

ABSTRACT
Apo2 ligand (Apo2L)/tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is unique to selectively induce apoptosis in tumor cells while sparing normal cells. Thus there is tremendous interest in Apo2L/TRAIL therapy; however, drug resistance is a serious limitation. Autophagy is a cellular housekeeping process that controls protein and organelle turnover, and is almost consistently activated in response to apoptosis-inducing stimuli, including Apo2L/TRAIL. Unlike apoptosis, autophagy leads to cell death or survival depending on the context. Various molecular mechanisms by which autophagy regulates Apo2L/TRAIL-induced apoptosis have been identified. Further, whether autophagy is completed (intact autophagic flux) or not could determine the fate of cancer cells, either cell survival or death. Thus, targeting autophagy is an attractive strategy to overcome Apo2L/TRAIL resistance. We present the current view of how these regulatory mechanisms of this interplay between autophagy and apoptosis may dictate cancer cell response to Apo2L/TRAIL therapy.
Keywords: Apoptosis, autophagosome, autophagy, Apo2L/TRAIL, flux, p62/SQSTM1, prostate cancer
Introduction
Apo-2 ligand (Apo2L) also called Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), was discovered in 1995 as a prototype of the TNF cytokine superfamily, based on 28% homology to Apo-2L, and 23% C-terminal homology to TNF.1,2 Unlike other members of the family, it has multiple receptors, which are highly expressed in various malignancies. Thus, its ability to induce cancer selective apoptosis, with no toxic side effects on normal cells, makes it an attractive candidate in the cancer treatment. Therefore, multiple clinical trials have been initiated using recombinant Apo2L/TRAIL or receptor agonists (reviewed in3,4). However, the success of Apo2L/TRAIL as an anticancer agent has been limited by resistance to Apo2L/TRAIL-induced apoptosis in cancer cells.3,4 Thus, understanding the Apo2L/TRAIL resistance mechanisms and investigation of the most “vulnerable” signaling pathway(s) in the context of resistance to Apo2L/TRAIL-induced apoptosis is critical to effectively exploit the Apo2L/TRAIL pathway in the clinic.
Autophagy is a cellular stress adaptation pathway of removal and recycling of damaged cellular components. By maintaining cellular homeostasis, it regulates cell survival in both normal and cancer cells.5 Numerous studies have reported extensive cross-talk between apoptosis and autophagy. In fact, apoptosis activating stimuli are almost invariably associated with activation of autophagy.6 While activation of autophagy can be pro-death or – survival depending on the cellular context, autophagy is more widely accepted as a mechanism of anticancer drug resistance. Indeed, inhibitors of autophagy have been recently used in combination with standard anticancer therapies in clinical trials in many cancers.7,8
Importantly, a large number of studies report that Apo2L/TRAIL can also activate autophagy, which, in turn, can contribute to increased sensitivity or resistance to Apo2L/TRAIL, in a context-dependent manner. Here, we review our current understanding of the regulation and effect on Apo2L/TRAIL sensitivity of the cross-talk between Apo2L/TRAIL-induced apoptosis and autophagy.
Apo2L/TRAIL and its receptors
Physiologically, Apo2L/TRAIL occurs either as a type 2 transmembrane protein, expressed mainly on the surface of immune cells, or as a soluble 24-kDa peptide formed by proteolytic cleavage of the extracellular portion of the transmembrane form. Even though both forms are biologically active, the transmembrane form is more potent as a ligand. Apo2L/TRAIL binds to its receptor as a trimer, such that one receptor molecule binds the crevice formed by two monomers of the trimer. Thus, the Apo2L/TRAIL trimer can engage the three receptors simultaneously.3,9-11
Apo2L/TRAIL interacts with four transmembrane receptors, TRAIL receptors 1 through 4 (TRAIL-R1-4): (1) TRAIL-R1 (also known as death receptor 4 (DR4), (2) TRAIL-R2 (DR5), (3) TRAIL-R3 (decoy receptor 1; DcR1), and (4) TRAIL-R4 (DcR2), and one soluble receptor, osteoprotegerin (OPG; TRAIL-R5).12-18 TRAIL-R1 (DR4) and TRAIL-R2 (DR5) contain functional cytoplasmic death domains (DD), which transmit an apoptotic signaling cascade upon Apo2L/TRAIL binding to the receptor. On the contrary, TRAIL-R3 (DcR1) and OPG completely, and TRAIL-R4 (DcR2) partially, lack the functional DD, and hence are unable to transmit apoptotic signaling. In fact, they are known to inhibit Apo2L/TRAIL-induced apoptosis by scavenging the ligand, hence the name decoy receptors.19,20
Apo2L/TRAIL signal transduction
