

ABSTRACT
Diffuse large B-cell lymphoma (DLBCL) is the most common type of adult lymphoma. It is a group of malignant tumors with a large number of clinical manifestations and prognoses. Therefore, it is necessary to explore its unknown potential therapeutic targets. Histone deacetylase inhibitor (HDACi) is a novel drug for the treatment of DLBCL, however pan-HDACis cannot be ignored because of their clinical efficacy. By contrast, specific HDACi is well-tolerated, and LMK-235 is a novel HDACi that is a specific inhibitor of HDAC4 and HDAC5. In this study, we investigated the up-regulation of BCLAF1 through NF-κB signaling pathways in LMK-235, mediating the apoptosis of two diffuse large B-cell lymphoma cell lines, OCI-LY10 and OCI-LY3. Further studies showed that BCLAF1 expression was increased in DLBCL cells after treatment with the NF-κB inhibitor Bay11-7082. The combination of Bay11-7082 and siRNA si-HDAC4 significantly increased BCLAF1 expression and further increased apoptosis. These results indicate that BCLAF1 plays an important role in LMK-235-mediated apoptosis and may be a potential target for the treatment of diffuse large B-cell lymphoma.
KEYWORDS: LMK-235, BCLAF1, HDAC4, NF-kB, apoptosis, diffuse large B cell lymphoma
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin's lymphoma (NHL), accounting for about 30% of all adult NHLs in Western countries1. Diffuse large B cell lymphoma is a group of heterogeneous tumors2, with differences in terms of gene alterations, clinical features, morphological manifestations, responses to treatment and prognoses3-5. Although most patients with DLBCL can be cured with 6–8 cycles of R-CHOP chemotherapy, there remain 10% -15% of patients with DLBCL who have primary resistance and 20%-30% of patients suffer recurrences6. Therefore, to find new treatments for DLBCL, a program is required to control the progression of the disease, possibly constituting a new strategy for the treatment of cancer.
Histone deacetylases (HDACs) are enzymes that play a role in the regulation of epigenetic genes through chromatin modification7. Histone deacetylase inhibitors (HDACis) are novel drugs used in the treatment of hematological malignancies; they increase histone acetylation, inhibit proliferation of tumor cells, and induce apoptosis and differentiation. They have a wider range of anti-tumor effects8-11. The pan-HDAC activity inhibitor vardinostat (HDACi, SAHA) was approved by the FDA as a drug for the treatment of relapsed and resistant T-cell lymphomas (CTCL)12, and pabitastat (HDACi, Panobinostat) has been used to treat multiple myeloma (MM)13, 14. However, pan-HDACis have side effects that should not be overlooked. By contrast, specific HDACi is well-tolerated, and LMK-235 is a novel HDACi with HDAC isoform selectivity that is a specific inhibitor of HDAC4 and HDAC515,16. In addition, previous reports have investigated the effects of vadarnota (SAHA) on diffuse large B-cell lymphoma17, 18, however, there is currently no study on the effect of specific HDACi LMK-235 on DLBCL.
Bcl-2-associated transcription factor 1 (BCLAF1) was originally identified as a protein partner of adenovirus bcl-2 homologue E1B19K19. Previous studies have shown that this protein acted as an inducer of apoptosis and transcription repression factors20, 21. Epigenetic studies have shown that BCLAF1 can act through an HDAC4-dependent pathway to regulate differentiation and/or apoptosis22. However, the role of BCLAF1 in DLBCL remains to be elucidated. Therefore, we analyzed the effect of BCLAF1 on apoptosis and proliferation inhibition of DLBCL cells induced by HDACi LMK-235. Nuclear factor kappa beta (NF-κB) is a pivotal transcription factor that promotes cell survival, proliferation and inhibits apoptosis23. In DLBCL cells, the target of NF- κB and its downstream genes can trigger apoptosis24, 25. Previous reports have confirmed that BCLAF1 was located directly downstream of NF-κB26. In the present study, we use a method involving knockdown of the target gene by siRNA and inhibition of the NF-κB signaling pathway by Bay11-7082, focusing on whether BCLAF1 overexpression plays an apoptotic role in DLBCL, and to explore its possible mechanism of action.
Results
LMK-235 induced apoptosis of DLBCL cells in a time- and dose-manner
We measured the effect of specific HDACi inhibitor LMK-235 on the apoptosis of the diffuse large B-cell lymphoma cell lines OCI-LY10 and OCI-LY3 by annexin-V-PI staining (supplementary fig.1A-B, fig.1A). We examined changes in apoptosis rates in OCI-LY10 and OCI-LY3 cells treated with LMK-235 at 12, 24, 36, and 48 hours. We found that apoptosis of LMK-235 cells was not substantial at 12 hours, and that the cell apoptosis rate began to increase significantly after 24 hours. The maximum effect was achieved at 48 hours. LMK-235 mediated apoptosis of DLBCL cells in a time- and dose-dependent manner.
Subsequently, we tested the activity of OCI-LY10 cells after LMK-235 treatment using a CCK8 assay. As expected, LMK-235 significantly inhibited the survival of the DLBCL cell line OCI-LY10 in a dose- and time-dependent manner (supplementary fig.2), consistent with the results of flow cytometry. Simultaneously, we measured protein expression levels of HDAC4 and HDAC5 after treatment with the same concentration of LMK-235 as above for the same period of time in DLBCL cells (Fig.1B). We observed that HDAC4 expression was decreased in both OCI-LY10 and OCI-LY3 cells, and that the HDAC5 trend was less pronounced than that of HDAC4.
Figure 1.
