observe serial changes in the TME by bulk analyzing RNA using gene chips.
In 2008, Quintana and colleagues injected a randomly selected single primary human melanoma cell into Foxn1nu /IL2Rγ−/− mice and found the xenograft efficiency was better than 25%.76 These mice had severely reduced T/B cells and no NK cells. The graft efficiency was a thousand to a million times more efficient than injecting human cancer cells into Foxn1nu (nude) mice. Even though Foxn1nu mice had severely reduced T/B cells, the number of NK cells were comparable to the number in the wild-type mice, thus illustrating the importance of NK cells and innate immunity in eliminating foreign cancer cells from mice. Further, in 2016, by injecting human cancer cells into Foxn1nu mice, and isolating cells that survived, Malladi and colleagues found LCCs.32 These LCCs escaped detection by NK cells. Thus, it seems possible that LCCs can survive in IFNγ −/− mice and wild-type mice treated with IFNγ −/− neutralizing agents.
Because LCCs or ex vivo assembled SMA-coated tumor capsules solicit almost no reaction from NK cells, it seems possible that they could be grafted onto wild-type mice treated with IFNγ neutralizing agents. For LCCs, adding DKK inhibitors could make them grow, and by withdrawing IFNγ neutralizing agents, perhaps the tumor developmental processes could be recreated in mice. If this were the case, the study of TMEs into almost all human cancer could be expanded. Once a mouse system to study human cancers' TME development has been established, it would be exciting to test how chemotherapeutics and radiation treatment could alter the TMEs, and find a way to restore lymphocytic infiltration. Lastly, because acquired mutations in the JAK2 gene are associated with cancer that becomes resistant to immune checkpoint therapies,77 JAK2 mutant cancer cells could be tested for the formation of tumors without infiltrating lymphocytes in this system as well.
Admittedly, findings from mouse models or human-mouse hybrid models are not always translatable for understanding the TME in humans. However, using bulk RNA analysis to determine tumor contents for comparing TMEs in mouse models and the clinical data, adjustments may be made to the mouse models so that they become reliably human-like.
Perspectives on the immunotherapy on solid tumors
Once lymphocytes find their way into a tumor, they will face many more hurdles before they can lyse cancer cells, such as navigating through immune checkpoints and regulatory T cells. Thus, successful infiltration of immune cells into tumors should not be the only benchmark for effective immunotherapy on solid tumors. Among all immunotherapies being developed, perhaps the most versatile is the chimeric antigen receptor therapy (CAR T). CAR T therapies can easily meet challenges such as AICD by having CAR T cells over-express c-FLIP, and the checkpoint activation by adding PD-1 antibodies. The most difficult challenge, however, is for immune cells to react only with cancer cells. For example, in one chimeric antigen receptor therapy (CAR T), anti-MAGE-3 antibody fused with TCR was intended to target melanoma and esophageal cancer, found its way into the brain of patients where previously unrecognized expression of MAGE-A12 was located. Their brain cells reacted with CAR T causing Parkinson-like syndrome in one patient that persisted for four weeks, and induced comas and death in two other patients.78 Sadly, adverse effects from unintended targeted cancer destruction often cause the death of patients.79 This is because CAR T kills any cell recognized by its chimeric receptor, not just those cells overexpressing antigens, or those cells vulnerable to the inhibition of antigen-associated activities. On the other hand, the CAR T targeting of B cells with anti-CD19 or CD20 antibodies has been hugely successful.80,81 However, we must note that in these therapies, the whole population of B cells are targeted, cancerous and otherwise, and the success of these therapies are based on the surprising fact that this elimination seemed to have only minor adverse effects.82 For targeting solid tumors, a more sophisticated approach of using multiple antibodies fused with TCR and its regulators is being considered. For example, adding a cancer-specific antibody fused with TCR to a healthy-specific antibody fused with its regulator would add a safety measure that could stop the killing of unintended healthy cells.83 These ideas are still in development. Thus, even when ways are found to restore lymphocyte infiltration into a tumor, there are many more problems to solve before immunotherapies will successfully treat solid tumors. However, the first step in developing a successful immune therapy appears to be the discovery of a way to restore lymphocyte infiltration of tumors.
