

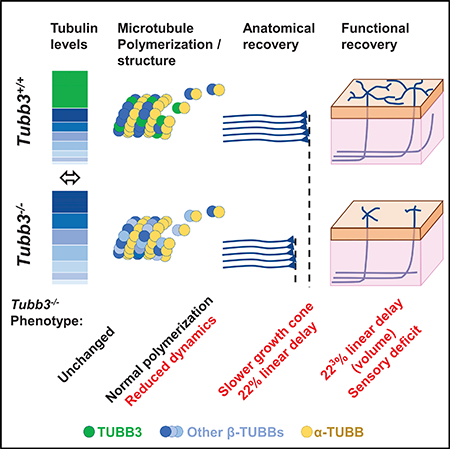
SUMMARY
We generated a knockout mouse for the neuronalspecific β-tubulin isoform Tubb3 to investigate its role in nervous system formation and maintenance. Tubb3−/− mice have no detectable neurobehavioral or neuropathological deficits, and upregulation of mRNA and protein of the remaining β-tubulin isotypes results in equivalent total b-tubulin levels in Tubb3−/− and wild-type mice. Despite similar levels of total β-tubulin, adult dorsal root ganglia lacking TUBB3 have decreased growth cone microtubule dynamics and a decreased neurite outgrowth rate of 22% in vitro and in vivo. The effect of the 22% slower growth rate is exacerbated for sensory recovery, where fibers must reinnervate the full volume of the skin to recover touch function. Overall, these data reveal that, while TUBB3 is not required for formation of the nervous system, it has a specific role in the rate of peripheral axon regeneration that cannot be replaced by other β-tubulins.
In Brief
Latremoliere et al. show that the neuronal-specific tubulin isoform TUBB3 is not required for normal development and function of the nervous system. Lack of TUBB3 decreases the dynamics of microtubules in growth cones, and this reduces axonal growth after peripheral nerve injury and strongly delays functional recovery.
Graphical Abstract
INTRODUCTION
Mice and humans encode multiple α- and β-tubulin isotypes that heterodimerize and polymerize to form microtubules (MTs). Tubb3 encodes the only β-tubulin isoform constitutively expressed in all neurons and not in radial or mature glia, making anti-TUBB3 antibody (Tuj1) the classic neuronal marker and leading to the general belief that Tubb3 has a critical role in neuronal structure and function (Ferreira and Caceres, 1992; Jiang and Oblinger, 1992). A role for TUBB3 in the formation of the central (CNS) and peripheral (PNS) nervous system is also suggested by evidence showing that human TUBB3 heterozygous missense mutations cause severe brain and nerve malformations. One series of TUBB3 mutations cause primary misguidance of cranial nerves accompanied, in some instances, by peripheral nerve degeneration (Chew et al., 2013; Tischfield et al., 2010; Whitman et al., 2016), and a second series results in primary malformations of cortical development and pontocerebellar hypoplasia (Bahi-Buisson et al., 2014; Oegema et al., 2015; Poirier et al., 2010). Either can also cause corpus callosum hypoplasia/agenesis and basal ganglia malformations. These mutations result in amino acid substitutions that alter heterodimer formation and incorporation into growing MTs, as well as MT dynamics and motor protein transport (Minoura et al., 2016; Niwa et al., 2013; Ti et al., 2016; Tischfield et al., 2010; Tischfield and Engle, 2010), and thus the phenotypes could reflect loss of function, gain of function, or altered TUBB3 function.
To determine TUBB3’s role in the nervous system, we generated and studied Tubb3−/− mice. Remarkably, we find that Tubb3−/− mice are viable and display no apparent neuroanatomical or behavioral defects under normal conditions or physiological stress. The deletion of Tubb3 leads to an upregulation of other b-tubulin isoform transcripts during early development, resulting in equivalent pan-b-tubulin protein levels in Tubb3−/− and wild-type (WT) mice. We find, however, that MTs in Tubb3−/− mice are less dynamic, and this is associated with a reduced growth rate of Tubb3−/− dorsal root ganglion (DRG) axons in vitro. After sciatic nerve crush, Tubb3−/− mice show delayed nerve regeneration, despite comparable upregulation of tubulins, and this leads to an exacerbated sensory recovery deficit, especially in the skin, where fibers need to occupy a volume to be functional.
Overall, our findings demonstrate that development of the nervous system and neuronal function does not require this neuronal specific β-tubulin isoform, but illuminate an important role of the TUBB3 isotype in MT dynamics and timely and adequate peripheral axon regeneration.
RESULTS
Tubb3−/− Mice Have No Apparent Neuroanatomical or Behavioral Defects
To formally demonstrate if TUBB3 is necessary for neuronal structureandfunction, wegeneratedTubb3−/− micebyflanking the isoform-specific exon 4, which represents more than 80% of the Tubb3 gene, with flox sequences and crossed these animals with germline Ella-Cre-driver mice. In the resultant Tubb3−/− mice, TUBB3 proteinwas undetectable by western blot and immunostaining in both embryonic and adult brains (Figures 1A and S1A). Remarkably, Tubb3−/− mice were born with normal Mendelian ratios; were viable; and had normal size, weight, and appearance (Figure 1B). Furthermore, while Tubb3R262C/R262C mice have aberrant CNS and cranial nerve axon guidance (Tischfield et al., 2010) and small hairpin RNA (shRNA) inhibition of TUBB3 expression causes a cortical neuronal migration delay (Saillour et al., 2014), the general histology of adult Tubb3−/− mice was comparable to WT littermates, including the anterior commissure (AC), corpus callosum (CC), basal ganglia, hippocampus, cerebellum, cortex, and hippocampal interneurons (Figures 1C, S1B, and S1C). Cortical cytoarchitecture and lamination at P2 also appeared normal (Figure 1D). To further assess structural anatomy and CNS white matter organization, we used magnetic resonance (MRI) and diffusion tensor imaging (DTI) (Pajevic and Pierpaoli, 2000) and detected no defects in adult Tubb3−/− brains (Figures 1E, S1D, and S1E). Similarly, cranial and spinal nerve anatomy of Tubb3−/− mice assessed at embryonic day (E) 11.5 was indistinguishable from that of wild-type embryos (Figure 1F). Finally, sciatic nerve anatomy of adult Tubb3−/− mice also appeared normal, with identical size distribution of myelinated fibers (Figures 1G and S1F), normal organization of MTs with the expected 25-nm ring structure, and presence of various organelles including mitochondria and vesicles, suggesting normal axonal transport (Figures 1H and S1G–S1P). In support of this, quantification of the amounts of conventional kinesin heavy chain (KHC), Kif21a, and dynein intermediate chain (DIC) molecular motors in sciatic nerve lysates showed no difference between Tubb3−/− and wild-type mice (Figure 1I).
Figure 1. Tubb3−/− Mice Are Viable with No Major Anatomical Abnormalities.

(A) Genetic construct to produce Tubb3 knockout mice. Mice were crossed with Ella-cre mice. Bottom: detection of TUBB3 in brain of E14.5 wild-type, Tubb3+/−, and Tubb3−/− mice by western blot and quantification (N = 9).
(B) Left: wild-type and Tubb3−/− littermates at 14 months of age. Middle: Mendelian birth ratio in WT (N = 14), Tubb3+/− (N = 26), and Tubb3−/− mice (N = 15). Right: weight gain of same mice at 3 months of age.
(C) Adult wild-type and Tubb3−/− mice brains stained with Luxol fast blue for myelin and Nissl counterstain. Coronal sections at the level of the anterior commissure (left) and hippocampus (middle), and sagittal sections of cerebellum (right) reveal no detectable abnormalities in architecture of corpus callosum (CC), anterior commissure (AC), cortex (CX), striatum (St), hippocampus (Hi), corticospinal tract (CT), lateral ventricle (LV), and cerebellum in Tubb3−/− mice compared to wild-type. Absence of the wild-type optic nerve at the level of the AC resulted from detachment during processing. Scale bars, 400 (left and middle) and 100 mm (right). N = 5.
(D) Cortical layers in wild-type (top, N = 5) and Tubb3−/− (bottom, N = 4) P2 mice. Brain sections stained for Reelin (layer I), Cux-1 (layers II–IV), Ctip2 (layer V), and Trb1 (layer VI).
(E) Directionally encoded color maps from diffusion tensor imaging of adult wild-type (top) and Tubb3−/− (bottom) mice to examine white matter tracts. A group template was generated for each strain and compared to identify potential anatomical abnormalities in the mutant template. Side-by-side slice views in each of the orthogonal orientations are shown at the same level in both the wild-type and Tubb3−/− templates. Red, left-right; green, dorsal-ventral; and blue, rostralcaudal. CG, cingulum; PF, pontine fibers; OT, optic tract; EC, external capsule; FI, fimbria; for other abbreviations refer to (C). N = 4+/+, 3–/–.
(F) Anatomy of cranial and spinal nerves stained with NF-M in E11.5 whole-mount embryos of wild-type and Tubb3−/− mice. I–XI: corresponding cranial nerves. N = 6 for each genotype.
(G) Top: representative bright-field cross-sectional images of sciatic nerve from wild-type and Tubb3−/− mice stained with toluidine blue. Scale bar, 100 mm. Bottom: distributions of myelinated fibers of different diameters in wild-type and Tubb3−/− mice. N = 3.
(H) Ultrastructure of myelinated large, medium, and small diameter fibers in cross-sectional images of adult wild-type and Tubb3−/− sciatic nerves. 49,0003. Inset: magnification highlights cytoskeletal MTs and neurofilaments. N = 3. Scale bar, 100 nm.
(I) Top: representative western blot and quantification of motor protein levels in sciatic nerve tissue lysates of adult wild-type and Tubb3−/− mice. Bottom: quantification (n = 3).
See also Figure S1.
Behaviorally, Tubb3−/− mice displayed no obvious defects when assessed with SHIRPA (Table S1), and daily locomotor activity, sleep-wake patterns and sleep architecture were indistinguishable from wild-type littermates (Figures 2A, 2B, S2A, and S2B). General sensory function and motor skills, including olfactory discrimination, visual acuity, thermal and tactile sensitivities, grip strength, and beam-walking assays were also comparable to those of wild-type mice (Figures 2C–2F). While a subset of human TUBB3 mutations causes a progressive polyneuropathy with onset between 5–25 years (Chew et al., 2013; Tischfield et al., 2010), 6-month-old Tubb3−/− mice had no signs of degeneration and had normal electromyographic conduction velocities and amplitudes (Figure 2G). Similarly, responses to innocuous and noxious stimuli were unaltered in mice tested at 6, 9, and 12 months (Figures S2C–S2E), suggesting normal maintenance of MTs along PNS nerves despite the absence of TUBB3.
Figure 2. Tubb3−/− Mice Have No Behavioral Defects.

(A) Actimetry of wild-type and Tubb3/ mice over a 4-day period (N = 6 or 6+/+, 6−/−).
(B) Wake, non-rapid eye movement (NREM), and rapid eye movement (REM) sleep amount expressed as percentage of the recording time in wild-type and Tubb3−/− mice (N = 4+/+, 4−/−).
(C) Olfactory discrimination (left) measured as seconds to nose touch with bacon-flavored softie. Visual acuity (right) assessed by eye tracking assay. (Olfaction: N = 8+/+, 8−/−; visual acuity: N = 10+/+, 10−/−).
(D) Number of brisk withdrawals out of 10 stimulations at various pressures applied onto the plantar surface of the hindpaw (left) or in the pinprick test (right) (N = 10+/+, 16−/−).
(E) Latency of withdrawal to contact heat at 49, 52 and 55C (left) and to a radiant heat source (middle). Time spent licking/flinching in the acetone test (right) (N = 10+/+, 16−/−).
(F) Balance beam assay for number of steps (columns 1 and 3) and slips (columns 2 and 4) (N = 10+/+, 13−/−).
(G) Representative compound muscle action potential (CMAP) recordings from intraplantar muscles evoked by sciatic nerve stimulation in wild-type and Tubb3−/− mice and quantification of CMAP intensity and velocity (N = 6+/+, 6−/−).
