

Abstract
Background
Prostate adenocarcinoma (AdPC) cells can undergo lineage switching to neuroendocrine cells and develop into therapy-resistant neuroendocrine prostate cancer (NEPC). While genomic/epigenetic alterations are shown to induce neuroendocrine differentiation via an intermediate stem-like state, RNA splicing factor SRRM4 can transform AdPC cells into NEPC xenografts through a direct neuroendocrine transdifferentiation mechanism. Whether SRRM4 can also regulate a stem-cell gene network for NEPC development remains unclear.
Methods
Multiple AdPC cell models were transduced by lentiviral vectors encoding SRRM4. SRRM4-mediated RNA splicing and neuroendocrine differentiation of cells and xenografts were determined by qPCR, immunoblotting, and immunohistochemistry. Cell morphology, proliferation, and colony formation rates were also studied. SRRM4 transcriptome in the DU145 cell model was profiled by AmpliSeq and analyzed by gene enrichment studies.
Findings
SRRM4 induces an overall NEPC-specific RNA splicing program in multiple cell models but creates heterogeneous transcriptomes. SRRM4-transduced DU145 cells present the most dramatic neuronal morphological changes, accelerated cell proliferation, and enhanced resistance to apoptosis. The derived xenografts show classic phenotypes similar to clinical NEPC. Whole transcriptome analyses further reveal that SRRM4 induces a pluripotency gene network consisting of the stem-cell differentiation gene, SOX2. While SRRM4 overexpression enhances SOX2 expression in both time- and dose-dependent manners in DU145 cells, RNA depletion of SOX2 compromises SRRM4-mediated stimulation of pluripotency genes. More importantly, this SRRM4-SOX2 axis is present in a subset of NEPC patient cohorts, patient-derived xenografts, and clinically relevant transgenic mouse models.
Interpretation
We report a novel mechanism by which SRRM4 drives NEPC progression via a pluripotency gene network.
Fund
Canadian Institutes of Health Research, National Nature Science Foundation of China, and China Scholar Council.
Keywords: SRRM4, RNA splicing, treatment-resistant mechanisms, transcriptome, lineage plasticity
Research in context.
Evidence before this study
The mechanisms by which prostate adenocarcinoma (AdPC) progress to aggressive therapy-resistant neuroendocrine prostate cancer (NEPC) are complicated. Previously, RNA splicing factor SRRM4 has been demonstrated to transform AdPC to NEPC through a direct transdifferentiation process.
Added value of this study
Here, we define a novel function of SRRM4 that can regulate a pluripotency network including SOX2 to induce a potential stem-like state of AdPC cells for NEPC development. This SRRM4-SOX2 axis is shown to be present in NEPC patients, patient-derived xenografts, and transgenic mouse models. Additionally, a new NEPC model, called DuNE, is established which recapitulates the phenotypes of clinical NEPC tumors with stem-like characteristics.
Implications of all the available evidence
These data, together with previous studies, demonstrate that an RNA splicing factor can drive the transformation of AdPC into NEPC tumors through either a neuroendocrine transdifferentiation pathway or pluripotency gene network. Whether a pluripotency gene network is initiated by SRRM4 likely relies on the epigenetic and genomic heterogeneity of AdPC cells. Furthermore, our new NEPC model adds to the small handful of available NEPC cell and tumor models.
Alt-text: Unlabelled Box
Abbreviations
| AdPC | prostate adenocarcinoma |
| NEPC | neuroendocrine prostate cancer |
| SRRM4 | serine/arginine repetitive matrix 4 (also known as nSR100) |
| SOX2 | sex determining region Y-box 2 |
| CRPC | castration-resistant prostate cancer |
| AR | androgen receptor |
| CRPC-Ad | castration-resistant prostate adenocarcinoma |
| t-NEPC | treatment-induced neuroendocrine prostate cancer |
| TP53 | tumor protein p53 |
| RB1 | retinoblastoma transcriptional corepressor 1 |
| PTEN | phosphatase and tensin homolog |
| NE | neuroendocrine |
| SC | stem cell |
| REST | RE1 silencing transcription factor |
| EZH2 | enhancer of zeste 2 polycomb repressive complex 2 |
| LnNE | SRRM4-transduced LNCaP cell-derived neuroendocrine prostate cancer model |
| PSA | prostate-specific antigen |
| VPC | Vancouver Prostate Centre |
| RISH | RNA in situ hybridization |
| IHC | immunohistochemistry |
| E-Cad | E-cadherin |
| SYP | synaptophysin |
| BrdU | bromodeoxyuridine |
| Doce | docetaxel |
| CPT | camptothecin |
| GSEA | gene set enrichment analysis |
| PDX | patient-derived xenograft |
| GEMM | genetically engineered mouse model |
| GEO | gene expression omnibus |
| WT | wild-type |
| SKO | single knock-out |
| DKO | double knock-out |
| TKO | triple knock-out |
| LE | luminal epithelial |
| c-PARP | cleaved poly(ADP-ribose) polymerase |
| GO | gene ontology |
| DuNE | SRRM4-transduced DU145 cell-derived neuroendocrine prostate cancer model |
1. Introduction
Tumor cell plasticity and heterogeneity create many challenges for prostate cancer disease management. To counteract anti-androgen therapies and become castration-resistant prostate cancers (CRPC), prostate adenocarcinoma (AdPC) cells can either enhance androgen receptor (AR) signaling in the presence of castration levels of androgens, progress into castration-resistant AdPC (CRPC-Ad) tumors or, alternatively, become AR-indifferent or AR-negative and progress into treatment-induced neuroendocrine prostate cancers (t-NEPC) [[1], [2], [3]]. While the molecular mechanisms by which AdPC cells transform into t-NEPC remain elusive, recent studies have shown that the combination of AR-inhibition with genomic inactivation of TP53, RB1, and PTEN confers AdPC cells lineage plasticity to gain basal, mesenchymal, or neuroendocrine (NE) phenotypes and subsequently the development of t-NEPC tumors [[4], [5], [6], [7]]. These studies demonstrate that this transition from AdPC to t-NEPC can be through an intermediate pluripotent stem cell (SC)-like state. During this state, there are elevated expressions of a network of pluripotency genes including the SOX family members such as SOX2 and SOX11 which are well known for their roles in early embryogenesis, embryonic SC pluripotency, and neurogenesis [[3], [4], [5], 7, 8]. Given the genomic heterogeneity of prostate tumor cells, these findings highlight that AdPC cells containing certain genomic features may be prone to undergo this lineage switching to develop into t-NEPC via a pluripotency gene network.
However, whole-exome sequencing has revealed that patient t-NEPC and AdPC tumors have similar gene mutation landscapes [2, 3, 9, 10]. In vitro, AdPC cell models were shown to undergo an AdPC-to-NE cell lineage switch through a transdifferentiation mechanism to initiate t-NEPC development. This NE transdifferentiation process is shown to be mediated by dysregulations of master transcriptional repressor of neuronal genes, REST [[11], [12], [13]], epigenetic modulators, such as EZH2 [9, 14, 15], and microenvironment factors (e.g. cAMP, IL-6, and hypoxia) [12, [16], [17], [18], [19]]. These results emphasize that multiple non-genomic factors also play important roles during t-NEPC establishment.
