

Abstract
Executive function is an umbrella term that includes cognitive processes such as decision-making, impulse control, attention, behavioral flexibility, and working memory. Each of these processes depends largely upon monoaminergic (dopaminergic, serotonergic, and noradrenergic) neurotransmission in the frontal cortex, striatum, and hippocampus, among other brain areas. Traumatic brain injury (TBI) induces disruptions in monoaminergic signaling along several steps in the neurotransmission process – synthesis, distribution, and breakdown – and in turn, produces long-lasting deficits in several executive function domains. Understanding how TBI alters monoamingeric neurotransmission and executive function will advance basic knowledge of the underlying principles that govern executive function and potentially further treatment of cognitive deficits following such injury. In this review, we examine the influence of TBI on the following measures of executive function – impulsivity, behavioral flexibility, and working memory. We also describe monoaminergic-systems changes following TBI. Given that TBI patients experience alterations in monoaminergic signaling following injury, they may represent a unique population with regard to pharmacotherapy. We conclude this review by discussing some considerations for pharmacotherapy in the field of TBI.
Keywords: behavioral flexibility, dopamine, human, impulsivity, norepinephrine, serotonin, working memory
Introduction
Traumatic brain injury (TBI) occurs when an external force (e.g. mechanical deformation, rapid deceleration, blast wave) is applied to the brain and affects more than 200 per 100 000 people each year globally (Bryan-Hancock and Harrison, 2010). While many people recover with minimal complications, a significant portion goes on to develop chronic behavioral and cognitive deficits, resulting in an estimated 1–2% of people with TBI-related disabilities (Thurman et al., 1999; Zgaljardic et al., 2015), and accounting for a staggering $76.5 billion USD economic burden as of 2010, with 10-year costs averaging $270 000 per patient (Corso et al., 2006; Coronado et al., 2012; Ponsford et al., 2013). Brain injury is considered to be a major risk factor in the development of neurodegenerative disorders, including Parkinson’s and Alzheimer’s diseases (Semchuk et al., 1993; Plassman et al., 2000), and is associated with increased rates of depression, anxiety, attention-deficit disorders, suicidality, and substance abuse following such central nervous system damage (Moor et al., 2006; Vaishnavi et al., 2009; Rao et al., 2010; Konrad et al., 2011; Reeves and Panguluri, 2011; Zgaljardic et al., 2015). Notably, these disorders impair a wide variety of behaviors commonly considered under the umbrella of ‘executive function,’ including memory, behavioral flexibility, impulsivity, and decision-making (Alves et al., 2014; Bredemeier and Miller, 2015; Day et al., 2015; Sharp et al., 2015; Kingdon et al., 2016).
Given the considerable difficulty of determining the causes of executive function deficits in clinical TBI patients, several injury models have been developed for inducing experimental TBIs in nonhuman animals, that map on to clinical injuries and severities (Morganti-Kossmann et al., 2010). These injury types may be induced using various methods (e.g. controlled-cortical impact, fluid percussion, weight drop, blast) and at a spectrum of severities, each with unique pathological characteristics. Focal injuries result largely in localized contusion and cell death while being highly reproducible in the animal laboratory. In contrast, concussive and blast injuries generate a diffuse pattern of injury with axonal shearing that are less reproducible than focal injuries. In animal models, severity of injury is directly related to the amount of force applied to the animal’s brain and strongly tied to neurological outcome (Xiong et al., 2013), while clinical measurements rely on neurological outcomes such as the Glasgow Coma Scale to assess severity (Teasdale and Jennett, 1974). Regardless of primary injury type, cascades of secondary damage are initiated, which may include neurochemical and ionic disequilibrium in neurons, depletion of cellular energy reserves, mitochondrial dysfunction, lipid peroxidation and DNA damage, upregulated neuroinflammation, and compromised glial and endothelial support networks, that coalesce to contribute to enduring dysfunction after TBI (Prins et al., 2013).
While various animal models have been used for several decades to study the pathology of brain injury, only recently has more attention been given to behavioral assessment of higher-order function. For many decades, the primary assessment of cognitive function after such trauma has been the Morris water maze (MWM), a task designed to measure hippocampal function (Morris, 1984). While this has proven to be very effective in rapidly assessing the effects of TBI on spatial learning and reference memory in animals, it is difficult to ascertain changes associated with a variety of other executive functions using this task. More recent studies have begun to make use of a wider variety of paradigms to measure cognition, including several methods based on standard operant techniques, which are the focus of this review.
To date, no pharmacological agents have been approved for the treatment of TBI, either in the acute or the chronic period. In particular, it is unclear whether patients with injury-induced psychiatric conditions represent a special population with regard to pharmacotherapies, and the degree to which commonly prescribed drugs are appropriate for patients with a TBI. Thus, the purpose of the present review is to (i) survey the literature on executive function assessments following human and animal brain injury; (ii) describe monoaminergic-systems changes following TBI that may influence how deficits in executive function respond to pharmacological intervention; and (iii) discuss considerations for behavioral pharmacology and pharmacotherapies in TBI.
Executive function following traumatic brain injury
Impulsivity
Impulsivity is a multifaceted concept describing actions with the potential to provide short-term gain at a cost to long-term benefits. The construct can be subdivided into motor impulsivity (i.e. failure to inhibit responding), choice impulsivity (i.e. inability to wait for delayed reinforcers or issues with delayed gratification), and sometimes risk-taking (i.e. choosing larger outcomes that are uncertain over smaller outcomes that are certain). Human studies suggest that deficits involving impulsivity following TBI are particularly problematic because of their wide impact on daily function (Rochat et al., 2010; James et al., 2014) and animal models with high translational validity have been developed within each realm of impulsive behavior.
Motor impulsivity
Motor impulsivity, also known as response disinhibition, refers broadly to acting without thinking. Response inhibition can be further divided into ‘stopping’ and ‘waiting’ impulsivity (Schachar et al., 2007; Robinson et al., 2009), in which ‘stopping’ impulsivity refers to the inability to stop an action that has already been initiated, while ‘waiting’ impulsivity refers to the inability to inhibit responding for an extended duration (i.e. premature responding). One of the clinical manifestations of increased motor impulsivity is impulsive aggression, which is also correlated with criminal behavior after TBI (Alderman, 2003; Dyer et al., 2006; Wood and Thomas, 2013). Impulsive aggression is operationalized as a quick, impulsive, aggressive response following minimal provocation (Barratt et al., 1997), and in its verbal form is relatively common during acute recovery post-TBI (35–38%; according to Dyer et al., 2006), although physical aggression may also be present (Dyer et al., 2006; Rao et al., 2009). Thus, deficits in motor impulsivity post-TBI have the potential to drastically impact quality of life for individuals with TBI, and their close relations.
Clinical evidence: There are several procedures for assessing motor impulsivity in humans, including go/no-go tasks (Donders, 1969), stop-signal tasks (SST; Logan, 1994), and continuous performance tasks (CPT; Rosvold et al., 1956). For humans, reinforcers are presented typically in the form of ‘points’ such as those delivered in a game.
During go/no-go tasks, response inhibition is evaluated by pairing a cue (e.g. tone, symbol) with each type of trial – ‘go’ and ‘no-go’ trials – to aid in discrimination between the two alternatives. In general, if responding is inhibited during the ‘no-go’ signal, a reinforcer is delivered, while responses are punished with a timeout. The opposite occurs during ‘go’ signals, with reinforcement of responses and punishment of nonresponding. The SST is a more complex version of go/no-go, where response inhibition is tested further as participants must inhibit an action that is already in progress. A rapid response sequence is trained on two different options to receive a reinforcer. At random intervals within the session, a stimulus is presented (typically a tone), which signals that response inhibition will be reinforced. Stopping difficulty is increased by parametrically manipulating the delay to presentation of the ‘stop’ trial signal, and the signal is not given until a motor action is already in progress. Specifically, the task is designed to measure the longest delay under which an already-initiated action can be inhibited. Those with poorer inhibitory control will require shorter delays and have increased stop-signal reaction times (Logan, 1994). Nativ et al. (1994) and Rochat et al. (2013) used the SST to assess stopping impulsivity of patients following moderate-to-severe TBIs as compared with healthy controls matched for handedness or age. Stimuli were different colored lights and participants were instructed to respond as quickly as possible in the presence of one color and to stop responding in the presence of the alternative color. During this procedure, TBI patients had significantly longer latencies to inhibit responding relative to controls, which is indicative of poorer response inhibition and resistance to performance interference following injury.
CPTs are a popular tool for assessing sustained attention and waiting impulsivity in various clinical populations (Riccio et al., 2002). Such tasks require subjects to respond when target stimuli are present (i.e. a specific tone or light) and to inhibit responding when alternative, distractor stimuli are present (Rosvold et al., 1956). Duncan et al. (2005) and Chen et al. (2012) used visual versions of the CPT to assess sustained attention and motor impulsivity at 2 years and 1 month postmild concussive TBI, respectively, as compared with age-matched, sex-matched, and education-matched controls. Together, results from these studies indicate that TBI patients have omission errors, comission errors, and reaction times that are comparable to non-TBI participants, and that regardless of whether tested during acute or chronic recovery, mild concussive TBI does not affect performance on visual CPTs.
Preclinical evidence: Given the significance of motor impulsivity problems in the clinical population, analogs of the tasks described above have been designed to assess motor impulsivity in preclinical animal models. Many of the tasks map onto the human assessments quite directly; however, reinforcers are typically delivered in the form of food.
Similar to the clinical evidence discussed above, preclinical evidence using rats and mice suggests that mild concussive experimental TBI produces deficits in stopping impulsivity in the go/no-go task, indicated by increased latencies to inhibit responding when no-go stimuli are presented (Hehar et al., 2015; Mychasiuk et al., 2015). In addition to a significant main effect of TBI on response inhibition, both of these studies also found that deficits for male rats were significantly more pronounced than those for female rats, suggesting that sex may play a prominent role in how TBI affects response inhibition. A similar deficit in latency to stop responding has been shown when utilizing the SST in rats. In particular, following severe focal TBI to the bilateral medial frontal cortex, response inhibition was impaired in rats when assessed using a SST, indicated by increased stop-signal reaction times and these deficits were exacerbated in rats that underwent voluntary exercise during acute recovery (Crane et al., 2012).
In contrast to the stop-signal and go/no-go tasks, differential reinforcement of low-rate responding (DRL) schedules of reinforcement (Ferster and Skinner, 1957) are free-operant arrangements and do not have designated ‘stop’ trials. Such reinforcement schedules are designed to evaluate waiting impulsivity and include a temporal duration in which lack of responding is reinforced. Specifically, if the time between two consecutive responses is greater than some duration (set by the experimenter), reinforcement occurs. If responding occurs before the duration elapses, the timer is reset. For example, on a DRL 20 s schedule, subjects must space two consecutive responses by at least 20 s to receive reinforcement. Severe focal injuries to the bilateral frontal cortex in rats impaired performance on a DRL 20 s schedule and deficits persisted until 11 months postinjury (Lindner et al., 1998), with TBI animals showing an ~15% reduction in efficiency.
Another behavioral task developed for studying motor impulsivity of nonhuman animals is the five-choice serial reaction time task (5CSRTT), which is considered an analog of human CPTs (Carli et al., 1983). During this procedure, subjects must wait a period of time until the brief illumination of one response option (commonly 0.5 s duration), after which a response to that alternative is reinforced. Because of the brief presentation of the stimulus, combined with punishment for incorrect or omitted responses, the task sets up a prepotent motor response, which is required to be inhibited for some duration (commonly 5 s, although longer and variable versions exist). Following reinforcer delivery, the next trial may be initiated immediately. Failure to inhibit leads to a timeout. Because each session is limited in duration, excessive premature responding leads to delays in reinforcement or even reduced total reinforcement. In one study utilizing the 5CSRTT in rats, mild, moderate, and severe focal TBI to the bilateral frontal cortex resulted in a significant increase in premature responding, and deficits persisted for 14 weeks postinjury (Vonder Haar et al., 2016).
Summary: While there are relatively few clinical and preclinical studies on response inhibition following TBIs, the existing evidence suggests that motor impulsivity (both, stopping and waiting impulsivity) increases following even mild injuries. The use of a variety of procedures for assessing motor impulsivity, as well as injury models, suggests that this is a robust phenomenon in the preclinical literature. Further work is needed to dissociate the mechanisms by which TBI reduces response inhibition in an effort to develop treatment strategies specific to TBI-induced motor impulsivity.
Choice impulsivity
Choice impulsivity is a term that captures decisions that result in immediate gains at the cost of long-term benefits. Commonly, this is referred to as ‘discounting’ the value of future reinforcers/rewards, which is thought to be because of the cost associated with waiting. One of the most common ways for choice impulsivity to be measured is through tasks that manipulate delays and reinforcer magnitudes parametrically to determine individual levels of temporal discounting, and impulsive choice is operationalized as preference for smaller, immediate reinforcers over larger, delayed reinforcers. Large-scale increases in choice impulsivity may result in overall poor decision-making, which can cause financial difficulties, physical health problems, and overall adverse life outcomes (Hamilton and Potenza, 2012; Boyle et al., 2013).
Clinical evidence: Impulsive choice is assessed primarily using various delay discounting procedures. In general, such procedures involve discrete-trials choices between a smaller, more immediate reinforcer and a larger, delayed reinforcer. For humans, reinforcers are hypothetical monetary rewards in most cases. Although the basic premise of delay discounting procedures is the same, several variations exist, which differ in the way that delays and/or reinforcer amounts are presented to subjects (Vanderveldt et al., 2016). For example, during the ‘adjusting-amount procedure’ (Du et al., 2002), the magnitude of the smaller reinforcer is titrated during each trial until a point of indifference is calculated, which is defined as the magnitude at which the subject chooses each magnitude/delay alternative with equal frequency. In general, greater choice for the smaller, more immediate reinforcer is considered impulsive and results in indifference points at smaller magnitudes.
Evidence from clinical TBI studies suggests that patients display increased impulsive choice following TBI, reporting that TBI patients choose smaller, immediate reinforcers over larger, delayed reinforcers more than healthy controls matched for age, sex, and educational status (Dixon et al., 2005; McHugh and Wood, 2008; Sellitto et al., 2010; Wood and McHugh, 2013). Such deficits in choice impulsivity have been reported following assessment on adjusting-amount procedures using various hypothetical monetary amounts as well as following mild, moderate, and severe concussive TBI to the frontal cortex (Dixon et al., 2005; McHugh and Wood, 2008; Sellitto et al., 2010; Wood and McHugh, 2013). Indeed, it seems as though deficits in choice impulsivity may be frontally mediated given that patients with TBI outside of the frontal lobe show performance similar to that of healthy controls (Sellitto et al., 2010). However, given the cross-sectional nature of clinical data in TBI patients, a temporal pathway between occurrence of TBI and choice impulsivity cannot be discerned.
Preclinical evidence: Despite the high translational validity of delay discounting procedures for assessing choice impulsivity, only one study has used it in the field of experimental TBI, using the Evenden and Ryan (1996) procedure. During this procedure, delays to the larger reinforcer are systematically increased across blocks of trials within each session while reinforcer magnitudes are held constant. In general, choices are between one food pellet delivered immediately and three food pellets delivered after a delay. During the first block of trials, the delay to both reinforcer options is 0 s, and the delay to the larger reinforcer increases systematically across blocks within sessions. Choice for the smaller, immediate reinforcer is considered impulsive in conditions where reinforcement can be maximized by maintaining exclusive choice for the larger, delayed reinforcer. Following severe or mild focal TBI to the bilateral frontal cortex in rats, impulsive choice increased, indicated by significantly more choice for the smaller, immediate reinforcer for TBI rats relative to shams (Vonder Haar et al., 2017). Not surprisingly, deficits in choice impulsivity were initially quite pronounced following severe TBI, but actually recovered to sham levels. Surprisingly, while mild TBI animals had relatively small increases in impulsivity relative to shams, their deficits persisted for the 8 weeks of testing. These results suggest that pure tissue damage may not be a primary mechanism driving impulsivity after TBI.