(H) Distance traveled in the visible, hidden and reversal phases of the water Morris maze assay (N = 8+/+, 13−/−). Inset: representative heatmap of the time spent during the hidden phase.
(I) Heatmap representation of time spent in closed/open arms in the elevated plus maze assay and quantification (s) (N = 16+/+, 17−/−).
(J) Representative traces of the time course of SWA (slow wave activity; spectral electroencephalogram (EEG) power in the 0.5- to 4.0-Hz range) during the first2.5 h of sleep opportunity in wild-type (top) and Tubb3/ (bottom) mice. Right: NREMS SWA computed over 4 hr (11:00–15:00) expressed as a percentage of the 24-hr baseline mean (N = 4+/+, 4−/−).
(K) Time spent licking/flinching the paw after intraplantar injection of formalin (1%) (N = 6+/+, 6−/−).
Error bars indicate SEM. See also Figure S2.
Next, we asked whether lack of TUBB3 impaired the ability of mice to respond to physiological challenges. We tested anxietyrelated behaviors in the elevated plus-maze, learning ability in the Morris water navigation assay, response to novelty in the novel object assay, homeostatic sleep rebound caused by sleep deprivation, and response to a sustained nociceptive input caused by intraplantar injection of formalin and, for each, the Tubb3−/− mice remained indistinguishable from their wild-type littermates (Figures 2H–2K and S2F).
Thus, despite comprehensive examination, we found that mice lacking the neuronal-specific β-tubulin isoform TUBB3 do not display any major neuroanatomical or behavioral deficits, even under physiological stress.
Tubb3−/− Mice Upregulate Other β-Tubulin Isoforms’ Transcripts and Have Indistinguishable Total β-Tubulin Protein Levels
We measured pan-b-tubulin protein levels in embryonic and adult Tubb3−/− mice and found them to be equivalent to wildtype littermates in both brain and sciatic nerve (Figure 3A), indicating a secondary compensation that has already occurred by E14 (Figure S3A). Immunoprecipitation with anti-TUBB3 antibody acutely depleted TUBB3 and reduced pan-β-tubulin protein levels by ~30% in both brain and sciatic nerve of adult wild-type mice, whereas these levels were unchanged in Tubb3−/− mice, confirming compensation by other tubulin isoforms (Figure 3B). This proportion of TUBB3 relative to pan-β-tubulin protein levels was consistent with the relative proportion of transcript levels assessed by qPCR (Figure S3B) (Jiang and Oblinger, 1992). To determine which b-tubulin isoforms increased transcriptional activity in absence of TUBB3, we assessed the relative proportion of each isoform by qPCR in adult Tubb3−/− mice and found a ~10%–20% increase in transcript level for most β-tubulin isoforms compared to wild-type (Figure 3C), indicating a general rather than an isoform-specific compensation.
Figure 3. Tubb3−/− Mice Compensate to Have Normal Pan-tubulin Levels.

(A) Representative western blot (left) and quantification (right) of tubulin protein levels in adult brain (top) and sciatic nerve (bottom) tissue lysates (n = 3+/+, 3−/−). (B) Left: schematic of TUBB3 depletion experiment. TUBB3 was immunoprecipitated away from adult brain and sciatic nerve (DRG/SN) tissues lysates, with antibIII tubulin antibody to determine the relative amount of TUBB3 compared to total pan-b-tubulins. Middle: representative western blots from brain (top) and sciatic nerve (bottom). Right: quantifications (n = 3+/+, 3−/−).
(B) Relative proportion of different b-tubulin isoform transcript levels in the adult brain (top) and DRG (bottom) of Tubb3−/− mice compared to wild-type animals assessed by quantitative qPCR (n = 6+/+, 6−/−).
(C) Left: Schematic of MT polymerization experiment. Middle: representative western blots and quantification of MT polymerization using adult brain (top) andsciatic nerve (bottom) tissue lysates (n = 3+/+, 3−/−).
(D) Representative western blot (left) and quantification (right) of tubulin post-translational modifications in adult brain (top) and sciatic nerve (bottom) tissue lysates (n = 3+/+, 3/).
Error bars indicate SEM. See also Figure S3.
Together, these data show that deleting the Tubb3 gene leads to compensation by other β-tubulin isoforms during development and adulthood, which results in normal levels of total tubulins and absence of any major neurological defects under basal and challenging conditions.
Next, we examined MT polymerization and post-translational modifications. Tubulin from lysates of adult wild-type and Tubb3−/− brain and sciatic nerve polymerize into MTs equally well (Figure 3D). MTs of adult Tubb3−/− mice, however, displayed higher levels of several markers associated with increased stability: MTs in the brain were more polyglutamylated and acetylated, whereas MTs in the sciatic nerve were more polyglutamylated and detyrosinated, with higher levels of delta-2-modified tubulin (Figure 3E).
TUBB3 Is Critical for Growth Cone Function
MTs undergo periods of growth (polymerization) and shrinkage (catastrophe), termed dynamic instability, and the presence of markers of increased stability in MTs from Tubb3−/− mice suggested that dynamics were potentially impaired (Rochlin et al., 1996; Tanaka et al., 1995). To formally assess this, we adapted an embryonic spot culture protocol (Cho et al., 2015) to adult DRG neurons. We overexpressed the GFP-fused tip protein EB3, which specifically binds to the plus end of a growing MT and tracks its growth, detaching when the MT pauses or undergoes catastrophe (Figures 4A and4B). We acquired 5-min live-imaging recordings of infected wild-type and Tubb3−/− adult DRG growth cones to detect individual EB3-GFP growth tracks and quantified three major parameters of MT dynamics (Figure 4B). EB3-GFP plus-end velocity, reflecting the speed of MT polymerization, was decreased by 7% (wildtype vs. Tubb3−/− = 15.22 ± 0.43 vs.13.95 ± 0.44 μm/min). EB3-GFP plus-end lifetime, reflecting the duration in seconds of MT polymerization prior to pause or catastrophe, was increased by 14% (wild-type vs. Tubb3/ = 9.58 ± 0.31 vs. 10.84 ± 0.30 s). By contrast, EB3-GFP plus-end displacement, reflecting the distance the MT grows prior to pause or catastrophe (Stout et al., 2014), was not changed (wild-type vs. Tubb3−/− = 2.06 ± 0.05 vs. 2.12 ± 0.05 mm) (Figure 4C). These data demonstrate, in a cell-based assay, that DRG neurons’ growth cones lacking TUBB3 have decreased MT dynamics, consistent with previous reports using cell-free assays (Pamula et al., 2016; Panda et al., 1994; Vemu et al., 2017).
Figure 4. TUBB3 Is Critical for Growth Cone Function.

(A) Representative image of EB3-GFP expression in a spot-cultured adult wild-type DRG growth cone on DIV6, infected by pAAV-EB3-GFP on DIV1. EB3-GFP specifically labels the plus ends of growing MTs shown as comets (green); growth cone genetically labeled with vGlut2-Tdtomato (red). Scale bar, 20 μm. (B) Left: representative frame from a 5-min duration (2-s interval) time-lapse live-cell imaging of EB3-GFP in spot cultured DRG growth cone from adult wild-type mice on DIV6 using a total internal reflection fluorescence (TIRF) microscope with 100x TIRF lens. Right: EB3-GFP tracking (red lines) over 5min recording using PlusTipTracker software. Scale bar, 20 μm.
(B) Quantifications of (B). Growth cone MT growth velocity (left), lifetime (middle), and length (right) in wild-type versus Tubb3−/− DRGs. Analysis of 100–200 comets per growth cone and 10 growth cones per experiment. n = 3 independent experiments. *p < 0.05; **p < 0.01. ns, not significant by unpaired, two-tailed Student’s t test between wild-type and Tubb3−/−mice for each condition. Scale bar, 20 μm.
(C) Left: representative images of last frame of a 1-hr duration (interval, 1 min) time-lapse live-cell imaging of growth cones of spot cultured vGlut2-Tdtomatopositive DRG neurons from wild-type and Tubb3−/− mice on DIV6 (left). Colored lines track growth cone trajectories over the 1-hr recording. Scale bar, 100 mm. Middle: quantification of growth cone area. Right: percentage of growth cones retracted (R), stationary (S), and forward moving (M). n = 6 (three pairs of wild-type and Tubb3/ littermates, two spot cultures per mouse, ~25 growth cones analyzed per culture).
(D) (E and F) Quantification (left) and histograms (right) of the displacement (E) and distance (F) of forward moving wild-type and Tubb3−/− growth cones from (D).
**p < 0.01 by unpaired, two-tailed Student’s t test between wild-type and Tubb3−/− mice for each condition. Error bars indicate SEM. Scale bar, 100 μm.
Next, we found that Tubb3−/− DRG growth cones were significantly larger than wild-type, consistent with slower growth, and so we tracked Tubb3−/− DRG growth cones and analyzed their trajectories over 60 min (Figure 4D). Comparing the relative positions of a given growth cone at the start and end of the recording, we placed it into one of three categories: retracted, in which the final position was more than one growth cone diameter behind its starting position; stationary, in which the final position was within one growth cone diameter of its starting position; or moving forward, in which the final position was more than one growth cone diameter ahead of its starting position (Cheng et al., 2014) (Figure 4D). While both genotypes had a similar percentage of retracted growth cones, forward moving Tubb3−/− DRG neurons had a significant reduction in total displacement (the linear distance between the location of the growth cone at the start and end of the recording), and a mild reduction in overall distance traveled (the entire trajectory of the growth cone over the duration of the recording), indicating a “wandering-like” behavior that resulted in a net reduction of forward movement compared to wild-type (Figures 4D–4F). Together, these results demonstrate that TUBB3 is necessary for MT dynamics and appropriate adult DRG growth cone behavior.
Lack of TUBB3 Causes a Linear Growth Defect in DRG Axons
To test if altered MT dynamics are critical for neurite outgrowth, we monitored the same population of adult DRG neurons in the spot culture over 8 days and found that absence of TUBB3 caused a consistent growth defect, which led to a cumulative 22% delay in neurite outgrowth (Figures 5A and S4). This growth defect was not due to an abnormal response to environmental cues, as indicated by measuring neurite outgrowth of both naive (DRG neurons from uninjured mice) and pre-conditioned (DRG neurons pre-injured by sciatic nerve crush 5 days before culture) neurons in both permissive (PDL) and non-permissive (CSPG) milieus. While pre-conditioned Tubb3−/− DRG neurons had significantly increased axonal growth on both substrates compared to naive Tubb3−/− DRG neurons, they consistently displayed reduced growth compared to wild-type DRG neurons (Figure 5B). This indicates that Tubb3−/− neurons can produce a similar pre-conditioning response to their wild-type counterparts regardless of their environment (wild-type PDL: +46%; Tubb3−/− PDL: +35%; wild-type CSPG: +59%; Tubb3−/− CSPG: +40%) and suggests that their growth defect is intrinsic. Finally, quantile-quantile plot (QQ plot) analysis of Tubb3−/− and wildtype dissociated DRG neurons on laminin for 24 hr also revealed a linear 22% defect in Tubb3/ neurites compared to wild-type, confirming that every DRG Tubb3−/− sensory neuron has the same growth defect (Figure 5C).
Figure 5. Axonal Growth Is Linear and Reduced by 20% in Absence of TUBB3.

(A) Left: high magnification of neurite outgrowth in Tubb3−/− versus wild-type spot cultures at DIV4 and DIV8. Scale bar, 50 mm. Right: quantification of Sholl analysis. The black and green lines represent WT and Tubb3−/− growth, respectively. n = 8. Scale bar, 50 μm.
(B) Quantifications of growth of naive versus pre-conditioned wild-type and Tubb3−/− DRG neurons on poly-D-lysine neutral substrate and on CSPG inhibitory substrate. *p < 0.05. One-way ANOVA, post hoc Sidak’s. n = 12 wells per condition.
(C) Quantile-quantile curve of Tubb3−/− neurite growth normalized to wild-type samples. Green dots represent neurite growth for each Tubb3−/− Tubb3−/− DRG neuron calculated by interpolation. Dashed black line represents WT growth. Dashed-red line is the linear regression of Tubb3/ neurite outgrowth. n = 486 neurons.