In fact, we have recently shown that RNA splicing mechanisms, mediated by the RNA splicing factor SRRM4, drive this NE transdifferentiation of AdPC cells to t-NEPC. The upregulation of SRRM4 is associated with t-NEPC and predominately establishes a NEPC-unique RNA splicing program distinctive from AdPC tumors [3, 13, 20, 21]. SRRM4 can transform LNCaP AdPC cells, which express functional TP53 and RB1, into NEPC xenografts under androgen-deprived conditions [21]. These SRRM4-transduced LNCaP cells also exhibit global transcriptome and RNA splicing signatures similar to that of t-NEPC tumors in patients. Subcutaneously inoculating these cells into castrated nude mice and serially passaging the tumors for five generations over 18 months, we have reported the establishment of a series of t-NEPC models, called LnNE [20]. LnNE tumors express strong NE markers and present a NEPC morphology. Tumors at later passages grow more aggressively and become androgen-insensitive and PSA-negative. These features mimic AdPC progression to t-NEPC in patients. Further dissection of SRRM4-mediated signaling has revealed that SRRM4 can not only compromise the functions of REST and FOXA1 to promote NE transdifferentiation [3, 13, [21], [22], [23]], but also regulate RNA splicing of other genes such as MEAF6 and Bif-1 to accelerate cell proliferation and tumorigenesis [24, 25]. Altogether, these findings indicate that SRRM4 is a driver of t-NEPC through the NE transdifferentiation mechanism.
SRRM4 is a master regulator of neural-specific exon networks that are required for the differentiation of embryonic SC and neural progenitor cells during embryonic development [3, [26], [27], [28]]. It exerts its actions by promoting the inclusion of neural-specific exons into the coding regions of target genes, therefore generating alterative protein isoforms necessary for remodeling protein-protein interactions networks and signaling pathways that are essential for neurogenesis and neuronal maturation and function [3, 29]. The developmental roles of SRRM4 in the differentiation of stem/neural progenitor cells leads us to hypothesize that SRRM4 may also regulate a pluripotency gene network in AdPC cells with specific genetic and epigenetic backgrounds during t-NEPC progression.
In this study, we report that SRRM4 induces a global NEPC-specific RNA splicing signature in a panel of prostate and prostate cancer cell models. However, SRRM4 induces heterogeneous transcriptomes and phenotypes among these cell models tested. The SRRM4-transduced DU145 cells are unique in which a pluripotency gene network including SOX2 is activated. This discovery characterizes another SRRM4-driven mechanism underlying lineage plasticity distinct from the LnNE model. This model, so called the DuNE model, recapitulates the molecular signatures of a subset of patient t-NEPC tumors expressing stem-like molecular features, emphasizing its clinical relevance and application in investigating the complex molecular mechanisms of t-NEPC development.
2. Materials and methods
2.1. Cell lines and cell culture
293 T, LNCaP, 22Rv1, PC-3, and DU145 cell lines have previously been described [24, 25]. WPMY-1 were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). RWPE-1 cells were generously provided by Dr. Michael Cox from the Vancouver Prostate Centre (VPC; BC, Canada). The BPH-1 and hTERT-Myo cell lines were provided by Dr. Simon Hayward (Vanderbilt University, TN, USA) and Dr. Jennifer Condon (Wayne State University, MI, USA), respectively. hTERT-Myo cells were cultured in Dulbecco Modified Eagle Medium/High Glucose (DMEM; Hyclone; Logan, UT, USA) with 10% fetal bovine serum (FBS; Gibco Life Technologies; Waltham, MA, USA), whereas BPH-1 cells were cultured in RPMI-1640 medium (Hyclone) with 10% FBS. Under experimental conditions, LNCaP cells were cultured in 5% charcoal stripped serum (Hyclone) and phenol-free RPMI-1640 medium (Hyclone). WPMY-1 cells were maintained in DMEM medium containing 10% FBS and 1% penicillin-streptomycin (Hyclone). RWPE-1 cells were grown in Keratinocyte-SFM (1×) media with human recombinant epidermal growth factor 1–53 and bovine pituitary extract supplements (Gibco Life Technologies). Cells were all incubated in 5% CO2 at 37 °C. All cell lines used were tested negative for mycoplasma contamination and authenticated by short tandem repeat assays.
2.2. Construction of cell models by lentiviral approaches
Lentiviral expression vectors (pFUGWBW) encoding Flag-SRRM4 and empty control vector were used as previously created and published [21]. Vectors were used to package lentivirus in 293 T cells and transduce target cells (i.e. LNCaP, 22Rv1, PC-3, DU145, WPMY-1, RWPE-1, BPH-1, hTERT-Myo) as previously described [21, 24, 25, 30, 31]. Transduced cells were selected by blasticidin (Gibco Life Technologies). Expression of SRRM4 was confirmed by real-time qPCR and immunoblotting assays (Fig. S1).
2.3. Real-time qPCR and immunoblotting assays
Real-time qPCR and immunoblotting assays were performed as previously described [21, 24, 25, 30, 31]. Real-time qPCR assays were carried out using three technical replicates and three independent biological replicates, and all immunoblotting assays were repeated in three independent experiments with one representative blot shown. Heatmaps were constructed based on the results of the qPCR. Primer information is listed in Supplementary Table 1. Antibody information is listed in Supplementary Table 2.
2.4. Human prostate cancer xenografts, RISH, and IHC assays
To construct xenografts, 1 × 106 cells of the established DU145, PC-3, LNCaP, and 22Rv1 cell models overexpressing SRRM4 or empty vector were implanted subcutaneously in bilateral flanks of 6–8-week old male NOD-SCID mice. All mice were surgically castrated prior to tumor implantation. All animal procedures were performed in accordance with the guidelines and regulations of the Canadian Council on Animal Care and approved by the Institutional Animal Care and Use Committee at the University of British Columbia. Tumors were harvested when they reached experimental endpoints (tumor volume of 1500 mm3), fixed in 10% paraformaldehyde, paraffin embedded, and cut for RNA in situ hybridization (RISH) to use an anti-sense probe to detect against SRRM4 (NM_194286.3; targeting 496-835 bp) or cut for immunohistochemistry (IHC) to stain against E-Cad (E-cadherin), pan-cytokeratin, synaptophysin (SYP), or Ki-67 (antibody information listed in Supplementary Table 2). RISH probes were designed by Advanced Cell Diagnostic (Hayward, CA, USA) and assays were performed using their BaseScope™ assay kit following manufacture's instruction. Scoring method and validation of the sensitivity and specificity of the probe has previously been described [24]. Positive RISH signals were presented as red dots under 10× and 40× magnifications. IHC was performed as previously described [20, 21, 24, 31, 32]. NCI-H660 xenografts as well as patient NEPC tumors were previously established and characterized [24]. IHC against SOX2 was performed using these tissue samples (antibody information listed in Supplementary Table 2).
2.5. Digital image analysis and Ki-67 scoring
All stained slides were digitalized with the SL801 autoloader and Leica SCN400 scanning system (Leica Microsystems; Concord, Ontario, Canada) at a magnification equivalent to 40× (scale bar, 100 μm). The images were subsequently stored in the SlidePath digital imaging hub (Leica Microsystems) of the VPC. Using the Aperio Image Analysis IHC menu (Leica Biosystems), we selected five areas of interest within the same core, defined the parameter, optimized the level of intensity and selected positive pixel Count Algorithm for Ki-67.
2.6. Cell morphology, proliferation, colony formation, and apoptosis assay
The morphology of the cells were imaged by Zeiss light microscope at 32× magnification or were fixed with 4% paraformaldehyde, treated in 0.25% Triton X-100 for 15 mins, incubated with F-actin conjugated to Phalloidin-iFluor 488 (Abcam; Cambridge, UK), and mounted with DAPI staining mount (Vector Labs; Burlingame, CA, USA). Cells were then imaged by confocal microscopy at 63× magnification. To calculate the length of the cell bodies, 50 cells were randomly selected, and the length was quantified by the Image J program. Cell proliferation assay methods were previously described [21, 25] where 2D-proliferation rates were measured every other day for 5 days post-seeding and 3D-proliferation rates measured 24-h post bromodeoxyuridine (BrdU) treatment. Colony formation assay techniques have been previously defined [25] where cells were stained with crystal violet after 10 days, and colony sizes of >100 μm were counted. Apoptosis assays have been previously detailed [24] where cells were treated with either docetaxel (Doce), camptothecin (CPT), UV irradiation, or vehicle (100% ethanol) for 48-h. Three independent biological replicates were performed for all the assays.