Acknowledgments
We thank Jon Massague of Memorial Sloan Kettering Cancer Center for answering our questions on LCC. The cost of publication was paid by JRC Medical Research Company in Osaka, Japan.
References
- 1.Ambrose J, Livitz M, Wessels D, Kuhl S, Lusche DF, Scherer A, Voss E, Soll DR. Mediated coalescence: a possible mechanism for tumor cellular heterogeneity. Am J Cancer. 2015;5(11):3485–504. [PMC free article] [PubMed] [Google Scholar]
- 2.Cassetta L, Noy R, Swierczak A, Sugano G, Smith H. Isolation of mouse and human tumor-associated macrophages. Adv Exp Med Biol. 2016;899:211–29. doi: 10.1007/978-3-319-26666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tommelein J, Verset L, Boterberg T, Demetter P, Bracke M, Wever O De. Cancer-associated fibroblasts connect metastasis- promoting communication in colorectal cancer. Front Oncol. 2015;5(March):1–11. doi: 10.3389/fonc.2015.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4(4):376–89. [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(May):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 6.Elpek KG, Cremasco V, Shen H, Harvey CJ, Wucherpfennig KW, Goldstein DR, Monach PA, Turley SJ. The tumor microenvironment shapes lineage, transcriptional , and functional diversity of infi ltrating myeloid cells. Cancer Immunol Res. 2014;2(7):1–14. doi: 10.1158/2326-6066.CIR-13-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joyce JA, Fearon D. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. [DOI] [PubMed] [Google Scholar]
- 8.Pepin F, Bertos N, Laferrière J, Sadekova S, Souleimanova M, Zhao H, Finak G, Meterissian S, Hallett MT, Park M. Gene-expression profiling of microdissected breast cancer microvasculature identifies distinct tumor vascular subtypes. Breast Cancer Res. 2012;14(4):R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saleh SMI, Bertos N, Gruosso T, Gigoux M, Souleimanova M, Zhao H, Omeroglu A, Hallett MT, Park M. Identification of interacting stromal axes in triple-negative breast cancer. Cancer Res. 2017;77(17):4673–84. doi: 10.1158/0008-5472.CAN-16-3427. [DOI] [PubMed] [Google Scholar]
- 10.Quail DF, Joyce JA. Perspective the microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–41. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma P, Allison J. The future of immune checkpoint therapy. Science. 2015;348(6230):55–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 12.Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, et al.. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing Microvesicles. JEM. 2002;195(10):1303–16. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motz GT, Santoro SP, Wang L, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–15. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O'Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 15.Molon B, Ugel S, Del Pozzo F, et al.. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. JEM. 2011;208(10):1949–62. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–86. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overmeire V, Ja VG. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim biophys acta. 2016;1865(1):23–34. doi: 10.1016/j.bbcan.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Leigh IM, Navsaria H, Purkis PE, Mckay IA, Bowden PE, Riddle PN. Keratins (Kl6 and Kl7) as markers of keratinocyte hyperproliferation in psoriasis. Br J Dermatol. 1995;133(4):6501–11. doi: 10.1111/j.1365-2133.1995.tb02696.x. [DOI] [PubMed] [Google Scholar]
- 19.Zarbo R, Gown A, Nagle R, Visscher D, Crissman J. Anomalous cytokeratin expression in malignant melanoma: one- and two- dimensional western blot analysis and immunohistochemical survey of 100. Mod Pathol. 1990;3(4):4949–501. [PubMed] [Google Scholar]
- 20.Brissette JL, Kumar NM, Gilula NB, Hall JE, Dotto GP. Switch in gap junction protein expression is associated with selective changes in junctional permeability during keratinocyte differentiation. Proc Natl Acad Sci U S A. 1994;91(July):6453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang YJ, Kim P, Lu YF, Feingold KR. PPARgamma activators stimulate aquaporin 3 expression in keratinocytes⁄epidermis. Exp Deramatology. 2011;20:595–9. doi: 10.1111/j.1600-0625.2011.01269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jefferson JJ, Leung CL, Liem RKH. Plakins : Goliaths that link cell junctions and the cytoskeleton PLAKINS : Goliaths that link cell junctions and the cytoskeleton. Nat Rev Mol Cell Bio. 2015;5(August 2004):542–53. doi: 10.1038/nrm1425. [DOI] [PubMed] [Google Scholar]