Error bars indicate SEM. See also Figure S4.
Tubb3−/− Mice Have Delayed Recovery after Sciatic Nerve Crush
Sciatic nerve crush in wild-type mice caused a progressive upregulation of Tubb3 mRNA in the DRG, consistent with previous reports (Hoffman and Luduena, 1996; Moskowitz and Oblinger, 1995; Moskowitz et al., 1993), which peaked at day 15 postinjury (Figure 6A); we also found Tubb2a, Tubb2b and Tubb6 to be upregulated, but with an earlier peak induction (d7) (Figure S5A). Transcriptional upregulation of non-Tubb3 β-tubulin isoforms in DRGs of Tubb3−/− mice 7 days after sciatic nerve crush was indistinguishable from wild-type mice (Figures 6B and S5B), suggesting the compensatory transcriptional upregulation of other b-tubulin isoforms was maintained during the axonal injury genetic response. In support of this, tubulin extracted from Tubb3−/− sciatic nerve distal to the injury site 7 days after crush did not show any differences in the amount of tubulin or its ability to polymerize compared to wild-type (Figures 6C and 6D). In the distal nerve segments from wild-type and Tubb3−/− mice, we found the same amount of tyrosinated MTs (Figure 6E), likely reflecting the presence of newly synthesized MTs in regenerating axons (Song et al., 2015). Altogether, these data show that injured peripheral axons can reform MTs even in the absence of TUBB3.
Figure 6. Lack of TUBB3 Delays Peripheral Nerve Regeneration.

(A) qPCR time course of Tubb3 fold increase in DRG after sciatic nerve crush (n = 9 per time point). *p < 0.05. One-way ANOVA, post hoc Dunnett’s.
(B) Fold ratio of the upregulation of transcript levels of different β-tubulin isoforms in DRG after sciatic nerve crush (n = 6+/+, 6−/−).
(C) Representative western blot (top) and quantification (bottom) ofα- and pan-β-tubulin levels from tissue lysates of segments of the sciatic nerve proximal and distal to the crush site 7 days after crush (n = 3+/+, 3−/−).
(D) Representative western blot (top) and quantification (bottom) of MT polymerization using proximal and distal segments of the sciatic nerve tissue lysates7 days after crush (n = 3+/+, 3−/−).
(E) Representative western blot (top) and quantification (bottom) of levels of polyglutamylated and tyronisated tubulin in proximal and distal segments of thesciatic nerve tissue lysates 7 days after crush (n = 3+/+, 3−/−).
(E) Pinprick assay scores over 24 days following sciatic nerve crush (N = 21+/+, 14−/−). *p < 0.05. Two-way RM ANOVA, post hoc Sidak’s.
(F) Brush assay scores over 24 days following sciatic nerve crush (N = 9+/+, 9−/−). *p < 0.05. Two-way RM ANOVA, post hoc Sidak’s (n = 9).
(G) Toes spread in % of pre-injury after sciatic nerve crush (N = 9+/+, 9−/−). *p < 0.05. Two-way RM ANOVA, post hoc Sidak’s.
(I–M) Drawing representing the skin region harvested and processed for immunostaining. Mice were tested for pinprick, given a score, and then the skin corresponding to the sural territory was harvested, fixed, unfolded, and processed for whole-mount staining against NF200 (I). Imaging was carried out from the dermis to the epidermis at 12 (J), 15 (K), 21 (L), and 30 (M) days after sciatic nerve crush. Pinprick score from each animal sampled is indicated in top-right corner of each picture. For score 3, the yellow star indicates the position of the last positive behavioral response. Scale bar, 500 μm.
Error bars indicate SEM. See also Figure S5.
Recovery of sensory function following crush was determined by assessing the ability of the injured mouse to respond to pinprick or brush stimulus at different locations on the hindpaw over time (Ma et al., 2011). For both modalities, Tubb3−/− mice displayed a significant delay, with full recovery at day 22 postinjury, compared to day 15 in wild-type littermates (Figures 6F and6G). Recovery of motor function was determined by return of toe spreading in walking Tubb3−/− mice, and we again found recovery in Tubb3−/− mice to be delayed compared to wild-type littermates 14 and 17 days after injury (Figure 6H). At day 21, toe spreading and the compound muscle action potentials of intraplantar muscles were similar between wild-type and Tubb3−/− mice, indicating full recovery (Figures 6H and S5C).
We confirmed that the delay in functional recovery was due to a growth defect by imaging the maximum-projection signal from whole-mount hindpaw skin immunostained for NF200, a marker of myelinated sensory fibers, in mice at different days after injury (Figure 6I). We found a strong correlation between the score obtained in the pinprick assay and the presence of fibers in the skin (Figures 6J–6M). Twelve days after sciatic nerve crush, wild-type mice had an average score of 3, corresponding to the detection of the pin applied up to the first plantar bump, an area where we could detect NF200+ fibers, while Tubb3−/− mice had a score of 0 and skin samples revealed no detectable fibers (Figure 6J). By day 15 after crush, wild-type mice had fully recovered pinprick sensation, while Tubb3−/− mice had only reached a score of 3, and their innervation profile was similar to that of wild-type mice 3 days earlier (12 days after crush; Figure 6K). At later time points, both wild-type and Tubb3/ mice had fully recovered pinprick responsiveness and their hindpaw skin was positive for NF200+ fibers, although the degree of innervation was less dense in Tubb3−/− samples both 21 and 30 days after crush (Figures 6L and 6M). These results indicate that Tubb3−/− axons regenerate more slowly than their wild-type counterparts despite identical production of total b-tubulin (see Figure 3) and that the regenerating fibers are functional immediately upon reaching their target tissues.
Functional Recovery Requires Reinnervation of a Volume, and This Exacerbates Delay in Tubb3−/− Mice
Because pinprick score correlates with the presence of fibers in the hindpaw skin, we transformed our score metric into the distance covered by regenerating fibers from the heel territory (Figure 7A), and this revealed near-perfect linear profiles for functional pinprick recovery after nerve crush in both wild-type and Tubb3−/− mice (Figure 7B). Moreover, in wild-type mice, the intersection of the linear regression of pinprick functional recovery on the y axis predicted the actual distance between the site of injury and the heel territory (25.305 mm) (Figure 7B), which we confirmed in vivo by dissection (25.4 ± 0.1 mm) (Figure 7C). Using the slope obtained from the wild-type pinprick recovery curve (2.58) (Figure 7B), we determined that thinly myelinated, pinprick-responsive sensory fibers grew at a rate of 2.58 mm per day after nerve injury in wild-type animals, much faster than previously reported (Buchthal and Kühl, 1979). Thus, the 22% decrease in speed of DRG neurite outgrowth found in Tubb3−/− mice in vitro would be predicted to correspond to an axon growth rate of 2 mm/day in vivo. If correct, regenerating Tubb3−/− DRG axons should reach the heel territory 12 days after sciatic nerve crush, 2 days later than in wild-type mice, which is precisely what we found experimentally using behavioral and immunological approaches (black and green arrowheads in Figure 7B; also see Figure 6). By contrast, while still displaying a linear profile, once sensory fibers reached the heel it took an additional 12 days to functionally reinnervate the entire 13.6-mm length of the hindpaw in Tubb3−/− mice, twice as long as the predicted 6 days based on a 22% decrease in growth rate (Figure 7B). We hypothesized that this additional delay may arise because, for functional recovery (transduction of the mechanical stimulus, i.e., high pressure on the skin), regenerating fibers must reoccupy their target space with the correct pattern/density within the skin.
Figure 7. Functional Recovery Requires Reinnervation of a Volume that Exacerbates Delay in Tubb3/ Mice.

(A) Functional recovery of pinprick response in wild-type and Tubb3−/− mice expressed as a distance from heel of regenerated pinprick-responsive fibers at various days after sciatic nerve crush. *p < 0.05. Two-way RM ANOVA, post hoc Sidak’s. (N = 21+/+, 14−/−).
(B) Linear regression of functional recovery of pinprick responses. In wild-type mice, this predicts the site of injury (intersection point on the y axis, black dot) and the rate of regeneration (2.58 mm per day). In Tubb3−/− mice, this linear regression cannot predict the site of injury (red dot). Blue dot represents the observed distance to the site of injury in both wild-type and Tubb3−/− mice.
(C) Confirmation of the distance between the site of injury in the sciatic nerve crush model and the beginning of the heel territory, where pinprick testing begins(N = 9+/+, 9−/−).
(D) Regenerating nerves stained with NF-H in whole-mount hindpaw skin of wild-type and Tubb3−/− mice after sciatic nerve crush (pinprick score 5 + 7 days). The stained fibers are pseudo-colored based on their depth within the skin tissue. Scale bar, 100 (left) and 25 mm (right), (n = 3+/+, 3−/− per time point).
(E) Model for functional recovery delay in Tubb3−/− mice compared to wild-type mice. Filled circles represent experimental points while the curves are extrapolated from a linear growth and a 22% delay in Tubb3/ mice. Predicted recovery of a 20 to 25% defect in growth compared to wild-type in 1D (yellow; length on a simple plane), 2D (pink; growth in an area), and 3D (green; growth in a volume). Dotted red line indicates a 22% defect in growth compared to wild-type in 3D; this is the same defect we observed in vitro.
(F) Consequences of a 22% slower regeneration rate on growth within the sciatic nerve and on functional recovery assessed by the pinprick test. Fibers need to occupy a volume for functional recovery within the hindpaw, which exacerbates the delay in Tubb3−/− mice.
(G) Time course of mechanical sensitivity for pinprick and 2-g, and 1-g pressures in wild-type and Tubb3−/−mice after peripheral nerve crush (N = 9+/+; 10−/− for vF, N = 8+/+; 8−/− for pinprick). The skin area tested is indicated by the red-dotted circle. *p < 0.05. Two-way RM ANOVA, post hoc Sidak’s.
Error bars indicate SEM.
To better understand the topography of regenerating fibers within the skin of the hindpaws of wild-type and Tubb3−/− mice, we pseudo-colored each z stack layer in NF200-stained whole-mount hindpaws from mice 7 days after they have reached full pinprick recovery (score 5 + 7 days). We could distinguish in situ the main nerve branches running deeper in the skin (orange), as well as the smaller nerve branches which arose from specific ramification points and extended into the most superficial layer of the skin (green; Figure 7D, left). We found that regenerating axons appeared to branch from the larger nerve and directly extend to the superficial layer of the skin prior to more extensively branching to cover the skin’s surface (Figure 7D, right). In Tubb3−/− mice, these most distal superficial branches were less abundant than those in wild-type mice with the same pinprick score, consistent with a slower growth rate (Figure 7D).
These results imply that the delay in functional recovery of Tubb3−/− mice within their paw may arise because their linear growth defect is amplified to reflect the volume of the skin. This is supported by the mathematical analysis of the relationship between the slopes of wild-type and Tubb3−/− functional recoveries, where the slope for Tubb3−/− mice is 1.22, compared with 2.58 in wild-type mice. Strikingly, the ratio of these slopes is 0.47, and, which means that the relationship between the slopes for the functional recovery of Tubb3−/− and wild-type mice is a linear defect of 22%, the same percent delay we found in in vitro assays of neurite outgrowth (see QQplot and spot cultures in Figures 3 and 4) and in in vivo growth from injury site to heel (see Figure 7A and 7B), but is now occurring within the volume of the paw. To test these findings, we ran predictive recovery curves to explain the defect we observed within the paws of Tubb3−/− mice compared to wild-type, and confirmed that a linear 22% growth defect to fill up a volume best fit our experimental results (Figure 7E).