Summary: Impulsivity is a major issue for individuals living with TBI. Notably, high levels of impulsivity could also be a contributing risk factor to TBI in clinical populations. However, at least in one study, choice impulsivity was increased following experimental TBI, lending support for a directional relationship between clinical TBI and enhanced impulsive decision-making. Despite this, preclinical data suggest that the persistence of such deficits may be dependent upon the severity of injury. Given that only one study has examined effects of experimental TBI on choice impulsivity, further research is needed to expand upon these findings using alternative delay discounting procedures and injury models.
Risk-taking
Risk-taking behaviors may be somewhat controversial to include with impulsivity, as impulsivity is defined commonly to include actions/choices that will have long-term detrimental effects. By contrast, risky decisions only probably lead to long-term problems. However, they are part of a cluster of symptoms identified in TBI patients, have considerable overlap with impulsive behaviors, and can lead to similar negative outcomes.
Clinical evidence
Numerous procedures exist for assaying risk-taking behaviors in humans, including probability-discounting tasks, various gambling tasks, the Balloon-Analog Risk Task (BART), and others (Bechara et al., 1994; Rogers et al., 1999; Lejuez et al., 2002). The most common assessment for use with clinical populations is the Iowa Gambling Task (IGT; Bechara et al., 1994). During the IGT, 100 discrete-trials choices are presented between four decks of cards, two of which are relatively ‘safe’ and two of which are ‘risky’. The two ‘safe’ options are set up to return the highest overall rate of reinforcement ($50 with frequent penalties ranging from $25 to $75, or $50 with infrequent penalties of $250), while the ‘risky’ options give large amounts, but low overall rates of reinforcement ($100 with frequent penalties ranging from $100 to $350, or $100 with infrequent penalties of $1250).
MacPherson et al. (2009) and Cotrena et al. (2014) found no differences in choice on the IGT between frontal TBI patients and healthy controls matched for age and education status during the first 60 trials of the task (both groups were largely indifferent between alternatives). However, between trials 61 and 100, TBI patients remained indifferent between options while controls chose the two ‘safe’ options significantly more, suggesting that control participants may have been more sensitive to the contingencies associated with each alternative. Xiao et al. (2013) observed a similar pattern of results with frontal TBI patients, although deficits in risky decision-making were observed as early as trial 21 out of 100.
An alternative assessment for risk-taking behavior is the Cambridge Gambling Task (CGT; Rogers et al., 1999), which has the major advantage of presenting known probabilities, and thus potentially achieving a more pure measurement of risk preference. During this task, participants are presented with 10 boxes, some of which are red and some blue, representing the probability for that trial. There is a token hidden in one box and participants must wager an amount on the box color in which they believe the token is located. Participants ‘win’ the wagered amount if they were correct or ‘lose’ if it is in the other color box. The primary risk-taking outcome measure is ‘risk adjustment’ and is calculated as the percentage of points risked across more-certain to less-certain probabilities. In addition, the latency between trial presentation and when participants place a bet is measured, and reduced deliberation times are reflective of enhanced motor impulsivity. When matched for age and education status, TBI patients had higher risk adjustment on the CGT at 4 and 6 months postinjury (Salmond et al., 2005; Newcombe et al., 2011). Brain injury patients also showed reduced deliberation times, lending additional support for impaired response inhibition following frontal TBI.
An alternative means for assessing risk tolerance is the BART (Lejuez et al., 2002), in which discrete trials are presented that begin with an un-inflated balloon on a computer screen. Participants click a button to inflate the balloon and gain two points per click. If the balloon is over-inflated, it may burst and participants lose all points acquired during the trial. The burst point varies unpredictably across trials such that participants cannot develop a rule for how many times to click before the balloon bursting. At any point during the trial, they may click an alternative button to ‘cash out’ their acquired points and move on to the next trial. During this procedure, a balance is needed between tolerating some risk, but not too much, to maximize returns. The primary dependent measure is risk tolerance, which is calculated as the average number of clicks to inflate the balloon per trial, excluding trials in which the balloon burst: more clicks are indicative of greater risk tolerance and greater risk-taking behavior. In the single study utilizing the BART with TBI patients, adolescents at least 12 months postinjury were assessed and compared with healthy control participants matched for handedness, age, sex, race/ethnicity, and maternal education status (Chiu et al., 2012). During this task, no differences in risk tolerance were observed between TBI and controls. The differences in findings between the BART and IGT/CGT are interesting, and may be because of differences in clinical samples (adolescent vs. adult), but also suggest that different risk-taking assessments may tap into alternative forms of risk-based decision-making.
Preclinical evidence
Risk-taking behavior has been largely unstudied in animal models of TBI. Recently, our laboratory has performed one such study using a rodent analog of the IGT, referred to as the rodent gambling task (RGT; Zeeb et al., 2009). The primary difference between the IGT and RGT is that the rodent version has a distinct, most-optimal option, while on the IGT, the two ‘risky’ choices and the two ‘safe’ choices are equivalent to one another. As in the IGT, rats make choices among four options, two ‘safe’ and two ‘risky’. Each choice is associated with a different number of sucrose pellets for ‘wins’ (1–4), and different penalties for ‘losses’ (5–40 s timeout), with the two-pellet option conferring the highest overall rate of reinforcement, followed by the one-pellet option (Winstanley and Clark, 2016). Following severe focal TBI to the bilateral frontal cortex of rats, there was a significant shift in choice away from the most-optimal option (Shaver TK, Ozga JE, Zhu B, Anderson KG, Martens KM, Vonder Haar C, unpublished data). Notably, this shift in choice preference reflected an increased preference for both the safer, slightly suboptimal one-pellet option, as well as the riskier three-pellet and four-pellet options, highlighting a complex decision-making phenotype. These deficits persisted for 12 weeks postinjury, and occurred even if rats were extensively pretrained on the task before injury.
Summary
The clinical literature emphasizes risk-taking as a major concern for patients with TBI. However, phenotypes are complex, as illustrated by differences in the BART and other gambling tasks. Further, preclinical evidence is quite limited, and identifies a more generalized deficit that may not be selective to risk-taking, but instead, may be more reflective of a generalized change in reinforcement/punishment sensitivity. Further research will be needed to identify which aspects of cognitive-behavioral therapies may be directed toward these patients.
Behavioral flexibility
Behavioral flexibility refers to the ability to adapt to changing reinforcement contingencies and is often contrasted with perseveration. It is typically assessed with discrimination-reversal procedures of varying complexity, several of which have been used in the fields of clinical and experimental TBI (Berg, 1948; Daum et al., 1989; Sherer et al., 2003; Hashimoto and Toshima, 2005; Myers et al., 2006; Martens et al., 2012; Martens et al., 2013; Bondi et al., 2014; Vonder Haar et al., 2014a; Chou et al., 2016).
Clinical evidence
In simple discrimination-reversal learning, subjects are trained to discriminate between two response alternatives that are distinguished by set stimuli (e.g. lights, shapes) and that discrimination is subsequently reversed. During the task, discrete trials are presented in which responding associated with the ‘correct’ stimulus is reinforced, while responses to other alternatives are on extinction. Once discrimination occurs reliably, the contingencies are reversed and the number of trials required to shift to a predefined mastery criterion is taken as a measure of behavioral flexibility, in which more trials are indicative of less flexibility. During simple discrimination-reversal assessments using lights, tones, or shapes as discriminative stimuli, TBI patients require more trials to reach mastery criteria relative to non-TBI patients matched for age and handedness (Daum et al., 1989; Hashimoto and Toshima, 2005; Myers et al., 2006) and never meet such criteria in some cases (Daum et al., 1989; Hashimoto and Toshima, 2005).
Another flexibility measurement, the Wisconsin Card Sorting Task (Berg, 1948), is commonly used in neuropsychiatric testing. In the task, participants are given several cards in a deck and asked to sort them, but are not told how the cards should be sorted. There are multiple classification systems (e.g. color, shape, number,) and each time the participant sorts a card, the experimenter tells them whether the classification is correct or not. However, after 10 cards are sorted correctly, the classification system changes and the number of trials required for participants to adapt to the new classification is taken as a measure of behavioral flexibility. On this task, patients with moderate-to-severe TBI are impaired and require more trials to meet mastery criterion (Sherer et al., 2003). However, this task is not sufficient in and of itself to delineate those with the worst TBI-related deficits (Greve et al., 2002, 2009).
Preclinical evidence
Animal models largely mirror the clinical tasks with regard to assessing flexibility, with stimuli (e.g. lights, odors, tones) and response options (e.g. lever press, nose poke) that are relevant for the animal. Severe focal TBI to the frontal cortex impairs behavioral flexibility using simple discrimination-reversal tests in rats and mice. Specifically, TBI animals show a range of deficits from small (additional trials or sessions to criterion) to major (never achieving criterion) during postacute and early chronic recovery periods (2–5 weeks postinjury; Martens et al., 2012; Bondi et al., 2014; Vonder Haar et al., 2014a, 2014b; Chou et al., 2016). In addition to acute impairments, Chou et al. (2016) suggest that deficits persist for 5.5 months postinjury.
Additional complexity can be added to discrimination-reversal tasks in nonhuman animals by utilizing complex multimodal discriminative stimuli (e.g. odor+texture, light+location), and shifting the discrimination only along one dimension of discriminability. This procedure, known as attentional set-shifting (Birrell and Brown, 2000), is roughly analogous to the Wisconsin Card Sorting Task used with humans. This results in a subtler shift in reinforcement contingencies (50% reinforced rather than extinction), which can make for a more difficult task. This form of learning is also impaired following unilateral parietal focal injury in rats, although only at higher injury severities (Bondi et al., 2014), but unaffected by unilateral frontal focal injury (Chou et al., 2016).
Summary
Impairments in flexibility affect an individual’s ability to appropriately change in response to their environment in a fundamental way, and may contribute to a poorer quality of life for individuals with TBI who report these symptoms. This phenomenon has been replicated in the preclinical literature, but only after relatively severe injuries, potentially limiting its applicability to the clinical condition. However, these data may serve as a starting point for the evaluation of pharmacotherapies, and could be extended to more complex discrimination-reversal procedures and more mild injury models.
Working memory
Working memory (WM) refers to the ability to remember a given stimulus over a relatively short time frame (commonly seconds to minutes). The assessment of WM typically involves presenting a stimulus, removing it for a given delay, and then testing for recall.
Clinical evidence
Numerous procedures exist for assessing WM in humans using verbal, visual, and spatial modalities (Dunning et al., 2016). Two common procedures that have been used in the field of clinical TBI are the digit-span and n-back tasks (Asloun et al., 2008; Levin et al., 2002; Chen et al., 2012). The digit-span task measures verbal WM and presents a sequence of numbers as the stimulus, which subjects are then asked to reproduce after a brief delay. Sequence length is gradually increased with each correct response. The primary dependent measure in this task is the number of digits correctly remembered in sequence, with fewer digits indicating impaired WM. Digit span is commonly included as part of the Weschler Adult Intelligence Test battery, and so has been fairly extensively tested. While it is relatively common to find some level of impairment post-TBI (Scherwath et al., 2011; Woods et al., 2011), there are studies that report no effect after milder TBI (Demery et al., 2010; Chen et al., 2012). In addition, some researchers have noted that this task may be confounded by the motivation of the individual, potentially exacerbating measurements in TBI populations (Clark et al., 2014; West et al., 2011).
Similar to the digit-span task, the n-back task presents a sequence of stimuli (commonly numbers or letters), one at a time. During each stimulus presentation, the participant is instructed to identify whether the current stimulus is the same as the one presented n trials (commonly 2 or 3) before the current stimulus. Thus, if n=3, the participant must identify when the current stimulus matches the one presented three trials before the current stimulus, typically with a mouse click or button press. Task difficulty increases as n increases and thus, the primary dependent measure is typically the proportion of correct responses at 2-back or 3-back, with smaller proportions indicating impaired WM. Levin et al. (2002) assessed WM of adolescents five years postmild or postsevere TBI compared with healthy controls, matched for age and parental education status, using 1-back, 2-back, and 3-back tasks. Adolescent patients with brain injuries demonstrated reduced WM performance on all three task levels. Similarly, adult patients with severe or mild TBI showed reduced WM performance on 1-back, 2-back, and 3-back tasks compared with matched control participants (Asloun et al., 2008; Chen et al., 2012).
Preclinical evidence
Despite the potential impact on daily function in clinical populations, memory impairments following experimental TBI have primarily been studied using the MWM, and largely focused on reference memory as opposed to WM. However, alternative measures for assessing WM deficits include variants of the MWM as well as delayed match-to-sample (DMTS) tasks. Here, we limit our discussion to these, given that they are highly dependent upon executive function relative to the traditional reference MWM.
Morris water maze variants
The traditional form of the MWM is ubiquitous in the field of experimental TBI. In this task, animals are placed in a tank of water, and may escape by locating a submerged platform. Animals gradually reduce their latency to escape using visual cues located around the room, typically considered a measure of reference memory that is highly hippocampal-dependent (Morris, 1984). Several variations of the MWM have been implemented aimed at assessing WM. These variations include a moving platform wherein the animals’ ability to track changing locations of the platform is assessed (Hamm et al., 1996; Hoane et al., 2003). During testing, the platform is submerged in a new quadrant of the MWM tank. After the platform is placed in its new location, animals are placed into the tank and given a brief period of time to locate the platform. If the platform is not located by the animal after the time interval elapses, the experimenter guides the animal to the platform. Following the first trial, considered an ‘information’ trial, additional trials are conducted with several minutes separating the start of each trial, and latencies to find the platform are taken as a measure of WM function. Because of the length of this delay, these procedures are considered different from delayed match-to-sample parameters described below, although there is considerable overlap. Longer latencies on this procedure have been indicative of WM deficits following moderate bilateral parietal (Hamm et al., 1996), severe frontal (Hoane et al., 2003, 2004, 2005; Kokiko et al., 2006) and severe unilateral parietal TBIs (Quigley et al., 2009; Swan et al., 2011) during acute recovery. Evaluation of chronic deficits has been limited, but unpublished data suggest that WM impairments do not resolve after focal, frontal TBI (Fig. 1).
Fig. 1.
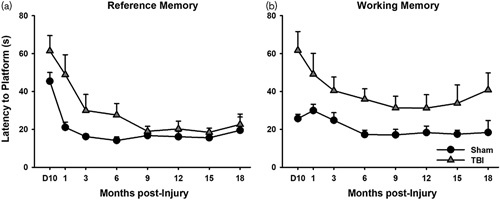
Long-term, selective deficits in working memory after traumatic brain injury (TBI) (Martens KM, Vonder Haar C, Swan AA, Emery MA, Clayton ER, Peterson TC, Hoane MR, unpublished data). Rats (3 months of age) received a bilateral frontal, focal TBI and were tested up to 18 months postinjury. (a) TBI caused deficits in the reference version of the Morris water maze (P=0.012), which resolved by 9 months postinjury. (b) TBI also caused deficits in a working memory version of the task where the platform was moved to a novel location daily, but these never resolved (P=0.004).