Binding of the Apo2L/TRAIL trimer to the DR4 and/or DR5 causes receptor trimerization. The cytoplasmic DDs of the activated receptors, in turn, recruit the adaptor protein Fas-associated DD (FADD) by interacting with the DD of FADD. This recruits pro-caspase-8 and pro-caspase-10 downstream of FADD via homotypic interactions between the death-effector domains (DED) of FADD and the DED of procaspase-8 and −10. Thus, a multi-protein complex comprising: TRAIL-Rs, FADD, and caspase-8/10 is formed, which is called the death-inducing signaling complex (DISC). Recruitment of pro-caspases-8 and −10 to the DISC complex results in their homodimerization, which, in turn, induces a conformational change and exposes their active sites. This causes auto-activation and subsequent engagement of additional pro-caspase-8 and −10 molecules, ultimately resulting in full caspase activation at the DISC.21-23
Induction of apoptosis by Apo2L/TRAIL
Apo2L/TRAIL can activate both intrinsic (mitochondrial) and extrinsic (death receptor) branches of the apoptosis pathway. In “type I” cells adequate caspase-8 activation takes place at the DISC, such that caspase-8 dependent activation of effector caspase-3 is sufficient to induce apoptosis. However, in “type II” cells caspase‐8 activation at the DISC is not sufficient, therefore, the mitochondrial intrinsic pathway is required to elicit an apoptosis response. In the latter case, active caspase‐8 cleaves Bid to form the active, truncated Bid (tBid), which, in turn, activates the mitochondrial intrinsic pathway of apoptosis. This involves mitochondrial outer membrane permeabilization (MOMP), followed by release of cytochrome c and Smac/DIABLO (second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI) from mitochondrial intermembrane space into the cytosol, where cytochrome c causes activation of caspase-9 by the formation of the apoptosome, a multimeric complex comprising cytochrome c, Apaf‐1, and caspase‐9. Activated caspase‐9 is the initiator caspase which, in turn, cleaves and activates the effector caspases, such as caspase‐3 and caspase‐7. Release of Smac further favors apoptosis signaling by blocking the inhibitor of apoptosis (IAP) proteins, a family of antiapoptotic proteins that bind to and inhibit effector caspases (reviewed in24).
Induction of necroptosis and proinflammatory response by Apo2L/TRAIL
Depending on the cell type, and most often in the context of apoptosis-deficiency, Apo2L/TRAIL signaling can also elicit a non-apoptotic response, such as: (1) necroptosis, (2) proinflammatory pathways, mediated by NF-κB, Akt, MAPK, and JNK activation, and (3) prosurvival signaling cascades that favor enhanced cancer cell proliferation, migration, invasion, and/or tissue metastasis.25-27 The activation of these pathways depends on the formation of a secondary complex, comprising the DISC components, i.e. TRAIL-Rs, FADD, caspase-8, and cFLIP, in addition to receptor interacting protein 1 (RIP1), TNF receptor-associated factor 2 (TRAF2), and the NF-κB essential modulator (NEMO).28 RIP1 acts as an important dual function regulatory component for activation of NF–κB, as well as caspase-8 at the DISC. Its function is determined by ubiquitination and phosphorylation.29
Potential and limitations of Apo2L/TRAIL in anticancer therapy
Due to its unique ability to induce apoptotic cell death in cancer cells, with no toxic side-effects on the normal cells, there has been tremendous clinical interest in using Apo2L/TRAIL in cancer treatment. Consequently, clinical trials targeting TRAIL-R1 or – R2 using soluble recombinant human rhuApo2L/TRAIL (Apo2L.0 or AMG-951/Dulanermin) as well as by using agonistic antibodies have been pursued.30,31 Although they are well tolerated, TRAIL-R agonists have proven largely ineffective in clinical trials unless selected cells are targeted.31-33
Clinical ineffectiveness of Apo2L/TRAIL has been largely attributed to inherent or acquired resistance of tumors to Apo2L/TRAIL-induced apoptosis.31,32,34-37 In addition, non-apoptotic signaling induced by Apo2L/TRAIL may contribute to Apo2L/TRAIL-resistance by favoring undesirable growth promoting events. Thus, in a pancreatic adenocarcinoma xenograft model, Apo2L/TRAIL treatment was shown to promote the development of liver metastasis, underscoring the potentially deleterious effect of non-apoptotic signaling by Apo2L/TRAIL monotherapy.38 Understanding and targeting the factors and/or cellular pathways contributing to inherent/acquired Apo2L/TRAIL resistance in tumors in combination with Apo2L/TRAIL is expected to overcome the undesirable and/or limited efficacy of Apo2L/TRAIL monotherapy.