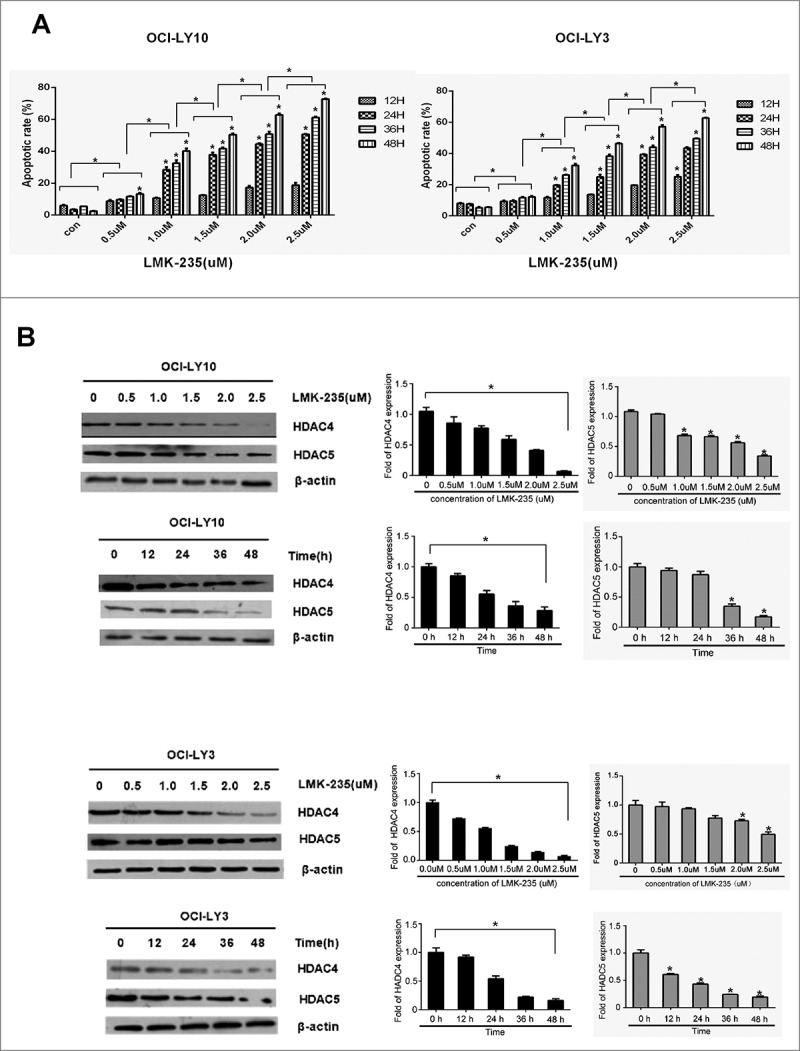
. LMK-235 induces apoptosis of DLBCL cells in a time and dose manner. (A) The DLBCL cell lines OCI-LY10 and OCI-LY3 cells were plated in triplicate and treated with 0, 0.5, 1.0, 1.5, 2.0 and 2.5 μmol/l LMK-235 for 12 hours, 24 hours, 36 hours and 48 hours, respectively. Flow cytometry was used to determine apoptosis.(B) OCI-LY10 and OCI-LY3 cells were treated with different concentration of LMK-235 (0.5, 1.0, 1.5, 2.0 and 2.5 μmol/l) for 24 hours,24 hours, 36 hours and 48 hours, and HDAC4 and HDAC5 protein levels were examined by Western blot .All experiments were conducted three times. Data are represented as mean±SD.*P<0.05,**P<0.01 versus the control group.
LMK-235 upregulated BCLAF1 expression
Due to the apoptotic changes caused by LMK-235 treatment of DLBCL cell lines, we investigated whether there were changes in the expression of related genes in DLBCL cells. BCLAF1 has previously been shown to function in an HDAC4-dependent manner. Therefore, we used q-RT-PCR and Western blot to analyze the effect of different concentrations of LMK-235 on BCLAF1 expression. The mRNA levels of BCLAF1 after treatment with LMK-235 were altered. With the increase of LMK-235 concentration, the expression of BCLAF1 in OCI-LY10 and OCI-LY3 cell lines was increased (Fig.2A). Western blotting also showed the same results.(Fig. 2C). At the same time, DLBCL cells were treated with the same concentration of LMK-235 (2 μmol/L) at different time points, and mRNA levels of BCLAF1 were analyzed by q-RT-PCR (fig.2B), and protein level expression was analyzed by Western blot (Fig.2C). Interestingly, BCLAF1 expression was not significantly increased after 12 hours of LMK-235 treatment but was significantly altered after 24 hours, with the greatest effect at 48 hours. When HDAC4 was significantly reduced in the cells, the induction of BCLAF1 gene expression was significantly increased (P < 0.05), and the time-dependent and concentration-dependent results were consistent.
Figure 2.