2.7. AmpliSeq Transcriptome Sequencing and GSEA
Transcriptome analyses were previously described [11]. Briefly, DU145(Ctrl) and DU145(SRRM4) cell models, were extracted using the mirVana RNA Isolation Kit (Ambion; Burlington, ON, Canada) according to the manufacturer's protocol and sent for AmpliSeq Transcriptome Sequencing. Three replicates were performed for each cell line. Library preparation, sequencing, and primary analyses were performed by the UBC-DMCBH Next Generation Sequencing Centre following the protocol described [33]. Transcriptome data is accessible under the accession number GSE118104 in the Gene Expression Omnibus (GEO) database.
Gene Set Enrichment Analysis (GSEA) (http://www.broadinstitute.org/gsea/index.jsp) was carried out to determine whether a defined set of genes showed concordant enrichment between two samples groups (e.g. SRRM4 vs Ctrl) or two clinical phenotypes (e.g. t-NEPC with high vs low SOX2 expression). The analyses were performed using the latest MSigDB database for each gene set collection or using gene sets curated based on published data. All GSEA analyses in this study used whole transcriptomic data with a p-value cut-off as 0.01. Heatmaps were constructed based on the results of the AmpliSeq data.
2.8. DNA and siRNA transfections
Cells were transfected with ON-TARGETplus SMARTpool (Dharmacon; Lafayette, CO, USA) siRNA targeting SRRM4 (catalogue #J-058651-10) or SOX2 (catalogue #L-011778-00-0005), or with non-targeting negative control siRNA using Lipofectamine 3000 (Invitrogen; Waltham, Massachusetts, USA) according to manufacturer's protocol. Lipofectamine 3000 was also used for SRRM4-expression plasmid (under a pCMV2 promoter) and empty control vector transfections. Plasmids and transfection protocols have previously been described [21, 24, 25].
2.9. Clinical, PDX, and GEMM datasets
Clinical cohorts used in this study includes the following: RNA-seq data for the Beltran et al. 2016 cohort (CRPC-Ad, n = 34; t-NEPC, n = 15) was from Weill Medical College of Cornell University [9]; RNA-seq data for the Robinson et al. 2015 cohort (CRPC, n = 118) [34] and microarray and clinical data for the Grasso et al. 2012 cohort (CRPC, n = 31) [35] were accessed through cBioPortal [36, 37]; microarray data for both the Varambally et al. 2005 (CRPC, n = 6) [38] and Kumar et al. 2016 cohorts (CRPC, n = 169) [39] were accessed from the GEO database, GSE3325 and GSE77930, respectively. Microarray data from the LTL331-331R castration time-series patient-derived xenografts (PDXs) were accessed by GSE59986 [40]. Sequencing data for the genetically engineered mouse models (GEMMs) including the Ku et al. 2017 (WT, wild-type, SKO, single knock-out, DKO, double knock-out, TKO, triple knock-out) [4] and Zou et al. 2017 (NPp53) cohorts [7] were accessed from GSE90891 and GSE92721, respectively.
2.10. Statistics
All results are expressed as the mean ± SD. Statistical analyses were done using GraphPad Prism (version 6). One-way ANOVA or Student t-tests were carried out to determine differences between groups. Correlation between two expression groups were measured by Pearson's r correlation coefficient, and overall survival was measured by Kaplan-Meier. The level of significance was set at p <0.05, p <0.01, p <0.001 denoted as *, **, and ***, respectively.
3. Results
3.1. SRRM4 induces heterogeneous t-NEPC transcriptomes in multiple cell models
Because of the genomic and epigenomic heterogeneous nature of prostate tumor cells, we applied lentivirus encoding SRRM4 to transduce multiple cell lines with no endogenous SRRM4 protein expression previously reported by Li et al., (2017) [21] including prostate cancer lines (LNCaP, PC-3, DU145, 22Rv1), benign prostate epithelial lines (BPH-1 and RWPE-1), prostate stromal line (WPMY-1), and uterine smooth muscle line (hTERT-Myo) (Fig. S1a). SRRM4 induced a previously reported NEPC-unique RNA splicing signature [21] in all of the eight cell models (Fig. 1a), suggesting that SRRM4 is functionally active as a RNA splicing factor in these cells. Furthermore, SRRM4 induced an overall increase of multiple NE markers including ASCL1, CHD2, SCG3, SYP, and SYT4 and decrease of luminal epithelial (LE) markers including CDH1 (E-cad), KRT8, and Nkx3.1 (Fig. 1b). However, the extent of the changes in NE and LE marker expression varied as shown by real-time qPCR and immunoblotting assays (Fig. 1b-c). These results indicate that although SRRM4 can activate a global NEPC-specific RNA splicing program, the subsequent induction of a NEPC-unique transcriptome is heterogeneous, which may be explained by the different genetic and epigenetic backgrounds of these cell models as previously reported [[41], [42], [43]].
Fig. 1.

SRRM4 induces heterogeneous t-NEPC transcriptomes in multiple cell models. Total RNA was extracted from the AdPC (LNCaP, PC-3, DU145, 22Rv1), benign prostate epithelial (BPH-1 and RWPE-1), prostate stromal (WPMY-1) and uterine smooth muscle (hTERT-Myo) generated stable cell lines overexpressing SRRM4 or empty vector control. Real-time qPCR was then performed to measure (a) the RNA splicing activity of SRRM4 in promoting the expression of neural-specific variants and decreasing expression levels of the respective constitutive variants and (b) the expression of LE and NE markers in SRRM4-overexpressing cells compared to that of the control cells. All heat maps represent the relative fold change in log10. (c) Protein lysates from all stable cell models were used to measure E-Cad, SYP, and vinculin protein levels by immunoblotting assays. Three independent biological replicates were performed for each experiment. AdPC, prostate adenocarcinoma; LE, luminal epithelial; NE, neuroendocrine; SYP, synaptophysin; E-Cad, E-cadherin.
3.2. Multifarious histological features of SRRM4-transduced prostate cancer xenografts
SRRM4 was incapable of inducing tumorigenesis of RWPE-1 cells that were inoculated into nude mice, indicating that SRRM4 is not essential for the oncogenesis of normal prostate cells—at least not within the 14-week duration of our experiment. However, SRRM4-transduced AdPC cell-derived xenografts presented differential phenotypes (Fig. 2). While all tumors were confirmed to express exogenous SRRM4 using RISH assays (Fig. S1b), since no commercially available antibodies effectively recognize SRRM4 via IHC, E-cad was reduced in the SRRM4-transduced DU145 and PC-3 tumors, but not the 22Rv1 and LNCaP tumors (Fig. 2a). Pan-cytokeratin levels were only robustly reduced in the SRRM4-transduced DU145, or DU145(SRRM4), tumors. In contrast, all xenografts strongly expressed the NE marker, SYP. The most striking difference that separated the DU145(SRRM4) tumors from the others was that the tumor cells grew in high density within the tumor mass with minimal stromal components (Fig. 2b). All DU145(SRRM4) tumor cells were strongly Ki-67 positive. The Ki-67 index in DU145(SRRM4) tumors were 3-fold higher than that of all other xenografts (Fig. 2c). This phenomenon is highly similar to NCI-H660 tumors, a known NEPC model. These findings suggest that SRRM4 confers DU145 cells strong anti-apoptotic and proliferative properties even under a hypoxic microenvironment with minimal stroma support and limited nutritional supply.
