

Abstract
Niche modification is a process whereby the activity of organisms modifies their local environment creating new niches for other organisms. This process can have a substantial role in community assembly of gut microbial ecosystems due to their vast and complex metabolic activities. We studied the postprandial diurnal community oscillatory patterns of the rumen microbiome and showed that metabolites produced by the rumen microbiome condition its environment and lead to dramatic diurnal changes in community composition and function. After feeding, microbiome composition undergoes considerable change in its phylogenetic breadth manifested as a significant 3–5-fold change in the relative abundance of methanogenic archaea and main bacterial taxa such as Prevotella, in a manner that was independent of individual host variation and diet. These changes in community composition were accompanied by changes in pH and methane partial pressure, suggesting a strong functional connection. Notably, cross-incubation experiments combining metabolites and organisms from different diurnal time points showed that the metabolites released by microbes are sufficient to reproduce changes in community function comparable to those observed in vivo. These findings highlight microbiome niche modification as a deterministic process that drives diurnal community assembly via environmental filtering.
Introduction
Given a constant regional species pool with no dispersal limitation, microbes can assemble into communities in a deterministic manner, creating the same compositional assemblage whenever environmental conditions repeat themselves. They can also create alternative assemblages given stochastic processes that act on the community’s assembly, such as the ones caused by random drift when there is low selection pressure [1–5]. Understanding the balance between these processes and the timing in which they act on a community is a standing question in the field. Furthermore, it is extremely important to our future ability to predict or synthetically create such communities. According to the niche concept as defined by Chase and Leibold [6] in which niche “is the environmental conditions that allow a species to satisfy its minimum requirements so that the birth rate of a local population is equal to or greater than its death rate along with the set of per capita effects of that species on these environmental conditions,” species are affected by and affect their niche. For example, a microbe producing lactic acid will decrease the environmental pH, which will subsequently affect the niche of the entire community of microorganisms. In this case, changes in microbial taxonomy will result in changes in the environment, thus modifying the niche and allowing its occupation by more phylogenetically distinct and functionally suitable lineages. Alternatively, species do not make alterations to the environmental factors within their niche, and therefore community assembly in this scenario is composed from organisms that are closer phylogenetically and are more similar in their requirements. We can therefore differentiate between niche modification and niche preemption in community assembly. These two mechanisms can be distinguished by the fact that, in niche modification, species phylogeny and functionality will change substantially, whereas in niche preemption, we can expect to have species with similar functionality and thus phylogenetic proximity [3]. Understanding these two mechanisms with regard to community assembly is important, as they substantially affect ecosystem function and are therefore central to our understanding of a given ecosystem. Microbial communities present a potent model to measure such community assembly mechanisms, specifically gut communities that are tightly connected to their host’s attributes and subjected to environmental disturbances that change their composition and push them to reassemble after every such event. Nevertheless, when inferring functionality from phylogenetic distances in microbial ecosystems one should take into account that microbes from different lineages can have similar functionality as well as exchange genes via horizontal gene transfer. Nonetheless, microbial horizontal gene transfer is limited by phylogenetic distances, therefore microbial phylogeny still associates with functional cohesiveness, and this association is strengthened with the increasing phylogenetic distance between the microbial lineages [7–9].
One intriguing microbial community is the one that resides in the upper digestive tract of ruminant animals, termed the rumen microbiome. This community sustains a strict obligatory relationship with its host, enabling the latter to digest its feed [10–12]. The degree of obligatory dependence of ruminants on their microbiome for feed digestion is extraordinary in nature and is therefore intriguing to study from both ecological and evolutionary perspectives. The confined nature of this ecosystem and our ability to control and monitor it enable studying overall changes in microbiome patterns and connecting them to specific microbial group functionalities [13]. We can also pinpoint, with high accuracy, specific factors that influence this ecosystem in a deterministic manner as a function of dietary disturbances [14]. These attributes enable us to explore important questions with regard to community assembly of the rumen microbiome, in particular its assembly during diurnal changes as a function of time after feeding. Diurnal changes in the microbial community of the mammalian gut have been recently discovered in humans and mice [15, 16]. The mammalian gut microbiome displays diurnal oscillations that are governed by consumption rhythmicity [15], as well as by type of diet and gender [16]. Moreover, changes in rumen environment characteristics following feed consumption have been known for decades [17]. During the feeding cycle, the ingestion of a large meal results in carbohydrate fermentation, followed by an increase in volatile fatty acid (VFA) concentration, declining pH, and an increase in rumen temperature [18]. Welkie et al. [19] used automated ribosomal intergenic spacer analysis to track changes in the microbial community during the feeding cycle of dairy cows. They experimented on two animals eating the same diet and found large changes in the associated liquid as well as in ruminal pH and VFA concentrations. Other in vitro studies have shown changes in microbiome composition after administration of plant materials, such as switchgrass and perennial ryegrass [20, 21]. To date, there have been very few studies on the stability and assembly of the rumen microbiome on these temporal scales. Furthermore, a standing question in the field of the rumen microbiome and gut microbiome in general is what drives these compositional changes as a function of time after feeding. Here we sought to understand the changes occurring in the rumen microbiome as a function of time after feeding and their consistency relative to other environmental factors, such as different diets or individual variation. We focused on the ecological processes that shape these communities’ assembly by examining their compositional and functional levels while focusing on the pivotal metabolic process of methanogenesis.
Materials and methods
Animal handling and sampling
The experimental procedures used in this study were approved by the Faculty Animal Policy and Welfare Committee of the Agricultural Research Organization (ARO) and were in accordance with the guidelines of the Israel Council for Animal Care.