Delayed match-to-sample
In general, DMTS procedures include a sample stimulus, followed by two comparison stimuli, and choice for one of the two comparison stimuli is recorded. Choice for the comparison stimulus that matches the sample stimulus is reinforced, while choice for the novel comparison stimulus results in extinction or timeout. Following accurate discrimination between the two comparison stimuli and correct ‘matching’ to some criterion, a delay is introduced such that the animal must remember which sample stimulus was presented for a period of time before the comparison stimuli are presented. A ‘non-match’ version of this procedure follows an opposite rule in which animals must identify the stimulus that is novel after the delay.
Various DMTS procedures have been used in the field of experimental TBI where stimuli are associated with spatial location. In one study that used an operant chamber for WM assessment in rats (Lindner et al., 1998), one lever was extended into the chamber and a response on that lever resulted in lever retraction (sample stimulus). Following an intertrial interval, which varied from 0 to 50 s, both levers were extended into the chamber (i.e. comparison stimuli) and animals were trained to respond on the lever that did not match the sample stimulus (non-match-to-sample). Following severe focal injuries to the lateral sensorimotor cortex, rats demonstrated a transient impairment, while those that received severe bilateral frontal cortex injuries, had deficits that lasted up to 11 weeks postinjury.
Although operant techniques represent a common procedure for testing WM, various maze assessments are more prevalent in experimental TBI (e.g. T-mazes and eight-arm radial mazes). In general, T-mazes have included a platform that allows animals to escape from a tub of water (Whiting and Hamm, 2006; Hoskison et al., 2009). During one variation of this procedure, a divider is placed on one side of the maze during forced-exposure trials, so that the animal is forced to enter that side and reach a platform to escape from the water (sample stimulus). Immediately or after a delay, a free-choice trial is given in which the divider is removed and the animal can enter either side of the maze (comparison stimuli). When the animal enters the same side of the maze as that presented during the forced-exposure trial, the choice is reinforced by escaping the water. Using this procedure, Whiting and Hamm (2006) found that when the delay between sample and comparison stimuli was relatively short (i.e. 15 s), matching of sham animals and those experiencing moderate lateral focal TBI reached ∼90% accuracy. Although performance was disrupted at relatively long delays (e.g. 30 and 120 s) for both groups (80 and 75% respectively for sham), it was disrupted to a larger degree following injury, with TBI animals displaying an ~15% reduction in accuracy relative to sham. Similarly, performance was disrupted at 5 and 10 s delays following unilateral parietal TBI in rats relative to sham animals (Kline et al., 2002; Hoskison et al., 2009; Dash et al., 2010; Kobori et al., 2011; Titus et al., 2016) and in one study, deficits persisted until at least 16 weeks postinjury (Hoskison et al., 2009; Dash et al., 2010; Kobori et al., 2011; Titus et al., 2016).
Similar to results from DMTS and T-maze tasks, animal studies suggest that WM, as measured by the eight-arm radial maze, is impaired after mild to severe focal unilateral TBI (Lyeth et al., 1990; Enomoto et al., 2005; Taylor et al., 2008; Sebastian et al., 2013; Shin et al., 2016). The typical radial maze paradigm involves delayed nonmatch to sample. Reinforcers are placed in four of the eight arms, and animals must remember locations they have already visited. Deficits, as measured by number of errors (repeated visits), on this task after TBI may be quite long lasting, as one study identified deficits even when testing began 6 weeks after injury (Sebastian et al., 2013).
Summary
Deficits in WM after TBI are robust in the preclinical literature and relatively common in clinical practice across many different approaches to testing. Most interesting is the animal finding that WM deficits happen regardless of location of injury. Given that WM is commonly considered to be frontal-dependent, this suggests that there is more at work than mere tissue loss, and that other factors, such as long-term alteration to neurotransmission, may play a role. Whereas motivational deficits are emphasized as a potential confound in clinical work, the animal research appears quite reliable in this regard, perhaps by using salient reinforcers such as escape from water or palatable sucrose pellets.
Monoaminergic system changes following traumatic brain injury
The brain regions that play prominent roles in executive function tasks discussed above include the frontal cortex, striatum (which includes the nucleus accumbens and caudate-putamen), and hippocampus (Baron et al., 1985; Hicks et al., 1993; McDonald et al., 2002; Chudasama and Robbins, 2006). These regions, and the circuits they form, are intimately dependent upon proper monoaminergic function. In the following sections, dopamine (DA), serotonin (5-hydroxytryptamine, 5-HT), and norepinephrine (NE) changes following TBI are discussed, with an emphasis on changes in brain regions known to be critical for executive function. It should be noted that although changes to monoaminergic systems are discussed in terms of disruptions, such changes may not necessarily be detrimental but rather, may reflect compensatory adaptations.
Dopamine
DA is a prominent signaling system with widespread effects and is critical for frontal-dependent, striatal-dependent, and hippocampal-dependent executive function (Beaulieu and Gainetdinov, 2011; Haber, 2014; Trantham-Davidson and Chandler, 2015). DA is heavily concentrated in striatal regions, and serves a strong signaling function within the prefrontal cortex. Changes to the DA system have been suggested to underlie chronic behavioral and cognitive dysfunction following TBI (Bales et al., 2009). This injury-induced disruption occurs directly, but also by indirect glutamatergic and GABAergic signaling alterations (Bales et al., 2009).
Clinical evidence
There are limited data available regarding the relationship between clinical TBI and DA transmission, and a small portion of such data are equivocal. Using positron emission and single-photon emission tomography, clinical imaging studies converge on moderate and severe TBI-related reductions in striatal DA transporter densities when compared with healthy control participants matched for age and educational status (Donnemiller et al., 2000; Wagner et al., 2014). However, effects of clinical TBI on DA receptor densities and metabolism are less clear. Studies suggest that striatal D2 receptor binding is altered following moderate or severe TBI, although there are reports of both up-regulation and down-regulation (Donnemiller et al., 2000; Wagner et al., 2014). Similarly, it seems as though DA turnover is significantly altered following severe TBI (both increases and decreases have been reported; Bareggi et al., 1975; Porta et al., 1975; Vecht et al., 1976; Majchrzak et al., 1979; Massucci et al., 2004). Such discrepant evidence may be because of contributing sex differences and/or genetic profiles (Wagner et al., 2007, 2014) in which DAergic transmission is more heavily impacted for female patients and for those with certain functional genetic variants (Wagner et al., 2007).
Preclinical evidence
Given the sparse and sometimes conflicting evidence for DA system changes following clinical TBI, as well as the cross-sectional nature of the data that precludes the detection of a causal relationship between TBI and DAergic transmission, the field of experimental TBI has focused on evaluating DA changes at each step in the process of neurotransmission – synthesis, distribution, and breakdown – using highly reproducible animal models of injury.
Tyrosine hydroxylase (TH) is the rate-limiting enzyme that is responsible for converting the amino acid l-tyrosine to l-DOPA. Given that l-DOPA is the immediate precursor for DA synthesis, changes in TH levels lead to alterations in DA signaling. Preclinical work suggests that TH may be influenced differentially across brain regions and time. Notably, disruptions in TH activity may not be apparent during the acute post-TBI phase (Huger and Patrick, 1979), but rather manifest themselves later during recovery (Yan et al., 2001, 2007; Shin and Dixon, 2011; Shin et al., 2011, 2012). Increased TH in the frontal cortex and substantia nigra have been observed following severe focal TBI in rats (Yan et al., 2001, 2007), but only at chronic time points (28+ days postinjury). In the same model of injury, Shin et al. (2011, 2012) determined the functional ability of TH to convert l-tyrosine to l-DOPA, with a similar lack of differences early postinjury, but decreases in TH function for TBI rats at 1 and 4 weeks postinjury. While the effect of TBI on TH activity has primarily been examined in the striatum and substantia nigra of rats (Yan et al., 2001, 2007; Shin and Dixon, 2011; Shin et al., 2011, 2012), mRNA levels for TH are elevated after mild blast injury in the locus coeruleus and raphe nucleus (Kawa et al., 2015), and protein levels and functional capacity of TH are elevated after moderate focal injury in the prefrontal cortex (Kobori et al., 2006). Together, the evidence suggests that TH function is disrupted following various experimental TBIs, but the nature of disruption may be dependent upon both the region of interest, and the time point at which it is measured (i.e. increase or decrease in activity). Vesicular storage of DA and other monoamines may also be altered after TBI as some alleles of the vesicular monoamine transporter are associated with cognitive dysfunction after TBI in patients (Myrga et al., 2016), and vesicular monoamine transporter is down-regulated after experimental TBI, albeit only in female rats (Xu et al., 2016).
Given that TH activity is disrupted post-TBI, it follows that DA release and basal DA concentrations also experience alterations. Indeed, evidence from high-performance liquid chromatography and western blot studies with rats suggests that there are initial increases in DA levels in the frontal cortex and striatum postmild blast or postsevere focal TBI that persists for at least 28 days (Massucci et al., 2004; Kobori et al., 2006; Kawa et al., 2015). While higher DA levels have been measured post-mortem after TBI, in-vivo recordings of DA release from presynaptic neurons using fast scan cyclic voltammetry or microdialysis show significantly lower levels for severe and moderate focal-TBI rats up until 8 weeks postinjury when compared with sham rats (McIntosh et al., 1994; Wagner et al., 2005; Shin and Dixon, 2011; Huang et al., 2014a; Chen et al., 2015, 2017). In turn, DA transporter down-regulation may be due to the significant, chronic reduction in frontal and striatal DA release following TBI, leading to a reduction in DA reuptake and clearance from the synapse (Yan et al., 2002; Wagner et al., 2005; Wilson et al., 2005; Huang et al., 2014a; Shimada et al., 2014). The effect of TBI on DA transporter down-regulation is true not only for the frontal cortex and striatum (Yan et al., 2002; Wagner et al., 2005; Wilson et al., 2005), but also for the midbrain (Shimada et al., 2014), and occurs even in the case of mild TBI (Yan et al., 2002; Wagner et al., 2005; Wilson et al., 2005; Huang et al., 2014a; Shimada et al., 2014). This is perhaps because of the action of DA as an excitotoxic agent, in which elevated levels in the acute post-TBI phase may lead to excitotoxicity and oxidative damage, resulting in lower levels of DA release at chronic time points and compensatory changes in DA transporter densities (Olney et al., 1990; Wagner et al., 2005). Moreover, DA transporter down-regulation appears to be a chronic effect and persists for at least 28 days postinjury for rats (), although additional time points have not be evaluated.
In rat models, some have reported transient (<24 h) reductions in D1 receptor density in the striatum after TBI (Henry et al., 1997), but another study observed a complex relationship at 24 days postinjury of decreased D1 receptor density in dorsal striatum, and increased D1 levels in the nucleus accumbens, but only after mild TBI (Vonder Haar et al., 2016). Moreover, no changes in striatal D2 receptor densities have been recorded across multiple time points (Henry et al., 1997; Wagner et al., 2005, 2008; Vonder Haar et al., 2018).
In addition to reuptake by transporters, the second mechanism by which DA is removed from the synapse is by enzymatic degradation; the enzymes catechol-O-methyl transferase (COMT) and monoamine oxidase (MAO) break down DA into its primary metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid. Although there have been no studies assessing effects of TBI on COMT or MAO directly, existing evidence suggests that DA metabolism may be affected acutely post-TBI. However, there is some discrepancy in the preclinical TBI field, in which some work suggests that there are heightened levels of DOPAC and homovanillic acid of rats in the short term, corresponding with elevated DA levels (Massucci et al., 2004; Shin and Dixon, 2011; Shin et al., 2012), while others report reduced levels of such metabolites in mice and rabbits during acute recovery (Edvinsson et al., 2009; Shen et al., 2011). In addition, DOPAC/DA ratios (a measure of DA turnover) are significantly altered in rats postinjury (both increases and decreases have been reported; Massucci et al., 2004; Shen et al., 2011), giving evidence for altered DA metabolism and not simply an up-regulation or down-regulation of DOPAC as a response to changes in DA levels. However, by 7 days postinjury, no differences were found between sham and injured rats, suggesting that DOPAC levels (and potentially COMT and/or MAO levels) become normative during the postacute recovery period (Shin and Dixon, 2011; Shin et al., 2012).
Summary
In addition to chronic, detrimental effects of TBI on DA synthesis, distribution, and breakdown, moderate focal TBI results in a significant, progressive loss of DA neurons in the substantia nigra (Wagner et al., 2009; Hutson et al., 2011; van Bregt et al., 2012), which may be representative of the established link between TBI and the development of Parkinson’s disease (Semchuk et al., 1993). Thus, TBI has widespread effects on the DAergic system, affecting all aspects of DA neurotransmission. Given the evidence from both experimental and clinical TBI fields, treatment has focused largely on providing DAergic pharmacotherapies (e.g. d-amphetamine, methylphenidate) for TBI patients to aid in symptom reduction following injury by increasing DA signaling (Bales et al., 2009), although these pharmacotherapies are not without potential pitfalls (see below: Special Considerations for Pharmacotherapy following TBI). Moreover, given the unique time course of changes to DA after TBI, the exact timing of pharmacotherapies will be of critical importance.
Norepinephrine
Of all the monoaminergic systems, NE has received the least attention in regard to alterations following TBI. DA is converted to NE by the rate-limiting enzyme, DA-β-hydroxylase. Therefore, the disruptions in TH or DA activity discussed above also contribute to alterations in NE activity.
Clinical evidence
The two clinical TBI studies that evaluated changes in NE signaling relative to non-TBI control participants suggest that brain injury increases NE metabolism, indicated by heightened levels of NE’s primary metabolite, 3-methoxy-4-hydroxyphenylglycol. However, this was only measured at between one and 12 days postinjury (Markianos et al., 1992, 1996). Although NE has not been a prime target of interest for clinical TBI research, selective NE reuptake inhibitors are a relatively common treatment for depression and anxiety, and may also improve attention, all of which suffer some impairment in TBI patients (Hibbard et al., 1998; Jorge and Robinson, 2003; Juengst et al., 2017). Thus, further clinical work investigating NE activity following clinical TBI is needed.
Preclinical evidence
Experimental work suggests that brain injury leads to significant increases in NE levels in the prefrontal cortex, hippocampus, cerebellum, and hypothalamus of rats (at 2 h, 7 days, and 14 days), which corresponds with increased TH levels at these same time points (Huger and Patrick, 1979; Kobori et al., 2006; Kawa et al., 2015). In addition, acute increases in NE occur specifically at the site of injury, followed by a return to levels comparable to those of sham rats by as early as one week postinjury (McIntosh et al., 1994; Levin et al., 1995; Dunn-Meynell et al., 1998; Fujinaka et al., 2003). Thus, the evidence suggests that relatively acute disruptions occur during NE synthesis and distribution/release following injury in experimental studies, although clinical work is needed to validate these findings in TBI patients. In addition to significant increases in basal NE, early studies suggested that α1A receptor subtypes are transiently reduced at the injury site and may persist until 30 days postinjury in rats (Prasad et al., 1992; Levin et al., 1995), while more current work reports increased α1A receptor mRNA in the medial prefrontal cortex following TBI (Kobori et al., 2011).