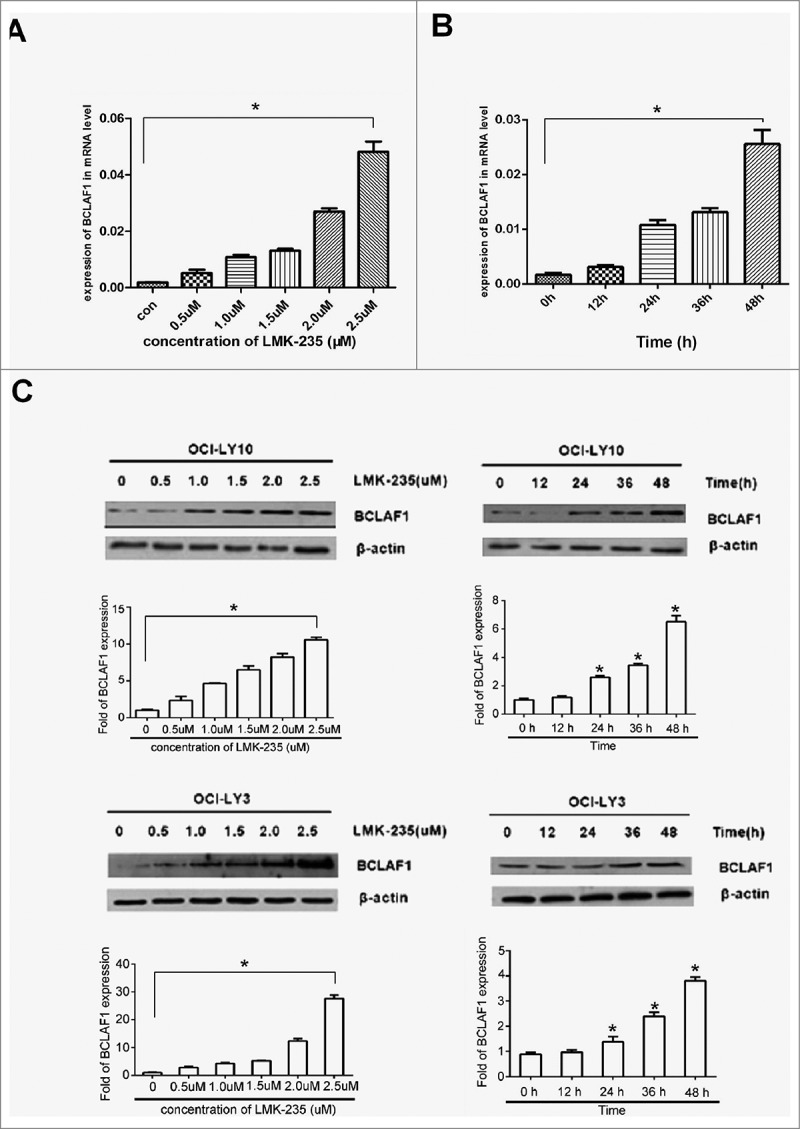
. LMK-235 upregulated BCLAF1 expression. (A) DLBCL cell lines OCI-LY10 were treated with different concentrations of LMK-235 (0, 0.5, 1.0, 1.5, 2.0, 2.5 μmol / l)for 24 hours or vehicle alone, and the mRNA expression of BCLAF1 was analyzed with q-PCR. Each value represents the average of three independent experiments. (B) DLBCL cell lines were treated with the indicated concentrations of LMK-235 at different times. The mRNA expression of BCLAF1 was analyzed with q-PCR. (C) DLBCL cell lines were treated with LMK-235, and the relative protein expression of the BCLAF1 were detected by Western blot. All experiments were performed three times. The data is expressed as mean ± SD. * P <0.05 compared with the control group.
LMK-235 inhibited the NF-kB signaling pathway
It has been confirmed that LMK-235 can cause apoptosis of DLBCL cells in a time- and dose-dependent manner; the next step was to investigate the mechanism that caused apoptosis in DLBCL cells. Previous studies have shown that NF-kB was constitutively activated in DLBCL and was associated with apoptosis of DLBCL. Therefore, we examined the effects of IκB-α and P65 on cells treated with various concentrations of LMK-235(Fig.3A). We found that the phosphorylation levels of IκB-α and P65 gradually decreased with increasing LMK-235 concentration, and that the total level of IκB-α and P65 did not change. Phosphorylation of IκB-α and P65 represented the activation of NF-kB. This indicated that LMK-235 inhibited NF-kB in a dose-dependent manner. Simultaneously, we used the same concentration of LMK-235 (2 μmol/L) to treat the OCI-LY10 cell line at various times after the observation of IκB-α and P65 phosphorylation (Fig. 3A). As time increased, the phosphorylation of IκB-α and P65 was also inhibited, and the total levels of IκB-α and P65 did not change. Similarly, another DLBCL cell line, OCI-LY3, showed the same change (Fig. 3B). These data demonstrated that LMK-235 inhibited the NF-kB signaling pathway.
Figure 3.
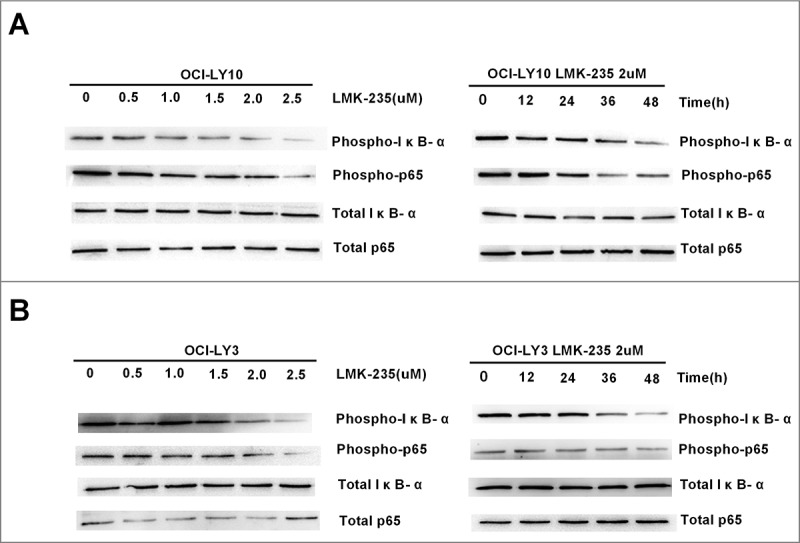
LMK-235 inhibited the NF-kB signaling pathway. (A)Western blot analysis showed that phosphorylation of IκB-α and p65 (p-p65 and pIκB-α) was inhibited after LMK-235 treatment in OCI-LY10 cells, but total-p65 or total-IκB- α were not affected. The cells were treated for the indicated drug concentration and sampled at the different time points with the same result.(B) The protein levels of p-p65 and pIκB-α in OCI-LY3 cellswere assessed by Western blot.