- 23.Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol. 2009;1:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi X, Jin L, Dang E, Chang T, Feng Z, Liu Y, Wang G. IL-17A upregulates Keratin 17 expression in keratinocytes through STAT1- and STAT3-dependent mechanisms. J Invest Dermatol. 2011;131(12):2401–8. doi: 10.1038/jid.2011.222. [DOI] [PubMed] [Google Scholar]
- 25.Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. 2015;21:3561–9. doi: 10.1158/1078-0432.CCR-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4(March):1–8. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan H, Hall P, Santos LL, Gregory JL, Fingerle-Rowson G, Bucala R, Morand EF, Hickey MJ. Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J Immunol. 2011;186:4915–24. doi: 10.4049/jimmunol.1003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, Chatterjee M, Schmid H, Beck S, Gawaz M. CXCL14 as an emerging immune and inflammatory modulator. J Inflamm. 2016;13((1)):1–8. doi: 10.1186/s12950-015-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanegashima K, Suzuki K, Nakayama Y, Tsuji K, Shigenaga A, Otaka A, Hara T. CXCL14 is a natural inhibitor of the CXCL12 – CXCR4 signaling axis. FEBS Lett. 2013;587(12):1731–5. doi: 10.1016/j.febslet.2013.04.046. [DOI] [PubMed] [Google Scholar]
- 30.Zhang XHF, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, Massagué J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154:1060–73. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, Wang L, Shifrin N, Raulet DH. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 1st ed. Elsevier Inc.; 2014;122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malladi S, Macalinao DG, Jin X, Zou Y, De Stanchina E, Massague J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165:45–60. doi: 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic b -catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 34.Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, et al.. Cutaneous cancer stem cell maintenance is dependent on b -catenin signalling. Nature. 2008;452(April):650–3. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 35.Enholm B, Paavonen K, Ristima A, Kumar V, Gunji Y, Klefstrom J, Kivinen L, Laiho M, Olofsson B, Joukov V, et al.. Comparison of VEGF , VEGF-B , VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14:2475–83. [DOI] [PubMed] [Google Scholar]
- 36.Velasquez S, Killian D, Schulte J, Sticht C, Thiel M, Lindner HA. Short term hypoxia synergizes with Interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. JBC. 2016;291(25):12960–77. doi: 10.1074/jbc.M116.721753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strengell M, Matikainen S, Sirén J, Foster D, Julkunen I, Sareneva T. IL-21 in synergy with IL-15 or IL-18 enhances IFN- γ production in human NK and T cells. J Immunol. 2018;170:5464–5469. doi: 10.4049/jimmunol.170.11.5464. [DOI] [PubMed] [Google Scholar]
- 38.Krzywinska E, Kantari-mimoun C, Kerdiles Y, Sobecki M, Isagawa T, Gotthardt D, Castells M, Haubold J, Millien C, Viel T, et al.. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. 2017;8:1597. doi: 10.1038/s41467-017-01599-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity : two sides of a coin. Front Immunol. 2015;6(March):1–7. doi: 10.3389/fimmu.2015.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor : expression by tumor cells and activation of NK cells and macrophages. Nat Immunolgy. 2000;1(2):119–26. [DOI] [PubMed] [Google Scholar]
- 41.Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, Meyer EM, Morel L, Petersen BE, Scott EW. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416(April):542–5. [DOI] [PubMed] [Google Scholar]
- 42.Noubissi FK, Ogle BM. Cancer cell fusion : Mechanisms slowly unravel. Int J Mol Sci. 2016;17:1587. doi: 10.3390/ijms17091587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sellappan S, Grijalva R, Zhou X, Yang W, Eli MB, Mills GB, Yu D. Lineage infidelity of MDA-MB-435 cells : Expression of melanocyte proteins in a breast cancer cell line. Cancer Resrearch. 2004;64:3479–85. [DOI] [PubMed] [Google Scholar]