Summary
While NE transmission is likely disrupted following TBI, research is lacking in specificity of the nature of those disruptions. It is likely that NE signaling disruption following TBI could influence executive function through the locus coeruleus innervation of the prefrontal cortex (Logue and Gould, 2014). In particular, whether densities of other receptor subtypes are altered, what changes occur at the NE transporter, and the degree to which these changes may contribute to behavioral dysfunction remain open questions. It may also be a clinical concern that elevated NE levels have been detected after TBI, yet selective NE reuptake inhibitors may be used clinically for a host of conditions in these patients. Overall, research regarding TBI and NE disruption is lacking and there is a critical need for both clinical and preclinical work.
Serotonin
Although more research has been conducted on 5-HT compared with NE, relatively little work has investigated the mechanisms by which TBI disrupts 5-HT signaling.
Clinical evidence
Clinical studies suggest that 5-HT transmission is indeed disrupted following TBI, revealed by significant changes in 5-HT metabolites in the cerebrospinal fluid of TBI patients (both increases and decreases have been reported; Porta et al., 1975; Vecht et al., 1976; Majchrzak et al., 1979; Markianos et al., 1992, 1996). Discrepancies in the nature of such changes in 5-HT metabolite levels may be explained, at least in part, by measurement time points, with 5-HT being elevated during the acute post-TBI phase and reduced during chronic recovery (Porta et al., 1975; Vecht et al., 1976; Majchrzak et al., 1979; Markianos et al., 1992, 1996). The mood-altering effects of 5-HT have been a primary area of focus, given that the development of mood disorders such as depression and anxiety are prevalent following brain injury (Hibbard et al., 1998; Jorge and Robinson, 2003; Juengst et al., 2017) and the most common treatment for such disorders are selective 5-HT reuptake inhibitors (SSRIs; Ciuna et al., 2004). Although effects have not been assessed directly, it is possible that 5-HT signaling disruption following TBI may also influence executive function (Cifariello et al., 2008). Indeed, some have reported significant inverse correlations between 5-HT signaling and impulsive behavior (Harrison et al., 1997; Dalley et al., 2002), and inverse relations have been identified between 5-HT metabolites and aggression levels, although these may also be mediated by DA signaling (Coccaro et al., 2010).
Preclinical evidence
Tryptophan hydroxylase (TPH) is the rate-limiting enzyme that is responsible for interacting with the amino acid tryptophan and converting it to 5-HT, which is then acted upon by DOPA decarboxylase to create 5-HT. Although TPH is significantly increased in the dorsal raphe nucleus and locus coeruleus in rats acutely following mild blast TBI (i.e. up until 24 h), by 48 h post-TBI, TPH levels return to those that are comparable to sham rats (Kawa et al., 2015). Similarly, acute changes have been observed in rat basal 5-HT levels post-TBI (10 min; Busto et al., 1997), suggesting that acute disruptions in TPH production directly affect 5-HT levels. However, there is some discrepancy as to whether 5-HT levels return to normal early during recovery. Some have reported a return to sham levels by seven days postinjury (Kawa et al., 2015), while others suggest that 5-HT levels are elevated until at least 2 weeks postinjury (Mustafa et al., 2017). Such discrepant findings may be because of the use of different injury models across studies, in which mild TBI induced by weight drop may affect 5-HT transmission longer into the recovery period than that induced by blast (Kawa et al., 2015; Mustafa et al., 2017). Given the limited and sometimes conflicting evidence, further preclinical work is needed to dissociate the mechanisms by which TBI affects 5-HT levels.
In addition to relative acute and transient disruptions in 5-HT synthesis and basal concentrations post-TBI, there are no changes in 5-HT2A receptor densities in the motor, prelimbic, agranular, sensory, or cingulate cortices of rats at 15 days postinjury (Dam et al., 2013), although more acute time points have not been assessed. In contrast, significant increases in 5-HT1A densities in the hippocampus have been shown at 15 days postinjury (Wilson and Hamm, 2002), which may contribute to memory deficits that are seen following TBI (Dale et al., 2016). Changes in other 5-HT receptors have not been evaluated after TBI, potentially because of a lack of strong radioligands for human patients, or because of the inherent complexities in studying the numerous receptors and their actions.
In terms of 5-HT deactivation, moderate or severe TBI produces significant, chronic reductions in 5-HT transporter densities in the frontal and cingulate cortices of rats (Abe et al., 2016). At the same time, moderate TBI leads to chronic increases in 5-HT transporter densities in the raphe nucleus while having no effect on densities in the hippocampus, thalamus, or amygdala of rats (Dam et al., 2007). Together, the evidence suggests that 5-HT reuptake is dysregulated following TBI, but the nature of such disruption may depend upon injury type and brain region. In addition to 5-HT deactivation by reuptake, 5-HT enzymatic degradation may be disrupted following TBI, which is in line with clinical evidence. Indeed, significant reductions in 5-HIAA in the rat hippocampus have been observed at 30 min post-TBI (Eschun et al., 1992), although additional time points have not been assessed.
Summary
Given the significant reduction in 5-HT transporters in the frontal cortex, a critical region for executive function (McDonald et al., 2002; Chudasama and Robbins, 2006), as well as disruptions in 5-HT metabolism, it is possible that disruptions in 5-HT breakdown and removal from the synapse contribute to behavioral impairments after TBI, although effects have not been assessed directly. In addition to replicating the results discussed above using alternative models of experimental TBI, the efficacy of commonly prescribed SSRIs (i.e. 5-HT transporter antagonists) on executive function following TBI should be assessed thoroughly given that a large portion of TBI patients experience postinjury depression and/or anxiety (Hibbard et al., 1998; Jorge and Robinson, 2003; Juengst et al., 2017) and are likely to be prescribed such pharmacotherapies.
Special considerations for pharmacotherapy following traumatic brain injury
Reduced sensitivity to reinforcement
TBI patients show significant impairments in executive function, which may be due, in part, to insensitivity to contingencies and natural reinforcement processes. Executive function tasks include reinforcing (and often concurrent punishing) consequences for appropriate responding, with optimal responding in these tasks producing higher rates of reinforcement. However, individuals with TBIs show reduced sensitivity to and awareness of reinforcement contingencies (Schlund and Pace, 2000; Schlund et al., 2001; Schlund, 2002a, 2002b; Larson et al., 2007), and are slower to adapt choices following changes in reinforcing contingencies compared to non-TBI controls (Schlund et al., 2001; Schlund, 2002a, 2002b). All of these processes are directly dependent on DAergic signaling, and a large literature has demonstrated the role of DA in primary reinforcement (Cameron et al., 2014; Shnitko and Robinson, 2015), reward expectation (Schultz et al., 1997; Cocker et al., 2016), and punishment salience (Tomer et al., 2014; van der Schaaf et al., 2014; Jean-Richard-Dit-Bressel et al., 2018). Despite these large effects in patients, sensitivity to contingencies may not be impacted at the most basic level. As such, one study examined responding under basic schedules of reinforcement (e.g. fixed ratio, fixed interval, variable ratio, variable interval) between sham and rats undergoing experimental TBI and found no notable deficits in TBI rats (Vonder Haar et al., 2016). However, in the same model of TBI, rats displayed substantial deficits in simple discriminations (Martens et al., 2012; Vonder Haar et al., 2014a, 2014b) and aberrant (but not purely disadvantageous) choice behavior on the RGT (Shaver TK, Ozga JE, Zhu B, Anderson KG, Martens KM, Vonder Haar C, unpublished data), suggesting that discrimination between concurrently available contingencies may be reduced, which may drive executive function deficits in TBI patients. Given the scope of this problem, augmented behavioral therapies may need to be developed specific to patients with brain injury in order to appropriately serve this population (Knight et al., 2002; Wood and Alderman, 2011). These foundational problems should also be considered when assessing executive function in animal models of TBI.
Altered pharmacology after traumatic brain injury
While pharmacotherapies to treat TBI remain a primary interest of the medical community, several concerns have been raised about factors that may alter the efficacy of these treatments. We have opted to focus primarily on mechanisms of monoaminergic dysfunction because of their suspected role in executive impairment; however, it should be noted that effects of TBI extend far beyond the monoamines. Notably, altered pharmacokinetics have been observed by multiple mechanisms after TBI and for many different drugs. In particular, hepatic cytochrome-P450 enzymes are upregulated, and protein-depot binding in blood may be reduced, resulting in faster metabolism of many substances (Empey et al., 2006; Anderson et al., 2015). Parsing these metabolic changes is further compounded by disruption of the blood–brain barrier (Stowe et al., 2000; Hay et al., 2015; Prakash and Carmichael, 2015), potentially resulting in higher than normal drug concentrations, and/or other interfering proteins reaching neural tissue as blood–brain barrier permeability shifts after injury. Together, these factors present challenges to both the experimental researcher and the clinician regarding concentration and frequency of dosing, and may help explain the numerous treatment failures experienced in the field of TBI.
While alterations to pharmacokinetics are concerning, fully understanding the changes in pharmacodynamics after TBI are an even more difficult challenge. Monoaminergic metabolism, as well as receptor and transporter densities are all altered at some point following TBI, leading to the question of whether drugs exert the same effects in TBI as non-TBI populations (McAllister et al., 2011a), or whether TBI patients represent a unique subgroup with regard to conventional pharmacotherapies. In addition, alterations in monoaminergic signaling are varied during acute versus chronic recovery, and thus, how pharmacotherapies affect executive function at different time points is also important for a full understanding of pharmacotherapy following TBI.
Dopaminergic therapies
DA pharmacotherapies, such as amantadine hydrochloride, bromocriptine, d-amphetamine, and methylphenidate (DA agonists), are reported to have cognitive-enhancing (i.e. attention and working memory) effects in TBI patients during chronic recovery (see Bales et al., 2009; Liepert, 2016 for reviews). Therefore, these and other pharmacotherapies (apiprazole, l-deprenyl, and methamphetamine) aimed at increasing DA signaling have been tested following experimental TBI. Several of these drugs show promise in reducing chronic signaling deficits following injury, such as increasing DA levels in the striatum and substantia nigra while also reducing neuronal death (Zhu et al., 2000; Wagner et al., 2008, 2009; Rau et al., 2012; Huang et al., 2014a, 2014b; Wang et al., 2014; Tan et al., 2015; Phelps et al., 2017). In turn, performance is improved acutely for TBI animals on traditional tasks for assessing cognitive deficits, such as the MWM and novel-object recognition tasks, following administration of such therapeutic agents (Zhu et al., 2000; Wagner et al., 2008, 2009; Rau et al., 2012; Huang et al., 2014a, 2014b; Wang et al., 2014; Tan et al., 2015; Leary et al., 2017; Phelps et al., 2017). At the same time, pharmacotherapies that reduce DA signaling, such as haloperidol and resperidone, exacerbate acute MWM deficits, in both TBI and non-TBI animals (Wilson and Hamm, 2002; Kline et al., 2007, 2008; Hoffman et al., 2008), giving support for a significant contribution of reduced DA signaling in cognitive dysfunction.
Most recently, work has focused on investigating how DA therapies may affect higher-order executive function using tasks such as those discussed earlier in this review. Similar to memory-related assessments, drugs that increase DA signaling (amantadine and d-amphetamine) reduce chronic deficits in motor impulsivity in TBI animals on the 5CSRTT (Vonder Haar et al., 2016), although effects of amantadine were accompanied by potential psychomotor slowing or motivational issues across groups. Perhaps most interesting was the fact that d-amphetamine selectively reduced impulsivity, but only in severely-injured rats; an effect that we have observed again recently (Fig. 2). This provides strong evidence that those with TBI may not respond to pharmacologic treatments in the same manner as those without such injury.
Fig. 2.
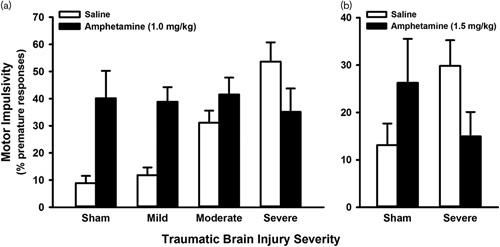
Differential effects of amphetamine challenge on motor impulsivity after traumatic brain injury (TBI). (a) High-dose amphetamine significantly reduced impulsivity on the five-choice serial reaction time task, but only for animals with a severe focal TBI (P=0.002) (adapted with permission from Vonder Haar et al., 2016, copyright 2016 American Chemical Society). (b); High-dose amphetamine significantly reduced impulsivity on the rodent gambling task in animals with a severe focal TBI (P=0.011) (Ozga JE, O'Hearn CM, Shaver TK, Lake AD, Vonder Haar C, unpublished data). Adaptations are themselves works protected by copyright. So in order to publish this adaptation, authorization must be obtained both from the owner of the copyright in the original work and from the owner of copyright in the translation or adaptation.
Indeed, human studies using functional MRI give evidence for less activation in various brain regions for TBI patients following acute dosing of bromocriptine when compared with healthy control participants (McAllister et al., 2011a). Thus, TBI patients may not respond in the same way as non-TBI patients to pharmacotherapy, both neurochemically or behaviorally. In addition, DA agonistic therapies may not have the same effect on all types of executive function, as evidenced by methylphenidate aiding in chronic recovery of working memory function in TBI patients (Liepert, 2016). These factors may become even more confounding when considering that mixed in with positive findings are studies claiming efficacy without achieving statistical significance (Kim et al., 2006), evidence for similar rates of recovery under placebo or nondrug conditions (Pavlovskaya et al., 2007), and differential drug effects between TBI and non-TBI animals (Vonder Haar et al., 2016; Shaver TK, Ozga JE, Zhu B, Anderson KG, Martens KM, Vonder Haar C, unpublished data), ), null drug effects (Wilson and Hamm, 2002; Ripley et al., 2014), as well as the need to consider natural aging- or sex-related changes in monoamine metabolism and drug responsiveness. When combined with studies that have identified altered DA pharmacology in TBI patients, it is clear that additional research is needed using animal models to determine whether current DAergics may be effective in treating complex cognitive dysfunction such as decision-making.
Noradrenergic therapies
Although NE signaling deficits have been observed following TBI, evaluations of cognitive performance following administration of pharmacotherapies targeting the NE system are relatively sparse and conflicting. Some have suggested that too much NE signaling is a cause for concern in the acute recovery phase of TBI patients, with α1 receptor antagonists (e.g. prazosin) improving MWM performance in rats with experimental TBI at 14 days postinjury (Kobori et al., 2011). However, others have suggested that too little NE may be the issue. In clinical assessments, guanfacine, an a2A receptor agonist, improved working memory deficits following mild TBI at one month postinjury (McAllister et al., 2011b), although preclinical assessments have reported no effect of guanfacine on MWM performance at 14 days postinjury (Kobori et al., 2011). There is also mixed evidence following administration of atomoxetine, a NE reuptake inhibitor, in which clinical assessments suggest no effect on attentional deficits at one year or more postinjury (Ripley et al., 2014), while preclinical work suggests improved working memory and response inhibition following atomoxetine treatment during both, acute and chronic recovery of TBI animals (i.e. 14 days and 12 weeks; Reid and Hamm, 2008; Vonder Haar et al., 2016). In addition to the sparse, and sometimes conflicting, evidence, atomoxetine has a low binding affinity for the DA transporter (Bymaster et al., 2002); thus, its effects on executive function may not be due to its action on NE per se but rather by its effects on DA.