HDAC4 gene silencing caused apoptosis of DLBCL cells and increased expression of BCLAF1
Previous experiments confirmed that LMK-235 induced apoptosis of DLBCL cells and increased BCLAF1 expression in cells, BCLAF1 had previously been shown to function in an HDAC4-dependent manner. To further verify these results, we used si-RNA to silence HDAC4. Before evaluating the effect of low expression of HDAC4 on DLBCL, we investigated whether the use of siRNA could effectively knock down the HDAC4 gene. We treated DLBCL cells with siRNA against HDAC4 (si-HDAC4) or used empty siRNA (NC siRNA) as a negative control to eliminate the side-effects of transfection or RNA incorporation into cells. DLBCL cells were treated with si-HDAC4 or NC siRNA for 48 hours. Silencing HDAC4 with siRNA significantly decreased gene and protein expression. However, NC siRNA hardly affected HDAC4 protein or gene expression (*P < 0.05, **P < 0.01) (Fig. 4A). Subsequently, we examined the effect of HDAC4 expression on BCLAF1 and NF-kB pathways in silencing and control groups by Western blot (Fig. 4B). We knocked down HDAC4 using si-RNA and used si-NC as a control. With the decrease of HDAC4, the expression of BCLAF1 was increased. HDAC4 knockdown inhibited the phosphorylation of IκB-α and P65, and the total levels of IκB-α and P65 did not change, indicating that HDAC4 affects the NF-kB pathway. We hypothesized that BCLAF1 overexpression in DLBCL cells could be regulated by NF-kB.
Figure 4.
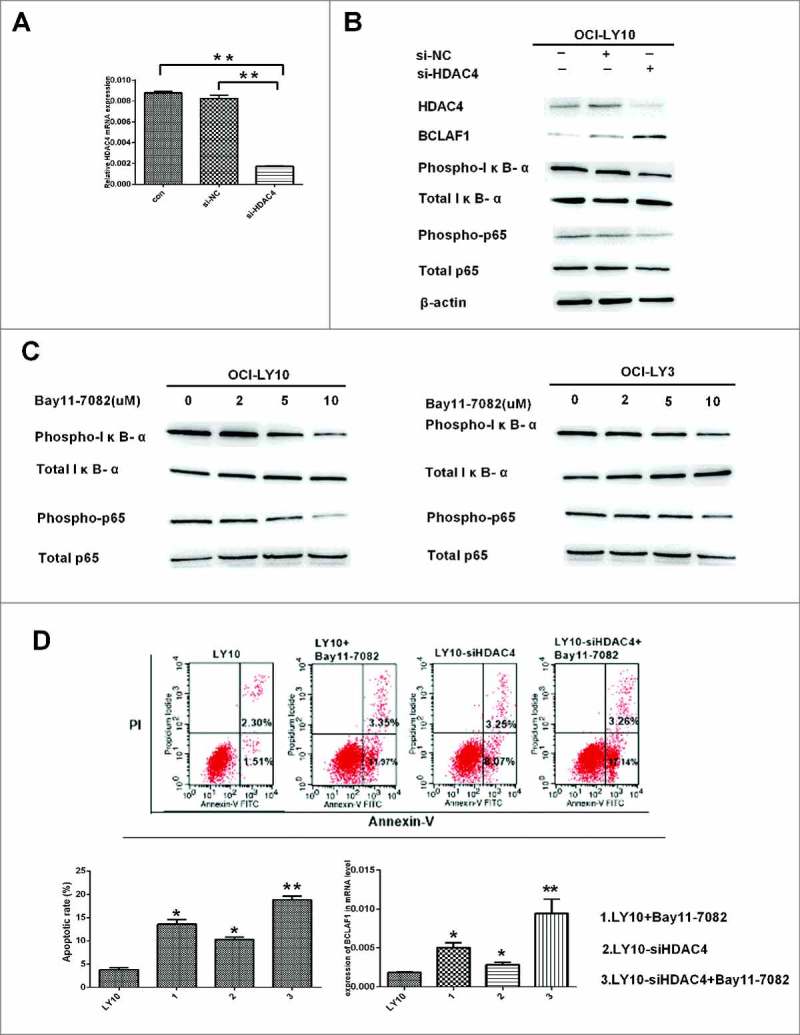
HDAC4 gene silencing caused apoptosis of DLBCL cells and increased expression of BCLAF1.(A) Relative HDAC4 mRNA expression in OCI-LY10 cells treated with or without si-NC and si-HDAC4.(B) The protein levels of HDAC4, BCLAF1, p-IκB-α and p-p65 were assessed by Western blot. (C)The protein levels of p-IκB-α and p-p65 were assessed by Western blot. (D)Cell apoptosis rates were tested by flow cytometry. The expression of BCLAF1 at the mRNA level was also analyzed by q-RT-PCR after si-HDAC4 and Bay11-7082 treatment. All experiments were performed three times.Data are represented as mean±SD.*P<0.05,**P<0.01 versus the control group.