- 44.Johnson-léger C, Aurrand-lions M, Imhof BA. The parting of the endothelium : miracle , or simply a junctional affair? J Cell Sci. 2000;113:921–33. [DOI] [PubMed] [Google Scholar]
- 45.Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, Montresor A, Bolomini-Vittori M, Feigelson SW, Kirchhausen T, et al.. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity. 2010;30(3):384–96. doi: 10.1016/j.immuni.2008.12.020.Lymphocyte. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shulman Z, Cohen SJ, Roediger B, et al.. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol. 2012;13(1):67–77. doi: 10.1038/ni.2173. [DOI] [PubMed] [Google Scholar]
- 47.Ren G, Zhao X, Zhang L, Zhang J, L'Huillier A, Ling W, Roberts AI, Le AD, Shi S, Shao C, et al.. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol. 2010;184:2321–8. doi: 10.4049/jimmunol.0902023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stocker CJ, Sugars KL, Harari OA, Landis C, Morley BJ, Haskard DO. TNF- α , IL-4, and IFN- γ regulate differential expression of P- and E-selectin expression by porcine aortic endothelial cells. J Immunol. 2000;164:3309–15. doi: 10.4049/jimmunol.164.6.3309. [DOI] [PubMed] [Google Scholar]
- 49.Nakajima C, Uekusa Y, Iwasaki M. A role of interferon- γ ( IFN- γ ) in tumor immunity : T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sitesin IFN- γ -deficient mice. Cancer Res. 2001:3399–405. [PubMed] [Google Scholar]
- 50.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFN g and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832, April):1107–11. [DOI] [PubMed] [Google Scholar]
- 51.Miller CP, Thorpe JD, Kortum AN, Coy CM, Cheng WY, Ou Yang TH, Anastassiou D, Beatty JD, Urban ND, Blau CA, et al.. JAK2 expression is associated with tumor-infiltrating lymphocytes and improved breast cancer outcomes : Implications for evaluating JAK2 inhibitors. Cancer Immunol Res. 2014;2(File 6643):301–7. doi: 10.7303/syn4517.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trivedi PM, Graham KL, Scott NA, Jenkins MR, Majaw S, Sutherland RM, Fynch S, Lew AM, Burns CJ, Krishnamurthy B, et al.. Repurposed JAK1/JAK2 inhibitor reverses established autoimmune insulitis in non-obese diabetic mice. Diabetes. 2017;66(October 2016):db161250. doi: 10.2337/db16-1250. [DOI] [PubMed] [Google Scholar]
- 53.Shea JJO, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii111–5. doi: 10.1136/annrheumdis-2012-202576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crispin MK, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, Chen JC, Cerise JE, Jabbari A, Winge MC, et al.. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1(15):e89776. doi: 10.1172/jci.insight.89776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology. 2013;2(12):e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1 / CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–70. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al.. Targeting CXCL12 from FAP-expressing carcinoma- associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. PNAS. 2013;110(50):20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ. Tumor-derived granulocytes-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pylayeva-gupta Y, Lee KE, Hajdu CH, Miller G, Bar-sagi D. Oncogenic kras-induced GM-CSF production promotes the development of pancreatic Neoplasia. Cancer Cell. 2012;21(6):836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garcia AJ, Ruscetti M, Arenzana TL, Tran LM, Bianci-frias D, Sybert E. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor. Mol Cell Biol. 2017;34(11):2017–28. doi: 10.1128/MCB.00090-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al.. Monocytic CCR2 þ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell In fi ltration into the tumor microenvironment. Cancer Res. 2012;72(4):876–87. doi: 10.1158/0008-5472.CAN-11-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tabbekh M, Franciszkiewicz K. Rescue of tumor-infiltrating lymphocytes from activation-induced cell death enhances the antitumor CTL response in CD5-deficient mice. J Immunol. 2018;187:102–9. doi: 10.4049/jimmunol.1004145. [DOI] [PubMed] [Google Scholar]
- 63.Brossard C, Semichon M, Trautmann A, Bismuth G. CD5 inhibits signaling at the immunological synapse without impairing its formation. J Immunol. 2018;170:4623–9. doi: 10.4049/jimmunol.170.9.4623. [DOI] [PubMed] [Google Scholar]