Serotonergic therapies
The majority of work with 5-HT agents has focused on improving disruptions in mood, such as depression and anxiety. However, executive function deficits are prevalent in patients with depressive disorders (Alves et al., 2014), and 5-HT agents may simultaneously improve executive function and mood (Gualtieri et al., 2006). Few studies have investigated how 5-HT pharmacotherapies may impact cognitive function directly either following clinical or experimental TBI. Similar to DA agonists, various 5-HT agonists (N-n-propyl-3-ethoxyquinoxaline-2-carboxamide, 8-OH-DPAT, repinotan, buspirone, and fluoxetine) produce increases in basal 5-HT levels as well as reduced neuronal cell loss and contusion volume following experimental TBI, corresponding with a reduction in acute and chronic anxiety-like (elevated plus maze, marble burying, and open field) and depressive-like (sucrose preference) behaviors (Kline et al., 2001, 2012; Cheng et al., 2007, 2008; Olsen et al., 2012; Monaco et al., 2014; Bhatt et al., 2017).
In addition to emotion-related behaviors, a variety of 5-HT1A receptor agonists improve acute and chronic MWM performance in TBI animals, suggesting that deficits in 5-HT signaling post-TBI contribute to cognitive deficits (Kline et al., 2001, 2012; Cheng et al., 2007, 2008; Olsen et al., 2012; Monaco et al., 2014; Bhatt et al., 2017). However, others have reported no effect on acute MWM performance in TBI animals following systemic fluoxetine administration, an SSRI that is prescribed commonly for depression and anxiety (Wilson and Hamm, 2002). Given the limited evidence for how 5-HT-enhancing drugs may affect cognitive function, in conjunction with a lack of work using higher-order executive function tasks, further work is needed to evaluate how common 5-HT pharmacotherapies (SSRIs, in particular) affect working memory, decision-making, and impulsivity.
Efficacy of pharmacotherapies
Reports showing mixed efficacy of pharmacotherapies in TBI populations (both human and nonhuman animal) are likely due to several factors, including timing of treatment during recovery. As an example, NE antagonists have shown promise in reducing hippocampally dependent cognitive deficits, while agonists have been shown to produce no improvements during acute recovery (Kobori et al., 2011). In contrast, during chronic recovery, NE agonists reduce cognitive function deficits (McAllister et al., 2011b; Vonder Haar et al., 2016), although the magnitude of effect is small. In addition to the timing of drug administration, alterations in monoaminergic pharmacodynamics may lead to differences in dosing guidelines needed to produce significant effects on executive function for TBI populations. Indeed, some have suggested that increased doses of DAergic therapies are needed to affect executive function following TBI in rats (Bondi et al., 2014; Vonder Haar et al., 2016; Leary et al., 2017; Shaver TK, Ozga JE, Zhu B, Anderson KG, Martens KM, Vonder Haar C, unpublished data), which is reflective of altered pharmacokinetics or pharmacodynamics following injury. Thus, the efficacy of traditional pharmacotherapies for reducing executive function deficits likely depends upon when during recovery and in what doses drugs are administered. Together, changes in pharmacodynamics and pharmacokinetics following TBI pose challenges to clinicians and researchers, and may explain the high frequency of treatment failures that exist in the field.
Deficits in executive function and monoaminergic signaling may also help to explain the heightened prevalence of alcohol, cocaine, opioid, marijuana, and amphetamine use disorders in TBI populations compared with those without such injuries (Walker et al., 2003; O’Phelan et al., 2008; Golub and Bennett, 2013; Singh et al., 2014; Ma et al., 2015; Ramesh et al., 2015). Given the large-scale alterations to monoaminergic systems following TBI, increased drug-seeking behaviors, particularly of psychostimulants, may represent an effort to ‘self-medicate’. While this simple pharmacological argument is attractive, it is unlikely to account for the entirety of substance abuse after TBI. In particular, research has showed that impulsivity is intimately linked to the development of substance abuse (Perry et al., 2005; Dalley et al., 2007), and thus may be a mediating factor. Further, deficits in behavioral flexibility are likely to promote continued drug dependence, even in the face of detrimental outcomes (Istin et al., 2017). Given that TBI patients demonstrate deficits in all of these domains, these are likely contributors to initiation, exacerbation, and maintenance of substance abuse following injury. However, data on increased incidence after TBI are a classic case of correlation (i.e. does substance abuse cause TBI or does TBI cause substance abuse?), with a lack of concrete prospective studies in humans. Importantly, a number of brain injuries occur while under the influence of various substances, particularly alcohol, with some studies ranging as high as 47% (Andelic et al., 2010). Despite this figure, some studies report no relationship between previous substance abuse and brain trauma (Lange et al., 2014) while others identify relationships only within injury subgroups (Andelic et al., 2010), and indeed, alcohol intoxication at the time of injury is associated with reduced injury severity in humans and animals (Andelic et al., 2010; Goodman et al., 2013; Kanbak et al., 2013; Raj et al., 2015).
The argument for TBI as causal to addiction is much stronger when considering the animal evidence. To date, few studies have examined voluntary drug self-administration after TBI. However, these studies largely converge on the conclusion that animals with injury self-administer more drug, or escalate their intake more quickly, across both alcohol and cocaine (Lim et al., 2015; Mayeux et al., 2015; Weil et al., 2016; Vonder Haar et al., 2018), although such self-administration may depend upon injury type, severity, and substance, as one group found no changes in cocaine intake after blast injury (Muelbl et al., 2018), while others have seen increased alcohol intake after blast (Lim et al., 2015), and increased cocaine after mild or severe focal injury (Vonder Haar et al., 2018). Moreover, studies have found interesting subgroup differences within TBI animals with regard to acquisition of drug self-administration (Lim et al., 2015; Vonder Haar et al., 2018), in which some TBI rats show faster acquisition of drug self-administration than others, suggesting potential biological or behavioral mediators of resilience that warrant investigation. In particular, both DA-related markers (e.g. DA-regulated and cAMP-regulated neuronal phosphoprotein) and multiple inflammatory markers (e.g. cytokines, glial activation) have been affiliated with increased intake (Vonder Haar et al., 2018) or greater sensitivity or preference for drugs of abuse (Lowing et al., 2014; Merkel et al., 2017a, 2017b). As research moves forward, a potential propensity to substance abuse should be a prime consideration when evaluating therapeutics, especially when considering implementation in clinical populations.
Conclusion
In this review, we have identified major changes in executive function following TBI, namely impulsivity, behavioral flexibility, and working memory (Fig. 3). These deficits are present in both human patients, and in animal models of brain injury. While the clinical effects are well-established, the animal literature lags behind. Much of the focus of the experimental TBI field has been on relatively simple assessments of learning and memory, such as the MWM, to the neglect of more complex cognitive function. Further studies are needed to characterize executive function deficits across the many existing injury models in order to generate a more clinically-relevant behavioral phenotype which may then be used to effectively screen treatment options.
Fig. 3.
Schematic representation of dysfunction after traumatic brain injury (TBI). Monoamine disruptions are common starting mere hours after injury, with some continuing for months or longer. Monoamines influence various types of executive function, and this dysregulation after TBI may account for some of the common behavioral disturbances that are observed. Finally, both altered monoamines and functional impairments may converge to explain several post-TBI complications, including numerous therapeutic failures, and increased propensity for substance abuse and dependence. Notably, the intertwined nature of altered functional and structural changes make it difficult to fully parse the separate factors that challenge clinical treatment of TBI. DA, dopamine; 5-HT, 5-hydroxytryptamine; NE, norepinephrine.
Moreover, these deficits in executive function may be driven by the major alterations in monoaminergic neurotransmission such as dopamine, norepinephrine, and serotonin (Fig. 3). However, the field is limited in its consensus on the exact nature of these changes, and there appear to be important factors related to injury type and severity, as well as time from injury. More research is needed at both the clinical and preclinical level to resolve major conflicts (e.g. up-regulation and down-regulation of DA receptors; increased NE despite cognitive impairments that may be alleviated by NE agonists) so that we may better understand the time course of these changes and target therapeutics appropriately.
Finally, these changes in both executive function and monoaminergic status may in turn complicate developing, administering, and achieving efficacious results not only with novel compounds, but also with traditional treatments for psychiatric-like symptoms experienced by these individuals. In particular, reduced sensitivity to contingencies surrounding the individual may limit the efficacy of pharmacotherapies or require an integrated approach with cognitive-behavioral therapies. In addition, altered doses, or less conventional drugs may be required to treat behavioral symptoms, accounting for monoaminergic alterations. Finally, the abuse potential for drugs, particularly psychostimulants which may remediate impulse control issues, needs to be carefully considered given links between substance abuse and TBI populations.
Acknowledgements
The authors thank Dr. Michael Hoane for providing unpublished data in support of this manuscript.
Conflicts of interest
There are no conflicts of interest.
References
- Abe K, Shimada R, Okada Y, Kibayashi K. (2016). Traumatic brain injury decreases serotonin transporter expression in the rat cerebrum. Neurol Res 38:358–363. [DOI] [PubMed] [Google Scholar]
- Alderman N. (2003). Contemporary approaches to the management of irritability and aggression following traumatic brain injury. Neuropsychol Rehabil 13:211–240. [DOI] [PubMed] [Google Scholar]
- Alves MR, Yamamoto T, Arias-Carrion O, Rocha NB, Nardi AE, Machado S, Cardoso A. (2014). Executive function impairments in patients with depression. CNS Neurol Disord Drug Targets 13:1026–1040. [DOI] [PubMed] [Google Scholar]
- Andelic N, Jerstad T, Sigurdardottir S, Schanke A-K, Sandvik L, Roe C. (2010). Effects of acute substance use and pre-injury substance abuse on traumatic brain injury severity in adults admitted to a trauma centre. J Trauma Manag Outcomes 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GD, Peterson TC, Vonder Haar C, Farin FM, Bammler TK, MacDonald JW, et al. (2015). Effect of traumatic brain injury, erythropoietin, and anakinra on hepatic metabolizing enzymes and transporters in an experimental rat model. AAPS J 17:1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asloun S, Soury S, Couillet J, Giroire J, Joseph P, Mazaux J, Azouvi P. (2008). Interactions between divided attention and working-memory load in patients with severe traumatic brain injury. J Clin Exp Neuropsychol 30:481–490. [DOI] [PubMed] [Google Scholar]
- Bales JW, Wagner AK, Kline AE, Dixon CE. (2009). Persistent cognitive dysfunction after traumatic brain injury: a dopamine hypothesis. Neurosci Biobehav Rev 33:981–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareggi SR, Porta M, Selenati A, Assael BM, Calderini G, Collice M, et al. (1975). Homovanillic acid and 5-hydroxyindole-acetic acid in the CSF of patients after a severe head injury. Eur Neurol 13:528–544. [DOI] [PubMed] [Google Scholar]
- Baron JC, Comar D, Zarifian E, Agid Y, Crouzel C, Loo H, et al. (1985). Dopaminergic receptor sites in human brain: positron emission tomography. Neurology 35:16–24. [DOI] [PubMed] [Google Scholar]
- Barratt ES, Stanford MS, Kent TA, Felthous A. (1997). Neuropsychological and cognitive psychophysiological substrates of impulsive aggression. Biol Psychiatry 41:1045–1061. [DOI] [PubMed] [Google Scholar]
- Beaulieu J, Gainetdinov RR. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio AR, Damasio H, Anderson SW. (1994). Insensitivity to future consequences following damage to human prefrontal cortex. Cognition 50:7–15. [DOI] [PubMed] [Google Scholar]
- Berg EA. (1948). A simple objective test for measuring flexibility in thinking. J Gen Psychol 39:15–22. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Mahesh R, Jindal A, Devadoss T. (2017). Neuropharmacological and neurochemical evaluation of N-n-propyl-3-ethoxyquinoxaline-2-carboxamide (6n): a novel serotonergic 5-HT3 receptor antagonist for co-morbid antidepressant- and anxiolytic-like potential using traumatic brain injury model in rats. J Basic Clin Physiol Pharmacol 28:93–100. [DOI] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ. (2000). Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci 20:4320–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi CO, Cheng JP, Tennant HM, Monaco CM, Kline AE. (2014). Old dog, new tricks: The attentional set-shifting test as a novel cognitive behavioral task after controlled cortical impact injury. J Neurotrauma 31:926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Gamble KJ, Bennett DA. (2013). Temporal discounting is associated with an increased risk of mortality among community-based older persons without dementia. PLoS ONE 8:e67376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredemeier K, Miller IW. (2015). Executive function and suicidality: a systematic qualitative review. Clin Psychol Rev 40:170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan-Hancock C, Harrison J. (2010). The global burden of traumatic brain injury: preliminary results from the Global Burden of Disease Project. Inj Prev 16:A17. [Google Scholar]
- Busto R, Dietrich WD, Globus MY-T, Alonso O, Ginsberg MD. (1997). Extracellular release of serotonin following fluid-percussion brain injury in rats. J Neurotrauma 14:35–42. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, et al. (2002). Atomoxetine increases extracellular levels of norepinephrine and dopmaine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27:699–711. [DOI] [PubMed] [Google Scholar]
- Cameron CM, Wightman RM, Carelli RM. (2014). Dynamics of rapid dopamine release in the nucleus accumbens during goal-directed behaviors for cocaine versus natural rewards. Neuropharmacology 86:319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carli M, Robbins TW, Evenden JL, Everitt BJ. (1983). Effects of lesions to ascending noradrenergic neurones on performance of a 5-choice serial reaction task in rats; implications for theories of dorsal noradrenergic bundle function based on selective attention and arousal. Behav Brain Res 9:361–380. [DOI] [PubMed] [Google Scholar]
- Chen C, Wu C, Liao Y, Hsu H, Tseng Y, Liu H, Chiu W. (2012). Working memory in patients with mild traumatic brain injury: functional MR imaging analysis. Radiology 264:844–851. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang EY, Kuo T, Ma H, Hoffer BJ, Tsui P, et al. (2015). Dopamine release impairment in striatum after different levels of cerebral cortical fluid percussion injury. Cell Transplant 24:2113–2128. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang EY, Kuo T, Hoffer BJ, Miller J, Chou Y, Chiang Y. (2017). Dopamine release in the nucleus accumbens is altered following traumatic brain injury. Neuroscience 348:180–190. [DOI] [PubMed] [Google Scholar]