Altogether, these data show how a consistent 22% reduction in linear axonal growth rate in Tubb3−/− mice translates into a reduced daily nerve regeneration rate of 2 mm/d. This causes a moderate delay in the time it takes for regenerating fibers to grow from the crush site to reach the heel. The effect of the reduced growth rate is exacerbated, however, once the axons reach the heel and must reinnervate the full volume of the hindpaw skin to allow for functional recovery. Thus, while sensory recovery is still linear, once axons reach the heel the slope of the functional recovery is strongly reduced in Tubb3−/− mice (Figure 7F). These data demonstrate that determining linear nerve growth (such as the distance regenerating axons have grown) can underestimate the actual defect in functional outcome, such as the ability to detect stimuli. Finally, this defect to fully reoccupy the skin in Tubb3−/− mice after sciatic nerve crush translates into a more prominent functional recovery deficit for milder punctate mechanical stimulus intensities (Figure 7G), because encoding of mild touch stimuli requires a greater amount of fine terminal innervation than does pinch stimuli.
DISCUSSION
The yeast genome encodes two α- and one β-tubulin isotype, whereas mouse and human genomes encode at least eight α-and eight β-tubulin isotypes (Luduena and Banerjee, 2008; Redeker, 2010). This increasing isotype diversity with species complexity, coupled with differential tissue expression during development, led to the hypothesis that tubulin genes evolved to allow functionally distinct isotypes to combine in various ratios to diversify MT function within and between cell types (Cleveland, 1987; Fulton and Simpson, 1976; Ludueña, 1993; Sullivan and Cleveland, 1986). While we estimate TUBB3 to represent ~30% of total tubulins in wild-type animals, we did not identify any structural or functional defects in naive Tubb3−/− adult mice despite using a wide array of anatomical and neurological tools (Crawley, 2007). Our results strongly suggest that the compensatory production of pan-β-tubulins in Tubb3−/− mice is sufficient for neurons to produce MTs that allow them to migrate to their correct cortical layer, develop axons that reach their expected targets, and produce functional tracks for motor proteins. These data indicate, therefore, that TUBB3 is not essential for mouse nervous system development and function.
Our findings, in conjunction with previous data showing that mice harboring a human TUBB3 missense mutation within the Tubb3 gene have cranial axon misguidance and corpus callosum and anterior commissure hypoplasia (Tischfield et al., 2010), suggest that the human TUBB3 tubulinopathies result from incorporation of mutant heterodimers that alter or disrupt specific MT functions rather than from a general loss of TUBB3 protein. By contrast, we found that the absence of TUBB3 causes a severe nerve regeneration delay after sciatic nerve crush. While the absence of TUBB3 can be compensated at the structural level to maintain the total pan-β-tubulin level in neurons, during the nerve regeneration process this substitution by other b-tubulin isoforms cannot completely replace TUBB3-specific function. Moreover, Tubb3−/− DRG neurons respond similarly to their wild-type counterparts when exposed to various permissive or non-permissive growth milieus in vitro, and display the stereotyped enhanced regeneration rate and acquire the ability to grow on myelin when subjected to a pre-conditioning injury (Neumann and Woolf, 1999). These findings support an intrinsic growth defect of Tubb3−/− DRG neurons rather than a defect in their ability to detect or respond to external factors in the environment and are consistent with the absence of neuronal migration or axonal guidance defects in Tubb3−/− mice. This linear reduction in the growth rate of all DRG sensory neurons following injury indicates that the replacement of TUBB3 by other tubulin isoforms cannot overcome the requirement for this β-tubulin isoform in peripheral nerve regeneration, supporting a context-dependent isotype-specific TUBB3 function.
Current strategies to enhance nerve regeneration seek to optimize the cellular response to axotomy by promoting the production of a limited number of regeneration-associated proteins (Chandran et al., 2016; Ma et al., 2011), while inhibiting deleterious elements, such as cell death or intracellular pathways, that are activated by growth inhibitory substrates (Belin et al., 2015; Nawabi et al., 2015; Sun et al., 2011). Technically, these strategies rely on the postulate that overexpression or inhibition of key factors identified during endogenous regeneration will be sufficient to drive faster axonal growth. In most experiments using this approach, the effects were much stronger in vitro than in vivo for PNS regeneration, leading to limited functional outcomes (Chandran et al., 2016; Ma et al., 2011; Seijffers et al., 2007).
Our results point to an alteration in MT dynamics in growth cones as the main cause for slower PNS regeneration in Tubb3−/− mice, rather than a defect in the genetic response to axotomy or insufficient total tubulin levels. Although recent reports indicate that increasing MT stability in CNS neurons promotes regeneration (Hellal et al., 2011; Mikhaylova et al., 2015; Ruschel et al., 2015), our results show that increased MT stability is associated with impaired peripheral nerve regeneration. The increase in CNS axon regeneration after spinal cord injury induced by very low doses of taxol (a MT stabilizer) more likely reflects removal of environmental growth barriers (glial scars) rather than enhancement of intrinsic axonal growth (Hellal et al., 2011). By contrast, PNS nerve regeneration occurs in a more permissive milieu, and enhanced intrinsic growth is likely to be the major driving force for axonal regeneration. Several lines of evidence support the modulation of MT dynamics as a promising strategy to promote PNS regeneration. Reduced glycogen synthase kinase 3 (GSK3) activity after nerve injury causes a reduction in phosphorylated MAP1B, which is required to maintain a pool of dynamic, tyrosinated MT in axons (Barnat et al., 2016; Trivedi et al., 2005). As a result, MT stability increases and axonal growth decreases (Gobrecht et al., 2016; Gobrecht et al., 2014). Genetic or pharmacologic promotion of GSK3 activity reduces MT detyrosination and promotes functional recovery in mice (Gobrecht et al., 2016). Similarly, altering the function of tubulin-tyrosine ligase (TTL) reduces the amount of tyrosinated MTs after injury and severely impedes axonal growth (Song et al., 2015), possibly by altering the MT dynamics required for growth cone function. Our study further highlights the complexity of the intrinsic regulatory processes in neurons that adjust MT dynamics in a context-dependent manner and are required for optimal and efficient growth cone elongation and, consequently, timely regeneration. Efforts to build new therapeutic strategies to promote PNS regeneration will need to determine the critical factors responsible for setting MT dynamics: MT composition of tubulin isoforms, post-translational modifications, MT-associated proteins, and their reciprocal interactions.
In clinical settings, sensory recovery, especially for light touch sensitivity, is very limited, and the reasons for this are not yet understood (Ciaramitaro et al., 2010; Ruijs et al., 2005). Our results show how a subtle, but constant, growth defect leads to a major functional sensory recovery defect, particularly for touch stimuli of lighter intensity, and this is likely caused by the time required to grow through and fully reoccupy the full volume of the skin. While the periphery is a growth-permissive milieu, the complexity—and volume—of the skin might prove a greater challenge for PNS regeneration than previously appreciated because the axonal growth required for functional recovery is “longer” than expected.
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GFP | Thermo Fisher Scientific | Cat#A-11122; RRID:AB_221569 |
| Mouse monoclonal antineurofilament (clone 2H3) | Developmental Studies Hybridoma Bank |
Cat#2H3; RRID:AB_531793 |
| Alexa Fluor 594 goat anti-mouse | Thermo Fisher Scientific | Cat#A-11032; RRID:AB_2534091 |
| Alexa Fluor 488 goat anti-mouse | Thermo Fisher Scientific | Cat#A-11001; RRID:AB_2534069 |
| Alexa Fluor 647 goat anti-rabbit | Thermo Fisher Scientific | Cat#A-21244; RRID:AB_10562581 |
| Mouse monoclonal anti-α-tubulin (clone DM1A) | Sigma-Aldrich | Cat#T6199; RRID:AB_477583 |
| Rabbit polyclonal anti-βIII-tubulin | Abcam | Cat#AB18207; RRID:AB_444319 |
| Mouse monoclonal anti kinesin heavy chain (clone H2) | EMD Millipore Corporation | Cat#MAB1614; RRID:AB_942284 |
| Rabbit polyclonal anti-Kif21a | Generated by our lab as described (PMID: 20074521) | N/A |
| Mouse monoclonal anti Dynein intermediate chain (clone 74.1) | EMD Millipore Corporation | Cat#MAB1618; RRID:AB_2246059 |
| Mouse monoclonal anti-pan β-tubulin (clone AA2) | EMD Millipore Corporation | Cat#05661; RRID:AB_309885 |
| Mouse monoclonal anti-acetylated tubulin (clone 6–11B-1) | Sigma-Aldrich | Cat#T7451; RRID:AB_609894 |
| Rat monoclonal anti-tyrosinated tubulin (clone YL1/2) | Abcam | Cat#ab6160; RRID:AB_305328 |
| Rabbit polyclonal anti-detyrosinated tubulin | EMD Millipore Corporation | Cat#AB3201; RRID:AB_177350 |
| Rabbit polyclonal anti-delta2 tubulin | EMD Millipore Corporation | Cat#AB3203; RRID:AB_177351 |
| Mouse monoclonal anti-polyglutamylation tubulin (clone GT335) | Adipogen | Cat#AG2080020C100; RRID:AB_177351 |
| Rabbit polyclonal anti-GAPDH | Santa Cruz Biotechnology | Cat#sc25778; RRID:AB_10167668 |
| Peroxidase-AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat#715035151; RRID:AB_2340771 |
| Peroxidase-AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | Cat#711035152; RRID:AB_10015282 |
| Peroxidase-AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson Immunoresearch | Cat#712035150; RRID:AB_2340638 |
| chicken anti-neurofilament heavy chain | Millipore | AB5539; RRID:AB_177520 |
| Goat anti-chicken | Jackson Immunoresearch | Cat#703–165-155; RRID:AB_2340363 |
| Bacterial and Virus Strains | ||
| pAAV2/8-EB3-GFP | Boston Children’s
Hospital Viral Core |
N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NGF 2.5S native mouse protein | Thermo Fisher Scientific | Cat#13257019 |
| GDNF | ProSpec | Cat#CYT-305 |
| Cytosine β-D-arabinofuranoside | Sigma-Aldrich | Cat#C1768 |
| StartingBlock PBS Blocking Buffer | ThermoFisher Scientific | Cat#37538 |
| Experimental Models: Organisms/Strains | ||
| Mouse: ROSA26::FLPe knock in:
129S4/SvJaeSor-Gt(ROSA) 26Sortm1(FLP1)Dym/J |
The Jackson Laboratory | JAX: 003946 |
| Mouse: 129S1/SvImJ | The Jackson Laboratory | JAX: 002448 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: E2a-Cre: B6.FVB-Tg(EIIa-cre)C5379Lmgd/J | The Jackson Laboratory | JAX: 003724 |
| Mouse: Ai14, Ai14D or Ai14(RCL-tdT)-D:
B6.Cg-Gt(ROSA) 26Sortm14(CAG-tdTomato)Hze/J |
The Jackson Laboratory | JAX: 007914 |
| Mouse: Vglut2-ires-cre: Slc17a6tm2(cre)Lowl/J | The Jackson Laboratory | JAX: 016963 |
| Mouse: ISLmn-GFP: SALK:SE1:FARN.ISL(mn):GFP-F | Samuel L Pfaff, The Salk Institute, |
SALK:SE1:FARN.ISL(mn):GFP-F |
| Tg(Isl1-EGFP*)1Slp/J | The Jackson Laboratory | JAX: 017952 |
| Mouse: 129S6/SvEvTac | Taconic | TACONIC: 129SVE |
| Oligonucleotides | ||
|
Tubb1- F/R primers
TCTTGGGTTCAAGGCTAGGAT/ CGCAGTCAATCCCATGTTC |
This paper | N/A |
|
Tubb2a- F/R primers
AACCAGATCGGCGCTAAGT/ TCCAGCTGCAAGTCACTGTC |
This paper | N/A |
|
Tubb2b- F/R primers
TCATCAGACCCACTGACACAG/ TTTCCAGTTGCAAATCACTGTC |
This paper | N/A |
|
Tubb2c- F/R primers
CTTCGGGCAGATCTTCAGAC/ ACGTCCAACACCGAG |
This paper | N/A |
|
Tubb3- F/R primers
GGTGGACTTGGAACCTGGAA/ TAAAGTTGTCGGGCCTGAATTCAA |
This paper | N/A |
|
Tubb4- F/R primers
TCTTTCGGCCAGACAACTTT/ CAGGACGGCATCCACTAACT |
This paper | N/A |
|
Tubb5- F/R primers
CCGTAGCCATGAGGGAAAT/ ATGCCATGTTCATCGCTTATC |
This paper | N/A |
|
Tubb6- F/R primers
CGGCCTGACAACTTCATCTT/ TGAAGACAGTCGCAATGCTC |
This paper | N/A |
| Genotyping | Transnetyx, Inc. | http://.transnetyx.com |
| Recombinant DNA | ||
| pAAV2/8-EB3-GFP | PMID: 26526391 | N/A |
| Software and Algorithms | ||
| PlusTipTracker | PMD: 25225829 | http://.utsouthestern.edu/labs/danuser/ |
| EthoVision ®XT | Noldus Information Technology | https://.noldus.com/animal-behavior-research/products/ethovision-xt |
| GraphPad Prism | GraphPad Softare | https://.graphpad.com/scientific-softare/prism/ |
| Adobe photoshop | Adobe photoshop | https://.adobe.com/ |
| Zen softare | Zeiss | https://.zeiss.com/microscopy/int/products/microscope-softare/zen-lite.html |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elizabeth C. Engle (elizabeth.engle@childrens.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The following transgenic mouse alleles were used in mixed-background mice: ROSA26::FLPe (JAX:003946), 129S1 (JAX:002448), C57BL/6J(JAX:000664), 129SVE (Taconic#129S6), E2a-Cre (JAX: 003724), TdTomato (JAX:007914), Vglut2-ires-cre (JAX:016963), ISLmn-GFP (JAX:017952) mice were originally donated by donated by Dr. Samuel L. Pfaff. This line is also available from The Jackson Laboratory.