Eighteen mature lactating Israeli Holstein dairy cows (second lactation and above) were housed in a dry lot barn at the ARO dairy farm in Rishon Lezion, Israel. The cows were grouped for similar physical conditions—age in months, body weight, and daily milk production—and were at the same stage of lactation. The cows were fed diets consisting of 30% forage and 70% concentrate that differed in their forage source and chop length, as described by Shaani et al. [22], and fed a total mixed ration ad libitum, provided once a day. Two cows belonging to two different groups were excluded from the experiment due to veterinary considerations.
Isolation of solide and liqued phase microbial fractions and DNA extraction from rumen samples
Thawed rumen fluids (50 ml) were homogenized in a blender and then centrifuged for 10 min at 10,000 × g. The pellet was suspended in extraction buffer (100 mM Tris–HCl, 10 mM EDTA, 150 mM NaCl pH 8.0, 0.15% v/v Tween-80) and incubated for 1 h at 48 °C to detach particle-associated microorganisms from the rumen content. Following slow centrifugation (500 × g) for 15 min at 48 °C, the microbiome-containing supernatant was filtered through eight layers of cheesecloth, centrifuged at 6000 × g for 10 min and resuspended in extraction buffer. The pellets were kept at −20 °C until DNA extraction, which was performed as described by Yu and Morrison [23] with modifications [14]. Briefly, the microbial cells were lysed using bead disruption and lysis buffer. The final supernatant was precipitated using ammonium acetate and isopropanol. The precipitate was then dissolved in TE buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA pH 8.0), checked for DNA concentration, diluted to 10 ng/ml, and stored at −20 °C.
16S rRNA amplicon sequencing
Amplification of 16S rRNA from the ruminal samples was performed according to Caporaso et al. [24] for the V4 region. Samples were sequenced using the Illumina MiSeq platform according to the protocols of Caporaso et al. [24]. Analyses were mostly performed using the Quantitative Insights into Microbial Ecology (QIIME) pipeline package [25]. The raw reads were assigned to their designated rumen sample using split library script in QIIME. The degree of similarity between sequences was set to a >97% identity in order to obtain operational taxonomic units (OTUs), which are commonly considered to represent species level. The OTUs were annotated using BLAST. OTUs that clustered to less than three reads were manually removed. We additionally used the DADA2 R package [26]. The filterAndTrim function was applied using the forward reads only, trimmed for the last 10 nucleotides (nts), according to the quality profile of the data. Removal of sequences with ambigues bases (Ns) or more than two expected errors. Dereplication and chimera removal of the sequences was done as in Callahan et al.
Ex-vivo methane measurement
Samples of rumen fluid (200 ml) were collected from each cow with a rumen vacuum sampler. The vacuum pump was turned on only after the special metal-coated sampler pipe had been inserted through the esophagus and located in the ventral sac of the rumen, to avoid contamination with saliva. The rumen pH values were immediately determined by a portable pH meter (PL 600, MRC, Holon, Israel). Rumen fluid samples used for the ex-vivo methane assay were taken immediately to an anaerobic chamber. Samples for the time series (1 ml) were incubated with 2 ml of buffer [27] in 5-ml rubber-sealed anaerobic GC vials (Supelco, Bellefonte, PA, USA). The vials were incubated for 48 h at 38 °C, and then 500 µl of the upper gas phase produced in each vial was injected into a gas chromatograph (GC 5890 Hewlett Packard, Palo Alto, CA, USA) equipped with a semi-capillary column (Supelco, molsieve 5A) and FID detector for quantitative methane measurement according to the method of Friedman et al. [14].
Microbiome modulation with premodified rumen fluid
For the crossover experiment, rumen fluid from the 06:00 and 17:00 samples was taken from each cow. The samples were centrifuged (12,000 × g) for 15 min at 25 °C. Buffer (30 ml; [27]) was added to the precipitate, thoroughly vortexed, and then centrifuged under the same conditions for 15 min. The supernatant from the first step was used to produce sterile rumen fluid through a series of filters. The filtration process was performed in an anaerobic chamber and consisted of filtration in 0.65-micron syringe-driven filter (Sigma-Aldrich, Rehovot, Israel), followed by 0.45-µm and then 0.22-µm filters. The sterile filtered rumen fluid and the microbiome from each cow were then mixed in triplicate for all four possible combinations of fluids and microbiomes from 06:00 and 17:00 samples in 5-ml GC vials and incubated for 48 h at 38 °C. Methane production was then measured.
Statistical analysis
Multivariate analysis was performed with the PAST 3.04 program [28]. Bacterial relative abundance, rumen metabolites, and function values of the PICRUST were analyzed using general linear model (GLM) of JMP (JMP® v13.0.0, SAS Institute Inc., Cary, NC) according to the following equation: Yijk = μ + Ci + Di + Tj + CTij + eijk, where Yijk was the observation, μ the overall mean, Ci the cow effect (i = 16), Di the diet effect (i = 3), Tj the sampling time effect (j = 4), CTij the interaction between cow and sampling time, and eijk the residual error. All P values from the GLM tests of relative abundance of bacterial taxa were corrected for a false discovery rate (FDR) of 0.05 using the Benjamini–Hochberg method, and FDR-corrected P values <0.05 (FDR < 0.05) were considered significant. If GLM tests indicated a significant difference between means, subsequent Tukey’s multiple range tests was done to determine which of the means differed from each other.