The autophagy pathway
“Macroautophagy”, hereafter referred to as “autophagy” is an evolutionary conserved, cellular “self-eating” process through which cellular contents (macromolecules and/or damaged organelles) undergo lysosomal degradation.39-41 It is the major pathway for turnover of damaged macromolecules and organelles, and ensures cellular homeostasis under stress conditions, such as starvation. It provides energy substrates and building blocks for cellular structures, such as formation of a special double-membrane structure called autophagosome that sequesters the cellular cargo to be degraded.39-41
The autophagy pathway comprises 6 different steps, representing different stages in the formation and maturation of autophagosome (reviewed in42). (1) Induction: The autophagy induction signal, the most well established being inhibition of mTORC1, marks the initiation of autophagy by activation of the ULK1 (Unc-like Kinase 1) complex (2) Nucleation of the phagophore: Initiation is followed by the assembly of autophagy-related proteins (ATGs) at the phagophore assembly site, where a coordinated fusion of membranes derived from plasma-membrane, Golgi, and endosomes gives rise to a cup-shaped initial membrane structure called phagophore that surrounds the autophagic target. Nucleation requires activation of Beclin-1 (BECN1)/VPS34 (BECN1/Vacuolar Protein Sorting 34) complex by ULK1. The BECN1/VPS34 complex has three forms, depending on the interacting partners of the core BECN1/VPS34/VPS15, ATG14L, and UVRAG association, which induces autophagy, whereas the Rubicon complex negatively regulates this complex. Further, anti-apoptotic family members, including BCL-2, BCL-xL and MCLl-1, have been shown to interact with BECN1 and inhibit its pre-autophagosomal assembly resulting in inhibition of autophagy.43 Conversely, proteins, such as High mobility group box 1 (HMGB1), a chromatin-associated nuclear protein and extracellular damage-associated molecular pattern molecule (DAMP) and BNIP3 that competitively displace BCL-2 or BECN1 and disrupt the complex are known to promote autophagy. (3) Elongation: The phagophore expands and ultimately closes at the two ends to give rise to a double-membrane structure called an autophagosome. Elongation of the phagophore requires two ubiquitination-like conjugation systems: ATG12-ATG5-ATG16, and LC3-PE. First, ATG7, an E1-like enzyme forms a thioester bond with and activates ATG12. Subsequently, ATG12 is transferred to ATG10, an E2-like enzyme, which is followed by conjugation of ATG12 to ATG5. The ATG12–ATG5 conjugate further interacts with ATG16 to give rise to ATG5-ATG12-ATG16, an E3-like enzyme needed for LC3 processing. ATG4, a protease, then cleaves LC3/ATG8 at its C terminus to form the cytosolic LC3-I. LC3-I is then modified to the phosphatidylethanolamine (PE)-conjugated LC3-II form by a ubiquitination-like reaction that requires ATG7 as the E1-like, ATG3 as the E2-like, and ATG5-ATG12-ATG16, as E3-like enzymes. The PE-conjugated form, known as LC3-II, gets incorporated into the autophagosomal membrane. (4) The production of PI3P: The BECN1-VPS34 complex recruits phospholipid-binding proteins, including ATG9, ATG2, WIPI1/2 (or ATG18), which function to recruit lipids to the phagophores. (5) Fusion: Autophagosomes ultimately fuse with the lysosomes and give rise to autolysosomes, which, along with their contents, are subsequently degraded and recycled. LAMP1/2, Rabs, and dynein are needed for the process. (6) Degradation: and recycling: Lysosomal hydrolases, Cathepsins -D, -L, and -B hydrolyse the autolysosomal content, and the products as well as the lysosomes are recycled.
Autophagy can be both non-selective, leading to bulk disintegration of cytoplasmic components or selective, targeting specific substrates, such as proteins, lipid droplets, and organelles. Mammalian cells possess cargo receptors, such as the sequestosome (SQSTM1, also called p62), which facilitate selective autophagic degradation of ubiquitinated targets. p62, by interacting with ubiquitin on target on one side, and with LC3, through LC3-interaction (LIR) domain on the other side, delivers the cargo to the autophagic machinery. During autophagic degradation of the target, the cargo receptors are also degraded, hence p62 degradation is often used as a marker of autophagic flux. Other such cargo receptors include Nix, Nuclear Dot Protein 52 (NDP52), and Neighbor of BRCA1 gene (NBR1).44
Autophagy in cancer
In normal cells, autophagy functions as a cell intrinsic tumor suppressor mechanism by removing damaged organelles, mitigating deleterious ROS, thus limiting chromosomal instability and mutations, and promoting oncogene-induced senescence.39,45-50 Thus, inhibition of autophagy is potentially oncogenic. However, in established tumors, cells hijack the autophagy pathway and use it for their own benefit, i.e. combating metabolic stress, hypoxia, ER stress, evading immune response, and promoting resistance against anticancer therapy.7,51-54 Multiple clinical trials using autophagy flux inhibitors, chloroquine and hydroxychloroquine, have been initiated for the treatment of multiple types of cancer.8
Activation of autophagy by Apo2L/TRAIL
Apoptosis and autophagy may lead to a different cellular fate, yet the two pathways do share common regulatory mechanisms and pathways. Therefore, a stimulus for induction of apoptosis, such as anticancer therapies, almost invariably alters autophagy signaling, and vice versa. Thus, the ultimate outcome of any anticancer therapy is determined by the cross-talk between these two pathways. Importantly, a better understanding of the molecular pathways that coregulate autophagy and apoptosis will help identify molecular determinants of the cellular death or resistance to Apo2L/TRAIL.
