

Abstract
The present review summarizes publications on the artemisinin peroxide fragment synthesis from 1983 to 2016. The data are classified according to the structures of a precursor used in the key peroxidation step of artemisinin peroxide cycle synthesis. The first part of the review comprises the construction of artemisinin peroxide fragment in total syntheses, in which peroxide artemisinin ring resulted from reactions of unsaturated keto derivatives with singlet oxygen or ozone. In the second part, the methods of artemisinin synthesis based on transformations of dihydroartemisinic acid are highlighted.
Keywords: artemisinin, artemisinic acid, organic peroxide, peroxidation, cycles
1. Introduction
In accordance with World Health Organization reports, malaria is one of the most widespread diseases, with 149–303 million of new malaria cases and 438,000 malaria deaths registered in 2015 all over the world. The largest number of new infection cases occur in the African Region (88%), the second one in Southeast Asia (10%), and the third in the Eastern Mediterranean (2%) [1].
Malaria is an infection caused by unicellular organisms of the Plasmodium type parasites. The growth and development of the parasites in red blood cells determine the pathology of malaria. In most cases, the disease is initiated by Plasmodium falciparum infection, although P. vivax, P. ovale, P. malariae, and P. knowlesi infection also cause malaria [2,3].
Traditionally, some drugs, such as quinine, chloroquine, mefloquine, doxycycline, and other antiparasitic ones, have been used for malaria treatment. However, the development of antibiotic resistance of Plasmodium significantly reduced the efficacy of therapy [4,5].
Much effort has been directed toward the treatment and prevention of malaria; as a result, the incidence of malaria (new cases of infection) reduced by 37% worldwide and by 42% in the African Region from 2000 to 2015. Over the same period, the rate of malaria deaths decreased by 60% worldwide and by 66% in the African Region [1]. Such impressive results have been achieved due to a complex of measures: insecticide-treated mosquito nets (ITNs), indoor residual spraying (IRS), rapid diagnostic testing (RDTs), and mainly artemisinin-based combination therapy (ACT) [6,7]. Artemisinin-based combination therapy (ACT) is highly efficient against malaria caused by P. falciparum, which is the most abundant and pathogenic type of Plasmodium for humans [8].
Artemisinin (Qinghaosu) (1) is a natural peroxide with high antimalarial activity [9,10]. It has been playing a key role in malaria treatment over the past several decades. Artemisinin was isolated from Artemisia annua for the first time in 1971 in the context of the scientific program “Project 523”, initiated by the Chinese government in 1967 [11,12,13]. The Nobel Prize in Physiology or Medicine 2015 was awarded to Chinese pharmaceutical chemist Tu Youyou “for her discoveries concerning a novel therapy against Malaria” [14,15,16].
The availability of artemisinin-based drugs is largely limited by their high cost [17,18]. Its isolation from Artemisia annua is still the main way for artemisinin production [19,20,21]. Full cycle time is 12–18 months, while overall yield of isolated artemisinin is usually less than 1 wt % [22,23].
As a result, the production of a sufficient amount of artemisinin with the use of only an isolation method remains an extremely challenging task. The problem of the sustainable manufacturing of artemisinin can be solved through a nontransgenic method [24] (selection, variation of nutrients and ambient conditions, and the use of test-tube cultures [25]), the use of transgenic A. annua [26], heterologous transgenic systems [27,28] (introduction of genes encoding the artemisinin biosynthesis into others cultures), semi-synthetic approaches based on available relative structures [29] and total synthesis [30] (Figure 1).
Figure 1.

Main strategies for artemisinin production.
Synthetic strategies for artemisinin production have been reported in several reviews [31,32,33,34,35]. The interdisciplinary approaches to the preparation of artemisinin were demonstrated by Abdin et al. [36] and Corsello et al. [37]. Semi-synthetic methods for artemisinin (1) production from artemisinic (3) and dihydroartemisinic (4) acids, more available metabolites of Artemisia annua, were shown in two other reviews [38,39].
1,2,4-Trioxolane cycle spiro-conjugated with lactone is the key pharmacophore fragment in artemisinin. The construction of this fragment is the most difficult stage both in synthetic and biological approaches to artemisinin production. The construction of peroxide fragment of artemisinin is carried out mainly in final synthetic steps with the use of one-pot peroxidation/cyclization reactions.
The present review is devoted to the strategies for construction of artemisinin peroxide fragment in synthetic and semi-synthetic approaches (Scheme 1). According to the published data, unsaturated keto derivatives 2 (route A) or dihydroartemisinic acid (4) (route B) serve as the precursor substance on final peroxidation/cyclization step in artemisinin synthesis. Synthetic methods where dihydroartemisinic acid (4) obtained from artemisinic acid (3) is directly oxidized into artemisinin (1) (route B) deserve special attention, due to the fact that these approaches form the basis of modern methods of artemisinin production [40]. In 2013, the Sanofi company launched the semi-synthetic artemisinin manufacturing based on photooxidation of dihydroartemisinic acid (4) [41].
Scheme 1.