Results
Ruminal pH, VFA, and methane concentrations show distinct patterns through the digestive stages that are not affected by dietary treatment
To examine the effect of digestive stage on microbiome composition and functionality, our experimental set-up included three groups of six lactating Holstein cows that were assigned to one of three different dietary treatments, two cows belonging to two groups were excluded from the trail due to veterinary issues. The rumen microbiome of these 16 cows was sampled at several time points during the day, representing different digestive stages. We sampled the rumen microbiome from each animal at two different occasions. In each of these occasions, after a night fast, four different time points relative to the feeding time were taken: 1 h prior (06:00), 4 h after (11:00), 7 h after (14:00), and 10 h after (17:00) feed delivery. Regardless of the dietary treatment or individual animal, we saw the same patterns of change in metabolite concentration, rumen pH, and methane production as the feeding cycle progressed. The change in relative abundance of the three main VFAs in the rumen followed the same pattern, whereas acetic and butyric acids were relatively stable, ranging from 54.4 to 54.2% for acetic acid, and 19.3 to 19.4% for butyric acid, respectively (Fig. 1a, c). Relative abundance of propionic acid increased from 20.9% in the morning to 23.9% in the evening (Fig. 1b). The concentration of the total VFA increased during the feeding cycle, reaching the peak 7 h after feeding time (Fig. 1d). Accordingly, rumen pH declined during the feeding cycle (Fig. 1e) from 7.1 ± 0.05 before feed delivery to 6.2 ± 0.07 at 10 h after feed delivery in all dietary treatments. Interestingly, methane production by the microbiome increased from 0.67 ± 0.047 µM before feed delivery to 1.59 ± 0.031 µM 10 h after feed delivery (Fig. 1f) in all dietary treatments.
Fig. 1.

Changes in the rumen environment along the feeding cycle in the three different dietary treatments show the same trend. Percentages represent the concentrations of the main volatile fatty acids in the rumen: a acetic, b propionic, and c butyric acid. d Total concentration of volatile fatty acids in the rumen, e Rumen pH, and f Ex-vivo production of methane over 48 h
Time after feeding is the dominant factor determining microbiome composition when comparing individual host variability and diet
We next sought to connect microbiome composition to the changing rumen metabolites and to understand how the different parameters of diet, individual variability, and time after feeding affect the rumen microbiome. Microbial composition of 64 rumen samples was sequenced for the V4 region of the 16S rRNA gene using the Illumina MiSeq platform. After size filtering, quality control, and chimera removal using the QIIME pipeline, 2,037,308 quality reads were generated with an average of 32,338 ± 1464 reads per sample. The overall number of OTUs detected by the analysis reached 14,561 based on ≥97% nt sequence identity between reads. When examining community structure, we found that, as a function of time relative to feed delivery, alpha-diversity decreased quadratically (P < 0.05) while community evenness increased in quadratic response (P < 0.05) (Figure S1), suggesting an increase in environmental selection that is connected to the reduction of species richness. Comparison of the rumen microbiome within each diet by non-metric multidimensional scaling (NMDS) using the Bray–Curtis similarity index revealed the same pattern of change along the feeding cycle in all dietary treatments (Fig. 2a). Specifically, we found that the 17:00 community was distinct from the 06:00 and 11:00 communities but not from the 14:00 community. Permutational analysis of variance test did not show any separation between the 06:00 and 11:00 communities, even though this represented the longest time interval between two samples (Figure S2). Iterative Unweighted Pair Group Method with Arithmetic Mean hierarchical clustering analysis revealed that the diurnal effects are the most dominant factor on microbiome composition compared to individual host variability or diet (Figure S3). This was also apparent in the NMDS analysis and confirmed by analysis of similarity (Fig. 2b and S4), which also showed significant clustering (P < 0.005) according to diurnal effects, except for 06:00 and 11:00, which showed only a trend (P = 0.087). We further compared the beta diversity of samples as a function of each of these parameters (Fig. 2c and Figure S5). The similarity between cows sampled at the same time (Bray–Curtis 0.86 ± 0.003, weighted Unifrac 0.81 ± 0.071) was significantly higher (P < 0.0001) than that between cows sampled at different time points (Bray–Curtis 0.74 ± 0.004, weighted Unifrac 0.70 ± 0.054). This was not the case when the diet effect was measured, as similarity between cows fed the same diet (Bray–Curtis 0.8 ± 0.004, weighted UniFrac 0.76 ± 0.059) was not significantly higher than that between cows fed different diets (Bray–Curtis 0.8 ± 0.004, weighted UniFrac 0.76 ± 0.065). Moreover, the similarity between different cows sampled at the same time was higher than that for the same cow at different time points on the same day (Bray–Curtis 0.83 ± 0.002, weighted UniFrac 0.77 ± 0.1). It is worth noting the fact that the Unifrac metric, which is sensitive to the changes in phylogenetic distances between the communities [29], shows the same fold change as the Bray–Curtis metric with regard to the measured parameters, which suggests that the differences in community stems from marked changes in phylogenetic distances between time points.
Fig. 2.