Fig. 2.

Multifarious histological features of SRRM4-transduced prostate cancer xenografts. DU145, PC-3, LNCaP, and 22Rv1 stable cell lines overexpressing SRRM4 or its respective control cells were inoculated in bilateral flanks subcutaneously in 6–8-week-old nude mice (n = 3 mice per cell line). Mice were euthanized when tumor burdens reached experimental endpoint (volume of 1500 mm3) and the tumors were harvested, fixed, paraffin embedded, and cut for IHC detection against (a) E-cad, pan-cytokeratin, SYP, and (b-c) Ki-67. (c) Previously processed NCI-H660 xenografts were also cut for IHC detection against Ki-67. Ki-67 was quantified by using the Aperio Image Analysis IHC menu (Leica Biosystems) and the results are represented on the dot plot. Results are presented as the mean ± SD (One-way ANOVA; n = 5; ***denotes p <0.001). Scale bars represent 100 μm. IHC, immunohistochemistry; E-Cad, E-cadherin; SYP, synaptophysin.
3.3. Differential cellular impacts of prostate cancer cell models by SRRM4
The cellular impacts of SRRM4 on multiple cell models were also studied. In contrast to PC-3, LNCaP, and 22Rv1 cells, only DU145(SRRM4) cells showed dramatic morphological changes (Fig. 3a). When cultured under 2D-conditions, a reduction of the cytoplasmic to nuclear ratio and 5-fold reduction in size of the cell bodies were observed in the DU145(SRRM4) cells compared to that of its respective control, DU145(Ctrl), cells. Dendritic projections similar to that of neuronal cells were also observed, exemplified by F-actin staining. Consistent with the high Ki-67 index of DU145(SRRM4) xenografts, SRRM4 stimulated proliferation of DU145 cells, but not the other cell models under 2D-conditions (Fig. 3b). For the reason that only the DU145(SRRM4) cell and xenograft models (designated as the DuNE models) have thus far shown unique characteristics of t-NEPC, we performed further functional assays on the DuNE cell model. DuNE cells formed spheroids on matrigel and showed stronger cell proliferation measured by BrdU incorporation assays when compared to that of the DU145(Ctrl) cells (Fig. 3c). Furthermore, they formed 3-times the number of colonies on soft agar compared to the DU145(Ctrl) cells (Fig. 3d). When challenged with apoptosis inducers including UV irradiation or chemotherapy reagents Doce and CPT, the DuNE cells showed stronger anti-apoptotic properties than that of their control cells, evidenced by the levels of cleaved PARP (c-PARP) (Fig. 3e). These results indicate that the DuNE cell model presents cellular phenotypes similar to that of t-NEPC tumor cells.
Fig. 3.

Differential cellular impacts of prostate cancer cell models by SRRM4. (a) The morphology of cells was imaged by Zeiss light microscope, where the scale bars represent 20 μm. Additionally, the DU145(SRRM4), or DuNE, cells and its control cells, DU145(Ctrl), were fixed for IF against F-actin using anti-F-actin antibody conjugated to Phalloidin-iFluor 488 and mounted with DAPI staining mount. Cells were then imaged by confocal microscopy, where the scale bar represents 10 μm. 50 cells were randomly selected to measure the average cell body length in μm in the Image J program and presented on the bar graph. (b) Cells were seeded in a 96-well plate for MTS assays to determine cell viability over a 5-day time course. The DuNE cells and its respective control cells, were seeded in (c) matrigel for 3D BrdU proliferation assays, (d) soft agar for 10-day-long colony formation assays (where colonies >100 μm were counted), or (e) 10cm2 dish and treated the next day with Doce (20 nM), CPT (60 nM), vehicle (100% ethanol), or 10 mJ/cm2 of UV irradiation for 48 h. The protein lysates were then used to measure c-PARP and vinculin protein levels by immunoblotting assays. Three independent biological replicates were performed for each experiment. All results are presented as the mean ± SD (Student t-test; n = 3; ***denotes p <0.001). IF, immunofluorescence; UV, ultra violet; CPT, camptothecin; Doce, docetaxel; c-PARP, cleaved PARP; OD, optical density.
3.4. DuNE cells possess a pluripotency gene network distinct from the LnNE model
To decipher signaling networks unique to DuNE, we performed Ion AmpliSeq Transcriptome analyses. There was a total of 3339 genes that were differentially upregulated and 2694 genes that were downregulated in the DuNE cells when compared to the DU145(Ctrl) cells. GSEA revealed that the DuNE transcriptome was significantly enriched with gene sets related to lineage plasticity, cell survival, neuron differentiation, and neuronal-specific cell morphogenesis (Fig. 4a). For example, SRRM4-target genes were enriched with gene ontology (GO) terms of stem cell differentiation, neuron differentiation, and cell morphogenesis in neuron differentiation, but were negatively associated with the hallmark apoptosis gene set. These findings indicate that SRRM4 can re-program the AdPC phenotype of DU145 cells towards a neuronal phenotype by activating a pluripotency gene network.
Fig. 4.

DuNE cells possess a pluripotency gene network distinct from the LnNE model. (a) Transcriptome data of the DuNE, or DU145(SRRM4), cell model and its respective control, DU145(Ctrl) (p-value cut-off as 0.01) were analyzed by GSEA based on the latest MSigDB database for each collection. GSEA revealed the enrichment of genes associated with lineage plasticity, cell survival, neural fate, and cell morphology in the DuNE cells. (b) DEGs (p-value cut-off as 0.01) caused by SRRM4 transduction were compared between the DuNE (n = 6063) and LnNE (n = 5925) cell models. GSEA show more enrichment of the lineage plasticity and cell survival-associated genes in distinct DEGs (n = 3883) mediated by SRRM4 in the DuNE model. (c) Thirteen SC- or pluripotency-related genes were selected from the leading-edge groups associated with the DuNE phenotype in the GSEA gene sets under the ‘lineage plasticity’ subgroup from subfigure 4A. The mRNA expression of these genes in the SRRM4-overexpressing DU145, PC-3, LNCaP, and 22Rv1 cell models were compared to that of their respective control cells via real-time qPCR. Heat maps represent the relative fold change in log10. GSEA, gene set enrichment analysis; DEGs, differential expression genes; GO, gene ontology; NES, normalized enrichment score; FDR, false discovery rate; CSS, charcoal-stripped serum; SC, stem cell.
Further analyses were performed to compare the transcriptomes of the DuNE and LnNE cell models (Fig. 4b). GSEA revealed that the 3883 genes unique to the DuNE cell model were uniquely enriched with gene sets related to SC differentiation, neuronal SC population maintenance, anti-apoptosis, and neural precursor cell proliferation. Furthermore, we extracted the leading-edge subsets of genes associated with DuNE phenotype from the three GSEA gene sets under the “lineage plasticity” group from Fig. 4A (Fig. S2). Of the 59 stem differentiation and SC-related genes, we validated the mRNA expressions of 13 genes including SOX2 in the SRRM4-transduced DU145, PC-3, LNCaP, and 22Rv1 cell models by real-time qPCR (Fig. 4c). These results demonstrate that SRRM4 regulates different transcriptomes to promote the transformation of DU145 and LNCaP AdPC cells into t-NEPC xenografts, and that SRRM4 regulates a pluripotency gene network in the DuNE model.