Peroxidation steps based on the formation of hydroperoxides in synthetic approaches to artemisinin (1).
In the peroxidation step, the unsaturated keto derivative 2 initially forms the hydroperoxyaldehyde 6, cyclizing into artemisinin (1) under the action of acids. Peroxidation of dihydroartemisinic acid (4) starts from the formation of intermediate 5, which was converted into artemisinin (1) via compound 6. Consequently, in the present review, the data are primarily classified according to the structures of final precursors 2 and 4 and secondly according to the peroxidation methods.
2. Construction of Artemisinin Peroxide Fragment in Total Synthesis
2.1. Using Singlet Oxygen
In 1983, Schmid and Hofheinz presented the first example of artemisinin (1) total synthesis (Scheme 2) [30]. The available terpene alcohol, (−)-isopulegol (7), was used as a starting compound. The ketone 8 was synthesized from (−)-isopulegol (7) in five steps with a 58% yield. Later, it was alkylated by Stork–Jung vinylsilane iodide with the formation of silylated ketone 9 with a 62% yield. The key intermediate 12 was synthesized from silylated ketone 9 in four steps. After the treatment of ketone 9 by excess of TMSCH(OMe)Li, the major diastereomer 10 was isolated with an 89% yield (ratio of diastereomers 8:1). The lactone 11 prepared by successive debenzylation and oxidation of alcohol 10 was transformed into enol ether 12 by TBAF (tetra-n-butylammonium fluoride). Ene reaction of enol ether 12 with 1O2 and subsequent acid-catalyzed cyclization of obtained hydroperoxide led to desired artemisinin (1) with a 30% yield. The total yield was 5% in 14 steps.
Scheme 2.

Total synthesis of artemisinin by Schmid and Hofheinz.
Zhou et al. demonstrated the second example of artemisinin total synthesis in 1986 (Scheme 3) [42]. R-(+)-Citronellal (13), chosen as a starting material, was initially transformed into dihydroxy compound 14. The benzylation and subsequent oxidation of 14 resulted in ketone 8 with a 51% yield. The reaction of deprotonated ketone 8 with silylated vinyl ketone afforded 1,5-diketone 15, which was transformed into unsaturated ketone 16 with a 62% yield. The reduction of unsaturated ketone 16 followed by oxidation by chromium trioxide led to ketone 17 with a 47% yield. The addition of MeMgI to ketone 17 followed by dehydration afforded the mixture of isomers from which the unsaturated product 18 was isolated with a 46% yield. It was transformed into methyl ester of dihydroartemisinic acid (19) in three steps with a 72% yield. Ozonolysis of 19 afforded ketoaldehyde 20. The ketone group of 20 was protected by 1,3-propanedithiol with the formation of intermediate 21, which was converted into the enol ether 22 with the use of trimethyl orthoformate. The deprotection of 22 afforded enol ether 23. The photooxidation of enol ether 23 in the presence of oxygen and Rose Bengal following by the treatment of 70% HClO4 resulted in artemisinin (1) with a yield of 0.2% in 21 steps.
Scheme 3.

Total synthesis of artemisinin by Zhou and co-workers.
A few years later, Ravindranathan et al. reported the approach to aldehyde 20, which reduced the synthetic pathway from 15 to 8 steps [43]. The synthesis of key precursor 20 was based on an intramolecular Diels–Alder reaction of triene 25 prepared from (+)-3-carene (24) in three steps (Scheme 4). The triene 25 was cyclized into unsaturated ether 26 under high temperature conditions. The epoxidation of 26 followed by reduction by LiAlH4 led to alcohol 27, which was transformed into key precursor 20 in five steps.
Scheme 4.

The synthesis of ketoaldehyde 20 through the Diels–Alder approach.
Yadav and colleagues used (+)-isolimonene (28) obtained by (+)-2-carene heating as a starting reagent for the synthesis of artemisinin. Hydroboration with subsequent oxidation of (+)-isolimonene (28) resulted in acid 29 (Scheme 5) [44]. Iodolactonization of acid 29 led to the separable mixture of iodolactones 30 and 31. The intermolecular radical reaction of iodolactone 30 with methyl vinyl ketone (MVK) resulted in the formation of diastereomers 32/33 mixture, which can be separable by the introduction of thioacetal fragment. Methyl ester 34 was prepared by saponification and consequent methylation of thioacetal-derivative. The oxidation of hydroxyl-group, the introduction of methoxymethyl fragment, and the removal of the thioacetal group led to the mixture of enol ether 23 isomers with a 32% yield. The construction of bicyclic peroxide fragment of artemisinin is a two-step process. The first step is photooxidation of the mixture of enol ether 23 isomers by 1O2, and the second one is acid-catalyzed cyclization. The total yield of artemisinin (1) was 0.3% in 12 steps.
Scheme 5.