To test this hypothesis, we treated DLBCL cells with Bay11-7082, an inhibitor of IκB-α phosphorylation. We chose to treat DLBCL cells at a concentration of 2µM,5µM and10 µM for 60 minutes and then perform Western blotting in both Bay11-7082-untreated and Bay11-7082-treated groups. As the Figure 4C shows, Bay11-7082 inhibited the phosphorylation of IκB-α and p65. Expression of BCLAF1 was increased in DLBCL cells treated with Bay11-7082, confirming that BCLAF1 overexpression was regulated by NF-kB.
Subsequently, we used flow cytometry to test the effect of low HDAC4 expression on apoptosis (Fig. 4D). First, after 48 hours of incubation with si-HDAC4 and si-NC siRNAs, the DLBCL cell line was treated with or without 10 μM Bay11-7082 for 24 hours. Apoptosis increased after siHDAC4 or Bay11-7082 alone. Interestingly, the combination of si-HDAC4 and Bay11-7082 significantly increased the rate of apoptosis (*P < 0.05, **P < 0.01). Similarly, BCLAF1 expression was increased after either si-HDAC4 or Bay11-7082 were used alone, and expression was further increased after the two were used in combination (Fig. 4D). HDAC4 may regulate BCLAF1 through the NF-kB pathway.
Down-regulation of HDAC4-induced upregulation of BCLAF1 was mediated through inhibition of NF-kB activity
To elucidate how LMK-235 affected BCLAF1 expression by specific pathways in DLBCL, we pretreated DLBCL cells (OCI-LY10 and OCI-LY3) with NF-κB inhibitor Bay11-7082 (2, 5, and 10 μM) for 60 minutes and then with or without LMK-235 (2 μM). Subsequently, protein expression of HDAC4, BClAF1 and p-p65 and p-IκB-α was measured by Western blot (Fig. 5). We showed that NF-κB activity was inhibited by LMK-235-mediated decrease of HDAC4, while BCLAF1 expression was increased. However, after the combination of Bay11-7082, the expression of HDAC4 was not affected.Treatment with Bay11-7082 had no effect on the expression of HDAC4 in the cells, indicating that HDAC4 was located upstream of NF-kB. With the increase of the concentration of Bay11-7082 used in combination, the expression of BCLAF1 was further increased. These results indicate that HDAC4 is upstream of NF-kB and increases BCLAF1 expression and induces apoptosis of DLBCL cells by inhibiting NF-kB activity.Therefore, overexpression of BCLAF1 may be responsible for these results in DLBCL,a combination of targets containing HDAC4 and BCLAF1 may provide new therapeutic options for DLBCL.
Figure 5.
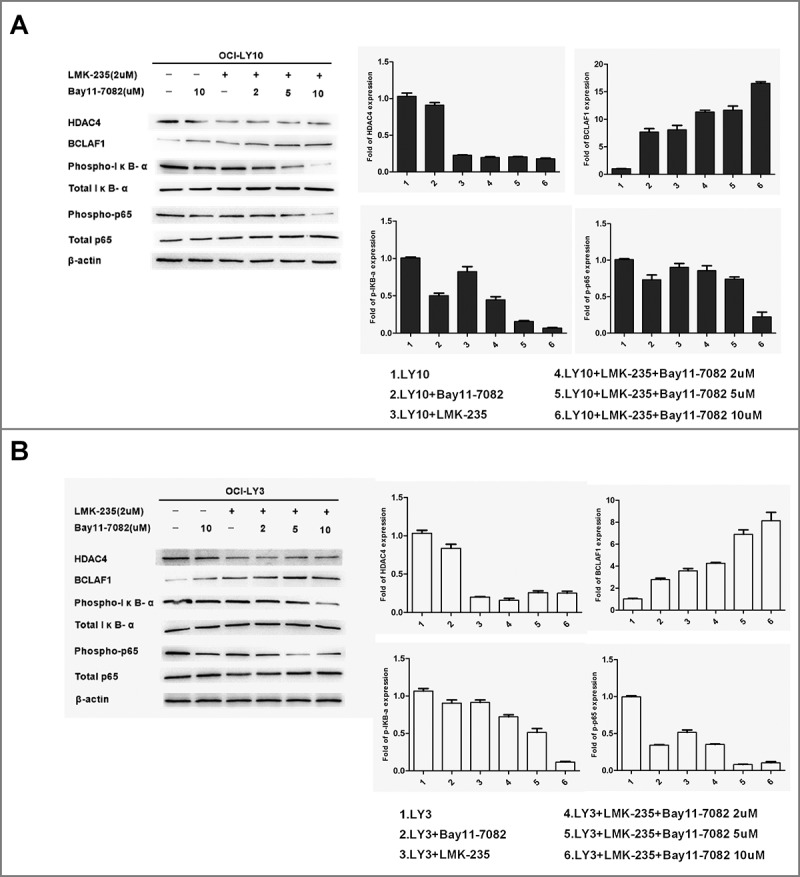
Down-regulation of HDAC4-induced upregulation of BCLAF1 was mediated through inhibition of NF-kB activity. Western blot detected the protein expression of HDAC4, BCLAF1, phosphorylated P65 and total P65, phosphorylated IκB-αS32 / S36 and total IκB-α. Western blot bands were quantified with Quantity One software. All experiments were repeated three times.
Discussion
Diffuse large B cell lymphoma (DLBCL) is the most common type of malignant lymphoma, with a high degree of heterogeneity in terms of clinical manifestations, pathological features and genetic features. With the use of rituximab, diffuse large B-cell lymphoma has a good prognosis, but more than half of patients still have a poor prognosis, and most patients eventually experience recurrence and drug resistance and eventually death. Therefore, it is necessary to seek potential therapeutic targets and new drugs for DLBCL1, 5, 27.