Apo2L/TRAIL has been explored for treatment of various tumors, including leukemia and prostate cancer.55 It has been found more effective particularly in combination with chemotherapeutic agents, such as the DNA-topoisomerase I inhibitor CPT-11.56 Apo2L/TRAIL treatment of cancer cells has been shown to activate autophagy by multiple mechanisms, involving common regulatory pathways that also regulate apoptosis. Apo2L/TRAIL treatment in lung, bladder, and prostate cancer cell lines resulted in TRAF2- and RIP1-dependent activation of MAPK8/JNK, which, in turn, led to degradation of BCL-2 and activation of BECN1-dependent prosurvival autophagy.57 On the other hand, Apo2L/TRAIL treatment in leukemic and pancreatic cancer cell lines cleaved and activated PARP1, which, in turn, led to poly-ADP-ribosylation of and cytoplasmic translocation of HMGB1 and subsequent HMGB1-BECN1 complex formation, resulting in activation of autophagy. Moreover, caspase-9 was found to be involved in regulating the initial steps of autophagosome formation. Caspase-9 formed a complex with ATG7, which inhibited the apoptotic potential of caspase-9 and at the same time increased LC3-II processing by ATG7.58 Co-regulation of prosurvival autophagy and apoptosis has been found in response to Apo2L/TRAIL by BECN1.59 cFLIP has been shown to downregulate both apoptosis and autophagy.60 In contrast, Apo2L/TRAIL treatment has been shown to inhibit autophagy. Melanoma cells that are inherently deficient in argininosuccinate synthetase (ASS) are addicted to autophagy to survive under arginine deprivation. Apo2L/TRAIL treatment resulted in inhibition of autophagy by caspase-8 dependent cleavage of BECN1 and ATG5.61 In another study, p62 dependent degradation of RIPK1 by autophagy was shown to switch necroptotic- to apoptotic cell death.62 Overall, these studies suggest that there are some regulatory molecules and pathways that potentially determine the overall outcome, cell death or survival, as a result of the cross-talk between Apo2L/TRAIL-induced autophagy and apoptosis. Better understanding of these regulatory mechanisms will help identify potential targets critical for predicting Apo2L/TRAIL resistance versus sensitivity in a given genomic/disease context.
The Thorburn lab has employed an innovative approach to identify the molecular signature to predict context-dependent role of autophagy.63 Autophagy flux along with cellular context (type 1 or type 2) could determine cell death or resistance by regulating Fap-1 protein phosphatase, a critical negative regulator of apoptotic cell death. A given cell population was separated into two subpopulations based on low-autophagy flux versus high autophagy flux by using flow cytometry. Cell death in response to Fas and Apo2L/TRAIL was investigated separately in these two subpopulations, in two different cellular contexts, i.e. type 1 and type 2 cells. Selective degradation of Fap1 by autophagy was found to cause cell death in type 1 cells in response to Fas in cells with high autophagy flux, which do not depend on the intrinsic apoptosis pathway for cell death, However, in Fas-treated type 2 cells, and Apo2L/TRAIL treated type1 or type 2 cells, high flux was associated with resistance. This study has been a major step forward in the understanding of context-dependent role of autophagy flux. It would be thus helpful to determine other molecular hallmarks of cancer cells, such as type 1 versus type 2 and Fap1, which could help explain the context-dependent role of autophagy in determining Apo2L/TRAIL resistance.
Autophagy and Apo2L/TRAIL resistance
The role of autophagy in Apo2L/TRAIL resistance is an area of active investigation. Below we will review various key mechanisms that have been identified by which autophagy confers Apo2L/TRAIL resistance (Figure 1).
Figure 1.
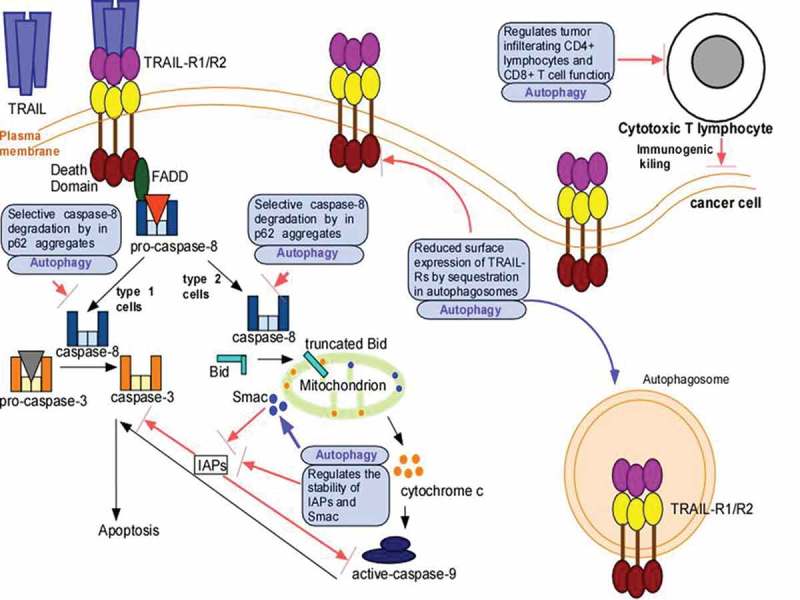
Mechanism(s) by which autophagy confers Apo2L/TRAIL resistance. Apo2L/TRAIL-binding to its receptors TRAIL-R1 and -R2 recruits FADD via homotypic interactions between the death domains of FADD and TRAIL-R which, in turn, converts pro-caspase-8 into active caspase-8. In ype 1 cells, caspase-8 directly cleaves pro-caspase-3 and this leads to apoptotic cell death. In type 2 cells, caspase-8 cleaves Bid to form truncated Bid, which associates with mitochondria and leads to release of cytochrome c and Smac. The cytochrome c release activates pro-caspase-9 to form caspase-9, which, in turn, ultimately leads to pro-caspase-3 cleavage and apoptotic cell death. Inhibitors of apoptosis (IAPs) inhibit caspase-9 and caspase-3, and antagonize apoptosis. Smac inhibits IAPs and favors cell death. Autophagy promotes Apo2L/TRAIL resistance by interfering with its apoptotic signaling in various ways such as: degradation of caspase-8, decrease in surface expression of TRAIL-Rs by autophagic degradation, and decrease in the levels of Smac and increasing that of IAPs. Autophagy can also suppress immunogenic killing by regulating T-cell recruitment and function.