Total synthesis of artemisinin by Yadav and co-workers (2003).
The synthesis of artemisinin (1) based on artemisinic acid (3) with a total yield of 37% via cyclic enol ether 35 was reported (Scheme 6) [45]. Photooxidation of 35 using 1O2 followed by the treatment with TMSOTf (trimethylsilyl trifluoromethanesulfonate), resulted in deoxoartemisinin (36). Oxidation of 36 by the RuC13–NaIO4 system afforded 1 with a 96% yield.
Scheme 6.

Synthesis of artemisinin from artemisinic acid (3) via enol ether 35.
An efficient partial synthesis of artemisinin (1) starting from artemisinic acid or arteannuin B (38) through enol ether 40 was reported by Lansbury and Nowak (Scheme 7) [46]. Artemisinic acid or arteannuin B were converted to lactone 37. Reduction of double bond of arteannuin B (38) followed by deoxygenation of formed saturated epoxide resulted in lactone (37) with a 44% yield. Ozonolysis of 37 and protection of obtained carbonyl group led to aldehyde 39 that was transformed into enol ether 40 by the cleavage of lactone ring with subsequent methylation. The construction of artemisinin peroxide fragment was realized by photooxidation with 1O2 followed by camphorsulfonic acid (CSA)-catalyzed cyclization. The total yield of artemisinin (1) from arteannuin B (38) was 10% in eight steps. Later, the authors proposed an alternative route from arteannuin B (38) to artemisinin (1) in six steps with a 5% yield [47].
Scheme 7.

The synthesis of artemisinin by Lansbury and Nowak.
A cost-effective total synthesis of artemisinin with a total yield of 9% in nine steps was reported by Cook and colleagues. They conducted a gram scale synthesis from cyclohexenone (41)—a widely available starting material (Scheme 8) [48]. The synthesis of α,β-unsaturated aldehyde 43 was performed by successive Michael-type methylation, allylation with the formation of ketone 42, and Shapiro-process. The unusual [4 + 2] reaction between α,β-unsaturated aldehyde 43 and a silyl ketene acetal provided ortho ester 44. The feature of proposed total synthesis of artemisinin lies in using of selective oxidation reactions in two final steps. The mild Wacker-type oxidation of internal olefin of 44 afforded enol olefin 45. The final oxidative rearrangement of the latter by singlet oxygen from the decomposition of H2O2 by ammonium molybdate resulted in artemisinin (1).
Scheme 8.

The gram scale total synthesis of artemisinin by Cook and co-workers.
2.2. Other Methods
Avery and colleagues developed quite a different precursor for the construction of bicyclic peroxide artemisinin fragment—vinyl silane 51 (Scheme 9) [49,50]. The synthesis started from the transformation of (R)-(+)-pulegone (46) into sulfoxide 47 with a 70% yield. The alkylation of sulfoxide 47 with LDA and 1,3-dioxane-derivative bromide followed by desulfurization with an aluminum amalgam afforded the ketone 48 with a 50% yield. The two-step Shapiro reaction converted ketone 48 into homologous aldehyde 49. The silylation and acylation of aldehyde 49 produced silane 50, which was transformed into vinyl silane 51 via Ireland–Claisen rearrangement. The reaction of vinyl silane 51 (the step of removing of acetal protecting group was in the first version of synthesis [49]) with ozone followed by the rearrangement of unstable peroxide intermediate provided artemisinin (1) with a 35% yield (the total yield was 3.5% in 14 steps).
Scheme 9.

The total synthesis of artemisinin by Avery and co-workers (1992).
Wu and colleagues reported the hydrogen peroxide-based access to artemisinin (1) with an overall yield of 14.5% in seven steps starting from aldehyde 39 converted into spiro epoxide 52 (Scheme 10) [51]. Addition of H2O2 to the highly hindered quaternary C-12a in precursor 52 was achieved through a facile perhydrolysis of a spiro epoxy ring with the help of molybdenum species (prepared from Na2MoO4 and glycine). Resulting β-hydroxyhydroperoxide 53 was further cyclized by p-TsOH with the formation of peroxide 54, followed by oxidation, which resulted in ketal 36. The artemisinin (1) was prepared by the oxidation of 36 by the RuCl3/NaIO4 system.
Scheme 10.