- Cheng JP, Aslam HA, Hoffman AN, Zafonte RD, Kline AE. (2007). The neurobehavioral benefit conferred by a single systemic administration of 8-OH-DPAT after brain trauma is confined to a narrow therapeutic window. Neurosci Lett 416:165–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JP, Hoffman AN, Zafonte RD, Kline AE. (2008). A delayed and chronic treatment regimen with the 5-HT1A receptor agonist 8-OH-DPAT after cortical impact injury facilitates motor recovery and acquisition of spatial learning. Behav Brain Res 194:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CYP, Tlustos SJ, Walz NC, Holland SK, Eliassen JC, Bernard L, Wade SL. (2012). Neural correlates of risky decision making in adolescents with and without traumatic brain injury using the Balloon Analog Risk Task. Dev Neuropsychol 37:176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou A, Morganti JM, Rosi S. (2016). Frontal lobe contusion in mice chronically impars prefrontal-dependent behavior. PLoS ONE 11:e0151418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. (2006). Functions of frontostriatal systems in cognition: comparative neuropsychopharmacological studies in rats, monkeys and humans. Biol Psychol 73:19–38. [DOI] [PubMed] [Google Scholar]
- Cifariello A, Pompili A, Gasbarri A. (2008). 5-HT7 receptors in the modulation of cognitive processes. Behav Brain Res 195:171–179. [DOI] [PubMed] [Google Scholar]
- Ciuna A, Andretta M, Corbari L, Levi D, Mirandola M, Sorio A, Barbui C. (2004). Are we going to increase the use of antidepressants up to that of benzodiazepines? Eur J Clin Pharmacol 60:629–634. [DOI] [PubMed] [Google Scholar]
- Clark AL, Amick MM, Fortier C, Milberg WP, McGlinchey RE. (2014). Poor performance validity predicts clinical characteristics and cognitive test performance of OEF/OIF/OND Veterans in a research setting. Clin Neuropsychol 28:802–825. [DOI] [PubMed] [Google Scholar]
- Coccaro EF, Lee R, Kavoussi RJ. (2010). Aggression, suicidality, and intermittent explosive disorder: serotonergic correlates in personality disorder and healthy control subjects. Neuropsychopharmacology 35:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocker PJ, Hosking JG, Murch WS, Clark L, Winstanley CA. (2016). Activation of dopamine D4 receptors within the anterior cingulate cortex enhances the erroneous expectation of reward on a rat slot machine task. Neuropharmacology 105:186–195. [DOI] [PubMed] [Google Scholar]
- Coronado VG, McGuire LC, Faul M, Sugerman D, Pearson W. (2012). The epidemiology and prevention of TBI brain injury medicine. New York, NY: Demos; 45–56. [Google Scholar]
- Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. (2006). Incidence and lifetime costs of injuries in the United States. Inj Prev 12:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrena C, Brancoa LD, Zimmermanna N, Cardosoa CO, Grassi-Oliveiraa R, Fonseca RP. (2014). Impaired decision-making after traumatic brain injury: the Iowa Gambling Task. Brain Inj 28:1070–1075. [DOI] [PubMed] [Google Scholar]
- Crane AT, Fink KD, Smith JS. (2012). The effects of acute voluntary wheel running on recovery of function following medial frontal cortical contusions in rats. Restor Neurol Neurosci 30:325–333. [DOI] [PubMed] [Google Scholar]
- Dale E, Pehrson AL, Jeyarajah T, Li Y, Leiser SC, Smagin G, et al. (2016). Effects of serotonin in the hippocampus: how SSRIs and multimodal antidepressants might regulate pyramidal cell function. CNS Spectr 21:143–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Eagle DM, Passetti F, Robbins TW. (2002). Deficits in impulse control associated with tonically-elevated serotonergic function in rat prefrontal cortex. Neuropsychopharmacology 26:716–728. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Fryer TD, Brichard L, Robinson ESJ, Theobald DEH, Lääne K, et al. (2007). Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science 315:1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam H, Mellerup ET, Plenge P, Winther R, Wortwein G. (2007). The serotonin transporter and 5HT2A reception in rat brain after localized lesions. Neurol Res 29:717–722. [DOI] [PubMed] [Google Scholar]
- Dam H, Mellerup ET, Plenge P, Winther R, Wortwein G. (2013). The serotonin transporter and 5HT2A receptor in rat brain after localized lesions. Neurol Res 29:717–722. [DOI] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Zhang M, Grill RJ, Pati S, Zhao J, Moore AN. (2010). Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS ONE 5:e11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum I, Channon S, Canavan AG. (1989). Classical conditioning in patients with severe memory problems. J Neurol Neurosurg Psychiatry 52:47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day AM, Kahler CW, Ahern DC, Clark US. (2015). Executive functioning in alcohol use studies: a brief review of findings and challenges in assessment. Curr Drug Abuse Rev 8:26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demery JA, Larson MJ, Dixit NK, Bauer RM, Perlstein WM. (2010). Operating characteristics of executive functioning tests following traumatic brain injury. Clin Neuropsychol 24:1292–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MR, Jacobs EA, Sanders S, Guercio JM, Soldner J, Parker-Singler S, et al. (2005). Impulsivity, self-control, and delay discounting in persons with acquired brain injury. Behav Interv 20:101–120. [Google Scholar]
- Donders FC. (1969). On the speed of mental processes. Acta Psychol (Amst) 30:412–431. [DOI] [PubMed] [Google Scholar]
- Donnemiller E, Brenneis C, Wissel J, Scherfler C, Poewe W, Riccabona G, Wenning GK. (2000). Impaired dopaminergic neurotransmission in patients with traumatic brain injury: A SPET study using 123 I-β-CIT and 123 I-IBZM. Eur J Nucl Med 27:1410–1414. [DOI] [PubMed] [Google Scholar]
- Du W, Green L, Myerson J. (2002). Cross-cultural comparisons of discounting delayed and probabilistic rewards. Psychol Rec 52:479–492. [Google Scholar]
- Duncan CC, Kosmidis MH, Mirsky AF. (2005). Closed head injury-related information processing deficits: an event-related potential analysis. Int J Psychophysiol 58:133–157. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Hassanain M, Levin BE. (1998). Norepinephrine and traumatic brain injury: a possible role in post-traumatic edema. Brain Res 800:245–252. [DOI] [PubMed] [Google Scholar]
- Dunning DL, Westgate B, Adlam A-LR. (2016). A meta-analysis of working memory impairments in survivors of moderate-to-severe traumatic brain injury. Neuropsychology 30:811–819. [DOI] [PubMed] [Google Scholar]
- Dyer KFW, Bell R, McCann J, Rauch R. (2006). Aggression after traumatic brain injury: Analysing socially desirable responses and the nature of aggressive traits. Brain Inj 20:1163–1173. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Owman C, Rosengren E, West KA. (2009). Brain concentrations of dopamine, noradrenaline, 5-hydroxytryptamine, and homovanillic acid during intracranial hypertension following traumatic brain injury in rabbit. Acta Neurol Scand 47:458–463. [DOI] [PubMed] [Google Scholar]
- Empey PE, McNamara PJ, Young B, Rosbolt MB, Hatton J. (2006). Cyclosporin A disposition following acute traumatic brain injury. J Neurotrauma 23:109–116. [DOI] [PubMed] [Google Scholar]
- Enomoto T, Osugi T, Satoh H, McIntosh TK, Nabeshima T. (2005). Pre-injury magnesium treatment prevents traumatic brain injury-induced hippocampal ERK activation, neuronal loss, and cognitive dysfunction in the radial-arm maze test. J Neurotrauma 22:783–792. [DOI] [PubMed] [Google Scholar]
- Eschun G, Parkinson D, Vriend J. (1992). Changes in hippocampal monoamine concentration following halothane anesthesia and concussion. Surg Neurol 37:101–105. [DOI] [PubMed] [Google Scholar]
- Evenden JL, Ryan CN. (1996). The pharmacology of impulsive behaviour in rats: the effects of drugs on response choice with varying delays of reinforcement. Psychopharmacology (Berl) 128:161–170. [DOI] [PubMed] [Google Scholar]
- Ferster CB, Skinner BF. (1957). Schedules of reinforcement. East Norwalk, CT: Appleton-Century-Crofts. [Google Scholar]
- Fujinaka T, Kohmura E, Yuguchi T, Yoshimine T. (2003). The morphological and neurochemical effects of diffuse brain injury on rat central noradrenergic system. Neurol Res 25:35–41. [DOI] [PubMed] [Google Scholar]
- Golub A, Bennett AS. (2013). Prescription opioid initiation, correlates, and consequences among a sample of OEF/OIF military personnel. Subst Use Misuse 48:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MD, Makley AT, Campion EM, Friend LA, Lentsch AB, Pritts TA. (2013). Preinjury alcohol exposure attenuates the neuroinflammatory response to traumatic brain injury. J Surg Res 184:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve KW, Bianchini KJ, Mathias CW, Houston RJ, Crouch JA. (2002). Detecting malingered performance with the Wisconsin Card Sorting Test: a preliminary investigation in traumatic brain injury. Clin Neuropsychol 16:179–191. [DOI] [PubMed] [Google Scholar]
- Greve KW, Heinly MT, Bianchini KJ, Love JM. (2009). Malingering detection with the Wisconsin Card Sorting Test in mild traumatic brain injury. Clin Neuropsychol 23:343–362. [DOI] [PubMed] [Google Scholar]
- Gualtieri CT, Johnson LG, Benedict KB. (2006). Neurocognition in depression: patients on and off medication versus healthy comparison subjects. J Neuropsychiatry Clin Neurosci 18:217–225. [DOI] [PubMed] [Google Scholar]
- Haber SN. (2014). The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 282:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton KR, Potenza MN. (2012). Relations among delay discounting, addictions, and money mismanagement: implications and future directions. Am J Drug Alcohol Abuse 38:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RJ, Temple MD, Pike BR, O’Dell DM, Buck DL, Lyeth BG. (1996). Working memory deficits following traumatic brain injury in the rat. J Neurotrauma 13:317–323. [DOI] [PubMed] [Google Scholar]
- Harrison AA, Everitt BJ, Robbins TW. (1997). Central 5-HT depletion enhances impulsive responding without affecting the accuracy of attentional performance: interactions with dopaminergic mechanisms. Psychopharmacology (Berl) 133:329–342. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Toshima T. (2005). Learning performance on the discrimination-shift task in patients with cortical and subcortical lesions. Appl Neuropsychol 12:158–168. [DOI] [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AMH, Smith DH, Stewart W. (2015). Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 74:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehar H, Yeates K, Kolb B, Esser MJ, Mychasiuk R. (2015). Impulsivity and concussion in juvenile rats: examining molecular and structural aspects of the frontostriatal pathway. PLoS ONE 10:e0139842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry JM, Talukder NK, Lee AB, Walker ML. (1997). Cerebral trauma-induced changes in corpus striatal dopamine receptor subtypes. J Invest Surg 10:281–286. [DOI] [PubMed] [Google Scholar]
- Hibbard MR, Uysal S, Kepler K, Bogdany J, Silver J. (1998). Axis I psychopathology in individuals with traumatic brain injury. J Head Trauma Rehabil 13:24–39. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, Lowenstein DH, Saint Marie R, McIntosh TK. (1993). Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J Neurotrauma 10:405–414. [DOI] [PubMed] [Google Scholar]
- Hoane MR, Akstulewicz SL, Toppen J. (2003). Treatment with vitamin B3 improves functional recovery and reduces GFAP expression following traumatic brain injury in rats. J Neurotrauma 20:1189–1199. [DOI] [PubMed] [Google Scholar]
- Hoane MR, Becerra GD, Shank JE, Tatko L, Pak ES, Smith M, Murashov AK. (2004). Transplantation of neuronal and glial precursors dramatically improves sensorimotor function but not cognitive function in the traumatically injured brain. J Neurotrauma 21:163–174. [DOI] [PubMed] [Google Scholar]
- Hoane MR, Wolyniak JG, Akstulewicz SL. (2005). Administration of riboflavin improves behavioral outcome and reduces edema formation and glial fibrillary acidic protein expression after traumatic brain injury. J Neurotrauma 22:1112–1122. [DOI] [PubMed] [Google Scholar]
- Hoffman AN, Cheng JP, Zafonte RD, Kline AE. (2008). Administration of haloperidol and risperidone after neurobehavioral testing hinders the recovery of traumatic brain injury-induced deficits. Life Sci 83:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskison MM, Moore AN, Hu B, Orsi S, Kobori N, Dash PK. (2009). Persistent working memory dysfunction following traumatic brain injury: evidence for a time-dependent mechanism. Neuroscience 159:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EY, Tsai T, Kuo T, Tsai J, Tsui P, Chou Y, et al. (2014a). Remote effects on the striatal dopamine system after fluid percussion injury. Behav Brain Res 267:156–172. [DOI] [PubMed] [Google Scholar]
- Huang EY, Tsui P, Kuo T, Tsai J, Chou Y, Ma H, et al. (2014b). Amantadine ameliorates dopamine-releasing deficits and behavioral deficits in rats after fluid percussion injury. PLoS ONE 9:e86354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huger F, Patrick G. (1979). Effect of concussive head injury on central catecholamine levels and synthesis rates in rat brain regions. J Neurochem 33:89–95. [DOI] [PubMed] [Google Scholar]
- Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet M. (2011). Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma 28:1783–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istin M, Thiriet N, Solinas M. (2017). Behavioral flexibility predicts increased ability to resist excessive methamphetamine self-administration. Addict Biol 22:958–966. [DOI] [PubMed] [Google Scholar]
- James LM, Strom TQ, Leskela J. (2014). Risk-taking behaviors and impulsivity among veterans with and without PTSD and mild TBI. Mil Med 179:357–363. [DOI] [PubMed] [Google Scholar]
- Jean-Richard-Dit-Bressel P, Killcross S, McNally GP. (2018). Behavioral and neurobiological mechanisms of punishment: implications for psychiatric disorders. Neuropsychopharmacology 43:1639–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge RE, Robinson RG. (2003). Mood disorders following traumatic brain injury. Int Rev Psychiatry 15:317–327. [DOI] [PubMed] [Google Scholar]
- Juengst SB, Kumar RG, Wagner AK. (2017). A narrative literature review of depression following traumatic brain injury: prevalence, impact, and management challenges. Psychol Res Behav Manag 10:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbak G, Kartkaya K, Ozcelik E, Guvenal AB, Kabay SC, Arslan G, Durmaz R. (2013). The neuroprotective effect of acute moderate alcohol consumption on caspase-3 mediated neuroapoptosis in traumatic brain injury: the role of lysosomal cathepsin L and nitric oxide. Gene 512:492–495. [DOI] [PubMed] [Google Scholar]
- Kawa L, Arborelius UP, Yoshitake T, Kehr J, Hökfelt T, Risling M, Agoston D. (2015). Neurotransmitter systems in a mild blast traumatic brain injury model: catecholamines and serotonin. J Neurotrauma 32:1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Ko M, Na S, Park S, Kim K. (2006). Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 20:24–30. [DOI] [PubMed] [Google Scholar]
- Kingdon D, Cardoso C, McGrath JJ. (2016). Research review: cxecutive function deficits in fetal alcohol spectrum disorders and attention-deficit/hyperactivity disorder: a meta-analysis. J Child Psychol Psychiatry 57:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AE, Yu J, Horváth E, Marion DW, Dixon CE. (2001). The selective 5-HT1A receptor agonist repinotan HCl attenuates histopathology and spatial learning deficits following traumatic brain injury in rats. Neuroscience 106:547–555. [DOI] [PubMed] [Google Scholar]