- 64.Tarakhovsky A, Muller W, Rajewsky K. Lymphocyte populations and immune responses in CD5- deficient mice. Eur J Immunol. 1994;24(7):1677–84. doi: 10.1002/eji.1830240733. [DOI] [PubMed] [Google Scholar]
- 65.Zhou R, He P, Ren Y, Wang WH, Zhou RY, Wan H, Ono S, Fujiwara H, Zuo JP. Myeloid suppressor cell-associated immune dysfunction in CSA1M fibrosarcoma tumor-bearing mice. Cancer Sci. 2007;98(6):882–9. doi: 10.1111/j.1349-7006.2007.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee E, Schultz KLW, Griffin DE. Mice deficient in interferon-gamma or interferon-gamma receptor 1 have distinct inflammatory responses to acute viral encephalomyelitis. PLoS One. 2013;8(10):1–13. doi: 10.1371/journal.pone.0076412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swiercz JM, Kuner R, Offermanns S. Plexin-B1 directly interacts with PDZ-RhoGEF / LARG to regulate RhoA and growth cone morphology. Neuron. 2002;35:51–63. [DOI] [PubMed] [Google Scholar]
- 68.Negishi-koga T, Shinohara M, Komatsu N, Bito H, Kodama T, Friedel RH, Takayanagi H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med. 2011;17(11):1473–80. doi: 10.1038/nm.2489. [DOI] [PubMed] [Google Scholar]
- 69.Evans EE, Paris M, Smith ES, Zauderer M. Immunomodulation of the tumor microenvironment by neutralization of Semaphorin 4D. Oncoimmunology. 2015;4(12):e1054599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Evans EE, Jonason AS Jr, Bussler H, Torno S, Veeraraghavan J, Reilly C, Doherty MA, Seils J, Winter LA, Mallow C, et al.. Antibody blockade of semaphorin 4D promotes immune infiltration into tumor and enhances response to other immunomodulatory therapies. Cancer Immunol Res. 2015;3(19):689–702. doi: 10.1158/2326-6066.CIR-14-0171. [DOI] [PubMed] [Google Scholar]
- 71.Delaire S, Billard C, Tordjman R, Chédotal A, Elhabazi A, Bensussan A, Boumsell L. Biological activity of soluble CD100. II. Soluble CD100, similarly to H-SemaIII, inhibits immune cell migration. J Immunol. 2001;166:4348–54. doi: 10.4049/jimmunol.166.7.4348. [DOI] [PubMed] [Google Scholar]
- 72.Shi W, Kumanogoh A, Watanabe C, Uchida J, Wang X, Yasui T, Yukawa K, Ikawa M, Okabe M, Parnes JR, et al.. The Class IV Semaphorin CD100 plays nonredundant roles in the immune system : Defective B and T cell activation in CD100-Deficient mice. Immunity. 2000;13:633–42. [DOI] [PubMed] [Google Scholar]
- 73.Ito T, Bai TAO, Tanaka T, Yoshida K, Ueyama T. Semaphorin 4D induces vaginal epithelial cell apoptosis to control mouse postnatal vaginal tissue remodeling. Mol Med Rep. 2015;11:829–36. doi: 10.3892/mmr.2014.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Man Y. Tumor cell budding from focally disrupted tumor capsules : a common pathway for all breast cancer subtype derived invasion ? J Cancer. 2010;1:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cho A, Reidy MA. Matrix Metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. 2002;2002(91):845–51. doi: 10.1161/01.RES.0000040420.17366.2E. [DOI] [PubMed] [Google Scholar]
- 76.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–8. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ribas A. Adaptive immune resistance : How cancer protects from immune attack. Cancer Discov. 2015;5(September):915–20. doi: 10.1158/2159-8290.CD-15-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morgan RA, Chinnasamy N, Abate-daga DD, et al.. Cancer regressions and neurologic toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2014;36(2):133–51. doi: 10.1097/CJI.0b013e3182829903.Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ErbB2. Mol Ther. 2010;18(4):843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer J. 2014;20(2):112–8. doi:doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang W, Wang Y, Guo Y, Dai HR, Yang QM, Zhang YJ, Zhang Y, Chen MX, Wang CM, Feng KC, et al.. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma : an early phase IIa trial report. Signal Transduct Target Ther. 2016;1(November 2015):1–9. doi: 10.1038/sigtrans.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen DR, Cohen PL. Living life without B cells: is repeated B-cell depletion a safe and effective long-term treatment plan for rheumatoid arthrits? Int J Clin Rheumtol. 2012;7(2):159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–40. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]