The hydrogen peroxide-based access to artemisinin.
3. Artemisinin Synthesis Based on Dihydroartemisinic Acid
3.1. Preparation of Dihydroartemisinic Acid
Currently, semi-synthetic methods of artemisinin (1) synthesis from available biosynthetic precursors such as artemisinic acid (3) have attracted an increasing amount of attention as cost-effective, environmentally friendly, and high-quality approaches to the production of artemisinin from reliable sources [40].
Artemisinic acid, also called arteannuic acid (3), is 8–10-fold more abundant in A. annua than 1 [39] and can be isolated without chromatography [52]. The developed biosynthetic methods allow for the production of artemisinic acid on a large scale with the use of other organisms [53,54,55]. In 2006, the fermentation process with engineered yeast to produce high titers of artemisinic acid was developed in Amyris Inc. and the University of California [56]. In 2013, Paddon, Newman, and co-workers reported the method for biological production of artemisinic acid with the help of genetically modified strains of Saccharomyces cerevisiae (baker’s yeast). This development avoids the increase in fermentation titers of produced artemisinic acid to 25 g/L [57].
One of the most critical steps during the production of artemisinin is the regio- and diastereoselective reduction of artemisinic acid (3) to diastereomeric dihydroartemisinic acid (4). The most perspective method in the context of industry is the reduction by hydrogen (22–46 atm) on Rh-based catalysts—(Ph3P)RhCl2 [57], RuCl2[(R)-DTBM-Segphos](DMF)2 (the full name of ligand (R)-DTBM-Segphos is (R)-(−)-5,5′-Bis[di(3,5-di-tert-butyl-4-methoxyphenyl) phosphino]-4,4′-bi- 1,3-benzodioxole) [58], the yield of dihydroartemisinic acid (4) was > 98% and 99%, respectively. In addition, NaBH4/NiCl2 [59] (98% yield) and LiBH4/NiCl2 [60] (quantitative yield) were used for the reduction of artemisinic acid. An attractive method toward the diastereoselective synthesis of dihydroartemisinic acid (4) from artemisinic acid (3) was realized with use of diimide as a reducing agent (generated by the reaction of hydrazine monohydrate and oxygen) in the pilot plant scale-up with a yield of >90% [61,62,63]. Moreover, dihydroartemisinic acid (4) or its methyl ester were prepared in 17 steps from (−)-β-pinene [64], in 9 steps from (−)-isopulegol [65], and in 8 steps from (R)-(+)-citronellal [66].
3.2. Transformation of Dihydroartemisinic Acid into Artemisinin via Hydroperoxidation and Rearrangements
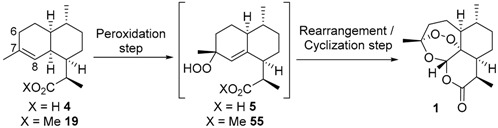
The first example of two-step conversion of dihydroartemisinic acid (4) into artemisinin (1) was reported by Roth and Acton in 1989 [29]. Dihydroartemisinic acid (4) was photooxidized into hydroperoxide 5, which in situ rearranged into artemisinin (1) by action of trifluoroacetic acid (TFA) (Scheme 11).
Scheme 11.

The transformation of dihydroartemisinic acid (4) into artemisinin (1).
Photooxygenation of dihydroartemisinic acid (4) with subsequent CH2N2 treatment provided corresponding ester 55 which was converted into peroxides 56 and 57 with a catalytic amount of Cu(OTf)2 (Scheme 12) [67]. The artemisinin (1) was prepared by p-TsOH-catalyzed cyclization of the crude mixture of peroxides 56 and 57. Additional methylation step did not significantly affect the yield of artemisinin. The total artemisinin (1) yield was 20% in four steps.
Scheme 12.

The transformation of dihydroartemisinic acid (4) into artemisinin (1) via hydroperoxyester 55.
Later, many attempts have been made to in situ transformation of dihydroartemisinic acid (4) to artemisinin (1) (Table 1); however, the yield of artemisinin (1) did not exceed 30%.
Table 1.
In situ transformation of dihydroartemisinic acid (4) or its ester 19 into artemisinin (1).
| No. | Substrate | Condition of Peroxidation Step | Condition of Rearrangement/Cyclization Step | Yield of 1, % | Reference |
|---|---|---|---|---|---|
| 1 | 4 | O2/hv, Methylene Blue, acetone, 0 °C, 30 min | TFA (cat.), petroleum ether, air, r.t., 4 days | 30 | [59] |
| 2 | crude mixture of 19 and isomer with double bond C6-C7 position | O2/hv, Methylene Blue, CH2Cl2, 20 °C, 90 min | TFA (cat.), petroleum ether, air, 20 °C, 4 days | 30 | [64] |
| 3 | crude mixture of 4 and isomer with double bond C6-C7 position | O2/hv, Methylene Blue, acetone, 0 °C, 30 min | TFA (cat.), petroleum ether, air, 20 °C, 24 h | 26 | [65] |
| 4 | 19 | O2/hv, Rose bengal, CH3CN, −30 °C, 6 h | (1) O2, Cu(OTf)2, CH3CN, −20 °C; | 25 | [66] |
| (2) p-TsOH (cat.), CH2Cl2, 20 °C, 4 h |
The mechanism of transformation of dihydroartemisinic acid (4) into artemisinin (1) was thoroughly studied [68,69,70,71,72,73]. The first step is peroxidation of 4 to form 5. Initially, it was suggested that conversion of hydroperoxide 5 into artemisinin (1) mainly proceeds through [2 + 2] cycloaddition of singlet oxygen to 5, with the formation of intermediate 58 followed by oxidation to intermediate 6 (Scheme 13) [74].
Scheme 13.