In apoptosis-deficient cells
Han et al. first described the role of autophagy in Apo2L/TRAIL resistance.64 Activation of autophagy by Apo2L/TRAIL-treatment was identified as a mechanism of Apo2L/TRAIL resistance in the apoptosis‐deficient tumor cells, as demonstrated using various experimental systems, including: (1) genetically engineered apoptotic deficiency, i.e. FLIP transfection or Bax knockout in HCT-116 or Jurkat cells, (2) cells with inherent Apo2L/TRAIL-resistance, i.e. colon cancer cell line, as well as (3) breast carcinoma cell lines selected for acquired drug resistance to cisplatin (MCF7) or VP16 (MDA-MB-231).65 In the presence of functional autophagy, apoptosis-deficient cells were resistant to Apo2L/TRAIL-induced cell death, despite caspase-8 processing. Inhibition of autophagy by various means, including knockdown of BECN1, UVRAG, Vps34, or ATG7, in these apoptosis deficient cells caused sensitization to Apo2L/TRAIL-induced cell death.64 Another independent study reported that MDA-MB-231 breast cancer cells with acquired resistance to chronic Apo2L/TRAIL treatment displayed upregulation of the autophagic activity compared to their sensitive counterparts. Targeting autophagosome formation could sensitize the resistant cells to Apo2L/TRAIL-induced apoptosis in the resistant cells.66 Apoptosis defects are often acquired during tumorigenesis either naturally, such as Bax frameshift mutations in colorectal carcinoma and certain hematological malignancies, or during the course of acquired drug resistance.43,67,68 Thus, targeting autophagy should be particularly relevant in such cancer settings.
Selective removal of caspase-8
The mechanism by which Apo2L/TRAIL‐induced autophagy counterbalances the Apo2L/TRAIL‐mediated cell death in cells with defects in apoptosis was recently delineated by showing how functional autophagy sequesters the large caspase‐8 subunit into autophagosomes, with its subsequent degradation in lysosomes.65 Similar observations were made in a recent study from our laboratory. Apo2L/TRAIL-resistant prostate cancer cells exhibited high autophagic flux, resulting in more efficient clearance of p62-aggregates. In contrast, autophagic flux was low in Apo2L/TRAIL-sensitive cells, leading to accumulation of p62-aggregates, leading to caspase-8 activation and cell death. In contrast, inhibition of autophagy induction prevented p62 accumulation and hence caspase-8 activation. Moreover, inhibition of autophagy induced caspase‐8 activation, thus suggesting the role of autophagy in regulating activation of caspase-8.69 PCa cells can be sensitized Apo2L/TRAIL with Bcl-2 family protein inhibitors.70 Interestingly, inhibition of autophagy in ABT-263-treated cells led to p62 accumulation, which co-localized with caspase-8 and promoted its self-aggregation, and hence apoptosis.71
Mislocalization of DR4 and DR5
Expression of death receptors DR4 and DR5 on the surface of cell membrane is a critical determinant of sensitivity of cancer cells to the rhuApo2L/TRAIL and its receptor agonistic antibodies. Mis-localization of these receptors in intracellular compartments has been shown to be an important resistance mechanism. Autophagy was shown to regulate the spatial dynamics of the Apo2L/TRAIL receptors in breast cancer cell lines. Apo2L/TRAIL resistant versus sensitive cell lines differed in basal accumulation of autophagosomes. Apo2L/TRAIL resistant (e.g. BT474 and AU565) cells had high basal autophagosome levels, which sequestered DR4 and DR5, as shown by their co-localization with LC3-II, resulting in their reduced surface expression. Inhibition of autophagy by genetic and pharmacological means to inhibit autophagy initiation restored surface expression of DR4 and DR5, leading to Apo2L/TRAIL sensitization in Apo2L/TRAIL-resistant cells. In contrast, Apo2L/TRAIL-sensitive (MDA-MB-231) cells lacked accumulated basal autophagosomes and consequently had high levels of surface expression of DR4 and DR5. Inhibition of autophagic flux by inhibiting lysosomal activity in these cells resulted in an accumulation of autophagosomes, which sequestered DR4 and DR5 and decreaseed their surface expression, resulting in Apo2L/TRAIL resistance.72
Stability of anti-apoptotic/pro-apoptotic factors
In glioma cells, BECN1 was shown to directly interact with and stabilize survivin, an IAP family member.59 In another study in lung, prostate, and bladder cancer cells, the inhibition of autophagy reduced the expression of antiapoptotic factors BIRC2/cIAP1, BIRC3/cIAP2, XIAP, and CFLAR/c-FLIP and sensitized these cells to Apo2L/TRAIL-induced cell death.57 In colorectal cancer cells, hypoxia (0.5% O2) led to activation of mitochondrial autophagy. The resulting loss of mitochondria decreased the availability of mitochondria-derived pro-apoptotic molecules, such as Smac and, therefore, compromised the signal amplification from intrinsic apoptosis pathway in Apo2L/TRAIL treated type-II cells, resulting in Apo2L/TRAIL resistance73.