3.5. SRRM4 enhances SOX2 expression in the DuNE cell model and in some GEMMs and PDX models
SOX2 was selected to further delineate the SRRM4-regulated pluripotency gene network as SOX2 was recently demonstrated to promote the lineage plasticity of AdPC cells during progression to t-NEPC [5]. SOX2 protein expression was only detected in the DuNE cells (Fig. 5a). We showed that the upregulation of SOX2 by SRRM4 was in both time- and dose-dependent manners when SRRM4-expressing plasmids were transiently transfected into DU145 cells (Fig. 5b). Moreover, SOX2 was downregulated when in DuNE cells were challenged with SRRM4-targeted siRNA (Fig. 5c), and SOX2 depletion resulted in the reduction of several pluripotency gene expressions and NE markers and induction of LE markers (Fig. 5d). The LTL331/331R PDXs that model the progression of AdPC to t-NEPC after castration surgery also showed positive correlations of SRRM4 with SOX2 and SYP, but negative correlations with AR expression (Fig. 5e). Furthermore, analyses of the recently reported WT/SKO/DKO/TKO GEMMs by Ku et al. (2017) [4] revealed that SOX2 was strongly and positively correlated with SRRM4 (Fig. 5f), and negatively associated with AR (Fig. S3a). This negative association of SOX2 with AR was also observed in four clinical CRPC patient cohorts (Fig. S3b). However, SRRM4 does not induce SOX2 expression in the NPp53 GEMMs by Zou et al. (2017) [7] (Fig. 5g), as well as the SRRM4-transduced PC-3, LNCaP, and 22Rv1 cell models, suggesting that the activation of the SRRM4-SOX2 axis does not apply to all prostate cancer models exhibiting t-NEPC features. Together our findings support that SOX2 is a major component of the pluripotency network regulated by SRRM4 in a subset of t-NEPC tumors.
Fig. 5.

SRRM4 enhances SOX2 expression in the DuNE cell model and in some GEMMs and PDX models. (a) Protein lysates from the SRRM4-overexpressing DU145, PC-3, LNCaP, and 22Rv1 cell models and their respective controls were used to measure SOX2 and vinculin protein levels by immunoblotting assays. (b) For the time-dependent studies, DU145 cells were transiently transfected with 4 μg of SRRM4 or control plasmids, and total RNA was extracted at times 0, 24, and 48 h. For the dose-dependent studies, DU145 cells were transiently transfected with 0, 2, and 4 μg of SRRM4 or control plasmids, and total RNA was extracted after 24 h. SOX2 expression was detected by real-time qPCR. (c-d) DuNE, or DU145(SRRM4), cells were transiently transfected with siRNA targeting SRRM4 (c), or SOX2 (d) and extracted for RNA following transfection to detect expression levels of indicated genes. All results are presented as the mean ± SD (Student t-test; n = 3; * denotes p <0.05, **p <0.01, and ***p <0.001). (e) The expressions of SRRM4, SOX2, AR, and SYP during the progression of AdPC (LTL331) to t-NEPC (LTL331R_NE) after castration in the PDX LTL331/331R model are shown. (c) SRRM4 and SOX2 expressions in WT/SKO/DKO/TKO GEMMs (Ku et al., 2017) are shown as well as the Pearson's r correlation coefficient between SRRM4 and SOX2 expressions. All results are presented as the mean ± SD (Student t-test; WT/SKO, n = 8; TKO/DKO, n = 19; *** denotes p <0.001). (g) SRRM4 and SOX2 expressions in different phenotypic subcategories of the NPp53 GEMMs (Zou et al., 2017) are shown as well as the Pearson's r correlation coefficient between SRRM4 and SOX2 expressions. All results are presented as the mean ± SD (One-way ANOVA; AdPC, n = 8; Focal NE, n = 3; Overt NE, n = 2; *** denotes p <0.001, and “ns” denotes not significant). GEMMs, genetically engineered mouse models; AdPC, prostate adenocarcinoma; WT, wild-type; SKO, single knock-out; DKO, double knock-out; TKO, triple knock-out; NE, neuroendocrine. PDX, patient-derived xenograft; AdPC-to-t-NEPC, prostate adenocarcinoma to treatment-induced neuroendocrine prostate cancer; d, day; wk., week; Cx, castration; NE, neuroendocrine.
3.6. The DuNE model represents a subset of clinical t-NEPC tumors with high SOX2 expression
To define the clinical relevance of SOX2, we found that high SOX2 expression was correlated with poor prognosis in patients after receiving hormone therapies in the Grasso et al. (2012) [35] CRPC cohort (Fig. 6a). When exploring the RNA-seq data of four patient cohorts reported by Beltran et al. (2016) [9], Robinson et al. (2015) [34], Kumar et al. (2016) [39], and Varambally et al. (2016) [38], we observed an overall positive association of SOX2 with SRRM4 expression (Fig. S4). However, we found markedly distinct expressions of SOX2 within the t-NEPC tumors from the Beltran et al. (2016) [9] cohort, the largest t-NEPC cohort containing pathological evaluation of each sample. There were 5 t-NEPC tumors with low SOX2 expression, t-NEPC(SOX2low), similar to that of CRPC-Ad tumors, while the remaining 10 t-NEPC tumors expressed a ~17.5-fold (log2) increase of SOX2 levels, t-NEPC(SOX2high) (Fig. 6b). These findings demonstrate that different subgroups of t-NEPC tumors exist, where a division of two subgroups is evident based a robust difference in SOX2 expression levels.
Fig. 6.

The DuNE model represents a subset of clinical t-NEPC tumors with high SOX2 expression. (a) Overall survival Kaplan-Meier between low and high expression of SOX2 in CRPC tumors from the Grasso et al. (2012) cohort. (b) SOX2 expression was compared among CRPC tumor samples from the Beltran et al. (2016) cohort and separated into three groups, CRPC-Ad (n = 34), t-NEPC expressing low level of SOX2, t-NEPC(SOX2low) (n = 5), and t-NEPC expressing high level of SOX2, t-NEPC(SOX2high) (n = 10). Results are presented as the mean ± SD (One-way ANOVA; *** denotes p <0.001, and “ns” denotes not significant). Expressions of SOX2 and SRRM4 in each patient sample (patient ID: “WCMC-”) are indicated as high, red, or low, blue, where the median value of the SRRM4 datasets define the cut-off. (c) GSEA revealed enrichment of the DU145(SRRM4), or DuNE, model with that of the differentially expressed top 500 genes in the 1) t-NEPC(SOX2high) tumors when compared with CRPC-Ad or 2) t-NEPC(SOX2low) and in the 3) t-NEPC(SOX2low) tumors compared with CRPC-Ad. (d) GSEA revealed the enrichment of the t-NEPC(SOX2high) tumors with well-defined gene sets named “Wong Embryonic stem cell core,” “Sarrio EMT up,” and “Benporath Proliferation,” where the leading-edge genes presented a consistent expression pattern in the DuNE cell model. (e) Six t-NEPC patient tumors were used to measure SRRM4 and SOX2 expression by applying RISH and IHC assays, respectively. Due to limited space, two SOX2-positive and two SOX2-negative tumor cores are shown. Scale bars represent 100 μm. CRPC-Ad, castration-resistant prostate adenocarcinoma; t-NEPC, treatment-induced neuroendocrine prostate cancer; GSEA, gene set enrichment analysis; NES, normalized enrichment score; FDR, false discovery rate; EMT, epithelial-to-mesenchymal transition; RISH, RNA in situ hybridization; IHC, immunohistochemistry; SYP, synaptophysin.