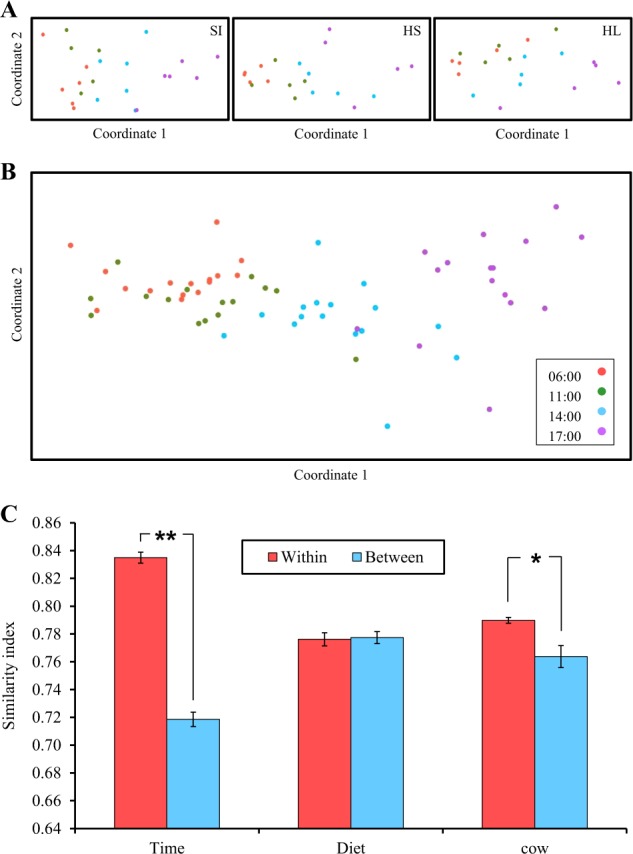
Community composition is determined by time after feeding and is independent of dietary regime and individual variability. a Similarity of bacterial communities within each dietary treatment. The distance between the samples is based on similarity in OTU composition (OTU similarity ≥0.97%) of each sample calculated using the Bray–Curtis similarity index and plotted using NMDS. Each point represents a different sample plotted according to their OTU composition and abundance. Confidence ellipses are plotted for each sampling–time group. b Similarity of the bacterial communities of all samples with no separation into dietary treatments. All parameters were calculated as in (a). c Average calculated Bray–Curtis similarity index for all samples relative to all of the others grouped as within or between the examined variance categories (*P < 0.05, **P < 0.001). The QIIME pipeline was used to compute weighted UniFrac metric similarity measurements. The y axis represents the degree of similarity, with 0 indicating no similarity between samples and 1 indicating that the samples are the same
Diurnal changes in community composition explain rumen methanogenesis
Following our findings, we next analyzed the diurnal effects on microbiome composition. We found that the four most abundant phyla in the rumen, making up >95% of the rumen microbiome, were significantly changed during the feeding cycle (Fig. 3a). Bacteroidetes abundance decreased from 43% at 06:00 (morning) to 13% at 17:00 (evening) (P < 0.001); on the other hand Firmicutes, Euryarchaeota, and Actinobacteria were increased significantly (P < 0.001) during the feeding cycle (Fig. 3a). In addition, the ratio of Firmicutes to Bacteroidetes, which has been previously shown to be strongly related to milk fat yield in dairy cows [10], increased six-fold from the morning to evening. Coriobacteriaceae, a family in the phylum Actinobacteria which part of its members obtain energy for growth strictly via anaerobic respiration processes [30], increased ten-fold, from 0.37% in the morning to 3.74% in the evening (P < 0.001) (Fig. 3b). The dramatic decrease in the Bacteroidetes phylum abundance (P < 0.001) was mostly seen in the genus Prevotella, accounting for 66% of the Bacteroidetes phylum, which was the most abundant genus in the morning (31.4%), decreased to only 9.2% in the evening. In agreement with the increase in methane production, the relative abundance of the methanogenic community belonging to the archaeal phylum of the Euryarchaeota increased nearly six-fold from morning to evening (Fig. 3a). The most dominant methanogen in our samples was the genus Methanobrevibacter, which made up to 95% of the methanogenic community along the feeding cycle, and Methanosphaera, which made up 4.5% (out of the 426 OTUs belonging to the Euryarchaeota, 386 belonged to the genus Methanobrevibacter and the rest to the genus Methanosphaera) (Fig. 3c). Methanobrevibacter and Methanosphaera showed a quadratic increase along the feeding cycle (P < 0.001). Methanobrevibacter increased from 0.79% of the entire microbiome in the morning to 4.6% in the evening. In addition to the aforementioned changes of substantial phylogenetic breadth, we also documented significant changes at the species level (97% sequence identity). Most of the OTUs that exhibited significant diurnal change did not have species-level annotation. Nevertheless, among the few annotated species were Ruminococcus flavefaciens, a major rumen fiber degrader that exhibited a significant decrease as a function of time after feeding, and Selenomonas ruminantium that exhibited almost 20-fold decrease from morning to evening (Supplementary Data File 1). The decrease in rumen pH to pH 6 seen in our results (Fig. 1) cannot solely explain this observation, since S. ruminantium can grow under very low pH conditions as low as pH 4.85 [31], while R. flavefaciens is more pH sensitive to this pH value [32]. Therefore, it is potentially other changing environmental conditions that affect S. ruminantium. Interestingly, previous experimental work has shown that S. ruminantium cannot degrade cellulose in isolation but can grow in the presence of R. flavefaciens, which supplies S. ruminantium with soluble sugars that are the byproducts of cellulose degradation [33]. It is interesting to speculate that R. flavefaciens abundance drops due to the diurnal decrease in pH, which in turn is followed by a decrease in S. ruminantium abundance, which depends on R. flavefaciens for soluble sugar availability. We additionally performed quantitative PCR measurements for both bacteria and archaea 16S rRNA gene counts (Figure S6). Our results demonstrate that in most animals there are little changes in counts between 06:00 until 14:00, with a slight decrease between 12:00 and 14:00. Here is, however, a sharp two-fold increase in counts between 14:00 to 17:00 that could be explained by increased metabolism and growth rate at the earlier time points that resulted in an increased number of cells at this time point.
Fig. 3.