It is known that Bcl-2 related transcription factor 1 (BCLAF1) was involved in various biological processes19. It can play a role in apoptosis and transcription control. In fact, the various functions of BCLAF1 have been described by different tissues, including lung development20, T cell activation28, Kaposi sarcoma-associated herpesvirus lytic infection29, and human cytomegalovirus infection30. The exact mechanism of BCLAF1 in DLBCL remains to be determined. Therefore, this study analyzed the role and potential mechanism of HDAC inhibitor (HDACi) LMK-235 mediated BCLAF1. In preclinical experiments, HDACi had antiproliferative and pro-apoptotic effects on DLBCL cell lines. LMK-235, a HDACi with a novel linker-linker region, exhibited specific inhibition of HDAC4 and HDAC515. Sinyi Kong et al. previously demonstrated that type III histone deacetylase Sirt1 inhibited BCLAF1 transcription by deacetylation of H3K56 in the promoter region, whereas BCLAF1 protein expression levels increased in Sirt1-deficient cells28. Similarly, previous articles reported a correlation between BCLAF1 and HDAC4 in AML22. Our study demonstrated that after treatment of DLBCL cells with the HDAC inhibitor LMK-235, BCLAF1 expression was increased and apoptosis rate was increased. To further verify these results, we used si-RNA to knock down HDAC4 expression. The down-regulation of BCLAF1 gradually increased. These results indicate that LMK-235 regulated the transcription of BCLAF1 and increased the expression of BCLAF1 by participating in the regulation of acetylation.
Our team previously demonstrated that DLBCL was characterized by constitutive activation of the TNF-α-mediated NF-kB pathway31. NF-κB is an important transcription factor that regulates various pathophysiological processes of cell survival and death, and specifically enhances tumor growth by inhibiting apoptosis during cancer progression and cancer progression32. To date, some studies have shown that BCLAF1 as associated with NF-κB, and A-W Shao et al. demonstrated that BCLAF1 was regulated by the ATM/Nemo/NF-κB pathway and that it was a direct target of p65 and c-Rel26, 28, 32. We found that both LMK-235 and si-HDAC4 inhibited the activation of NF-κB and enhanced BCLAF1 expression. However, although the NF-κB inhibitor Bay11-7082 increased the expression of BCLAF1, it did not alter HDAC4, suggesting that HDAC4 was located upstream of NF-κB and regulated BCLAF1 through NF-κB. Interestingly, the combination of LMK-235 or si-HDAC4 with Bay11-7082 increased BCLAF1 expression in DLBCL cells and further promoted apoptosis. It has been demonstrated that down-regulation of HDAC4-induced overexpression of BCLAF1 occurs via NF-κB.
The specific mechanism by which BCLAF1 promotes apoptosis in DLBCL cells requires further study. Our team focused on the study of heme oxygenase-1 (HO-1) and confirmed in previous studies that characteristic overexpression of HO-1 in DLBCL was mediated by constitutively activated NF-κB. It is known that HO-1 expression inhibited apoptosis and HO-1 silencing promoted apoptosis33, 34. BCLAF1 is a direct downstream of NF-κB, and whether it mediates apoptosis of DLBCL cells via HO-1 requires further exploration in future experiments. In conclusion, BCLAF1 is a potential epigenetic therapeutic target. These findings may provide valuable preclinical evidence to improve the sensitivity of DLBCL patients to HDACi.
Materials and methods
Reagents and antibodies
The following reagents were used: specific selective HDACi LMK-235 (MedChemExpress, USA); fetal bovine serum (gibco BRL); RPMi-1640 medium (gibco BRL); dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO,USA); annexin V-fluorescein isothiocyanate (fiTC)/propidium liodide (Pi) apoptosis detection kit (BD Biosciences, San Jose, CA, USA); primary antibodies such as p-p65 S536 and p-I κ B- α S32/S36 for western blot analysis (Cell Signaling Technology, Beverly, MA, USA); secondary antibodies (Li-Cor Corp., Lincoln, NE, USA); TRIzol reagent (Life Technologies); and VCR and DxM (Sigma-aldrich).
Cell lines and cell culture
Human DLBCL cell lines OCI-LY10, OCI-LY3 were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen. DLBCL cell lines OCI-LY10 and OCI-LY13 were cryopreserved in Guizhou Hematopoietic Stem Cell Transplantation Center Laboratory (Guiyang, China). These cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Gibco BRL, MD, USA), penicillin (100 units/mL) and streptomycin (100 mg/mL) at a temperature of 37°C and humidity of 5%. All experiments were conducted using logarithmically growing cells (3–6 × 105 cells/ml).
Cell viability assay
Cells were seeded in 96-well plates at a density of 5000/well. After overnight incubation, the cells were treated with different concentrations (0, 0.5, 1, 2, 2.5, 5, and 10μmol/l) of LMK-235 for 12, 24, 36, and 48 hours. The inhibitory effect of LMK-235 was determined using the cell counting kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology, Haimen, China). The experiment was repeated three times. The survival rate (SR) was determined using the following equation: SR (%) = (A Treatment /A Control) × 100%. The concentration that produced 50% cytotoxicity (IC50) was determined using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA).