To determine the clinical relevance of the DuNE model to the t-NEPC(SOX2high) subgroup, we applied GSEA to compare DuNE transcriptome to that of: 1) the top 500 differentially expressed genes between t-NEPC(SOX2high) and CRPC-Ad, 2) the top 500 differentially expressed genes between t-NEPC(SOX2high) and t-NEPC(SOX2low), and 3) the top 500 genes differentially expressed between t-NEPC(SOX2low) and CRPC-Ad (Fig. 6c). We observed strong enrichment of the DuNE transcriptome with that of the t-NEPC(SOX2high) subgroup. Moreover, GSEA revealed that the t-NEPC(SOX2high) subgroup was enriched with gene sets associated with pluripotency and proliferation, where the leading-edge genes presented a consistent expression pattern in the DuNE cell model (Fig. 6d). Furthermore, this SRRM4-SOX2 axis was also seen in t-NEPC patients that have been previously characterized [24] (Fig. 6e). In the six t-NEPC patient tumors, all were SRRM4 positive but only four were SOX2 positive. Collectively, these findings recognize two distinct subgroups of t-NEPC tumors classified by SOX2 expression. Although both subgroups have relatively high levels of SRRM4, our results support that SRRM4 may drive t-NEPC development through different mechanisms (Fig. 7). The DuNE model represents a clinically relevant model to study SRRM4-SOX2 axis during t-NEPC progression.
Fig. 7.
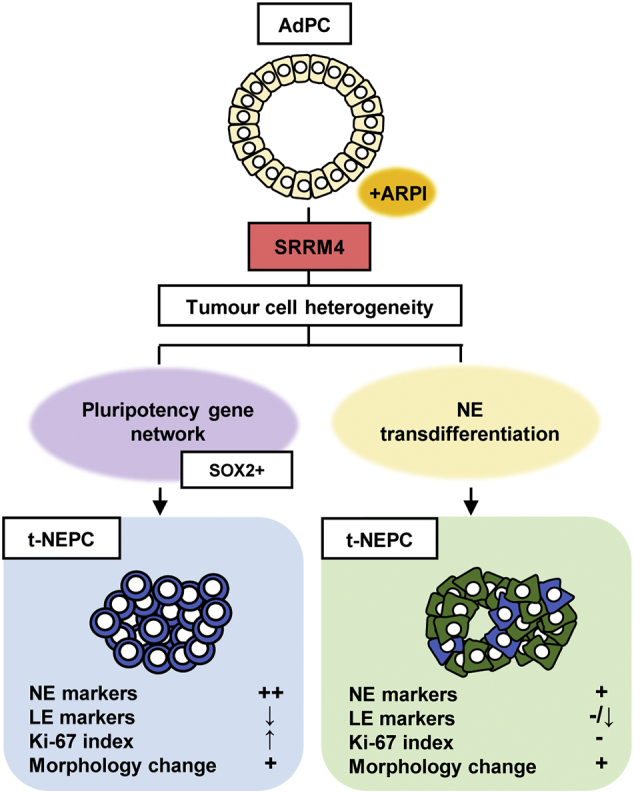
Proposed models for SRRM4 in driving t-NEPC development through different mechanisms. A schematic diagram illustrates two mechanisms of SRRM4 in driving the development of t-NEPC tumors. AdPC, prostate adenocarcinoma; t-NEPC, treatment-induced neuroendocrine prostate cancer; ARPI, androgen receptor pathway inhibitors; NE, neuroendocrine; LE, luminal epithelial.
4. Discussion
This study identifies a novel mechanism by which SRRM4 transforms AdPC cells into t-NEPC xenografts through a pluripotency gene network containing SOX2. Importantly, this newly discovered SRRM4-SOX2 signaling axis can be stratified from a subgroup of t-NEPC in patients, emphasizing its clinical significance. Additionally, we have established a new NEPC cell and xenograft model (DuNE), which recapitulates the phenotypes of clinical t-NEPC tumors expressing stem-like characteristics. Together with our previously reported LnNE model, we propose that SRRM4 can drive the transformation of AdPC into t-NEPC tumors through either a NE transdifferentiation pathway or a pluripotency gene network. Whether a pluripotency gene network is initiated by SRRM4 likely relies on the epigenetic and genomic heterogeneity of AdPC cells. These studies highlight the complexity of SRRM4 signaling in driving tumor progression to t-NEPC to develop therapy resistance.
Heterogeneous genomic features of cancer cells within an AdPC tumor may contribute to the activation of the SRRM4-SOX2 signaling pathway in the progression of t-NEPC. Upregulation of SRRM4 and SOX2 expressions only occurs in the Pten/Rb1 DKO and Pten/Rb1/Trp53 TKO GEMMs [4], but not in the WT and Pten SKO GEMMs (Fig. 5f), suggesting that Rb1-inactivation is necessary for the activation of the SRRM4-SOX2 signaling to confer the lineage plasticity of LE cells to a NE cell lineage. While the Pten/Trp53 DKO NPp53 GEMMs show either focal or overt NE differentiation [7], the upregulation of SRRM4 does not concur with SOX2 stimulation (Fig. 5g), indicating that the Trp53-inactivation is not sufficient in activating the SRRM4-SOX2 signaling axis. However, a recent study has shown that an increase in SOX2 expression was observed in PTEN-deficient LNCaP cells with TP53- and RB1-inactivations, while SRRM4 remained negative in these cells [5]. Although DU145 cells are also TP53- and RB1-deficient cells, RB1 silencing to SRRM4-transduced TP53-null PC-3 cells do not show similar phenotypes to the DuNE model (data not shown). Collectively, these findings demonstrate that the activation of the SRRM4-SOX2 signaling axis cannot be solely explained by loss-of-function of PTEN, TP53 and RB1. This suggests that other complex mechanisms in addition to the abovementioned genomic factors also contribute to the t-NEPC progression driven by a SRRM4-mediated pluripotency gene network.
Epigenetic mechanisms may be responsible for SRRM4 and SOX2 activation. CBX8 and EZH2 are the two most overexpressed epigenetic regulators in t-NEPC clinical samples and PDXs [2, 9, 14]. It is known that EZH2 is implicated in t-NEPC development [2, 15, 44]. Intensive H3K27Me3 covers the coding regions of the SRRM4 and SOX2 genes in LNCaP cells and prevents the transcription of these genes. Furthermore, it has been reported that LNCaP cells cultured in an androgen-free neural/neural crest stem medium can redirect the cells into a neural-fate possibly via EZH2-mediated epigenetic reprogramming [45], indicating the importance of the tumor microenvironment in regulating tumor cell plasticity [2, 45, 46]. The DU145 cell line is originally derived from an AdPC metastasis legion to the brain [47], suggesting that the neural microenvironment of the cell niche may have altered the epigenome of the DU145 cells to be predisposed to a NE lineage switch. In addition, although both DU145 and PC-3 cells exhibit stem-like phenotypes and properties such as expressions of SC markers and self-renewal capabilities [2, 48, 49], only the SRRM4-transduced DU145 cells undergo a robust NE differentiation via the activation of a pluripotency gene network containing SOX2. These findings indicate that the DU145 cells possess unique epigenetic features that may contribute to the SRRM4 and SOX2 signaling axis, although the connection between epigenetic factors and the SRRM4-SOX2 axis warrant further investigations.
Currently, there are limited t-NEPC cell and xenograft models that can be utilized to efficiently manipulate signaling pathways and study molecular mechanisms of t-NEPC development. The only NEPC cell line, NCI-H660, is very difficult to be transfected or transduced by lentivirus and has a doubling time of >100 h [50]. More importantly, it is unclear whether this cell line was derived from AdPC cells or primary NEPC cells. The five newly developed LnNE xenografts, replicate the prolonged transition process of AdPC progression to t-NEPC in an in vivo environment [20]. However, compared to the LnNE model, the DuNE model has several advantages. The DuNE cell model develops into xenografts faster than the LnNE model and exhibits more dramatic changes in LE and NE marker expressions. Additionally, the DuNE model is AR- and PSA-negative, TP53- and RB1-null, exhibits striking neuronal-like morphologies, and mimics the molecular signatures of a subset of clinical t-NEPC (i.e. small-cell prostatic carcinoma) that present stem-like characteristics as previously demonstrated [2, 51, 52]. Moreover, plasmid transfection efficiency can be achieved as high as 80%, and these cells have a doubling time of ~15–20 h in 2D-conditions. DuNE cells can be adapted into 3D-culture formats (such as petri dishes and hanging-drop systems) and studied as xenografts in immunocompromised mice, all systems of which are suitable for high throughout drug screening.