Distinct phylogenetic groups show marked changes as a function of time after feeding. a Box plot showing the relative abundance of the main bacterial phyla that showed marked changes as a function of time after feeding: Bacteroidetes, Firmicutes, Actinobacteria, and Euryarchaeota. b Firmicutes-to-Bacteroidetes ratio. c Bacterial families showing significant changes during the feeding cycle: Prevotellaceae, Ruminococcaceae, Lachnospiraceae, and Coriobacteriaceae. d The two methanogenic archaea genera exhibiting significant changes as a function of time after feeding: Methanobrevibacter and Methanosphaera. F values represent the significance of the quadratic change over time
Changes in functional coding capacity explain diurnal metabolic output
We next examined the diurnal functional changes in light of the coding capacity of the microbiome. Since the rumen microbiome shares many taxa with the human microbiome, it benefits from the large microbial isolation and genome-sequencing efforts in the human microbiome. Taking this into account, we used PICRUST software that predicts the functional composition of a metagenome using marker gene data and a database of reference genomes [34]. In agreement with our findings regarding the increase in methane production during the feeding cycle, this analysis revealed a large increase in methane metabolism genes (F < 0.0001) during the feeding cycle (Fig. 4). In the metabolic pathways (Fig. 4), a quadratic decrease in oxidative phosphorylation (F < 0.0001) and energy metabolism (F < 0.0001) was found but not in starch or sucrose metabolism (F < 0.0669). On the other hand, an increase in pyruvate and butyrate metabolism (F < 0.0001) occurred during the feeding cycle. An increase in cell motility functions (Fig. 4), such as flagellar assembly (F < 0.0001) and bacterial motility proteins (F < 0.0001), was found even though cell wall functions, such as peptidoglycan and lipopolysaccharide biosynthesis, also increased (F < 0.0001).
Fig. 4.

Marked functional changes of the coding capacity of the rumen microbiome as a function of time after feeding. Box plot of the microbiome functions that exhibited the most pronounced changes. Function values were calculated with the PICRUST program that uses an extended ancestral-state reconstruction algorithm to predict which gene families are present and then combines gene families to estimate the composite metagenome. F values represent the significance of the quadratic change over time
Rumen metabolic environment has a stronger effect than microbiome composition on methane production by the microbiome
In light of our observation of increased methane production by the microbiome as well as increased relative abundance of the methanogenic orders and the methanogenesis pathways as a function of time after feeding, we asked whether these changes are directly linked to one another. Specifically, we asked whether microbiome composition is solely and directly responsible for the increase in methane production or whether the rumen metabolic environment itself was modified by the microbiome and influenced this observed increase. To answer this, we conducted a crossover experiment in which we switched for each cow the rumen microbiome (after separating it from the rumen fluids) and the rumen fluids (after filtering them from the microbiome) from the morning samples (06:00), which showed lower methane production, and the evening samples (17:00), which showed increased methane production (Fig. 5), and incubated them for 48 h. We found that, regardless of the sample it was taken from, the microbiome incubated with rumen fluid from the evening exhibited higher methane production than that incubated with rumen fluid from the morning (40–50% higher). The difference was significant in the microbiome from the morning samples and was seen as a trend in microbiome from the evening samples (Fig. 5). This result suggests that the evening rumen fluid contains a different metabolic environment that promotes higher methane production, and the morning microbiome has better potential for methane production.
Fig. 5.
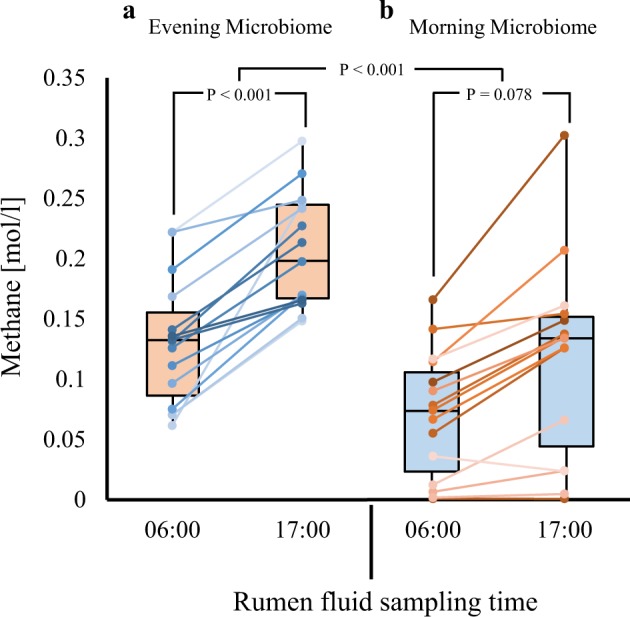
Microbiome functionality depends in modified metabolic landscape. The microbiome and rumen fluid were taken from each individual cow (n = 26), separated using washing and filtration as described in Materials and methods, and then switched. a Box plot showing ex-vivo methane production of microbiome from the morning sample incubated with either morning or evening filtered rumen fluid. b Box plot showing ex-vivo methane production of microbiome from the evening sample incubated with either morning or evening filtered rumen fluid
Discussion
The objective of this study was to understand the pattern of microbiome composition, assembly, and functionality during the feeding cycle and the forces that shape it. We designed our experiment to follow the main events in the feeding cycle of ruminants, which occur at dusk and dawn [35]. The changes in rumen pH (Fig. 1e) suggested that our feeding protocol creates the desired conditions in the rumen as it followed the previously described natural oscillations [36]. Typically, in cows fed once a day, ruminal pH decreases after feeding for a few hours and then increases again due to VFA removal, rumination, and salivation [37]. The diets used in our experiment are different from each other by their forage characteristic, which either consist of wheat silage or wheat hay of chopping length. All of these diets were characterized by a low forage-to-concentrate ratio (30:70), thus promoting rapid fermentation in the rumen, and larger amplitude of pH oscillations (Fig. 1e). This finding is consistent with the decrease in the alpha diversity estimate as a function of time after feeding, suggesting increased selection and habitat filtering of species (Figure S2). Moreover, we found that the similarity within a sampling time is higher (0.82) than the similarities within cow or diet (0.78 and 0.76, respectively). These results suggest that the selective power on microbiome composition as a function of time after feeding is stronger than the host or diet effect. Moreover, this force affects community assembly in a highly deterministic manner, making the microbiome composition of different hosts and diets converge into a similar phenotype (Fig. 3c). This suggests