Immunosuppressive-related chemo-resistance. In late, metastatic lung cancer tissues, Inhibition of autophagy resulted in upregulation of CD4+, Foxp3+ tumor infiltrating lymphocytes as well as decreased expression of PD-L1, a marker of tumor specific CD8+ T cell dysfunction and immune activaton. In addition, there was activation of Apo2L/TRAIL-dependent apoptosis mediated by caspase 8.74
Overall, all these studies suggest that autophagy has a very important role in determining the cellular response to Apo2L/TRAIL. Thus, activation or inhibition of autophagy by directly targeting the pathway seems an attractive combination approach with Apo2L/TRAIL monotherapy. Importantly, since autophagy activation occurs in response to most cell death inducing agents, considering the role of autophagy, prodeath or-survival, while designing any combination regimens with Apo2L/TRAIL is imperative. Selective targeting of mitochondrial HSP90 in cancer cells activated an organelle unfolded protein response (UPR), which, in turn, activated cytoprotective autophagy.75 Similarly, deferoxamine (DFO),76 and niacin77 were shown to activate autophagic flux and induce Apo2L/TRAIL resistance in human colon cancer cells. On similar lines, inhibition of autophagy would act synergistically with combination of ABT-263 and Apo2L/TRAIL.71 In contrast, in another study genistein, a major isoflavone compound, known to limit tumor cell proliferation, inhibited autophagic flux which, in turn, sensitized Apo2L/TRAIL-resistant A549 adenocarcinoma cells to Apo2L/TRAIL treatment.78 Interestingly, autophagy inhibition antagonized the cell death induced by combination of Apo2L/TRAIL with a chalcone derivative (Chal-24)79 or with metformin.80
Conclusion
Overall, extensive literature suggests that apoptosis and autophagy are interlinked through common regulatory networks. Thus, in response to Apo2L/TRAIL autophagy signaling is inseparable from apoptosis. Since autophagy can induce cell death or resistance in response to Apo2L/TRAIL in any given cancer setting, the context-dependent role of autophagy will contribute to the overall cell survival or death in response to Apo2L/TRAIL monotherapy. Evaluation of this contribution of autophagy will help strategizing effective combination therapy regimens with Apo2L/TRAIL for personalized cancer therapy. Therefore, understanding such cellular and molecular contexts will help identify critical determinants for predicting Apo2L/TRAIL response or resistance to it.
Funding Statement
This work was supported by the HHS | NIH | National Cancer Institute (NCI) [CA184137];
Acknowledgments
This work was supported by the National Institutes of Health (NCI) grant CA184137 to Alexandru Almasan.
Conflict of interest
No potential conflicts of interest were disclosed.
References
- 1.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A.. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 271;1996:12687–12690. [DOI] [PubMed] [Google Scholar]
- 2.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–4765. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- 4.Lim B, Allen JE, Prabhu VV, Talekar MK, Finnberg NK, El-Deiry WS. Targeting TRAIL in the treatment of cancer: new developments. Expert Opin Ther Targets. 2015;19:1171–1185. doi: 10.1517/14728222.2015.1049838. [DOI] [PubMed] [Google Scholar]
- 5.Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC Mammalian autophagy: how does it work?. Annu Rev Biochem. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 6.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nature Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016. doi: 10.1016/j.ebiom.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O'Connell M, Kelley RF, Ashkenazi A, de Vos AM Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell. 1999;4:563–571. [DOI] [PubMed] [Google Scholar]
- 10.Cha SS, Kim MS, Choi YH, Sung BJ, Shin NK, Shin HC, Sung YC, Oh BH 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity. 1999;11:253–261. [DOI] [PubMed] [Google Scholar]
- 11.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 14;2003:337–348. [DOI] [PubMed] [Google Scholar]
- 12.Pan G, O"Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–113. [DOI] [PubMed] [Google Scholar]
- 13.Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–830. [DOI] [PubMed] [Google Scholar]
- 14.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–14367. [DOI] [PubMed] [Google Scholar]
- 15.Marsters SA, Sheridan JP, Pitti RM, Huang A, Skubatch M, Baldwin D, Yuan J, Gurney A, Goddard AD, Godowski P, et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7:1003–1006. [DOI] [PubMed] [Google Scholar]
- 16.Wallach D, Boldin M, Varfolomeev E, Beyaert R, Vandenabeele P, and Fiers W. Cell death induction by receptors of the TNF family: towards a molecular understanding. FEBS Lett. 1997;410:96–106. [DOI] [PubMed] [Google Scholar]
- 17.Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999;59:2770–2775. [PubMed] [Google Scholar]
- 18.Wu GS, Burns TF, McDonald ER, 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nature Gen. 1997;17:141–143. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 19.Morizot A, Merino D, Lalaoui N, Jacquemin G, Granci V, Iessi E, Lanneau D, Bouyer F, Solary E, Chauffert B, et al. Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance at the DISC level. Cell Death Diff. 2011;18:700–711. doi: 10.1038/cdd.2010.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merino D, Lalaoui N, Morizot A, Schneider P, Solary E, Micheau O Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol. 2006;26:7046–7055. doi: 10.1128/MCB.00520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp J TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nature Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 22.Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, Schwabe JW, Cain K, Macfarlane M A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291–305. doi: 10.1016/j.molcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peter ME. 2000. The TRAIL DISCussion: it is FADD and caspase-8! Cell Death Diff. 7:759–760. doi: 10.1038/sj.cdd.4400735. [DOI] [PubMed] [Google Scholar]