- Kline AE, Massucci JL, Marion DW, Dixon CE. (2002). Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J Neurotrauma 19:415–425. [DOI] [PubMed] [Google Scholar]
- Kline AE, Massucci JL, Zafonte RD, Dixon CE, DeFeo JR, Rogers EH. (2007). Differential effects of single versus multiple administrations of haloperidol and risperidone on functional outcome after experimental brain trauma. Crit Care Med 35:919–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AE, Hoffman AN, Cheng JP, Zafonte RD, Massucci JL. (2008). Chronic administration of antipsychotics impede behavioral recovery after experimental traumatic brain injury. Neurosci Lett 448:263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AE, Olsen AS, Sozda CN, Hoffman AN, Cheng JP. (2012). Evaluation of a combined treatment paradigm consisting of environmental enrichment and the 5-HT1A receptor agonist buspirone after experimental traumatic brain injury. J Neurotrauma 29:1960–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight C, Rutterford NA, Alderman N, Swan LJ. (2002). Is accurate self-monitoring necessary for people with acquired neurological problems to benefit from the use of differential reinforcement methods? Brain Inj 16:75–87. [DOI] [PubMed] [Google Scholar]
- Kobori N, Clifton GL, Dash PK. (2006). Enhanced catecholamine synthesis in the prefrontal cortex after traumatic brain injury: implications for prefrontal dysfunction. J Neurotrauma 23:1094–1102. [DOI] [PubMed] [Google Scholar]
- Kobori N, Hu B, Dash PK. (2011). Altered adrenergic receptor signaling following traumatic brain injury contributes to working memory dysfunction. Neuroscience 172:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokiko ON, Murashov AK, Hoane MR. (2006). Administration of raloxifene reduces sensorimotor and working memory deficits following traumatic brain injury. Behav Brain Res 170:233–240. [DOI] [PubMed] [Google Scholar]
- Konrad C, Geburek AJ, Rist F, Blumenroth H, Fischer B, Husstedt I, et al. (2011). Long-term cognitive and emotional consequences of mild traumatic brain injury. Psychol Med 41:1197–1211. [DOI] [PubMed] [Google Scholar]
- Lange RT, Shewchuk JR, Rauscher A, Jarrett M, Heran MK, Brubacher JR, Iverson GL. (2014). A prospective study on the influence of acute alcohol intoxication versus chronic alcohol consumption on outcome following traumatic brain injury. Arch Clin Neuropsychol 29:478–495. [DOI] [PubMed] [Google Scholar]
- Larson MJ, Kelly KG, Stigge-Kaufman DA, Schmalfuss IM, Perlstein WM. (2007). Reward context sensitivity impairment following severe TBI: an event-related potential investigation. J Int Neuropsychol Soc 13:615–625. [DOI] [PubMed] [Google Scholar]
- Leary JB, Bondi CO, LaPorte MJ, Carlson LJ, Radabaugh HL, Cheng JP, Kline AE. (2017). The therapeutic efficacy of environmental enrichment and methylphenidate alone and in combination after controlled cortical impact injury. J Neurotrauma 34:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejuez CW, Read JP, Kahler CW, Richards JB, Ramsey SE, Stuart GL, et al. (2002). Evaluation of a behavioral measure of risk tasking: the Balloon Analogue Risk Task (BART). J Exp Psychol Appl 8:75–84. [DOI] [PubMed] [Google Scholar]
- Levin BE, Brown KL, Pawar G, Dunn-Meynell AA. (1995). Widespread and lateralized effects of acute traumatic brain injury on norepinephrine turnover in the rat brain. Brain Res 674:307–313. [DOI] [PubMed] [Google Scholar]
- Levin HS, Hanten G, Chang C, Zhang L, Schachar R, Ewing-Cobbs L, Max JE. (2002). Working memory after traumatic brain injury in children. Ann Neurol 52:82–88. [DOI] [PubMed] [Google Scholar]
- Liepert J. (2016). Update on pharmacotherapy for stroke and traumatic brain injury recovery during rehabilitation. Curr Opin Neurol 29:700–705. [DOI] [PubMed] [Google Scholar]
- Lim YW, Meyer NP, Shah AS, Budde MD, Stemper BD, Olsen CM. (2015). Voluntary alcohol intake following blast exposure in a rat model of mild traumatic brain injury. PLoS ONE 10:e0125130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner MD, Plone MA, Cain CK, Frydel B, Francis JM, Emerich DF, Sutton RL. (1998). Dissociable long-term cognitive deficits after frontal versus sensorimotor cortical contusions. J Neurotrauma 15:199–216. [DOI] [PubMed] [Google Scholar]
- Logan GD.Dagenbach D, Carr TH. (1994). On the ability to inhibit thought and action: a users’ guide to the stop signal paradigm. Inhibitory processes in attention, memory, and language. San Diego, CA: Academic Press; 189–239. [Google Scholar]
- Logue SF, Gould TJ. (2014). The neural and genetic basis of executive function: attention, cognitive flexibility, and response inhibition. Pharmacol Biochem Behav 123:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowing JL, Susick LL, Caruso JP, Provenzano AM, Raghupathi R, Conti AC. (2014). Experimental traumatic brain injury alters ethanol consumption and sensitivity. J Neurotrauma 31:1700–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, et al. (1990). Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res 526:249–258. [DOI] [PubMed] [Google Scholar]
- Ma L, Steinberg JL, Keyser-Marcus L, Ramesh D, Narayana PA, Merchant RE, et al. (2015). Altered white matter in cocaine-dependent subjects with traumatic brain injury: a diffusion tensor imaging study. Drug Alcohol Depend 151:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson SE, Phillips LH, Della Sala S, Cantagallo A. (2009). Iowa Gambling Task impairment is not specific to ventromedial prefrontal lesions. Clin Neuropsychol 23:510–522. [DOI] [PubMed] [Google Scholar]
- Majchrzak H, Kmieciak-Kołada K, Herman Z. (1979). Disorders of dopamine and 5-hydroxytryptamine metabolism in the early period after cranio-cerebral injuries. Neurol Neurochir Pol 13:289–293. [PubMed] [Google Scholar]
- Markianos M, Seretis A, Kotsou S, Baltas I, Sacharogiannis H. (1992). CSF neurotransmitter metabolites and short-term outcome of patients in coma after head injury. Acta Neurol Scand 86:190–193. [DOI] [PubMed] [Google Scholar]
- Markianos M, Seretis A, Kotsou A, Christopoulos M. (1996). CSF neurotransmitter metabolites in comatose head injury patients during changes in their clinical state. Acta Neurochir (Wien) 138:57–59. [DOI] [PubMed] [Google Scholar]
- Martens KM, Vonder Haar C, Hutsell BA, Hoane MR. (2012). A discrimination task used as a novel method of testing decision-making behavior following traumatic brain injury. J Neurotrauma 29:2505–2512. [DOI] [PubMed] [Google Scholar]
- Martens KM, Vonder Haar C, Hutsell BA, Hoane MR. (2013). The dig task: a simple scent discrimination reveals deficits following frontal brain damage. J Vis Exp 71:e50033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massucci JL, Kline AE, Ma X, Zafonte RD, Dixon CE. (2004). Time dependent alterations in dopamine tissue levels and metabolism after experimental traumatic brain injury in rats. Neurosci Lett 372:127–131. [DOI] [PubMed] [Google Scholar]
- Mayeux JP, Teng SX, Katz PS, Gilpin NW, Molina PE. (2015). Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav Brain Res 279:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister TW, Flashman LA, McDonald BC, Ferrell RB, Tosteson TD, Yanofsky NN, et al. (2011a). Dopaminergic challenge with bromocriptine one month after mild traumatic brain injury: altered working memory and BOLD response. J Neuropsychiatry Clin Neurosci 23:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister TW, McDonald BC, Flashman LA, Ferrell RB, Tosteson TD, Yanofsky NN, et al. (2011b). Alpha-2 adrenergic challenge with guanfacine one month after mild traumatic brain injury: altered working memory and BOLD response. Int J Psychophysiol 82:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BC, Flashman LA, Saykin AJ. (2002). Executive dysfunction following traumatic brain injury: neural substrates and treatment strategies. NeuroRehabilitation 17:333–344. [PubMed] [Google Scholar]
- McHugh L, Wood RL. (2008). Using a temporal discounting paradigm to measure decision-making and impulsivity following traumatic brain injury: a pilot study. Brain Inj 22:715–721. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Yu T, Gennarelli TA. (1994). Alterations in regional brain catecholamine concentrations after experimental brain injury in the rat. J Neurochem 63:1426–1433. [DOI] [PubMed] [Google Scholar]
- Merkel SF, Andrews AM, Lutton EM, Razmpour R, Cannella LA, Ramirez SH. (2017a). Dexamethasone attenuates the enhanced rewarding effects of cocaine following experimental traumatic brain injury. Cell Transplant 26:1178–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel SF, Razmpour R, Lutton EM, Tallarida CS, Heldt NA, Cannella LA, et al. (2017b). Adolescent traumatic brain injury induces chronic mesolimbic neuroinflammation with concurrent enhancement in the rewarding effects of cocaine in mice during adulthood. J Neurotrauma 34:165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco CM, Gebhardt KM, Chlebowski SM, Shaw KE, Cheng JP, Henchir JJ, et al. (2014). A combined therapeutic regimen of buspirone and environmental enrichment is more efficacious than either alone in enhancing spatial learning in brain-injured pediatric rats. J Neurotrauma 31:1934–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor E, Shohami E, Kanevsky E, Grigoriadis N, Symeonidou C, Kohen R. (2006). Impairment of the ability of the injured aged brain in elevating urate and ascorbate. Exp Gerontol 41:303–311. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Yan E, Bye N. (2010). Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury 41:S10–S13. [DOI] [PubMed] [Google Scholar]
- Morris R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60. [DOI] [PubMed] [Google Scholar]
- Muelbl MJ, Slaker ML, Shah AS, Nawarawong NN, Gerndt CH, Budde MD, et al. (2018). Effects of mild blast traumatic brain injury on cognitive- and addiction-related behaviors. Sci Rep 8:9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa G, Hou J, Nelson R, Tsuda S, Jahan M, Mohammad NS, et al. (2017). Mild closed head traumatic brain injury-induced changes in monoamine neurotransmitters in the trigeminal subnuclei of a rat model: mechanisms underlying orofacial allodynias and headache. Neural Regen Res 12:981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mychasiuk R, Hehar H, Esser MJ. (2015). A mild traumatic brain injury (mTBI) induces secondary attention-deficit hyperactivity disorder-like symptomology in young rats. Behav Brain Res 286:285–292. [DOI] [PubMed] [Google Scholar]
- Myers CE, DeLuca J, Hopkins RO, Gluck MA. (2006). Conditional discrimination and reversal in amnesia subsequent to hypoxic brain injury or anterior communicating artery aneurysm rupture. Neuropsychologia 44:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrga JM, Failla MD, Ricker JH, Dixon CE, Conley YP, Arenth PM, Wagner AK. (2016). A dopamine pathway gene risk score for cognitive recovery following traumatic brain injury: methodological considerations, preliminary findings, and interactions with sex. J Head Trauma Rehabil 31:E15–E29. [DOI] [PubMed] [Google Scholar]
- Nativ A, Lazarus JC, Nativ J, Joseph J. (1994). Potentials associated with the go/no-go paradigm in traumatic brain injury. Arch Phys Med Rehabil 75:1322–1326. [PubMed] [Google Scholar]
- Newcombe VFJ, Outtrim JG, Chatfield DA, Manktelow A, Hutchinson PJ, Coles JP, et al. (2011). Parcellating the neuroanatomical basis of impaired decision-making in traumatic brain injury. Brain 134:759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Phelan K, McArthur DL, Chang CWJ, Green D, Hovda DA. (2008). The impact of substance abuse on mortality in patients with severe traumatic brain injury. J Trauma Acute Care Surg 65:674–677. [DOI] [PubMed] [Google Scholar]
- Olney JW, Zorumski CF, Stewart GR, Price MT, Wang G, Labruyere J. (1990). Excitotoxicity of l-DOPA and 6-OH-DOPA: implications for Parkinson’s and Huntington’s diseases. Exp Neurol 108:269–272. [DOI] [PubMed] [Google Scholar]
- Olsen AS, Sozda CN, Cheng JP, Hoffman AN, Kline AE. (2012). Traumatic brain injury-induced cognitive and histological deficits are attenuated by delayed and chronic treatment with the 5-HT1A-receptor agonist buspirone. J Neurotrauma 29:1898–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovskaya M, Hochstein S, Keren O, Mordvinov E, Groswasser Z. (2007). Methylphenidate effect on hemispheric attentional imbalance in patients with traumatic brain injury: a psychophysical study. Brain Inj 21:489–497. [DOI] [PubMed] [Google Scholar]
- Perry JL, Larson EB, German JP, Madden GJ, Carroll ME. (2005). Impulsivity (delay discounting) as a predictor of acquisition of IV cocaine self-administration in female rats. Psychopharmacology (Berl) 178:193–201. [DOI] [PubMed] [Google Scholar]
- Phelps TI, Bondi CO, Mattiola VV, Kline AE. (2017). Relative to typical antipsychotic drugs, aripiprazole is a safer alternative for alleviating behavioral disturbances after experimental brain trauma. Neurorehabil Neural Repair 31:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. (2000). Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 55:1158–1166. [DOI] [PubMed] [Google Scholar]
- Ponsford JL, Spitz G, Cromarty F, Gifford D, Attwood D. (2013). Costs of care after traumatic brain injury. J Neurotrauma 30:1498–1505. [DOI] [PubMed] [Google Scholar]
- Porta M, Bareggi SR, Collice M, Assael BM, Selenati A, Calderini G, et al. (1975). Homovanillic acid and 5-hydroxyindole-acetic acid in the CSF of patients after a severe head injury. Eur Neurol 13:545–554. [DOI] [PubMed] [Google Scholar]
- Prakash R, Carmichael ST. (2015). Blood–brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol 28:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MR, Tzigaret CM, Smith DH, Soares H, McIntosh TK. (1992). Decreased α1-adrenergic receptors after experimental brain injury. J Neurotrauma 9:269–279. [DOI] [PubMed] [Google Scholar]
- Prins M, Greco T, Alexander D, Giza CC. (2013). The pathophysiology of traumatic brain injury at a glance. Dis Models Mech 6:1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley A, Tan AA, Hoane MR. (2009). The effects of hypertonic saline and nicotinamide on sensorimotor and cognitive function following cortical contusion injury in the rat. Brain Res 1304:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj R, Skrifvars MB, Kivisaari R, Hernesniemi J, Lappalainen J, Siironen J. (2015). Acute alcohol intoxication and long-term outcome in patients with traumatic brain injury. J Neurotrauma 32:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh D, Keyser-Marcus LA, Ma L, Schmitz JM, Lane SD, Marwitz JH, et al. (2015). Prevalence of traumatic brain injury in cocaine-dependent research volunteers. Am J Addict 24:341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V, Rosenberg P, Bertrand M, Sealehinia S, Spiro J, Vaishnavi S, et al. (2009). Aggression after traumatic brain injury: prevalence and correlates. J Neuropsychiatry Clin Neurosci 21:420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V, Rosenberg P, Miles QS, Patadia D, Treiber K, Bertrand M, et al. (2010). Neuropsychiatric symptoms in dementia patients with and without a history of traumatic brain injury. J Neuropsychiatry Clin Neurosci 22:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau TF, Kothiwal AS, Rova AR, Brooks DM, Poulsen DJ. (2012). Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe traumatic brain injury. J Trauma Acute Care Surg 73:S165–S172. [DOI] [PubMed] [Google Scholar]
- Reeves RR, Panguluri RL. (2011). Neuropsychiatric complications of traumatic brain injury. J Psychosoc Nurs Ment Health Serv 49:42–50. [DOI] [PubMed] [Google Scholar]
- Reid WM, Hamm RJ. (2008). Post-injury atomoxetine treatment improves cognition following experimental traumatic brain injury. J Neurotrauma 25:248–256. [DOI] [PubMed] [Google Scholar]