that metabolic processes occurring within the microbiome at a given time give rise to new habitats that select and shape microbiome composition according to available metabolites and physical environment conditions, such as pH. This suggestion assumes that the mechanism of this community-assembly process is driven by niche modification and not niche preemption, the latter describing an environment that stays similar while being inhabited by different species [3]. As already noted, niche modification, unlike niche preemption, affects the identity of the microbial lineages across clades and functional groups. This process changes the ecological niches, and subsequently, the organisms that occupy these niches have different functional capabilities to distinct phylogenetic groups that occupy these niches. Environmental factors that create niches (e.g., amount of undigested feed) and niche modification by other microorganisms can be distinguished by examining the functionality of the sequence of assembling microorganisms as well as the ecosystem function. This pattern is seen in our results both at the functional and taxonomic level of the microbiome as well as at the physical characteristics of the rumen fluid. In our study, pH values substantially decreased after feeding, a phenomenon that was previously reported and it is attributed to increase of microbial fermentation [37], at the same time we recorded increase in methane production (Fig. 1e, f). At the microbiome composition, we see extreme changes at the phylogenetic distances of the changing lineages going up to the taxonomic level of domain and phyla, which are clear distinct taxonomic groups that differ in their functionality (Fig. 3). We observed clear and significant increase in the abundance of Euryarchaeota belonging to the archaeal domain as well as Firmicutes and Actinobacteria belonging to the bacterial domain. This is accompanied by decrease in Bacteroidetes abundance, which decrease by more than three-fold. As expected, these changes at the taxonomic levels are also reflected at the functional capacity of the microbiome where dramatic changes at the relative abundance of the predicted gene families of the microbiome are seen (Fig. 4). Several of these gene families are directly related to microbial metabolism and might be interpreted based on the functional observations. For example, the glycolysis, pyruvate metabolism, and butyrate metabolism genes increased in abundance as a function of time after feeding (Fig. 4). This increase potentially reflects expansion in the abundance of functional microbial groups that are involved in the breakdown and fermentation of sugar compounds. This in turn might explain the pH drop due to the production of organic and short-chain fatty acids as previously reported by Aschenbach et al. [37]. This phenomenon has been recently described by Van Lingen et al. [38]: increase in short-chain fatty acids' production and methane emission during the feeding cycle. Interestingly, in their study the rate of short-chain fatty acids' production and methane emission after the meal was much faster, which might be related to either the feeding pattern of the cows or to the high sugar content relative to starch in the diets of the current study. Methanogenesis metabolism genes dramatically increased in their relative abundance as a function of time after feeding. This was also mirrored by the microbiome’s increased methane production (Fig. 1e).
Overall, the dramatic turnover of distinct microbial groups coding for a distinct genetic repertoire, as well as the changes in pH level and methane production, highlight modification of the ecological niche as the mechanism determining community assembly as a function of time after feeding. In this model, the niche is shaped by modification of the metabolic repertoire that directly affects the environmental landscape, which in turn filters specific functional taxonomic groups. The pH value could be one of the important dominant factors acting as an environmental filter and could explain the marked changes in the microbial community, as previously shown for soil microbial communities [39]. Nevertheless, despite the decrease in pH values, we observed an increase in the relative abundance of methanogenic archaea accompanied by an increase in methane production by the microbiome. As methanogenic archaea have been shown to be sensitive to decreases in pH values [40], this finding suggests that, although this group is negatively affected by the decrease in pH, other factors existing within the environment affect it and select it with time after feeding, such as redox potential that was shown to have dramatic effect on some methanogenic groups regardless of pH [14]. This enabled us to test the niche modification hypothesis and to use this microbial group as an indicator for niche modification as a function of time after feeding. By switching the metabolic environments and the microbiomes, we were able to show that the metabolic environment of the evening rumen fluid favors methanogenesis metabolism (Fig. 5). This was apparent as the microbiome of the morning samples produced significantly more methane when it was exposed to the evening vs. morning ruminal fluid environment (Fig. 5). This was not the case with the evening microbiome, which apparently had already been selected by this metabolic environment (Fig. 5). It should be noted that the morning microbiome produced more methane when exposed to both morning and evening ruminal fluid environment when compared to the evening microbiome, and this might be attributed to higher potency of this microbiome to produce available metabolites important for methanogenesis such as hydrogen and could be seen in the higher abundance of energy metabolism pathways existing in this microbiome composition (Fig. 4). These findings highlight niche modification with time after feeding as a selective deterministic process that shapes community assembly via environmental filtering. The fact that the metabolic environments were created by filtering the rumen metabolites from the rumen fluid and separating them from feed particles further supports that the diurnal effects are the outcome of the microbiome output metabolites and not of the diets themselves. Moreover, our results indicate that the attributed inhibitory effect of low