- 24.Mohamed MS, Bishr MK, Almutairi FM, and Ali AG. Inhibitors of apoptosis: clinical implications in cancer. Apoptosis. 2017;22:1487–1509. doi: 10.1007/s10495-017-1429-4. [DOI] [PubMed] [Google Scholar]
- 25.Morel J, Audo R, Hahne M, and Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280:15709–15718. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 26.Tran SE, Holmstrom TH, Ahonen M, Kahari VM, and Eriksson JE. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J Bioll Chem. 2001;276:16484–16490. doi: 10.1074/jbc.M010384200. [DOI] [PubMed] [Google Scholar]
- 27.Grunert M, Gottschalk K, Kapahnke J, Gundisch S, Kieser A, and Jeremias I. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-kappaB by TRAIL. Cell Death Dis. 2012;3:e414. doi: 10.1038/cddis.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, Ashkenazi A Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem. 2005;280:40599–40608. doi: 10.1074/jbc.M509560200. [DOI] [PubMed] [Google Scholar]
- 29.Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S, Liu ZG The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol Cell Biol. 2000;20:6638–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finnberg NK, Gokare P, Navaraj A, Lang Kuhs KA, Cerniglia G, Yagita H, Takeda K, Motoyama N, El-Deiry WS Agonists of the TRAIL death receptor DR5 sensitize intestinal stem cells to chemotherapy-induced cell death and trigger gastrointestinal toxicity. Cancer Res. 2016;76:700–712. doi: 10.1158/0008-5472.CAN-15-2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clinic Oncol. 2010;28:2839–2846. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 32.Soria JC, Smit E, Khayat D, Besse B, Yang X, Hsu CP, Reese D, Wiezorek J, Blackhall F Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J Clinic Oncol. 2010;28:1527–1533. doi: 10.1200/JCO.2009.25.4847. [DOI] [PubMed] [Google Scholar]
- 33.Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell J, Witt R, Hockstein N, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clinic Cancer Res. 2017;23:2942–2950. doi: 10.1158/1078-0432.CCR-16-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holland PM. 2014. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine Growth Factor Rev. 25:185–193. doi: 10.1016/j.cytogfr.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 35.Lemke J, Von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Diff. 2014;21:1350–1364. doi: 10.1038/cdd.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L, Fang B. 2005. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 12:228–237. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 37.Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL.. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene. 2013;32:1341–1350. doi: 10.1038/onc.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, Emme D, Roder C, Kalthoff H, Wajant H TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene. 2006;25:7434–7439. doi: 10.1038/sj.onc.1209719. [DOI] [PubMed] [Google Scholar]
- 39.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nature Cell Biol. 2013;15:713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Diff. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babcook MA, Sramkoski RM, Fujioka H, Daneshgari F, Almasan A, Shukla S, Nanavaty RR, Gupta S Combination simvastatin and metformin induces G1-phase cell cycle arrest and Ripk1- and Ripk3-dependent necrosis in C4-2B osseous metastatic castration-resistant prostate cancer cells. Cell Death Dis. 2014;5:e1536. doi: 10.1038/cddis.2014.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Current Op Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma A, Singh K, Mazumder S, Hill BT, Kalaycio M, Almasan A. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013;4:e628. doi: 10.1038/cddis.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 7;2011:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Youle RJ, Narendra DP. 2011. Mechanisms of mitophagy. Nature Rev Mol Cell Biol. 12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 47.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathew R, Kongara S, B Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park Y-E, Hayashi YK, Bonne G, Arimura T, Noguchi S, Nonaka I, Nishino I Autophagic degradation of nuclear components in mammalian cells. Autophagy. 2009;5:795–804. [DOI] [PubMed] [Google Scholar]
- 50.Singh K, Matsuyama S, Drazba JA, Almasan A. Autophagy-dependent senescence in response to DNA damage and chronic apoptotic stress. Autophagy. 2012;8:236–251. doi: 10.4161/auto.8.2.18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Viry E, Noman MZ, Arakelian T, Lequeux A, Chouaib S, Berchem G, Moussay E, Paggetti J, Janji B Hijacker of the antitumor immune response: autophagy is showing its worst facet. Front Oncol. 2016;6:246. doi: 10.3389/fonc.2016.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nature Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 54.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bucur O, Ray S, Bucur MC, Almasan A. APO2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in prostate cancer therapy. Front Biosci. 11;2006:1549–1568. [DOI] [PubMed] [Google Scholar]