In summary, our study reports a novel SRRM4-driven mechanism underlying lineage plasticity where lineage switching is primarily mediated by a pluripotency network including SOX2, a key regulator of dedifferentiation and neural lineage-specific fate. Furthermore, the newly established DuNE model can also serve as a useful t-NEPC model to study the complexity of AdPC progression to t-NEPC.
Availability of data and materials
The data generated and analyzed during this study are available upon reasonable request from the corresponding author.
Acknowledgments
Acknowledgements
The authors would like to thank Mary Bowden and Dr. Ladan Fazli for their technical expertise.
Funding sources
This study was supported by the Canadian Institutes of Health Research (MOP-137007 & PTJ156150; X. Dong), the National Nature Science Foundation of China (81571432; Y. Tang), and a visiting scholarship from the China Scholar Council (201606370204; Y. Gan).
Conflicts of interest
The authors declare no conflict of interest.
Authors contributions
ARL and YG were the lead co-investigators in the conception and design of the work. All authors (ARL, YG, YT, and XD) revised the work critically for important intellectual content and contributed to manuscript revision, read, and approved the submitted version.
Disclosure statement
The authors have nothing to disclose.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.08.011.
Contributor Information
Ahn R. Lee, Email: alee@prostatecentre.com.
Yu Gan, Email: ygan@prostatecentre.com.
Yuxin Tang, Email: tangyuxin@csu.edu.cn.
Xuesen Dong, Email: xdong@prostatecentre.com.
Appendix A. Supplementary data
Supplementary material: Fig. S1 Validation of SRRM4 expressions in the SRRM4-transduced stable cell and xenograft models. Lentiviral transduction to create SRRM4-overexpressing stable lines in hTERT-Myo, RWPE-1, WPMY-1, BPH-1, DU145, PC-3, LNCaP, and 22Rv1 parental cells were validated by (a) Western blot and real-time qPCR. Anti-flag antibody was used to detect against exogenous SRRM4 and anti-vinculin antibody was used to detect the loading control, vinculin. This validation was repeated three times, and only one representative repeat is presented. All results are presented as the mean ± SEM. (b) DU145, PC-3, LNCaP, and 22Rv1 stable cell lines overexpressing SRRM4 or its respective control cells were inoculated in bilateral flanks subcutaneously in 6–8-week-old nude mice. Tumors were harvested, fixed in 10% paraformaldehyde, paraffin embedded, and cut for RNA in situ hybridization using an anti-sense probe to detect against SRRM4 mRNA. Scale bars represents 100 μm.
Fig. S2 SRRM4 induces stem cell differentiation and neural fate factors in DU145 cells. GSEA enrichment plots shows the correlations of the DU145(SRRM4), DuNE, vs DU145(Ctrl) dataset (n = 6033) with the GSEA gene sets in the ‘lineage plasticity’ subgroup from subfigure 4a. Thirteen stem cell marker or stem cell-related genes were selected from the leading-edge groups associated with the DuNE phenotype in the three GSEA enrichment plots to validate in subfigure 4C. GSEA, gene set enrichment analysis; GO, gene ontology; NES, normalized enrichment score; FDR, false discovery rate.
Fig. S3 SOX2 and AR are negatively correlated in CRPC cohorts. (a-b) Pearson's r correlation coefficient between SOX2 and AR expressions obtained from GEMMs by Ku et al. (2017) (a), and human CRPC cohorts by Beltran et al. (2016), Robinson et al. (2015), Kumar et al. (2016), and Varambally et al. (2005) (b) are shown. GEMMs, genetically engineered mouse models; CRPC, castration-resistant prostate cancer.
Fig. S4 SRRM4 and SOX2 are positively correlated in CRPC cohorts. Pearson's r correlation coefficient between SRRM4 and SOX2 expressions obtained from human CRPC cohorts by Beltran et al. (2016), Robinson et al. (2015), Kumar et al. (2016), and Varambally et al. (2005) are shown. CRPC, castration resistant prostate cancer.
References
- 1.Bishop J.L., Davies A., Ketola K., Zoubeidi A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocr Relat Cancer. 2015;22(3):R165–R182. doi: 10.1530/ERC-15-0137. [DOI] [PubMed] [Google Scholar]
- 2.Davies A.H., Beltran H., Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol. 2018;15(5):271–286. doi: 10.1038/nrurol.2018.22. [DOI] [PubMed] [Google Scholar]
- 3.Lee A.R., Che N., Lovnicki J.M., Dong X. Development of Neuroendocrine Prostate Cancers by the Ser/Arg Repetitive Matrix 4-Mediated RNA Splicing Network. Front Oncol. 2018;8:93. doi: 10.3389/fonc.2018.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ku S.Y., Rosario S., Wang Y. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mu P., Zhang Z., Benelli M. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355(6320):84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Z., Flesken-Nikitin A., Corney D.C. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66(16):7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 7.Zou M., Toivanen R., Mitrofanova A. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017;7(7):736–749. doi: 10.1158/2159-8290.CD-16-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kregel S., Kiriluk K.J., Rosen A.M. Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PloS One. 2013;8(1) doi: 10.1371/journal.pone.0053701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beltran H., Prandi D., Mosquera J.M. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin D., Wyatt A.W., Xue H. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74(4):1272–1283. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
- 11.Chen R., Li Y., Buttyan R., Dong X. Implications of PI3K/AKT inhibition on REST protein stability and neuroendocrine phenotype acquisition in prostate cancer cells. Oncotarget. 2017;8(49):84863–84876. doi: 10.18632/oncotarget.19386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin T.P., Chang Y.T., Lee S.Y. REST reduction is essential for hypoxia-induced neuroendocrine differentiation of prostate cancer cells by activating autophagy signaling. Oncotarget. 2016;7(18):26137–26151. doi: 10.18632/oncotarget.8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X., Coleman I.M., Brown L.G. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clinical Cancer Research. 2015;21(20):4698–4708. doi: 10.1158/1078-0432.CCR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clermont P.L., Lin D., Crea F. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin Epigenetics. 2015;7:40. doi: 10.1186/s13148-015-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dardenne E., Beltran H., Benelli M. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell. 2016;30(4):563–577. doi: 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox M.E., Deeble P.D., Lakhani S., Parsons S.J. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res. 1999;59(15):3821–3830. [PubMed] [Google Scholar]
- 17.Mori S., Murakami-Mori K., Bonavida B. Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem Biophys Res Commun. 1999;257(2):609–614. doi: 10.1006/bbrc.1999.0515. [DOI] [PubMed] [Google Scholar]
- 18.Whitesell L., Mimnaugh E.G., De Costa B., Myers C.E., Neckers L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91(18):8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan T.C., Veeramani S., Lin F.F. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer. 2006;13(1):151–167. doi: 10.1677/erc.1.01043. [DOI] [PubMed] [Google Scholar]
- 20.Li Y., Chen R., Bowden M. Establishment of a neuroendocrine prostate cancer model driven by the RNA splicing factor SRRM4. Oncotarget. 2017;8(40):66878–66888. doi: 10.18632/oncotarget.19916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y., Donmez N., Sahinalp C. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur Urol. 2017;71(1):68–78. doi: 10.1016/j.eururo.2016.04.028. [DOI] [PubMed] [Google Scholar]
- 22.Kim J., Jin H., Zhao J.C. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene. 2017;36(28):4072–4080. doi: 10.1038/onc.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimojo M., Shudo Y., Ikeda M., Kobashi T., Ito S. The small cell lung cancer-specific isoform of RE1-silencing transcription factor (REST) is regulated by neural-specific Ser/Arg repeat-related protein of 100 kDa (nSR100) Mol. Cancer Res. 2013;11(10):1258–1268. doi: 10.1158/1541-7786.MCR-13-0269. [DOI] [PubMed] [Google Scholar]