pH on ruminal methanogenesis, which had been shown in in vitro studies [40, 41], plays only a minor role in the daily feeding cycle. This is evident from the higher methane production in the evening that was accompanied by an increase in methanogenic archaeal relative abundance and 16S rRNA gene counts despite the pH decrease, as well as an increase in methane production when the morning microbiome was exposed to the evening metabolic environment (Figs. 3–5, S6). To our understanding, these findings that highlight the microbiome as the main driver for its own oscillations have broader implications in other gut ecosystems. Diurnal microbiome oscillations have been documented in several other gut microbial ecosystems, including humans and mice, and were shown to affect metabolic homeostasis [15, 16, 42]. Interestingly, microbiome oscillations were shown to affect host transcriptional and metabolites levels [43, 44]. Therefore, it is very tempting to speculate that in these ecosystems microbiome niche modification has a cardinal role in shaping diurnal community assembly oscillations as well. From an applied perspective, our results suggest that different feeding frequencies could be used to reduce CH4 production by controlling available metabolites. By taking into account the effect of microbiome oscillations on host physiology and metabolism in human and mice together with the established connection between the rumen microbiome and its host physiology and energy balance [12, 45], it is reasonable to assume that the metabolic homeostasis of ruminants is also affected by these diurnal microbiome oscillations. Our findings also add an important aspect to our understanding of community assembly, as it has been shown over long timescales that, in rich and highly productive environments, stochastic and non-deterministic process are more likely to play a role in community assembly [46]. Here we show that, despite the high productivity of the rumen environment and its high species richness, the process that determines the assembly of community composition is deterministic and not stochastic. This apparent discrepancy could stem from the timescales over which these studies were conducted: our observation focusing on community assembly in the relatively short time frame of hours compared to the reported experiment that lasted for 7 years [46]. Hence, in our experiment, we might observe compositional cycles of community assembly [3] driven by deterministic niche modification, whereas in the aforementioned report with a longer time scale, stochastic processes govern highly enriched habitats. We previously reported that the rumen community assembles in a deterministic manner [10] as a function of age. Nevertheless, the effect of stochastic processes on its assembly has never been examined. Therefore, one cannot exclude a contribution of such processes to the developing rumen on the long timescale that could be driven by random dispersal of species early in life leading to historical contingency effects [3]. To reconcile these two observations from two different timescales, we should examine rumen community assembly over longer time periods and quantify the contribution of stochastic processes, such as priority effects. We propose that the diurnal community states created by niche modification as a function of time after feeding are in fact compositional cycles of community assembly defined by the metabolic landscape. These findings are extremely important from an applied perspective: when the same ruminal community is exposed to different metabolic landscapes, it changes its functionality (Fig. 5). In essence, this provides a proof of concept and a stepping stone to deterministically designing community composition and function by understanding the metabolic landscape that drives each of these community states.
Electronic supplementary material
Acknowledgements
The research described here was supported by grants from Israel Science Foundation (1313/13) and by the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement 640384).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0203-0) contains supplementary material, which is available to authorized users.
References
- 1.Amarasekare P. Interference competition and species coexistence. Proc Biol Sci. 2002;269:2541–50. doi: 10.1098/rspb.2002.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connell JH, Slatyer RO. Mechanisms of succession in natural communities and their role in community stability and organization. Am Nat. 1977;111:1119–44. doi: 10.1086/283241. [DOI] [Google Scholar]
- 3.Fukami T. Historical contingency in community assembly: integrating niches, species pools, and priority effects. Annu Rev Ecol Evol Syst. 2015;46:1–23. doi: 10.1146/annurev-ecolsys-110411-160340. [DOI] [Google Scholar]
- 4.Fukami T, Nakajima M. Community assembly: alternative stable states or alternative transient states? Ecol Lett. 2011;14:973–84. doi: 10.1111/j.1461-0248.2011.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinski TM, Stanish LF, et al. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev. 2013;77:342–56. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chase, J.M. and Leibold, M.A. Ecological Niches: Linking Classical and Contemporary Approaches. University of Chicago Press, Chicago; 2003. 10.7208/chicago/9780226101811.001.0001
- 7.Kamneva OK. Genome composition and phylogeny of microbes predict their co-occurrence in the environment. PLoS Comput Biol. 2017;13:e1005366. doi: 10.1371/journal.pcbi.1005366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kloesges T, Popa O, Martin W, Dagan T. Networks of gene sharing among 329 proteobacterial genomes reveal differences in lateral gene transfer frequency at different phylogenetic depths. Mol Biol Evol. 2011;28:1057–74. doi: 10.1093/molbev/msq297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loman NJ, Pallen MJ. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 2015;13:787–94. doi: 10.1038/nrmicro3565. [DOI] [PubMed] [Google Scholar]
- 10.Jami E, White BA, Mizrahi I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE. 2014;9:e85423. doi: 10.1371/journal.pone.0085423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizrahi I. Rumen symbioses. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F. (eds) The prokaryotes. Springer, Berlin, Heidelberg; 2013. p. 533–44.