The mechanistic possibilities for the conversion of artemisinic acid into artemisinin.
Later, it was shown that the key steps involve terminal protonation of hydroperoxide 5, Hock cleavage via intermediate 59, and reaction of enol 60 with triplet oxygen to form hydroperoxide 6, which is cyclized to artemisinin (1) (Scheme 14) [75]. The formation of possible by-products, such as five-membered lactone dihydro-epi-deoxyarteannuin B (37) and enol ester 61, was minimized by TFA catalysis.
Scheme 14.

Acid-catalyzed reaction steps in the formation of artemisinin and side products.
A continuous-flow process for the synthesis of artemisinin (1) from dihydroartemisinic acid (4), which does not require isolation and purification of intermediates, was proposed (Scheme 15) [76]. The key step is a photochemical transformation involving a singlet-oxygen induced ene reaction, Hock cleavage, and the addition of triplet oxygen, followed by cyclization with the formation of an endoperoxide group. Tetraphenylporphyrin (TPP) was used as photosensitizer. Isolated yield of artemisinin (1) was 39% (1.36 g).
Scheme 15.

Reaction conditions for continuous flow synthesis of artemisinin.
Later, the authors succeeded in optimizing this one-pot photochemical continuous-flow method for the synthesis of artemisinin from dihydroartemisinic acid (4). The optimization of reaction conditions was conducted (LED lamp emitting at 420 nm, temperature, solvent, TFA concentration); as a result, the isolated yield of artemisinin (1) increased to 57% [75].
The continuous synthesis of derivatives of artemisinin: dihydroartemisinin 62, artemether 63, arteether 64, and artesunate 65 was also realized (Scheme 16) [77]. The system consists of three linkage modules for photooxidation/cyclization (9,10-dicyanoanthracene (DCA) was chosen as photosensitizer), reduction, and derivatization steps. The purification of crude products was carried out by column chromatography in fourth module.
Scheme 16.

Flow-synthesis of artemisinin derivatives—dihydroartemisinin (62), artemether (63), arteether (64), and artesunate (65).
The Paddon group showed that chemical conversion of dihydroartemisinic acid (4) into artemisinin (1) can be performed without specific photochemical equipment (Scheme 17) [57]. The methylation of dihydroartemisinic acid (4) afforded methyl ester 19, which was oxidized by singlet oxygen, generated through the disproportionation of concentrated H2O2 under the action of molybdate catalyst.
Scheme 17.

Conversion of dihydroartemisinic acid to artemisinin using a chemical source of singlet oxygen.
Using the same Na2MoO4/H2O2 protocol, Yikang Wu also realized the gram-scale synthesis of artemisinin (1) without any special equipment (Scheme 18) [78]. Ene reaction of dihydroartemisinic acid (4) with singlet oxygen afforded intermediate 5, which was rearranged into artemisinin (1) by action of TFA and oxygen.
Scheme 18.

The gram-scale synthesis of artemisinin by Wu.
Sanofi company developed the large-scale process of artemisinin (1) production from artemisinic acid (3) using diastereoselective hydrogenation and photooxidation (Scheme 19, Figure 2) [58]. At 22 bar of H2, a mixture of enantiomers 4 and 4′ was obtained with the help of RuCl2[(R)-DTBM-Segphos](DMF)2 catalyst. The activated esters 66 and 66′ (mixed anhydride) were obtained from 4 and 4′ with the use of ethyl chloroformate/potassium carbonate. They were expected to facilitate the final ring closure. Mixed anhydrides were introduced to the Schenck ene reaction using tetraphenylporphyrin (TPP) in CH2Cl2 as a sensitizer. Cascade of several chemical steps (Hock cleavage, additional oxygenation, ring closure) initiated by TFA resulted in a high overall yield (55%) of isolated artemisinin (1) (370 kg) from artemisinic acid (3) used as the starting material (Scheme 19).
Scheme 19.

The production of semi-synthetic artemisinin (1) by Sanofi.
Figure 2.