Both male and female mice were included in anatomic studies and neuronal culture experiments. Behavioral experiments were conducted in male mice. All animal work was approved and performed in compliance with Boston Children’s Hospital Institutional Animal Care and Use Committee protocols (17–01-3359R).
METHODS DETAILS
Generation of Tubb3 knockout mouse model
Generation of the Tubb3R262C mutant mice using a construct containing loxP sites flanking exon 4 was previously reported (Tischfield and Engle, 2010). Tubb3R262C mice on a mixed 129/B6 background were crossed with an oocyte-active Cre line (Ella-Cre) to remove exon 4, which contains 79.8% of the coding region and all functional domains of Tubb3. Tubb3−/+Cre−/+ mice were then crossed with wild-type to generate Tubb3−/+Cre−/− mice.
Histological analysis of adult mouse brains
Mice were intracardially perfused with 4% PFA or 10% neutral formalin, the skull removed, and the brains carefully dissected. Brains were fixed in 4% PFA or 10% neutral formalin for 24–48 hr and then processed for embedding in paraffin. Serial sections at 5 mm thickness were collected and mounted on superfrost-coated slides. Sections were subsequently stained using the Nissl method with Luxol fast blue. Results were examined and images were acquired by light microscopy on an Olympus BX51 epifluorescence microscope (Olympus, Center Valley, Pennsylvania) or Nikon Eclipse-80i transmitted light microscope (Nikon, Melville, New York) with Spot Xplorer CCD camera and Spot Version 4.6 acquisition software (Diagnostic Instruments, Sterling Heights, Michigan).
Whole mount embryo fluorescent immunohistochemistry
Whole mount embryonic day (E) 11.5 embryos were prepared as previously described (Huber et al., 2005). Embryos were fixed overnight in 4% paraformaldehyde, dehydrated through a methanol series, fixed overnight in 1 part dimethyl sulfoxide: 4 parts methanol, then rehydrated through a methanol series. They were blocked in a solution of 5% goat serum and 20% DMSO in PBS for 3 hours, placed in primary antibodies diluted in blocking solution for five days, washed with blocking solution for 4 hours, then placed in secondary diluted in blocking solution for 2 days. Mouse anti-neurofilament primary antibody (1:500; clone 2H3, Developmental Studies Hybridoma Bank, Iowa City, Iowa) and Alexa Fluor 594 goat anti-mouse secondary antibody (1:1000; Thermo Fisher Scientific, Carlsbad, California) were used to stain prepared embryos. Once stained, embryos were dehydrated through a methanol series and cleared in a solution of 2 parts benzoic acid to 1 part benzyl benzoate. Cleared samples were mounted on coverslips with the benzoic acid-benzyl benzoate solution and imaged on a Zeiss LSM 700 series laser scanning confocal microscope. Images were acquired using Zen Software (Carl Zeiss MicroImaging GmbH, Göttingen, Germany) and uniformly adjusted for contrast and brightness using ImageJ software (NIH).
Whole mount hindpaw skin fluorescent immunohistochemistry
Mice were perfused with 4% paraformaldehyde and skin tissue corresponding to the sural territory (most lateral part of the hindpaw) was dissected and mounted on a silicone plated surface then kept under gentle agitation for 3–7 days in 4% paraformaldehyde/ 0.01% azide. Samples were then incubated with chicken anti-neurofilament primary antibody (AB5539, Millipore; 1:5000 dilution) in StartingBlock PBS Blocking Buffer (ThermoFisher Scientific #37538) + 1% Triton X-100 + 0.01% sodium azide for 14 days. After 3 washes in PBS +1% Triton X-100 for 1–2 hours, they were incubated with goat anti-chicken secondary antibody (Jackson Immunoresearch Laboratories #703–165-155; 1:500 dilution) in StartingBlock PBS Blocking Buffer + 1% Triton X-100/0.01% sodium azide with for 7 days. Samples were washed 3x in PBS + 1% Triton X-100 for 1–2 hours and incubated in Vectashield (Vectorlabs, USA) for several days before imaging by confocal microscopy.
Ex-vivo magnetic resonance imaging (MRI) and diffusion tensor imaging (DTI)
Perfusion fixed brain specimens from Tubb3+/+ (n = 4) and Tubb3−/− (n = 3) mice were prepared for MRI and contained in a 10mm NMR tube filled with Fluorinert (FC-3283 3M, St Paul, MN) during imaging using a 14T vertical bore microimaging system (Bruker, Bilirica MA). Anatomical images were acquired using a spin echo multi-slice pulse sequence with TE/TR = 30/3000ms and 100 micron isotropic spatial dimensions. Diffusion weighted images were acquired with the same spatial dimensions using a 3D EPI pulse sequence with TE/TR = 53/750ms, nex = 1 and 2 repetitions having opposite phase encode directions for use with “blip-up-blipdown” correction of geometric distortions (Irfanoglu et al., 2015). The DWI sampling scheme was b/#dirs = 250/6,500/6,1500/32 and 3000/32, total time for all MRI and DTI scans was 15.5 hours per brain.
DTI processing was performed offline using TORTOISE3 software for apparent motion correction, geometric distortion correction and tensor fitting. The diffusion tensor volumes from individual brains within each group were used to generate Tubb3+/+ and Tubb3/ DTI templates by diffeomorphic registration for tensor accurate alignment of anatomical structures. The resulting template DTI maps were systematically inspected for gross anatomical differences between Tubb3+/+ and Tubb3−/− groups with particular attention to the white matter tracts as visualized by directionally encoded color (DEC) maps (Pajevic and Pierpaoli, 2000).
In addition to qualitative inspection for group differences of the anatomic images, quantitative morphometric analysis was performed by the tensor based morphometry (TBM) technique (Ashburner and Friston, 2000). Following generation of a single template from all brains in the study using the ANTs affine and diffeomorphic registration tools (Avants et al., 2008), the Log of the determinant of the Jacobian of the deformation fields (LogJ maps) was generated for each brain. LogJ maps were then averaged within each group to provide scaled values of local volume differences between the template brain and each group, where positive/negative values indicate increased/decreased local volume of the group anatomical images compared with the template.
Electron microscopy and morphometric analysis
Adult wild-type and Tubb3−/− mutant littermates were perfused with fixation solution (2% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M cacodylate buffer) and sacrificed, and sciatic nerves were dissected away and collected. Sciatic nerve tissues were postfixed for 4 hr at RT, and processed for plastic embedding and electron microscopy. For morphometric analysis, tissue crosssections were stained with toluidine blue. Bright-field Images were acquired, regions containing cross-sectional myelinated axons were chosen for analysis, and the internal diameters of axons (not including the myelin sheath) were measured using “Thermo Scientific HCS Studio: Cellomics Scan Version 6.60.” Ultra-structural images were acquired at stated magnifications using a Tecnai G2 Spirit BioTWIN Transmission Electron Microscope. Results were obtained from three wild-type and three mutant mice littermates.
Protein extraction and western blotting
Brain or sciatic nerve tissues were dissected from E14 or adult (2–4 month) mice and homogenized in ice-cold tissue lysate buffer (50mM Tris.HCL, 150mM NaCl, 1mM EDTA, 1%NP40, pH6.8) supplemented with Halt™ protease inhibitor and phosphatase inhibitor (single-use cocktail 100x, Thermo Fisher Scientific, Inc, Rockford Illinois). Tissue lysates were clarified by centrifugation at 14,000 rpm for 15 mins at 4°C, and were denatured in LDS Sample Buffer (Thermal Fisher Scientific, Carlsbad, California). Lysate samples were loaded on NuPAGE 4%–12% Bis-Tris gels (Thermo Fisher Scientific, Carlsbad, California), and subjected to SDS-PAGE electrophoresis. Proteins were then transferred from gels to nitrocellulose membranes (Thermol Fisher Scientific, Carlsbad, California) and membranes were blocked with 5% non-fat-milk in 0.01M PBST (0.1%Tween20) at RT for 1 hr. Membranes were then incubated for 1–2 hr at RT with primary antibodies diluted in 0.01M PBST (0.1%Tween20) as follows: mouse anti-α-tubulin clone DM1A (1:10,000, Sigma, St. Louis, Missouri), rabbit anti-bIII-tubulin (1:15,000, Abcam, Cambridge, Massachusetts), mouse-anti acetylated tubulin clone 11B-1(1:20,000, Sigma, St. Louis, Missouri), mouse-anti kinesin heavy chain (KHC) clone H2 (1:3000, EMD Millipore Corporation, Billerica, Massachusetts), rabbit anti-Kif21a (1:5000, generated by our lab as described (PMID: 20074521), mouse-anti Dynein intermediate chain (DIC) clone 74.1 (1:1500, EMD Millipore Corporation, Billerica, Massachusetts), mouse anti-pan β-tubulin clone AA2 (1: 5000, EMD Millipore Corporation, Billerica, Massachusetts), rat-anti-tyrosinated tubulin clone YL1/2 (1:12,000, Abcam, Cambridge, Massachusetts), rabbit antidetyrosinated tubulin (1:2000, EMD Millipore Corporation, Billerica, Massachusetts), rabbit anti-delta2 tubulin (1:2000, EMD Millipore Corporation, Billerica, Massachusetts), mouse anti-polyglutamylation tubulin clone GT335 (1:2000, Adipogen), rabbit anti-GAPDH (1:475, Santa Cruz Biotechnology, Inc, Santa Cruz, California). Following several washes with PBST, membranes were incubated with Peroxidase-AffiniPure donkey anti-mouse, anti-rabbit or anti-rat IgG (H+L) secondary antibodies (1:10,000, Jackson Immunoresearch, West Grove, Pennsylvania) at RT for 1 hr. Following several additional washes with PBST, chemiluminescent substrate (Amersham ECL Plus, GE Healthcare Life Sciences, Pittsburgh, PA) was added, and images were captured real-time at 10–20 s intervals using a FujiFilm LAS-4000 with CCD camera (GE Healthcare Life Sciences, Pittsburgh, PA). Protein measurement and quantification were performed using the “analyze Gels” function of the ImageJ software (National Institutes of Health).