- 12.Shabat SKB, Sasson G, Doron-Faigenboim A, Durman T, Yaacoby S, Berg Miller ME, et al. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016;10:2958–72. doi: 10.1038/ismej.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman N, Jami E, Mizrahi I. Compositional and functional dynamics of the bovine rumen methanogenic community across different developmental stages. Environ Microbiol. 2017;19:3365–73. doi: 10.1111/1462-2920.13846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman N, Shriker E, Gold B, Durman T, Zarecki R, Ruppin E, et al. Diet-induced changes of redox potential underlie compositional shifts in the rumen archaeal community. Environ Microbiol. 2017;19:174–84. doi: 10.1111/1462-2920.13551. [DOI] [PubMed] [Google Scholar]
- 15.Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom control of microbiome diurnal oscillations promotes metabolic homeostasis. Cell. 2014;159:514–29. doi: 10.1016/j.cell.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 16.Zarrinpar A, Chaix A, Yooseph S, Panda S. Article diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014;20:1006–17. doi: 10.1016/j.cmet.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant AM. Variations in the pH and volatile fatty acid concentration within the bovine reticulo-rumen. NZ J Agric Res. 1964;7:694–706. doi: 10.1080/00288233.1964.10416398. [DOI] [Google Scholar]
- 18.Leedle JA, Bryant MP, Hespell RB. Diurnal variations in bacterial numbers and fluid parameters in ruminal contents of animals fed low- or high-forage diets. Appl Environ Microbiol. 1982;44:402–12. doi: 10.1128/aem.44.2.402-412.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welkie DG, Devenson DM, Weimer PJ. ARISA analysis of ruminal bacterial community dynamics in lactating dairy cows during the feeding cycle. Anaerobe. 2010;16:94–100. doi: 10.1016/j.anaerobe.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Huws SA, Edwards JE, Creevey CJ, Rees Stevens P, Lin W, Girdwood SE, et al. Temporal dynamics of the metabolically active rumen bacteria colonizing fresh perennial ryegrass. FEMS Microbiol Ecol. 2016;92:1–12. doi: 10.1093/femsec/fiv137. [DOI] [PubMed] [Google Scholar]
- 21.Piao H, Lachman M, Malfatti S, Sczyrba A, Knierim B, Auer M, et al. Temporal dynamics of fibrolytic and methanogenic rumen microorganisms during in situ incubation of switchgrass determined by 16s rRNA gene profiling. Front Microbiol. 2014;5:1–11. doi: 10.3389/fmicb.2014.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaani Y, Nikbachat M, Yosef E, Mizrahi I, Miron J. Effect of feeding long or short wheat hay vs wheat silage in the ration of lactating cows on intake, milk production and digestibility. Animal. 2017;11:2203–10. doi: 10.1017/S1751731117001100. [DOI] [PubMed] [Google Scholar]
- 23.Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36:808–12. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]
- 24.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tilley JMA, Terry RA. A two-stage technique for the in vitro digestion of forage crops. Grass Forage Sci. 1963;18:104–11. doi: 10.1111/j.1365-2494.1963.tb00335.x. [DOI] [Google Scholar]
- 28.Hammer Ø, Harper DAT, Ryan PD. PAST: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica. 2001;4:9. http://palaeo-electronica.org/2001_1/past/issue1_01.htm
- 29.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson RC, Rasmussen MA, Jensen NS, Allison MJ. Denitrobacterium detoxificans gen. nov., sp. nov., a ruminal bacterium that respires on nitrocompounds. Int J Syst Evol Microbiol. 2000;50:633–8. doi: 10.1099/00207713-50-2-633. [DOI] [PubMed] [Google Scholar]
- 31.Russell J, Dombrowski DB. Effect of pH on the efficiency of growth by pure cultures of rumen bacteria in continuous culture. Appl Environ Microbiol. 1980;39:604–10. doi: 10.1128/aem.39.3.604-610.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyazaki K, Hino T, Itabashi AH. Effects of extracellular pH on the intracellular pH and membrane potential of cellulolytic ruminal bacteria, Ruminococcus albus, Ruminococcus flavefaciens, and Fibrobacter succinogenes. J Gen Appl Microbiol. 1992;38:567–73. doi: 10.2323/jgam.38.567. [DOI] [Google Scholar]
- 33.Scheifinger CC, Wolin MJ. Propionate formation from cellulose and soluble sugars by combined cultures of Bacteroides succinogenes and Selenomonas ruminantium. Appl Microbiol. 1973;26:789–95. doi: 10.1128/am.26.5.789-795.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dulphy JP, Remond MT. Ingestive behaviour and related activities in ruminants. In: Ruckebusch Y, Thivend P, editors. Digestive physiology and metabolism in ruminants. MTP Press Limited, Springer Netherlands; 1980. p. 103–22.
- 36.Palmonari A, Stevenson DM, Mertens DR, Cruywagen CW, Weimer PJ. pH dynamics and bacterial community composition in the rumen of lactating dairy cows. J Dairy Sci. 2010;93:279–87. doi: 10.3168/jds.2009-2207. [DOI] [PubMed] [Google Scholar]
- 37.Aschenbach JR, Penner GB, Stumpff F, Gäbel G. Ruminant Nutrition Symposium: Role of fermentation acid absorption in the regulation of ruminal pH. J Anim Sci. 2011;89:1092–107. doi: 10.2527/jas.2010-3301. [DOI] [PubMed] [Google Scholar]
- 38.Van Lingen HJ, Edwards JE, Vaidya JD, van Gastelen S, Saccenti E, van den Bogert B, et al. Diurnal dynamics of gaseous and dissolved metabolites and microbiota composition in the bovine rumen. Front Microbiol. 2017;8:425. doi: 10.3389/fmicb.2017.00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauber CL, Hamady M, Knight R, Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol. 2009;75:5111–20. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ann J, Van Kessel S, Russell JB. The effect of pH on ruminal methanogenesis. FEMS Microbiol Ecol. 1996;20:205–10. doi: 10.1111/j.1574-6941.1996.tb00319.x. [DOI] [Google Scholar]
- 41.Lana RP, Russell JB, Van Amburgh ME. The role of pH in regulating ruminal methane and ammonia production. J Anim Sci. 1998;76:2190–6. doi: 10.2527/1998.7682190x. [DOI] [PubMed] [Google Scholar]
- 42.Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care. 2010;33:2277–84. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leone V, Gibbons SM, Martinez K, Hutchison AL, Huang EY, Cham CM, et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015;17:681–9. doi: 10.1016/j.chom.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thaiss CA, Levy M, Korem T, Dohnalová L, Shapiro H, Jaitin DA, et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell. 2016;167:1495.e12–510.e12. doi: 10.1016/j.cell.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Sasson G, Kruger Ben-Shabat S, Seroussi E, Doron-Faigenboim A, Shterzer N, Yaacoby S, et al. Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow’s capacity to harvest energy from its feed. mBio. 2017;8:E00703-17. doi: 10.1128/mBio.00703-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chase JM. Stochastic community assembly causes higher biodiversity in more productive environments. Science. 2010;328:1388–91. doi: 10.1126/science.1187820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.