Photooxidation process in semi-synthetic artemisinin (1) production by Sanofi (Reprinted with permission from [58] Copyright 2016 American Chemical Society).
Rossen, Poliakoff, and George with colleagues reported two green chemistry approaches to artemisinin (1) from dihydroartemisinic acid (4) [79]. The first strategy lies in the use of liquid CO2 as a solvent and photocatalyst (commercially available porphyrins—TPP and TPFPP) immobilized onto the sulfonated ion-exchange resin Amberlyst-15; 460 mg of artemisinin (1) was isolated (Scheme 20).
Scheme 20.

Synthesis artemisinin (1) with use of dual solid catalyst.
The second strategy [79] is based on an application of alcohol/water mixtures. The major advance of this method is the possibility of recycling of solvents, photocatalyst (tris(bipyridine)ruthenium(II) chloride), and aqueous acid (Scheme 21).
Scheme 21.

The synthesis of artemisinin (1) in an aqueous condition.
The isolated yield of artemisinin (1) after three cycles is 37% (2.094 g). The authors [79] attempted to explain the modest yield of artemisinin (1). In slightly changed reaction conditions (Scheme 21, the lower equation), artemisinin (1) (50% NMR yield) and the stable minor hydroperoxide 68 (10% NMR yield) were detected and isolated. Hydroperoxide 68 is likely formed as a by-product in the ene reaction of dihydroartemisinic acid (4) with singlet oxygen. After column chromatography of the reaction mixture residue with high polar solvents (EtOAc and EtOH) the oily yellow oligomers were isolated.
The moderate yields of artemisinin are the result of a balance between factors stabilizing the artemisinin structure and the ease of side reactions. On the one hand, it was shown [80] that the strong anomeric nO → σ*CO interaction stabilizes artemisinin. On the other hand, in the peroxidation/cyclization step, highly reactive compounds can be easily oxidized, polymerized, and rearranged.
4. Conclusions
Analysis of the published data shows that synthetic approaches to the construction of peroxide ring in artemisinin has attracted increasing interest in recent times (Scheme 22). In total syntheses, various unsaturated keto derivatives 12, 23, 40, 45, or 51 were used as key precursors for the construction of artemisinin via hydroperoxyaldehyde 6. The less common strategy lies in the construction of peroxide fragment based on precursors 35 or 52 followed by the subsequent formation of the artemisinin lactone cycle. In the past two decades, significant progress has been made in semi-synthetic methods. Compared with other routes to artemisinin, its semi-synthesis is still less studied, despite the fact that it could satisfy the demand for the cheap production of sufficient amounts of artemisinin. The semi-synthesis of artemisinin based on artemisinic acid seems to be more efficient and environmentally friendly because reliable sources are used in production and high-quality chemical methods (catalytic hydrogenation and photooxidation) are applied. Nowadays, artemisinin construction in semi-synthetic methods is performed through precursors 4, 19, or 66.
Scheme 22.