Apoptosis assay
Apoptosis was determined by double staining of annexin-V–FITC and propidium iodide (PI) according to the manufacturer's instructions (7Sea Biotech, Shanghai, China). The DLBCL cells were treated with fresh drug formulation and culture medium for 12 hours, 24 hours, 36 hours and 48 hours, respectively, washed with PBS, and resuspended in 100 μl of binding buffer containing 5 μl of annexin-V (BD Pharmingen, San Diego, California, USA). The cells were analyzed by flow cytometry after the addition of 7 μl of PI and data were collected on an FACSCalibur flow cytometry (BD Biosciences). The results represented the mean of three independent experiments.
siRNA transfection
siRNA against HDAC4 (si-HDAC4) was used to inhibit endogenous HDAC4 expression. Scrambled siRNA (si-NC) was used as a negative control. The siRNA sequences were designed and synthesized by TranSheep Bio-Tech Co., Ltd. (Shanghai, China).
si-HDAC4:5′-GCGUCUUAUUGAACUUAUUTTAAUAAGUUCAAUAAGACGCTT-3′
The transfection concentration of each siRNA was 50 nM. Cells were electroporated using transfection buffer and Soly-fecter according to the manufacturer's instructions (TranSheep Bio-Tech Co., Ltd.,Shanghai, China). 24 hours after the transfection, the cells were collected for quantitative real-time polymerase chain reaction (qRT-PCR) or reagent treatment. Western blot was performed after the transfected cells were cultured for 48h.
RNA isolation and qRT-PCR
Total RNAs from cell lines were extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. qRT-PCR was performed using the SYBR Green PCR Master Mix (TianGen Biotech, Beijing, China) and the PRISM 7500 real-time PCR detection system (ABI, USA). The levels of gene expression relative to that of β-actin gene transcript were analyzed.
The sequences of qRT-PCR were:
BCLAF1-F 5′–ATCCATTTCCAACAGAACCAG-3′
BCLAF1-R 5′–TCCTTCACCTATTGCTACACC-3′
β-actin-F 5′-GAGACCTTCAACACCCCAGC-3′
β-actin-R 5′-ATGTCACGCACGATTTCCC-3′
The cDNA samples were mixed with a total volume of 20ml of primer and SYBR Master Mix. The thermal cycling conditions used in the protocol were 94°C for 1 minute, and then 94°C for 10s and 60°C for 15s for 40 cycles.
Western blot analysis
Cells from different groups were collected. After two washes in ice-cold PBS, the cells were lysed by sonication in RIPA buffer (150 mM NaCl, 50mM Tris-HCl, 1% Triton ´100, 2% SDS, and 1% sodium deoxycholate) containing 1mM phenylmethanesulfonyl fluoride (Solarbio Science & Technology, Beijing, China). The lysate was transferred into EP tubes and centrifuged at 12,000 ´g for 10min at 4°C. The supernatant was collected and mixed with loading buffer. The final solution was boiled for 10min and aliquots were stored at −80°C until use. Equal amounts of protein (50–100mg) were added and separated by 10% SDS-PAGE, and transferred to a PVDF membrane (Millipore Corporation, Milford, MA, USA) which was then blocked in 5% skim milk in Tris buffer overnight at 4°C. The membrane was blotted with related antibodies for 2h. After washing, the blots were incubated for 1h at room temperature with secondary antibodies (HRP-conjugated goat anti-rabbit or anti-mouse; Beyotime, Shanghai; China). The protein bands were observed on the membrane by enhanced chemiluminescence (7sea Biotech, Shanghai, China) following the protocol of the manufacturer. Quantity one 4.6.2 of the image processing software was used to quantify the intensity of protein bands in each treatment at least three times.
Statistical analysis
GraphPad Prism 5.0 software (Graphpad Software, Inc, USA) was used to statistically analyze the data. All data are expressed as mean ± standard deviation. Statistical analysis was performed using analysis of variance and t-test. A P value of less than 0.05 was considered statistically significant.
Supplementary Material
Funding Statement
This study was supported by the National Science Foundation of China(No.81660616). There have no conflicts of interest among the authors.
Acknowledgements
We thanked the Guizhou Medical University hematopoietic stem cell laboratory for providing diffuse large B-cell lymphoma cell lines.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nature reviews Clinical oncology 2014; 11:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li S, Young KH, Medeiros LJ. Diffuse large B-cell lymphoma. Pathology 2018; 50:74–87. [DOI] [PubMed] [Google Scholar]
- 3.Guo L, Lin P, Xiong H, Tu S, Chen G. Molecular heterogeneity in diffuse large B-cell lymphoma and its implications in clinical diagnosis and treatment. Biochimica et biophysica acta 2018; 1869:85–96. [DOI] [PubMed] [Google Scholar]
- 4.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. . Genetic and functional drivers of diffuse large B cell Lymphoma. Cell 2017;171:481-94 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nowakowski GS, Blum KA, Kahl BS, Friedberg JW, Baizer L, Little RF, Maloney DG, Sehn LH, Williams ME, Wilson WH, et al. . Beyond RCHOP: A blueprint for diffuse Large B cell lymphoma research. J Natl Cancer Inst. 2016;108:djw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuno Y, Tsukamoto T, Kawata E, Uoshima N, Uchiyama H, Yokota I, Maegawa S, Takimoto T, Tanba K, Matsumura-Kimoto Y, et al. . Chromosomal abnormality variation detected by G-banding is associated with prognosis of diffuse large B-cell lymphoma treated by R-CHOP-based therapy. Cancer Med. 2018;7(3):655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SH, Yoo C, Im S, Jung JH, Choi HJ, Yoo J. Expression of histone deacetylases in diffuse large B-cell lymphoma and its clinical significance. Int J Medical Sci. 2014;11:994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mensah AA, Kwee I, Gaudio E, Rinaldi A, Ponzoni M, Cascione L, Fossati G, Stathis A, Zucca E, Caprini G, et al. . Novel HDAC inhibitors exhibit pre-clinical efficacy in lymphoma models and point to the importance of CDKN1A expression levels in mediating their anti-tumor response. Oncotarget. 2015;6:5059–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganai SA. Histone deacetylase inhibitor givinostat: the small-molecule with promising activity against therapeutically challenging haematological malignancies. J Chemother. 2016;28:247–54. [DOI] [PubMed] [Google Scholar]