TUBB3 depletion from tissue lysates by immunoprecipitation with anti-bIII-tubulin antibody
Adult mouse brains or sciatic nerves were dissected and homogenized in ice-cold tissue lysate buffer (50mM Tris.HCl, 150mM NaCl, 1Mm EDTA, 1% NP40, pH 6.8) supplemented with Halt™ protease inhibitor and phosphatase inhibitor (single-use cocktail 100x, Thermo Fisher Scientific, Inc, Rockford, Illinois). Tissue lysates were clarified by centrifugation at 14,000 rpm for 15 mins at 4°C. 1ml clarified lysate at 0.5mg/ml protein concentration was incubated with 6~8μg mouse anti-bIII-tubulin (Thermo Fisher Scientific, Inc, Rockford, Illinois) or normal mouse IgG (Santa Cruz Biotechnology, Inc, Santa Cruz, California) respectively for 2 hr at 4°C with gentle rotation, then with 50μl protein-G agarose beads (Invitrogen, Carlsbad, California) for an additional 6–8hrs.The tissue lysates containing the protein-G agarose beads were then centrifuged at 500 g for 5 mins at 4C, the supernatants collected and incubated for a second time with antibody and protein-G as described above, recentrifuged at 500 g for 5 mins at 4°C and collected. These final supernatants, depleted of βIII-tubulin, were denatured and analyzed by SDS-PAGE and western blot as described previously. Protein measurements and quantifications were performed using the “analyze Gels” function of the ImageJ software (National Institutes of Health). Three independent experiments were performed.
Microtubule polymerization and co-sedimentation assay
Brain or sciatic nerve tissues were dissected from E14 or adult (2–3 months) mice and homogenized in ice-cold BRB80 buffer (80 mM PIPES / KOH, 1mM EGTA, 1mM MgCl2, pH 6.8) supplemented with Halt™ protease inhibitor and phosphatase inhibitor (single-use cocktail 100x, Thermo Fisher Scientific, Inc, Rockford, Illinois). Crude tissue lysates were clarified by centrifugation at 14,000 rpm for 15 mins at 4C, and then the clarified lysates were centrifuged at 55,000 rpm for 35 mins at 4°C. 400μl of final supernatants (S1 fraction) at equal protein concentrations (about 1mg/ml) were added at final concentration with 1 mM GTP (Sigma, St. Louis, Missouri) and 20% glycerol. The mixtures were incubated for 1hr at 37C to allow tubulin to polymerize into microtubules, and then were centrifuge at 67,000rpm for 40 mins at 25°C. This yielded a pellet containing microtubules and associated proteins (P2 fraction) and a soluble supernatant (S2 fraction). The P2 fraction was rinsed twice by lysate buffer, re-suspended, and depolymerized completely by ice-cold BRB80 buffer on ice. All fractions were denatured with 1x loading buffer and analyzed by SDS-PAGE and western blot. The ability of motor proteins to bind to microtubules was represented as the ratio of the amount of motor protein present in P2 fraction to the total amount of motor protein present in both P2 and S2 fractions. Three independent experiments were performed for each study. Statistical analyses were performed by using a two-tailed paired Student’s t test for two-group comparison experiments, and by one-way ANOVA with post hoc Tukey t test for multiple comparisons experiments.
Semiquantitative real-time PCR
The L3/L4 DRGs were harvested and RNA extracted by acid phenol extraction (TRIzol reagent, Invitrogen). First-stranded cDNA synthesis (1 μg of total RNA per reaction) was performed with SuperScript III Reverse Transcriptase per the manufacturer’s instructions (Invitrogen). Quantitative real-time PCR was performed using the Sybr green detection system with primer sets designed on Primer Express. Specific PCR product amplification was confirmed using dissociation protocol. Transcript regulation was determined using the relative standard curve method per the manufacturer’s instructions (Applied Biosystems). Relative loading was determined before RT with RNA spectrophotometry followed by gel electrophoresis, and after RT by amplification of glyceraldehyde-3-phosphate dehydrogenase. Primers were as follows:
Tubb1- F/R: TCTTGGGTTCAAGGCTAGGAT / CGCAGTCAATCCCATGTTC; Tubb2a- F/R: AACCAGATCGGCGCTAAGT / TCCAGCTGCAAGTCACTGTC; Tubb2b- F/R: TCATCAGACCCACTGACACAG / TTTCCAGTTGCAAATCACTGTC; Tubb2c- F/R: CTTCGGGCAGATCTTCAGAC / ACGTCCAACACCGAGTCAA; Tubb3- F/R: GGTGGACTTGGAACCTGGAA / TAAAGTTGTCGGGCCTGAAT; Tubb4- F/R: TCTTTCGGCCAGACAACTTT / CAGGACGGCATCCACTAACT; Tubb5- F/R: CCGTAGCCATGAGGGAAAT / ATGCCATGTTCATCGCTTATC; Tubb6- F/R: CGGCCTGACAACTTCATCTT / TGAAGACAGTCGCAATGCTC
Adult DRG dissociated and spot cultures
DRGs were dissected from adult vGLUT2:Tdtomato Tubb3−/− and Tubb+/+ mice and digested with collagenase (5mg/ml) (Worthington Biochemical Corporation, Lakewood, New Jersey) and dispase (1mg/ml) (Sigma, St. Louis, Missouri) for 2 hours at 37°C. Neurons were then dissociated by mechanical trituration using a Pasteur pipette. Dissociated neurons were loaded over a 10% BSA (Sigma, St. Louis, Missouri) cushion and centrifuged at 1000 g for 10 mins. The pellet of dissociated neurons was resuspended and plated on PDL/laminin (Thermo Fisher Scientific, Carlsbad, California) coated culture plates and cultured in neural basal medium supplemented with B27 (Thermo Fisher Scientific, Carlsbad, California), L-glutamine, streptavidin/ampicillin and 50 ng/ml of NGF (Thermo Fisher Scientific, Carlsbad, California), 2 ng/ml GDNF (ProSpec, East Brunswick, New jersey) and 0.1mM Cytosine β-D-arabinofuranoside (Sigma, St. Louis, Missouri). For dissociated cultures, a neuronal suspension of 2×104 cells in 2ml culture medium was plated. For spot cultures, a neuronal suspension of 2×104 cells/7 mL from each mouse was placed as a spot in a defined region of the 35mm plate, and the culture medium was added 90mins after plating. For both culture conditions, half of the culture medium was changed every two days.
Adult DRG culture immunocytochemistry and neurite outgrowth analyses
After being cultured for the length of time indicated for each experiment, cultures were fixed by 4% PFA/0.01 M PBS for 20 mins at room temperature (RT) and washed x 3 with 0.01M PBS. DRG neurons were then permeabilized with 0.1% Triton X-100/0.01M PBS, incubated in blocking solution (5% normal goat serum, 1% BSA, 0.1% Triton X-100 in 0.01M PBS) for 1 hr at RT, and then incubated overnight at 4°C with two primary antibodies, rabbit anti-bIII-tubulin (1:1000, Abcam, Cambridge, Massachusetts) and mouse antiNeurofilament-medium chain (1:500; clone 2H3, Developmental Studies Hybridoma Bank, Iowa City, Iowa). The following day, the neurons were washed x3 in 0.01M PBS, and incubated with Alexa Fluor 647 and Alexa Fluor 488 conjugated goat anti-rabbit and goat anti-mouse IgG secondary antibodies (1:1000, Thermo Fisher Scientific, Carlsbad, California). Both primary and secondary antibodies were diluted in blocking solution. After several washes in 0.01M PBS, images were acquired on an inverted Nikon Eclipse Ti epifluorescence microscope with Coolsnap HQ2 camera (Photometrics, Tucson, Arizona), Nikon Elements software (Nikon, Melville, New York) and a 10X Plan Apo objective.
Neurite length and number were analyzed from cultures of dissociated neurons using the software “Thermo Scientific HCS Studio: Cellomics Scan Version6.60.” Neurite outgrowth was analyzed from spot cultures using the Sholl analysis and ImageJ software with a written macro (National Institutes of Health). Growth cone behavior was analyzed from DIV6 spot cultures by capturing 1 hour time-lapse (interval = 1min) images of the distal axon and growth cone of vGLUT2:Tdtomato positive neurons. The trajectory of the movement of growth cones was traced using the “Manual Tracking” function of ImageJ software (National Institutes of Health).
EB3-GFP tracking of microtubule dynamics in DRG growth cones
Adult DRG neuron spot cultures were infected with purified AAV8-EB3-GFP one day after plating. 4 days after infection, 5mins time-lapse images were acquired on an inverted Nikon Eclipse Ti epifluorescence microscope with a Total Internal Reflection Fluorescence (TIRF) microscope with a 100X 1.45 N.A. oil immersion TIRF objective (Olympus, Center Valley, Pennsylvania). Microtubule dynamics in axonal growth cones, including microtubule growth velocity, growth lifetime, and growth length, were analyzed using the software plusTipTracker. The experiment was repeated three times and, for each experiment, at least 10 to 15 growth cones from each condition were quantified. Statistical significance was determined by a two-tailed paired Student’s t test for comparison between Tubb3−/− and WT mice.
Behavioral studies
All animal procedures were approved by the Boston Children’s Hospital Animal Care and Use Committee and conducted in a blinded fashion in a quiet room (temperature 22 ± 1°C) between the hours of 9 AM to 6 PM. Only males were used for behavioral studies (3 to 6 months, unless otherwise stated. Mice were housed with their littermates (2 to 5 mice per cage based on the litters) in OptiMICE cages with food and water ad libitum. All animals were maintained under the same conditions (22 ± 1°C, 50% relative humidity, 12-h light/dark cycle). Randomization was achieved through the breeding: at the time of weaning mice were separated based on their sex and placed in their new home cage. Only cages with a mixed representation of transgenic mice and their littermates were used for behavioral experiments. All experiments used at least 2 independent litters and were duplicated. Behavioral assays and genotyping were performed by different experimenters.
General behavioral assays
Actimetry
Ambulatory locomotion was monitored with an infrared detector (InfraMot-Activity System; TSE Systems Inc, Chesterfield, MO, USA). Mice were placed individually into their evaluation cage (445 cm2) for 2 days for habituation. The number of ambulatory events was then collected over 4 days and computed by bins of 1hr.
Social interaction
Mice in their home cages were habituated to the testing room for 30 minutes prior to testing. The testing apparatus was a rectangular black acrylic box divided equally into (3) separate chambers. The two inner dividers were clear acrylic and contained sliding doorways to allow mice access into each individual chamber. A video camera was mounting above the testing chamber to record mouse during the test. Each 10-minute test consisted of two phases: Novel mouse versus Empty cage and Familiar mouse versus new Novel mouse. Prior to adding the novel mouse, each test mouse was allowed to explore the (3) chambers with full access to each partition containing small empty cages made of acrylic bars. Following the habituation phase the test mouse was placed in the center chamber with the doorways closed. An unfamiliar mouse was place on one of the empty acrylic cages (alternating left and right). Both doorways were then opened simultaneously to allow the test mouse to once again explore each chamber for 10 minutes. The test mouse was then removed from the test chamber and an unfamiliar mouse was added to the second holding cage opposite from the now familiar mouse. The test mouse was then placed in the center chamber. The doorways were opened simultaneously to allow the test mouse to explore each chamber containing the familiar and new novel mouse.
Elevated plus maze
The elevated plus maze (Noldus Information Technology Inc., Leesburg, VA) consisted of two opaque closed arms and two open arms joined by a center opening. A video camera was mounted above the center of the plus maze. Mice were habituated in their home cages in the testing room 30 minutes prior to testing and then each mouse was placed in the center of the plus maze toward the closed arm furthest away from investigator and allowed to freely explore the maze. Video was recorded for a 5-minute trial per animal and the time spent in closed and open arms were calculated using Noldus Ethovison XT.
Water Morris
Mice were habituated in their home cages in testing room for 30 minutes prior to testing. The Morris water maze test consisted of one test per day for 5 days. Day 1, visual platform: mice were tested with a visual platform raised above the water with a visible lollipop stand placed in center of the platform. Two visual trials were conducted, one trial with 4 start points. Day 2, hidden platform 1: Platform was moved 180 degrees and submerged under 1cm of water. Two hidden trials were conducted. Day 3, hidden platform 2: Platform remained submerged in the same location as the previous day and three hidden trials were conducted. Day 4, probe trial: platform was removed and mice were given a 60 s swimming trial. Day 5, reversal: Platform was relocated 180 degrees and submerged under 1 cm of water. Three reversal trials were conducted. A video camera was mounted above the large tank and tracking data was recording using Noldus Ethovison XT.