Summary of key-precursors in different approaches.
The drawback of the majority of the available methods for the synthetic approaches to the artemisinin, which limits their application, is the low to moderate yield in the final peroxidation/cyclization step. In all published studies, the yield of artemisinin never exceeds 57%. Moreover, the synthesis often requires UV-irradiation equipment.
The main goals in the development of the synthetic strategies for the construction of a peroxide ring of artemisinin are as follows: (1) a development of methods for artemisinin precursor synthesis (artemisinic and dihydroartemisinic acids); (2) an investigation of the mechanisms of the peroxidation/cyclization step in order to predict optimal conditions for efficient synthesis; (3) the search for selective one-pot methods of peroxidation/cyclization; (4) applying green chemistry approaches to artemisinin production.
Acknowledgments
The authors are grateful to the Russian Foundation for Basic Research (Grant 16-29-10678) for financial support.
Author Contributions
All authors have contributed substantially: A.O.T., V.A.V., and A.I.I. wrote the manuscript, I.A.Y. conducted the research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.World Health Organization World Malaria Report 2015. [(accessed on 8 December 2016)]. Available online: http://www.who.int/malaria/publications/world-malaria-report-2015/en/
- 2.Roberts D.J., Chitnis C.E. Molecular Hematology. Wiley-Blackwell; Oxford, UK: 2010. Molecular pathogenesis of malaria; pp. 196–207. [Google Scholar]
- 3.Lin Chua C.L., Ataíde R., Umbers A.J., Boeuf P. Human Emerging and Re-Emerging Infections. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2015. Pathogenesis of malarial parasites in humans; pp. 393–422. [Google Scholar]
- 4.Meshnick S.R., Dobson M.J. The history of antimalarial drugs. In: Rosenthal P.J., editor. Antimalarial Chemotherapy: Mechanisms of Action, Resistance, and New Directions in Drug Discovery. Humana Press; Totowa, NJ, USA: 2001. pp. 15–25. [Google Scholar]
- 5.Petersen I., Eastman R., Lanzer M. Drug-resistant malaria: Molecular mechanisms and implications for public health. FEBS Lett. 2011;585:1551–1562. doi: 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 6.Gunn A., Pitt S.J. Parasitology. John Wiley & Sons, Ltd.; Chichester, UK: 2012. Parasite treatment and control; pp. 339–373. [Google Scholar]
- 7.Mutabingwa T.K. Artemisinin-based combination therapies (acts): Best hope for malaria treatment but inaccessible to the needy! Acta Trop. 2005;95:305–315. doi: 10.1016/j.actatropica.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Thwing J., Eisele T.P., Steketee R.W. Protective efficacy of malaria case management and intermittent preventive treatment for preventing malaria mortality in children: A systematic review for the lives saved tool. BMC Public Health. 2011;11:S14. doi: 10.1186/1471-2458-11-S3-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho W.E., Peh H.Y., Chan T.K., Wong W.S.F. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014;142:126–139. doi: 10.1016/j.pharmthera.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Li Y. Qinghaosu (artemisinin): Chemistry and pharmacology. Acta Pharmacol. Sin. 2012;33:1141–1146. doi: 10.1038/aps.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao W., Zhang Y., Zhang A. Case Studies in Modern Drug Discovery and Development. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2012. Discovery of antimalarial drug artemisinin and beyond; pp. 227–256. [Google Scholar]
- 12.Tu Y. The discovery of artemisinin (qinghaosu) and gifts from chinese medicine. Nat. Med. 2011;17:1217–1220. doi: 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- 13.White N.J., Hien T.T., Nosten F.H. A brief history of qinghaosu. Trends Parasitol. 2015;31:607–610. doi: 10.1016/j.pt.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Nobel Prize in Physiology or Medicine 2015. Oct 17, 2016. [(accessed on 17 November 2016)]. Nobelprize.Org. Nobel Media ab 2014. Web. Available online: http://www.Nobelprize.Org/nobel_prizes/medicine/laureates/2015/
- 15.Tu Y.Y., Ni M.Y., Zhong Y.R., Li L.N., Cui S.L., Zhang M.Q., Wang X.Z., Liang X.T. Studies on the constituents of Artemisia annua L. (author’s transl.) Acta Pharm. Sin. 1981;16:366–370. [PubMed] [Google Scholar]
- 16.Miller L.H., Su X. Artemisinin: Discovery from the chinese herbal garden. Cell. 2011;146:855–858. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White N.J. Qinghaosu (artemisinin): The price of success. Science. 2008;320:330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 18.Enserink M. Source of new hope against malaria is in short supply. Science. 2005;307:33. doi: 10.1126/science.307.5706.33. [DOI] [PubMed] [Google Scholar]
- 19.Haynes R.K. From artemisinin to new artemisinin antimalarials: Biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006;6:509–537. doi: 10.2174/156802606776743129. [DOI] [PubMed] [Google Scholar]
- 20.Wallaart T.E., van Uden W., Lubberink H.G.M., Woerdenbag H.J., Pras N., Quax W.J. Isolation and identification of dihydroartemisinic acid from Artemisia annua and its possible role in the biosynthesis of artemisinin. J. Nat. Prod. 1999;62:430–433. doi: 10.1021/np980370p. [DOI] [PubMed] [Google Scholar]