- 24.Gan Y., Li Y., Long Z. Roles of Alternative RNA Splicing of the Bif-1 Gene by SRRM4 During the Development of Treatment-induced Neuroendocrine Prostate Cancer. EBioMedicine. 2018;31:267–275. doi: 10.1016/j.ebiom.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee A.R., Li Y., Xie N. Alternative RNA splicing of the MEAF6 gene facilitates neuroendocrine prostate cancer progression. Oncotarget. 2017;8(17):27966–27975. doi: 10.18632/oncotarget.15854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calarco J.A., Superina S., O'Hanlon D. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138(5):898–910. doi: 10.1016/j.cell.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 27.Quesnel-Vallieres M., Irimia M., Cordes S.P., Blencowe B.J. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 2015;29(7):746–759. doi: 10.1101/gad.256115.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raj B., Irimia M., Braunschweig U. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol Cell. 2014;56(1):90–103. doi: 10.1016/j.molcel.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irimia M., Weatheritt R.J., Ellis J.D. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159(7):1511–1523. doi: 10.1016/j.cell.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L.L., Xie N., Sun S., Plymate S., Mostaghel E., Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33(24):3140–3150. doi: 10.1038/onc.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Y., Liu L., Xie N. Expression and function of the progesterone receptor in human prostate stroma provide novel insights to cell proliferation control. J Clin Endocrinol Metab. 2013;98(7):2887–2896. doi: 10.1210/jc.2012-4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan M., Li H., Bao L., Li M., Lye S., Dong X. In Vivo Evidence of the Androgen Receptor in Association With Myometrial Cell Proliferation and Apoptosis. Reprod Sci. 2016;23(2):264–271. doi: 10.1177/1933719115602771. [DOI] [PubMed] [Google Scholar]
- 33.Li W., Turner A., Aggarwal P. Comprehensive evaluation of AmpliSeq transcriptome, a novel targeted whole transcriptome RNA sequencing methodology for global gene expression analysis. BMC Genomics. 2015;16:1069. doi: 10.1186/s12864-015-2270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson D., Van Allen E.M., Wu Y.M. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grasso C.S., Wu Y.M., Robinson D.R. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerami E., Gao J., Dogrusoz U. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J., Aksoy B.A., Dogrusoz U. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varambally S., Yu J., Laxman B. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8(5):393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Kumar A., Coleman I., Morrissey C. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akamatsu S., Wyatt A.W., Lin D. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015;12(6):922–936. doi: 10.1016/j.celrep.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Ehrich M., Turner J., Gibbs P. Cytosine methylation profiling of cancer cell lines. Proc Natl Acad Sci U S A. 2008;105(12):4844–4849. doi: 10.1073/pnas.0712251105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massie C.E., Mills I.G., Lynch A.G. The importance of DNA methylation in prostate cancer development. J Steroid Biochem Mol Biol. 2017;166:1–15. doi: 10.1016/j.jsbmb.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 43.van Bokhoven A., Varella-Garcia M., Korch C. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57(3):205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 44.Kleb B., Estecio M.R., Zhang J. Differentially methylated genes and androgen receptor re-expression in small cell prostate carcinomas. Epigenetics. 2016;11(3):184–193. doi: 10.1080/15592294.2016.1146851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nouri M., Caradec J., Lubik A.A. Therapy-induced developmental reprogramming of prostate cancer cells and acquired therapy resistance. Oncotarget. 2017;8(12):18949–18967. doi: 10.18632/oncotarget.14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imamura T., Uesaka M., Nakashima K. Epigenetic setting and reprogramming for neural cell fate determination and differentiation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014;369 doi: 10.1098/rstb.2013.0511. (1652) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone K.R., Mickey D.D., Wunderli H., Mickey G.H., Paulson D.F. Isolation of a human prostate carcinoma cell line (DU 145) Int J Cancer. 1978;21(3):274–281. doi: 10.1002/ijc.2910210305. [DOI] [PubMed] [Google Scholar]
- 48.Pfeiffer M.J., Schalken J.A. Stem cell characteristics in prostate cancer cell lines. Eur Urol. 2010;57(2):246–254. doi: 10.1016/j.eururo.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 49.Rybak A.P., He L., Kapoor A., Cutz J.C., Tang D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochim Biophys Acta. 2011;1813(5):683–694. doi: 10.1016/j.bbamcr.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 50.Cowley G.S., Weir B.A., Vazquez F. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Scientific data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith B.A., Sokolov A., Uzunangelov V. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc Natl Acad Sci U S A. 2015;112(47):E6544–E6552. doi: 10.1073/pnas.1518007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang D., Park D., Zhong Y. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun. 2016;7:10798. doi: 10.1038/ncomms10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material: Fig. S1 Validation of SRRM4 expressions in the SRRM4-transduced stable cell and xenograft models. Lentiviral transduction to create SRRM4-overexpressing stable lines in hTERT-Myo, RWPE-1, WPMY-1, BPH-1, DU145, PC-3, LNCaP, and 22Rv1 parental cells were validated by (a) Western blot and real-time qPCR. Anti-flag antibody was used to detect against exogenous SRRM4 and anti-vinculin antibody was used to detect the loading control, vinculin. This validation was repeated three times, and only one representative repeat is presented. All results are presented as the mean ± SEM. (b) DU145, PC-3, LNCaP, and 22Rv1 stable cell lines overexpressing SRRM4 or its respective control cells were inoculated in bilateral flanks subcutaneously in 6–8-week-old nude mice. Tumors were harvested, fixed in 10% paraformaldehyde, paraffin embedded, and cut for RNA in situ hybridization using an anti-sense probe to detect against SRRM4 mRNA. Scale bars represents 100 μm.
Fig. S2 SRRM4 induces stem cell differentiation and neural fate factors in DU145 cells. GSEA enrichment plots shows the correlations of the DU145(SRRM4), DuNE, vs DU145(Ctrl) dataset (n = 6033) with the GSEA gene sets in the ‘lineage plasticity’ subgroup from subfigure 4a. Thirteen stem cell marker or stem cell-related genes were selected from the leading-edge groups associated with the DuNE phenotype in the three GSEA enrichment plots to validate in subfigure 4C. GSEA, gene set enrichment analysis; GO, gene ontology; NES, normalized enrichment score; FDR, false discovery rate.
Fig. S3 SOX2 and AR are negatively correlated in CRPC cohorts. (a-b) Pearson's r correlation coefficient between SOX2 and AR expressions obtained from GEMMs by Ku et al. (2017) (a), and human CRPC cohorts by Beltran et al. (2016), Robinson et al. (2015), Kumar et al. (2016), and Varambally et al. (2005) (b) are shown. GEMMs, genetically engineered mouse models; CRPC, castration-resistant prostate cancer.
Fig. S4 SRRM4 and SOX2 are positively correlated in CRPC cohorts. Pearson's r correlation coefficient between SRRM4 and SOX2 expressions obtained from human CRPC cohorts by Beltran et al. (2016), Robinson et al. (2015), Kumar et al. (2016), and Varambally et al. (2005) are shown. CRPC, castration resistant prostate cancer.
Data Availability Statement
The data generated and analyzed during this study are available upon reasonable request from the corresponding author.