Olfaction
Mice were habituated to the testing food, Bacon Softies S3580–1 (Bio-Serv, Flemington, NJ 08822), in their home cage for three days prior to olfaction testing. On the day of testing, mice were first habituated in their home cages in the testing room for 30 minutes, and then each test mouse was placed in a clean pie shaped OptiMICE cage containing 1/4 inch recycled paper bedding with the cage lid closed. Mice were allowed to explore the cage for 5 minutes. Subsequently, forceps were used to place a 1/2 inch piece of bacon softie on the cage filter located in the OptiMICE cage cover. The duration of time taken by each mouse from softie placement to onset of seeking behavior (head positioning, whisker twitching), and from onset of seeking behavior to when the mouse’s nose touched the filter that held the bacon softie were recorded for each mouse. Mice were not food restricted at any point during the study, and were rewarded with the softie once the food object was found.
Visual Acuity
Mice were habituated in their home cage in the testing room for 30 minutes prior to start of testing. Mice were also habituated to the Mouse OptoMotry testing chamber (Cerebral Mechanics) for 10 minutes prior to start of testing. The testing chamber consisted of a square box with a video camera mounted in the top of the hatch lid. Four video monitors comprised the walls of the chamber, and were used to display visual stimuli to the test mouse. A round platform mounted in the center of the floor the box was used as a pedestal for the mouse to stand on during testing. The unrestrained test mouse was placed on the pedestal and presented with visual stimuli in the form of vertical bars of varying frequencies rotating steadily across the monitors in a clockwise or counterclockwise direction. A positive response was recorded as the mouse fixating on one of the bars and rotating the head with the bar. A negative response was recorded as failure to fix on bar and rotate the head as the bar moved across the monitor. Frequencies of 0.078, 0.228 and 0.303 Hz (cyc/deg) were measured (Prusky et al., 2004).
Nociception assays
Pinprick test
Mice were placed onto a mesh grid (5 × 5 mm) under an upside-down 500 mL beaker, and an Austerlitz pin (000; Fine Scientific Tools, USA) was gently applied onto the sural territory of the paw. The number of withdrawals out of 10 stimulations was measured. The same male experimenter tested all conditions.
Von Frey filaments (static punctate mechanical stimuli)
Mice were placed on a mesh grid (5 × 5 mm) under an upside-down 500 mL beaker, and mechanical sensitivity was determined with a graded series of seven von Frey filaments that produced a bending force of 0.07, 0.16, 0.4, 0.6, 1, 1.4 and 2 g. The stimuli were applied within the sciatic nerve territory of the mice for 1–3 s. Each filament was tested ten times in order of increasing force, starting with the filament producing the lowest force. Successive von Frey filament applications were separated by at least 5 s after the mice had returned to their initial resting state. The same male experimenter tested all conditions.
Acetone test (response to cold)
Mice were placed onto a mesh grid (5 × 5 mm) under an upside-down 500 mL beaker, and a small volume of acetone (5 ml) was applied to the plantar surface of the hindpaw using a 1 mL syringe. The time spent flinching or licking the paw was recorded for 1 min with a stopwatch. Mice were tested twice per session. The same male experimenter tested all conditions.
Radiant heat pain (Hargreaves test)
Before testing, each mouse was placed on an elevated glass surface set at 30°C and habituated in their individual cage for 1 h. Then a radiant heat source (beam intensity set to cause baseline latencies in C57Bl6/j mice~15 s) was targeted at the paw and the latency to withdraw was measured. Radiant heat apparatus was from IITC Life Science Inc. (USA).
Contact heat pain (hot plate test)
Mice were placed on a metallic plate heated at 52°C within an acrylic container (Bioseb, France), and the latency to flinching, licking one of the hindpaws or jumping was measured. Mice were habituated to the procedure by placing them on a plate set at 30°C, and several baseline measurements were taken to reduce the risk of habituation or sensitization. Only one measurement was performed per day. The same male experimenter tested all conditions.
Formalin assay
Mice were gently handled and a solution containing 1% formalin dissolved in water was injected intraplantarly. Immediately after injection, mice were placed under an upside-down 500 mL beaker and the time spent flinching or licking the injected paw was measured. Results are presented by 3 minutes bin. The same female experimenter tested all mice.
Sciatic nerve crush surgery
Sciatic nerve crush surgery (SNcrush) was performed under 3% induction / 2% maintenance with isoflurane on adult mice (8 to 12 weeks old). The left sciatic nerve was exposed just below the sciatic notch under sterile conditions and then crushed using Dumont #5/45 forceps for 30 s under a dissection microscope. After the nerve injury, the incision wound was sutured and animals were allowed to recover on heated pads before being returned to their home cage. The surgeon who performed the SNI was blinded to the genotype.
Sensory functional recovery following sciatic nerve crush
High threshold mechano-sensitivity (pinprick test)
Mice were placed on wire mesh cages and tested after a 20-minute habituation period, or when they were idle. An Austerlitz pin (000; Fine Scientific Tools, USA) was then gently applied to the plantar surface of the paw without moving the paw or penetrating the skin. For measuring regeneration rate, the most lateral part of the plantar surface of the hind paw (sensory field of the sciatic nerve) was divided into 5 areas. The pinprick was applied from the most lateral toe to the heel. A response was considered positive when the animal briskly removed its paw, and the animal was graded 1 for this area, and then tested for the next one. If none of the applications elicited a positive response, the overall grade was 0. In that case, the saphenous territory of the same paw was tested as a positive control, which always elicited a positive response. For sensitivity, the pin was applied ten times in the same area and the number of brisk withdrawals was measured.
Low-threshold mechano sensitivity (brush test)
Mice were placed onto a mesh grid (5 × 5 mm) under an upside-down 500 mL beaker, and three successive gentle touch stimuli were applied with a round-head paintbrush with a diameter of 2 mm (Princeton Brush Co.) onto the sural territory of the paw separated into 2 halves. Each series of stimulations started at the distal part of the paw to its middle and lasted less than 1 s. For regeneration studies, the brush was applied twice in each territory and the mouse was scored 1 per territory if it withdrew its paw upon stimulation. For sensory sensitivity, the brush was applied 10 times across the whole hindpaw and the number of withdrawals and time spent flinching/liking was recorded. The same male experimenter tested all conditions.
Von Frey filaments (static punctate mechanical stimuli)
Mice were placed on a mesh grid (5 × 5 mm) under an upside-down 500 mL beaker, and mechanical sensitivity after sciatic nerve crush was determined using von Frey filaments that produced a bending force of 1 and 2 g. The stimuli were applied within the sciatic nerve territory (#2) of the mice for 1–3 s ten times and the number of brisk withdrawals was measured. Mice were tested daily from d7 to d24 after sciatic nerve crush. The same male experimenter tested all conditions.
Motor functional recovery following sciatic nerve crush
Toes spreading (TS)
Mice were recorded walking (18 cm/sec) using DigiGait apparatus (Mouse Specifics, MA, USA) and TS was calculated using imageJ to calculate the distance between the most lateral hindpaw toes at the time of flexion. In a separate series of experiments, toes spreading was evaluated manually by gently covering mice with a piece of cloth and lifting them by the tail, uncovering the hind paws for clear observation. Under this condition, the digits spread, maximizing the space between them (the toe spreading reflex). The reappearance of this reflex results from reinnervation of the small muscles of the foot and was scored: 0, no spreading; 1, intermediate spreading with all toes; and 2, full spreading. The results were comparable to the scoring of the DigiGait signal. All mice were tested before injury and had a score of 2.
Compound muscle action potentials (CMAP)
Mice were deeply anesthetized by an intra-peritoneal injection of urethane (200 mg/kg). Sciatic nerves were exposed bilaterally and a steel bipolar stimulating electrode placed under the sciatic nerve at the proximal level of sciatic notch. The recording electrode was placed in the intraplantar muscle, a reference electrode was placed distal to the recording electrode and a ground electrode was placed in the lower thigh. CMAP evoked by supramaximal square-wave stimulus pulses were recorded using a MEB-9400A-L-T EMG/NCV/EP system (Nihon Kohden, Foothill Ranch, CA). Amplitude, area, duration of compound muscle action potentials and the distal motor latency were measured.
Sleep recording surgery
Mice were anesthetized with ketamine–xylazine (100 mg and 10 mg per kg bodyweight, i.p.) and placed in a stereotaxic apparatus (model 1900, David Kopf Instruments). Two stainless steel screws were implanted for ipsilateral frontoparietal EEG recordings (1.5 mm lateral to the right of the sagittal suture, 1 mm anterior to the bregma and 2 mm anterior to the lambda). Two flexible EMG electrodes (multistranded stainless steel wire; AS131, Cooner Wire) were inserted into the neck extensor muscles. All electrodes were attached to a 2 ×3 2 microstrip connector affixed to the animal’s head with dental cement, and the scalp wound was closed with surgical sutures. Mice were given meloxicam (5 mg per kg bodyweight, i.p.) before they regained consciousness and then daily for 2 d and were housed singly after surgery. Ten days later, they were transferred to individual recording cages in a sound-attenuated chamber and connected by a flexible tether to a commutator (Crist Instrument Co.) for 5 d before their baseline sleep–wake and sensory behaviors were measured.
EEG and EMG acquisition and analysis
EEG and EMG signals were amplified ( × 5,000) using a Grass Instruments model 12 amplifier (West Warwick, RI) and were filtered as follows: high-pass filter at 0.3 Hz and low-pass filter at 1,000 Hz. Signals were sampled and stored at 128 Hz, digitally filtered (EEG, 0.3–30 Hz; EMG, 10–100 Hz), and semi-automatically scored in 10 s epochs as wake, NREMS or REMS using SleepSign for Animal (Kissei Comtec). This preliminary scoring was visually inspected by a trained experimenter and corrected when appropriate. The percentage of time spent in wake, NREMS and REMS, as well as the mean duration and number of behavioral state bouts, was calculated for each condition. NREMS sleep attempts were defined as NREMS bouts of < 5 s. NREMS latency was defined as the time elapsed between the beginning of the recovery period (immediately after the mice returned to their home cages and were reconnected to their tethers) and the first NREMS episode lasting ≥ 30 s.
EEG power spectra were computed by a fast Fourier transformation routine (using a Hanning window) for each 10 s epoch between 0.5 and 64.0 Hz with 0.25-Hz resolution. Epochs containing movement artifacts, predominantly during active wake, were excluded from spectral analysis. SWA (EEG power in the band at 0.5–4.0 Hz) during NREMS was computed over the first 5 h of the sleep opportunity period (2:00 p.m.–7:00 p.m.) after sleep deprivation in absolute values and expressed as the percentage of the baseline SWA mean value during NREMS measured over 24 h for each mouse. This transformation allowed for correction for individual differences in absolute power. Next, SWA was calculated over 360 NREMS epoch intervals (first, second and third hours of NREMS) to plot the time course of SWA decay expressed as the percentage of the baseline SWA mean value during NREMS measured over 24 h. Changes in SWA were analyzed within an individual NREMS episode by selecting all NREMS bouts lasting at least 60 s and preceded by at least 10 s of wake that occurred during the first 3 h of the sleep opportunity period and the corresponding baseline time interval for each animal. For normalization, all 10 s epochs were expressed relative to the baseline SWA mean value during NREMS measured over 24 h.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed as indicated using GraphPad Prism7 or Microsoft Excel for analyses and graphical representations. Quantitative data are presented as mean ± SEM. All experiments were independently repeated at least three times. Statistical details of experiments and statistical significance can be found in the figure legends.
Supplementary Material
High Lights.