- 56.Ray S, Almasan A. Apoptosis induction in prostate cancer cells and xenografts by combined treatment with Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand and CPT-11. Cancer Res. 63;2003:4713–4723. [PubMed] [Google Scholar]
- 57.He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, Gou X, Lin Y Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 2012;8:1811–1821. doi: 10.4161/auto.22145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han J, Hou W, Goldstein LA, Stolz DB, Watkins SC, Rabinowich H. 2014. A complex between Atg7 and caspase-9: a novel mechanism of cross-regulation between autophagy and apoptosis. J Biol Chem. 289:6485–6497. doi: 10.1074/jbc.M113.536854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niu TK, Cheng Y, Ren X, Yang JM. Interaction of Beclin 1 with survivin regulates sensitivity of human glioma cells to TRAIL-induced apoptosis. FEBS Lett. 2010;584:3519–3524. doi: 10.1016/j.febslet.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JS, Li Q, Lee JY, Lee SH, JH Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, et al. FLIP-mediated autophagy regulation in cell death control. Nature Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.You M, Savaraj N, Kuo MT, Wangpaichitr M, Varona-Santos J, Wu C, Nguyen DM, Feun L TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol Cell Biochem. 2013;374:181–190. doi: 10.1007/s11010-012-1518-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–349. doi: 10.1016/j.devcel.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nature Cell Biol. 2014;16:47–54. doi: 10.1038/ncb2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, Rabinowich H Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem. 2008;283:19665–19677. doi: 10.1074/jbc.M710169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lv S, Wang X, Zhang N, Sun M, Qi W, Li Y, Yang Q Autophagy facilitates the development of resistance to the tumor necrosis factor superfamily member TRAIL in breast cancer. Internat J Oncol. 2015;46:1286–1294. doi: 10.3892/ijo.2014.2812. [DOI] [PubMed] [Google Scholar]
- 67.Molenaar JJ, Gérard B, Chambon-Pautas C, Cavé H, Duval M, Vilmer E, Grandchamp B Microsatellite instability and frameshift mutations in BAX and transforming growth factor-β RII genes are very uncommon in acute lymphoblastic leukemia in vivo but not in cell lines. Blood. 1998;92:230–233. [PubMed] [Google Scholar]
- 68.Ionov Y, Yamamoto H, Krajewski S, Reed JC, Perucho M. Mutational inactivation of the proapoptotic gene BAX confers selective advantage during tumor clonal evolution. Proc Nat Acad Sci USA. 2000;97:10872–10877. doi: 10.1073/pnas.190210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh K, Sharma A, Mir MC, Drazba JA, Heston WD, Magi-Galluzzi C, Hansel D, Rubin BP, Klein EA, Almasan A Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer. 2014;13:70. doi: 10.1186/1476-4598-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ray S, Bucur O, Almasan A. Sensitization of prostate carcinoma cells to Apo2L/TRAIL by a Bcl-2 family protein inhibitor. Apoptosis. 2005;10:1411–1418. doi: 10.1007/s10495-005-2490-y. [DOI] [PubMed] [Google Scholar]
- 71.Huang S, Okamoto K, Yu C, Sinicrope FA.. p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. J Biol Chem. 288;2013:33654–33666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Di X, Zhang G, Zhang Y, Takeda K, Rivera Rosado LA, Zhang B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget. 2013;4:1349–1364. doi: 10.18632/oncotarget.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Knoll G, Bittner S, Kurz M, Jantsch J, Ehrenschwender M. 2016. Hypoxia regulates TRAIL sensitivity of colorectal cancer cells through mitochondrial autophagy. Oncotarget. 7:41488–41504. doi: 10.18632/oncotarget.9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zarogoulidis P, Petanidis S, Domvri K, Kioseoglou E, Anestakis D, Freitag L, Zarogoulidis K, Hohenforst-Schmidt W, Eberhardt W Autophagy inhibition upregulates CD4+ tumor infiltrating lymphocyte expression via miR-155 regulation and TRAIL activation. Molec Oncol. 2016;10:1516–1531. doi: 10.1016/j.molonc.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, Ross AH, Plescia J, Altieri DC Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clinic Investig. 2011;121:1349–1360. doi: 10.1172/JCI44855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moon JH, Jeong JK, Park SY. Deferoxamine inhibits TRAIL-mediated apoptosis via regulation of autophagy in human colon cancer cells. Oncol Rep. 2015;33:1171–1176. doi: 10.3892/or.2014.3676. [DOI] [PubMed] [Google Scholar]
- 77.Kim SW, Lee JH, Moon JH, Nazim UM, Lee YJ, Seol JW, Hur J, Eo SK, Lee JH, Park SY Niacin alleviates TRAIL-mediated colon cancer cell death via autophagy flux activation. Oncotarget. 2016;7:4356–4368. doi: 10.18632/oncotarget.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nazim UM, Park SY. 2015. Genistein enhances TRAIL-induced cancer cell death via inactivation of autophagic flux. Oncol Rep. 34:2692–2698. doi: 10.3892/or.2015.4247. [DOI] [PubMed] [Google Scholar]
- 79.Xu J, Xu X, Shi S, Wang Q, Saxton B, He W, Gou X, Jang JH, Nyunoya T, Wang X, et al. Autophagy-mediated degradation of IAPs and c-FLIP(L) potentiates apoptosis induced by combination of TRAIL and Chal-24. J Cell Biochem. 2016;117:1136–1144. doi: 10.1002/jcb.25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nazim UM, Moon JH, Lee JH, Lee YJ, Seol JW, Eo SK, Lee JH, Park SY Activation of autophagy flux by metformin downregulates cellular FLICE-like inhibitory protein and enhances TRAIL- induced apoptosis. Oncotarget. 2016;7:23468–23481. doi: 10.18632/oncotarget.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]