- 10.Adams CM, Hiebert SW, Eischen CM. Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016;76:736–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grant S, Dai Y. Histone deacetylase inhibitors and rational combination therapies. Adv Cancer Res. 2012;116:199–237. [DOI] [PubMed] [Google Scholar]
- 12.Xue K, Gu JJ, Zhang Q, Mavis C, Hernandez-Ilizaliturri FJ, Czuczman MS, Guo Y. Vorinostat, a histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents. J Cancer Res Clin Oncol. 2016;142:379–87. [DOI] [PubMed] [Google Scholar]
- 13.Rajkumar SV, Kumar S. Multiple Myeloma: Diagnosis and treatment. Mayo Clinic Proc. 2016;91:101–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng T, Grasse L, Shah J, Chandra J. Panobinostat, a pan-histone deacetylase inhibitor: rationale for and application to treatment of multiple myeloma. Drugs Today (Barce). 2015;51:491–504. [DOI] [PubMed] [Google Scholar]
- 15.Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, Kurz T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem. 2013;56:427–36. [DOI] [PubMed] [Google Scholar]
- 16.Li A, Liu Z, Li M, Zhou S, Xu Y, Xiao Y, Yang W. HDAC5, a potential therapeutic target and prognostic biomarker, promotes proliferation, invasion and migration in human breast cancer. Oncotarget. 2016;7:37966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Persky DO, Li H, Rimsza LM, Barr PM, Popplewell LL, Bane CL, Von Gehr A, LeBlanc M, Fisher RI, Smith SM, et al. . A phase I/II trial of vorinostat (SAHA) in combination with rituximab-CHOP in patients with newly diagnosed advanced stage diffuse large B-cell lymphoma (DLBCL): SWOG S0806. Am J Hematol. 2018;93:486–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Z, Fang Q, Ma D, Zhe N, Ren M, Cheng B, Li P, Liu P, Lin X, Tang S, et al. . Silencing heme oxygenase-1 increases the sensitivity of ABC-DLBCL cells to histone deacetylase inhibitor in vitro and in vivo. Oncotarget. 2017;8:78480–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q, Han J, Wu G, Fang J, Feng Y. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun. 2014;5:4581. [DOI] [PubMed] [Google Scholar]
- 20.McPherson JP, Sarras H, Lemmers B, Tamblyn L, Migon E, Matysiak-Zablocki E, Hakem A, Azami SA, Cardoso R, Fish J, et al. . Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ. 2009;16:331–9. [DOI] [PubMed] [Google Scholar]
- 21.Sarras H, Alizadeh Azami S, McPherson JP. In search of a function for BCLAF1. ScientificWorldJ. 2010;10:1450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dell'Aversana C, Giorgio C, D'Amato L, Lania G, Matarese F, Saeed S, Di Costanzo A, Belsito Petrizzi V, Ingenito C, Martens JHA, et al. . miR-194-5p/BCLAF1 deregulation in AML tumorigenesis. Leukemia. 2018;32:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. . Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, et al. . A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–10. [DOI] [PubMed] [Google Scholar]
- 25.Lam LT, Davis RE, Pierce J, Hepperle M, Xu Y, Hottelet M, Nong Y, Wen D, Adams J, Dang L, et al. . Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin Cancer Res. 2005;11:28–40. [PubMed] [Google Scholar]
- 26.Shao AW, Sun H, Geng Y, Peng Q, Wang P, Chen J, Xiong T, Cao R, Tang J. Bclaf1 is an important NF-kappaB signaling transducer and C/EBPbeta regulator in DNA damage-induced senescence. Cell Death Differ. 2016;23:865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarkozy C, Traverse-Glehen A, Coiffier B. Double-hit and double-protein-expression lymphomas: aggressive and refractory lymphomas. Lancet Oncol. 2015;16:e555–e67. [DOI] [PubMed] [Google Scholar]
- 28.Kong S, Kim SJ, Sandal B, Lee SM, Gao B, Zhang DD, Fang D. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J Biol Chem. 2011;286:16967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziegelbauer JM, Sullivan CS, Ganem D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nature genetics 2009; 41:130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SH, Kalejta RF, Kerry J, Semmes OJ, O'Connor CM, Khan Z, Garcia BA, Shenk T, Murphy E. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc Natl Acad Sci U S A. 2012;109:9575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang J, Guo P, Ma D, Lin X, Fang Q, Wang J. Overexpression of heme oxygenase-1 induced by constitutively activated NF-kappaB as a potential therapeutic target for activated B-cell-like diffuse large B-cell lymphoma. Int J Oncol. 2016;49:253–64. [DOI] [PubMed] [Google Scholar]
- 32.Qu Y, Qu B, Wang X, Wu R, Zhang X. Knockdown of NF-kappaB p65 subunit expression suppresses growth of nude mouse lung tumour cell xenografts by inhibition of Bcl-2 apoptotic pathway. Cell Biochem Function. 2015;33:320–5. [DOI] [PubMed] [Google Scholar]
- 33.Liu P, Ma D, Yu Z, Zhe N, Ren M, Wang P, Yu M, Huang J, Fang Q, Wang J. Overexpression of heme oxygenase-1 in bone marrow stromal cells promotes microenvironment-mediated imatinib resistance in chronic myeloid leukemia. Biomed Pharmacother. 2017;91:21–30. [DOI] [PubMed] [Google Scholar]
- 34.Chau LY. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci. 2015;22:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.