Tubb3−/− mice are viable and have no detected neuroanatomical or behavioral defects
Tubb3−/− mice compensate to have wild-type pan-b-tubulins levels
Tubb3−/− growth cones have less dynamic microtubules and a 22% reduction in growth rate
Tubb3−/− dorsal root ganglia axon regeneration is slowed by 22% and recovery is delayed
ACKNOWLEDGMENTS
We thank Laura Anne Lowrey and Garrett Cammarata for technical assistance, the Assay Development & Screening Facility, the Neurodevelopmental Behavior, and the Viral Core of the Boston Children’s Hospital IDDRC (NIH; 1U54HD090255), the Histology and Electron Microscopy Cores of Harvard Medical School, the NIH Intramural Program, the Section for Quantitative Imaging and Tissue Sciences (NICHD/NIH), and the NIH Mouse Imaging Facility. This work was supported by the HHMI Medical Fellows Program and Harvard Medical School (to S.C.); the HHMI EXROP Program (to S.G.); the Harvard College Research Program (to A.S.); a Congressionally Directed Medical Research Programs (CDMRP) Award (W81XWH-13–2-0019) (to E.B.H.); the NIH (5T32HL110852–05 to S.-H.S.; K08EY027850 to M.C.W.); a Children’s Hospital Ophthalmology Foundation Faculty Discovery Award (to M.C.W.); and the NIH (R01DE022912) and the Dr. Miriam and Sheldon M. Adelson Medical Research Foundation (to C.J.W.). E.C.E. is a Howard Hughes Medical Institute Investigator.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.029.
DATA AND SOFTWARE AVAILABILITY
Software plusTipTracker was freely downloaded from https://www.utsouthwestern.edu/labs/danuser/.
REFERENCES
- Ashburner J, and Friston KJ (2000). Voxel-based morphometry–the methods. Neuroimage 11, 805–821. [DOI] [PubMed] [Google Scholar]
- Avants BB, Epstein CL, Grossman M, and Gee JC (2008). Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med. Image Anal 12, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, Hully M, Bianco CF, Boddaert N, Elie C, et al. ; LIS-Tubulinopathies Consortium (2014). The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 137, 1676–1700. [DOI] [PubMed] [Google Scholar]
- Barnat M, Benassy MN, Vincensini L, Soares S, Fassier C, Propst F, Andrieux A, von Boxberg Y, and Nothias F (2016). The GSK3-MAP1B pathway controls neurite branching and microtubule dynamics. Mol. Cell. Neurosci 72, 9–21. [DOI] [PubMed] [Google Scholar]
- Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, Schorle H, Uncu C, Woolf CJ, He Z, and Steen JA (2015). Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 86, 1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchthal F, and Kühl V (1979). Nerve conduction, tactile sensibility, and the electromyogram after suture or compression of peripheral nerve: a longitudinal study in man. J. Neurol. Neurosurg. Psychiatry 42, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L, et al. (2016). A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89, 956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Desai J, Miranda CJ, Duncan JS, Qiu W, Nugent AA, Kolpak AL, Wu CC, Drokhlyansky E, Delisle MM, et al. (2014). Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 82, 334–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew S, Balasubramanian R, Chan WM, Kang PB, Andrews C, Webb BD, MacKinnon SE, Oystreck DT, Rankin J, Crawford TO, et al. (2013). A novel syndrome caused by the E410K amino acid substitution in the neuronal b-tubulin isotype 3. Brain 136, 522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, and Cavalli V (2015). Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 161, 691. [DOI] [PubMed] [Google Scholar]
- Ciaramitaro P, Mondelli M, Logullo F, Grimaldi S, Battiston B, Sard A, Scarinzi C, Migliaretti G, Faccani G, and Cocito D; Italian Network for Traumatic Neuropathies (2010). Traumatic peripheral nerve injuries: epidemiological findings, neuropathic pain and quality of life in 158 patients. J. Peripher. Nerv. Syst 15, 120–127. [DOI] [PubMed] [Google Scholar]
- Cleveland DW (1987). The multitubulin hypothesis revisited: what have we learned? J. Cell Biol 104, 381–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley J (2007). What’s Wrong with My Mouse? Behavioral Phenotyping of Transgenic and Knockout Mice (John Wiley & Sons; ). [Google Scholar]
- Ferreira A, and Caceres A (1992). Expression of the class III beta-tubulin isotype in developing neurons in culture J. Neurosci. Res 32, 516–529. [DOI] [PubMed] [Google Scholar]
- Fulton C, and Simpson PA (1976). Selective synthesis and utilization of flagellar tubulin In The Multitubulin Hypothesis, Goldman R, Pollard T, and Rosenbaum J, eds. (Cold Spring Harbor Publications; ), pp. 987–1005. [Google Scholar]
- Gobrecht P, Leibinger M, Andreadaki A, and Fischer D (2014). Sustained GSK3 activity markedly facilitates nerve regeneration Nat. Commun 5, 4561. [DOI] [PubMed] [Google Scholar]
- Gobrecht P, Andreadaki A, Diekmann H, Heskamp A, Leibinger M, and Fischer D (2016). Promotion of functional nerve regeneration by inhibition of microtubule detyrosination. J. Neurosci 36, 3890–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, et al. (2011). Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331, 928–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PN, and Luduena RF (1996). Changes in the isotype composition of beta-tubulin delivered to regenerating sensory axons by slow axonal transport. Brain Res. 742, 329–333. [DOI] [PubMed] [Google Scholar]
- Huber AB, Kania A, Tran TS, Gu C, De Marco Garcia N, Lieberam I, Johnson D, Jessell TM, Ginty DD, and Kolodkin AL (2005). Distinct roles for secreted semaphoring signaling in spinal motor axon guidance. Neuron 48, 949–964. [DOI] [PubMed] [Google Scholar]
- Irfanoglu MO, Modi P, Nayak A, Hutchinson EB, Sarlls J, and Pierpaoli C (2015). DR-BUDDI (diffeomorphic registration for blip-up blip-down diffusion imaging) method for correcting echo planar imaging distortions. Neuroimage 106, 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YQ, and Oblinger MM (1992). Differential regulation of beta III and other tubulin genes during peripheral and central neuron development. J. Cell Sci 103, 643–651. [DOI] [PubMed] [Google Scholar]
- Ludueña RF (1993). Are tubulin isotypes functionally significant. Mol. Biol. Cell 4, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luduena RF, and Banerjee A (2008). The Isotypes of Tubulin (Humana Press; ). [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latrémoliére A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, et al. (2011). Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J. Clin. Invest 121, 4332–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova M, Cloin BM, Finan K, van den Berg R, Teeuw J, Kijanka MM, Sokolowski M, Katrukha EA, Maidorn M, Opazo F, et al. (2015). Resolving bundled microtubules using anti-tubulin nanobodies. Nat. Commun 6, 7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoura I, Takazaki H, Ayukawa R, Saruta C, Hachikubo Y, Uchimura S, Hida T, Kamiguchi H, Shimogori T, and Muto E (2016). Reversal of axonal growth defects in an extraocular fibrosis model by engineering the kinesin-microtubule interface. Nat. Commun 7, 10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz PF, and Oblinger MM (1995). Sensory neurons selectively upregulate synthesis and transport of the beta III-tubulin protein during axonal regeneration. J. Neurosci 15, 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz PF, Smith R, Pickett J, Frankfurter A, and Oblinger MM (1993). Expression of the class III beta-tubulin gene during axonal regeneration of rat dorsal root ganglion neurons. J. Neurosci. Res 34, 129–134. [DOI] [PubMed] [Google Scholar]
- Nawabi H, Belin S, Cartoni R, Williams PR, Wang C, Latremolière A, Wang X, Zhu J, Taub DG, Fu X, et al. (2015). Doublecortin-like kinases promote neuronal survival and induce growth cone reformation via distinct mechanisms. Neuron 88, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, and Woolf CJ (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91. [DOI] [PubMed] [Google Scholar]
- Niwa S, Takahashi H, and Hirokawa N (2013). β-tubulin mutations that cause severe neuropathies disrupt axonal transport. EMBO J. 32, 1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema R, Cushion TD, Phelps IG, Chung SK, Dempsey JC, Collins S, Mullins JG, Dudding T, Gill H, Green AJ, et al. (2015). Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum. Mol. Genet 24, 5313–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajevic S, and Pierpaoli C (2000). Color schemes to represent the orientation of anisotropic tissues from diffusion tensor data: application to white matter fiber tract mapping in the human brain. Magn. Reson. Med 43, 921. [DOI] [PubMed] [Google Scholar]
- Pamula MC, Ti SC, and Kapoor TM (2016). The structured core of human b tubulin confers isotype-specific polymerization properties. J. Cell Biol 213, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D, Miller HP, Banerjee A, Ludueña RF, and Wilson L (1994). Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proc. Natl. Acad. Sci. USA 91, 11358–11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Saillour Y, Bahi-Buisson N, Jaglin XH, Fallet-Bianco C, Nabbout R, Castelnau-Ptakhine L, Roubertie A, Attie-Bitach T, Desguerre I, et al. (2010). Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet 19, 4462–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, and Douglas RM (2004). Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest. Ophthalmol. Vis. Sci 45, 4611–4616. [DOI] [PubMed] [Google Scholar]
- Redeker V (2010). Mass spectrometry analysis of C-terminal posttranslational modifications of tubulins. Methods Cell Biol. 95, 77–103. [DOI] [PubMed] [Google Scholar]
- Rochlin MW, Wickline KM, and Bridgman PC (1996). Microtubule stability decreases axon elongation but not axoplasm production. J. Neurosci 16, 3236–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijs AC, Jaquet JB, Kalmijn S, Giele H, and Hovius SE (2005). Median and ulnar nerve injuries: a meta-analysis of predictors of motor and sensory recovery after modern microsurgical nerve repair. Plast. Reconstr. Surg 116, 484–494, discussion 495–486. [DOI] [PubMed] [Google Scholar]
- Ruschel J, Hellal F, Flynn KC, Dupraz S, Elliott DA, Tedeschi A, Bates M, Sliwinski C, Brook G, Dobrindt K, et al. (2015). Axonal regeneration. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science 348, 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saillour Y, Broix L, Bruel-Jungerman E, Lebrun N, Muraca G, Rucci J, Poirier K, Belvindrah R, Francis F, and Chelly J (2014). Beta tubulin isoforms are not interchangeable for rescuing impaired radial migration due to Tubb3 knockdown. Hum. Mol. Genet 23, 1516–1526. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, and Woolf CJ (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci 27, 7911–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Cho Y, Watt D, and Cavalli V (2015). Tubulin-tyrosine ligase (TTL)-mediated increase in tyrosinated α-tubulin in injured axons is required for retrograde injury signaling and axon regeneration. J. Biol. Chem 290, 14765–14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout A, D’Amico S, Enzenbacher T, Ebbert P, and Lowery LA (2014). Using plusTipTracker software to measure microtubule dynamics in Xenopus laevis growth cones. J. Vis. Exp 91, e52138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KF, and Cleveland DW (1986). Identification of conserved isotypedefining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proc. Natl. Acad. Sci. USA 83, 4327–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, and He Z (2011). Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 480, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka E, Ho T, and Kirschner MW (1995). The role of microtubule dynamics in growth cone motility and axonal growth. J. Cell Biol 128, 139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ti SC, Pamula MC, Howes SC, Duellberg C, Cade NI, Kleiner RE, Forth S, Surrey T, Nogales E, and Kapoor TM (2016). Mutations in human tubulin proximal to the kinesin-binding site alter dynamic instability at microtubule plus- and minus-ends. Dev. Cell 37, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, and Engle EC (2010). Distinct alpha- and beta-tubulin isotypes are required for the positioning, differentiation and survival of neurons: new support for the ‘multi-tubulin’ hypothesis. Biosci. Rep 30, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL, et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, and Gordon-Weeks PR (2005). Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell Sci 118, 993–1005. [DOI] [PubMed] [Google Scholar]
- Vemu A, Atherton J, Spector JO, Moores CA, and Roll-Mecak A (2017). Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 28, 3564–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman MC, Andrews C, Chan WM, Tischfield MA, Stasheff SF, Brancati F, Ortiz-Gonzalez X, Nuovo S, Garaci F, MacKinnon SE, et al. (2016). Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am. J. Med. Genet. A 170A, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.