- Riccio CA, Reynolds CR, Lowe P, Moore JJ. (2002). The continuous performance test: a window on the neural substrates for attention? Arch Clin Neuropsychol 17:235–272. [PubMed] [Google Scholar]
- Ripley DL, Morey CE, Gerber D, Harrison-Felix C, Brenner LA, Pretz CR. (2014). Atomoxetine for attention deficits following traumatic brain injury: results from a randomized controlled trial. Brain Inj 28:1514–1522. [DOI] [PubMed] [Google Scholar]
- Robinson ESJ, Eagle DM, Economidou D, Theobald DEH, Mar AC, Murphy ER, et al. (2009). Behavioural characterisation of high impulsivity on the 5-choice serial reaction time task: specific deficits in ‘waiting’ versus ‘stopping’. Behav Brain Res 196:310–316. [DOI] [PubMed] [Google Scholar]
- Rochat L, Beni C, Billieux J, Azouvi P, Annoni J, Van der Linden M. (2010). Assessment of impulsivity after moderate to severe traumatic brain injury. Neuropsychol Rehabil 20:778–797. [DOI] [PubMed] [Google Scholar]
- Rochat L, Beni C, Annoni J, Vuadens P, Van der Linden M. (2013). How inhibition relates to impulsivity after moderate to severe traumatic brain injury. J Int Neuropsychol Soc 19:890–898. [DOI] [PubMed] [Google Scholar]
- Rogers RD, Everitt BJ, Baldacchino A, Blackshaw AJ, Swainson R, Wynne K, et al. (1999). Dissociable deficits in the decision-making cognition of chronic amphetamine abusers, opiate abusers, patients with focal damage to prefrontal cortex, and tryptophan-depleted normal volunteers: evidence for monoaminergic mechanisms. Neuropsychopharmacology 20:322–339. [DOI] [PubMed] [Google Scholar]
- Rosvold HE, Mirsky AF, Sarason I, Bransome ED, Jr, Beck LH. (1956). A continuous performance test of brain damage. J Consult Psychol 20:343–350. [DOI] [PubMed] [Google Scholar]
- Salmond CH, Menon DK, Charfield DA, Pickard JD, Sahakian BJ. (2005). Deficits in decision-making in head injury survivors. J Neurotrauma 22:613–622. [DOI] [PubMed] [Google Scholar]
- Schachar R, Logan GD, Robaey P, Chen S, Ickowicz A, Barr C. (2007). Restraint and cancellation: multiple inhibition deficits in attention deficit hyperactivity disorder. J Abnorm Child Psychol 35:229–238. [DOI] [PubMed] [Google Scholar]
- Scherwath A, Sommerfeldt DW, Bindt C, Nolte A, Boiger A, Koch U, Petersen-Ewert C. (2011). Identifying children and adolescents with cognitive dysfunction following mild traumatic brain injury--preliminary findings on abbreviated neuropsychological testing. Brain Inj 25:401–408. [DOI] [PubMed] [Google Scholar]
- Schlund MW. (2002a). Effects of acquired brain injury on adaptive choice and the role of reduced sensitivity to contingencies. Brain Inj 16:527–535. [DOI] [PubMed] [Google Scholar]
- Schlund MW. (2002b). The effects of brain injury on choice and sensitivity to remote consequences: deficits in discriminating response–consequence relations. Brain Inj 16:347–357. [DOI] [PubMed] [Google Scholar]
- Schlund MW, Pace G. (2000). The effects of traumatic brain injury on reporting and responding to causal relations: an investigation of sensitivity to reinforcement contingencies. Brain Inj 14:573–583. [DOI] [PubMed] [Google Scholar]
- Schlund MW, Pace GM, McGready J. (2001). Relations between decision-making deficits and discriminating contingencies following brain injury. Brain Inj 15:1061–1071. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. (1997). A neural substrate of prediction and reward. Science 275:1593–1599. [DOI] [PubMed] [Google Scholar]
- Sebastian V, Diallo A, Ling D, Serrano P. (2013). Robust training attenuates TBI-induced deficits in reference and working memory on the radial 8-arm maze. Front Behav Neurosci 7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellitto M, Ciaramelli E, di Pellegrino G. (2010). Myopic discounting of future rewards after medial orbitofrontal damage in humans. J Neurosci 30:16429–16436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semchuk KM, Love EJ, Lee RG. (1993). Parkinson’s disease: a test of the multifactorial etiologic hypothesis. Neurology 43:1173–1180. [DOI] [PubMed] [Google Scholar]
- Sharp PB, Miller GA, Heller W. (2015). Transdiagnostic dimensions of anxiety: neural mechanisms, executive functions, and new directions. Int J Psychophysiol 98:365–377. [DOI] [PubMed] [Google Scholar]
- Shen H, Harvey BK, Chiang Y, Pick CG, Wang Y. (2011). Methamphetamine potentiates behavioral and electrochemical responses after mild traumatic brain injury in mice. Brain Res 1368:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer M, Nick TG, Millis SR, Novack TA. (2003). Use of the WCST and the WCST-64 in the assessment of traumatic brain injury. J Clin Exp Neuropsychol 25:512–520. [DOI] [PubMed] [Google Scholar]
- Shimada R, Abe K, Furutani R, Kibayashi K. (2014). Changes in dopamine transporter expression in the midbrain following traumatic brain injury: an immunohistochemical and in situ hybridization study in a mouse model. Neurol Res 36:239–246. [DOI] [PubMed] [Google Scholar]
- Shin SS, Dixon CE. (2011). Oral fish oil restores striatal dopamine release after traumatic brain injury. Neurosci Lett 496:168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SS, Bray ER, Zhang CQ, Dixon CE. (2011). Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res 1369:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SS, Bray ER, Dixon CE. (2012). Effects of nicotine administration on striatal dopamine signaling after traumatic brain injury in rats. J Neurotrauma 29:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MS, Park HK, Kim TW, Ji ES, Lee JM, Choi HS, et al. (2016). Neuroprotective effects of bone marrow stromal cell transplantation in combination with treadmill exercise following traumatic brain injury. Int Neurourol J 20:S49–S56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnitko TA, Robinson DL. (2015). Regional variation in phasic dopamine release during alcohol and sucrose self-administration in rats. ACS Chem Neurosci 6:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Venkateshwara G, Nair KPS, Khan M, Saad R. (2014). Agitation after traumatic brain injury and predictors of outcomes. Brain Inj 28:336–340. [DOI] [PubMed] [Google Scholar]
- Stowe CD, Lee KR, Storgion SA, Phelps SJ. (2000). Altered phenytoin pharmacokinetics in children with severe, acute traumatic brain injury. J Clin Pharmacol 40:1452–1461. [PubMed] [Google Scholar]
- Swan AA, Chandrashekar R, Beare J, Hoane MR. (2011). Preclinical efficacy testing in middle-aged rats: nicotinamide, a novel neuroprotectant, demonstrates diminished preclinical efficacy after controlled cortical impact. J Neurotrauma 28:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Ge H, Tang J, Fu C, Duanmu W, Chen Y, et al. (2015). Amantadine preserves dopamine level and attenuates depression-like behavior induced by traumatic brain injury in rats. Behav Brain Res 279:274–282. [DOI] [PubMed] [Google Scholar]
- Taylor AN, Rahman SU, Sanders NC, Tio DL, Prolo P, Sutton RL. (2008). Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J Neurotrauma 25:311–323. [DOI] [PubMed] [Google Scholar]
- Teasdale G, Jennett B. (1974). Assessment of coma and impaired consciousness. A practical scale. Lancet 2:81–84. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. (1999). Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil 14:602–615. [DOI] [PubMed] [Google Scholar]
- Titus DJ, Wilson NM, Freund JE, Carballosa MM, Sikah KE, Furones C, et al. (2016). Chronic cognitive dysfunction after traumatic brain injury is improved with a phosphodiesterase 4B inhibitor. J Neurosci 36:7095–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer R, Slagter HA, Christian BT, Fox AS, King CR, Murali D, et al. (2014). Love to win or hate to lose? Asymmetry of dopamine D2 receptor binding predicts sensitivity to reward versus punishment. J Cogn Neurosci 26:1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, Chandler LJ. (2015). Alcohol-induced alterations in dopamine modulation of prefrontal activity. Alcohol 49:773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi S, Rao V, Fann JR. (2009). Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics 50:198–205. [DOI] [PubMed] [Google Scholar]
- van Bregt DR, Thomas TC, Hinzman JM, Cao T, Liu M, Bing G, et al. (2012). Substantia nigra vulnerability after a single moderate diffuse brain injury in the rat. Exp Neurol 234:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaaf ME, van Schouwenburg MR, Geurts DEM, Schellekens AFA, Buitelaar JK, Verkes RJ, Cools R. (2014). Establishing the dopamine dependency of human striatal signals during reward and punishment reversal learning. Cereb Cortex 24:633–642. [DOI] [PubMed] [Google Scholar]
- Vanderveldt A, Oliveira L, Green L. (2016). Delay discounting: pigeon, rat, human: does it matter? J Expe Psychol Anim Learn Cogn 42:141–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecht CJ, van Woerkom TCAM, Teelken AW, Minderhoud JM. (1976). On the nature of brain stem disorders in severe head injured patients. Acta Neurochir (Wien) 34:11–21. [DOI] [PubMed] [Google Scholar]
- Vonder Haar C, Maass WR, Jacobs EA, Hoane MR. (2014a). Deficits in discrimination following experimental frontal brain injury are mediated by motivation and can be improved by nicotinamide administration. J Neurotrauma 31:1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Smith TR, French EJ, Martens KM, Jacobs EA, Hoane MR. (2014b). Simple tone discriminations are disrupted following experimental frontal traumatic brain injury in rats. Brain Inj 28:235–243. [DOI] [PubMed] [Google Scholar]
- Vonder Haar C, Lam FCW, Adams WA, Riparip L, Kaur S, Muthukrishna M, et al. (2016). Frontal traumatic brain injury in rats causes long-lasting impairments in impulse control that are differentially sensitive to pharmacotherapeutics and associated with chronic neuroinflammation. ACS Chem Neurosci 7:1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Martens KM, Riparip L, Rosi S, Wellington CL, Winstanley CA. (2017). Frontal traumatic brain injury increases impulsive decision making in rats: a potential role for the inflammatory cytokine interleukin-12. J Neurotrauma 34:2790–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Ferland JN, Kaur S, Riparip L, Rosi S, Winstanley CA. (2018). Cocaine self-administration is increased after frontal traumatic brain injury and associated with neuroinflammation. Eur J Neurosci. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Sokoloski JE, Ren D, Chen X, Khan AS, Zafonte RD, et al. (2005). Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. J Neurochem 95:457–465. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Ren D, Conley YP, Ma X, Kerr ME, Zafonte RD, et al. (2007). Sex and genetic associations with cerebrospinal fluid dopamine and metabolite production after severe traumatic brain injury. J Neurosurg 106:538–547. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Drewencki LL, Chen X, Santos FR, Khan AS, Harun R, et al. (2008). Chronic methylphenidate treatment enhances striatal dopamine neurotransmission after experimental traumatic brain injury. J Neurochem 108:986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Sokoloski JE, Chen X, Harun R, Clossin DP, Khan AS, et al. (2009). Controlled cortical impact injury influences methylphenidate-induced changes in striatal dopamine neurotransmission. J Neurochem 110:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Scanion JM, Becker CR, Ritter AC, Niyonkuru C, Dixon CE, et al. (2014). The influence of genetic variants on striatal dopamine transporter and D2 receptor binding after TBI. J Cereb Blood Flow Metab 34:1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R, Hiller M, Staton M, Leukefeld CG. (2003). Head injury among drug abusers: an indicator of co-occurring problems. J Psychoactive Drugs 35:343–353. [DOI] [PubMed] [Google Scholar]
- Wang T, Huang X, Van KC, Went GT, Nguyen JT, Lyeth BG. (2014). Amantadine improves cognitive outcome and increases neuronal survival after fluid percussion traumatic brain injury in rats. J Neurotrauma 31:370–377. [DOI] [PubMed] [Google Scholar]
- Weil ZM, Karelina K, Gaier KR, Corrigan TED, Corrigan JD. (2016). Juvenile traumatic brain injury increases alcohol consumption and reward in female mice. J Neurotrauma 33:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West LK, Curtis KL, Greve KW, Bianchini KJ. (2011). Memory in traumatic brain injury: the effects of injury severity and effort on the Wechsler Memory Scale-III. J Neuropsychol 5:114–125. [DOI] [PubMed] [Google Scholar]
- Whiting MD, Hamm RJ. (2006). Traumatic brain injury produces delay-dependent memory impairment in rats. J Neurotrauma 23:1529–1534. [DOI] [PubMed] [Google Scholar]
- Wilson MS, Hamm RJ. (2002). Effects of fluoxetine on the 5-HT1A receptor and recovery of cognitive function after traumatic brain injury in rats. AmJ Phys Med Rehabil 81:364–372. [DOI] [PubMed] [Google Scholar]
- Wilson MS, Chen X, Ma X, Ren D, Wagner AK, Reynolds IJ, Dixon CE. (2005). Synaptosomal dopamine uptake in rat striatum following controlled cortical impact. J Neurosci Res 80:85–91. [DOI] [PubMed] [Google Scholar]
- Winstanley CA, Clark L. (2016). Translational models of gambling-related decision-making. Curr Topics Behav Neurosci 28:93–120. [DOI] [PubMed] [Google Scholar]
- Wood RL, Alderman N. (2011). Applications of operant learning theory to the management of challenging behavior after traumatic brain injury. J Head Trauma Rehabil 26:202–211. [DOI] [PubMed] [Google Scholar]
- Wood RLl, McHugh L. (2013). Decision making after traumatic brain injury: a temporal discounting paradigm. J Int Neuropsychol Soc 19:181–188. [DOI] [PubMed] [Google Scholar]
- Wood RLl, Thomas RH. (2013). Impulsive and episodic disorders of aggressive behaviour following traumatic brain injury. Brain Inj 27:253–261. [DOI] [PubMed] [Google Scholar]
- Woods DL, Herron TJ, Yund EW, Hink RF, Kishiyama MM, Reed B. (2011). Computerized analysis of error patterns in digit span recall. J Clin Exp Neuropsychol 33:721–734. [DOI] [PubMed] [Google Scholar]
- Xiao L, Wood SMW, Denburg NL, Moreno GL, Hernandez M, Bechara A. (2013). Is there a recovery of decision-making function after frontal lobe damage? A study using alternative versions of the Iowa Gambling Task. J Clin Exp Neuropsychol 35:518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. (2013). Animal models of traumatic brain injury. Nat Rev Neurosci 14:128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Cao S, Chao H, Liu Y, Ji J. (2016). Sex-related differences in striatal dopaminergic system after traumatic brain injury. Brain Res Bull 124:214–221. [DOI] [PubMed] [Google Scholar]
- Yan HQ, Kline AE, Ma X, Hooghe-Peters EL, Marion DW, Dixon CE. (2001). Tyrosine hydroxylase, but not dopamine beta-hydroxylase, is increased in rat frontal cortex after traumatic brain injury. Neuroreport 12:2323–2327. [DOI] [PubMed] [Google Scholar]
- Yan HQ, Kline AE, Ma X, Li Y, Dixon CE. (2002). Traumatic brain injury reduces dopamine transporter protein expression in the rat frontal cortex. Neuroreport 13:1899–1901. [DOI] [PubMed] [Google Scholar]
- Yan HQ, Ma X, Chen X, Li Y, Shao L, Dixon CE. (2007). Delayed increase of tyrosine hydroxylase expression in rat nigrostriatal system after traumatic brain injury. Brain Res 1134:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeb FD, Robbins TW, Winstanley CA. (2009). Serotonergic and dopaminergic modulation of gambling behavior as assessed using a novel rat gambling task. Neuropsychopharmacology 34:2329–2343. [DOI] [PubMed] [Google Scholar]
- Zgaljardic DJ, Seale GS, Schaefer LA, Temple RO, Foreman J, Elliott TR. (2015). Psychiatric disease and post-acute traumatic brain injury. J Neurotrauma 32:1911–1925. [DOI] [PubMed] [Google Scholar]
- Zhu J, Hamm RJ, Reeves TM, Povlishock JT, Phillips LL. (2000). Postinjury administration of l-deprenyl improves cognitive function and enhances neuroplasticity after traumatic brain injury. Exp Neurol 166:136–152. [DOI] [PubMed] [Google Scholar]