- 21.Laughlin J.C. Agricultural production of artemisinin—A review. Trans. R. Soc. Trop. Med. Hyg. 1994;88:21–22. doi: 10.1016/0035-9203(94)90465-0. [DOI] [PubMed] [Google Scholar]
- 22.Hale V., Keasling J.D., Renninger N., Diagana T.T. Microbially derived artemisinin: A biotechnology solution to the global problem of access to affordable antimalarial drugs. Am. J. Trop. Med. Hyg. 2007;77:198–202. [PubMed] [Google Scholar]
- 23.Jain D.C., Mathur A.K., Gupta M.M., Singh A.K., Verma R.K., Gupta A.P., Kumar S. Isolation of high artemisinin-yielding clones of Artemisia annua. Phytochemistry. 1996;43:993–1001. doi: 10.1016/S0031-9422(96)00369-X. [DOI] [Google Scholar]
- 24.Weathers P.J., Arsenault P.R., Covello P.S., McMickle A., Teoh K.H., Reed D.W. Artemisinin production in Artemisia annua: Studies in planta and results of a novel delivery method for treating malaria and other neglected diseases. Phytochem. Rev.: Proc. Phytochem. Soc. Eur. 2011;10:173–183. doi: 10.1007/s11101-010-9166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nair M.S.R., Acton N., Klayman D.L., Kendrick K., Basile D.V., Mante S. Production of artemisinin in tissue cultures of Artemisia annua. J. Nat. Prod. 1986;49:504–507. doi: 10.1021/np50045a021. [DOI] [PubMed] [Google Scholar]
- 26.Brown G.D. The biosynthesis of artemisinin (qinghaosu) and the phytochemistry of Artemisia annua L. (qinghao) Molecules. 2010;15:7603–7698. doi: 10.3390/molecules15117603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arsenault P.R., Wobbe K.K., Weathers P.J. Recent advances in artemisinin production through heterologous expression. Curr. Med. Chem. 2008;15:2886–2896. doi: 10.2174/092986708786242813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng Q., Qiu F., Yuan L. Production of artemisinin by genetically-modified microbes. Biotechnol. Lett. 2008;30:581–592. doi: 10.1007/s10529-007-9596-y. [DOI] [PubMed] [Google Scholar]
- 29.Roth R.J., Acton N. A simple conversion of artemisinic acid into artemisinin. J. Nat. Prod. 1989;52:1183–1185. doi: 10.1021/np50065a050. [DOI] [PubMed] [Google Scholar]
- 30.Schmid G., Hofheinz W. Total synthesis of qinghaosu. J. Am. Chem. Soc. 1983;105:624–625. doi: 10.1021/ja00341a054. [DOI] [Google Scholar]
- 31.Zhou W.-S., Xu X.-X. Total synthesis of the antimalarial sesquiterpene peroxide qinghaosu and yingzhaosu A. Acc. Chem. Res. 1994;27:211–216. doi: 10.1021/ar00043a005. [DOI] [Google Scholar]
- 32.Kim B.J., Sasaki T. Recent progress in the synthesis of artemisinin and its derivatives. Org. Prep. Proced. Int. 2006;38:1–80. doi: 10.1080/00304940609355981. [DOI] [Google Scholar]
- 33.Cook S.P. Artemisinin: A case study in the evolution of synthetic strategy. Synlett. 2014;25:751–759. doi: 10.1055/s-0033-1340627. [DOI] [Google Scholar]
- 34.Kumar V., Mahajan A., Chibale K. Synthetic medicinal chemistry of selected antimalarial natural products. Bioorgan. Med. Chem. 2009;17:2236–2275. doi: 10.1016/j.bmc.2008.10.072. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z., Yang L., Yang X., Zhang X. Advances in the chemical synthesis of artemisinin. Synth. Commun. 2014;44:1987–2003. doi: 10.1080/00397911.2014.884225. [DOI] [Google Scholar]
- 36.Abdin M.Z., Israr M., Rehman R.U., Jain S.K. Artemisinin, a novel antimalarial drug: Biochemical and molecular approaches for enhanced production. Planta Med. 2003;69:289–299. doi: 10.1055/s-2003-38871. [DOI] [PubMed] [Google Scholar]
- 37.Corsello M.A., Garg N.K. Synthetic chemistry fuels interdisciplinary approaches to the production of artemisinin. Nat. Prod. Rep. 2015;32:359–366. doi: 10.1039/C4NP00113C. [DOI] [PubMed] [Google Scholar]
- 38.Haynes R.K., Vonwiller S.C. From qinghao, marvelous herb of antiquity, to the antimalarial trioxane qinghaosuand some remarkable new chemistry. Acc. Chem. Res. 1997;30:73–79. doi: 10.1021/ar950058w. [DOI] [Google Scholar]
- 39.Kong J., Yang Y., Wang W., Cheng K., Zhu P. Artemisinic acid: A promising molecule potentially suitable for the semi-synthesis of artemisinin. RSC Adv. 2013;3:7622–7641. doi: 10.1039/c3ra40525g. [DOI] [Google Scholar]
- 40.Dhaintaut J., Dlubala A., Guevel R., Medard A., Oddon G., Raymond N., Turconi J. Photochemical Process for Producing Artemisinin. US20110065933 A1. U.S. Patent. 2011 Mar 17;
- 41.Pantjushenko E., Lemonde-San F. Sanofi and Path Announce the Launch of Large-Scale Production of Semisynthetic Artemisinin against Malaria. [(accessed on 20 November 2016)]. Available online: http://en.sanofi.com/Images/32474_20130411_ARTEMISININE_en.pdf.
- 42.Xu X.-X., Zhu J., Huang D.-Z., Zhou W.-S. Total synthesis of arteannuin and deoxyarteannuin. Tetrahedron. 1986;42:819–828. [Google Scholar]
- 43.Ravindranathan T., Anil Kumar M., Menon R.B., Hiremath S.V. Stereoselective synthesis of artemisinin. Tetrahedron Lett. 1990;31:755–758. doi: 10.1016/S0040-4039(00)94621-5. [DOI] [Google Scholar]
- 44.Yadav J.S., Satheesh Babu R., Sabitha G. Stereoselective total synthesis of (+)-artemisinin. Tetrahedron Lett. 2003;44:387–389. doi: 10.1016/S0040-4039(02)02500-5. [DOI] [Google Scholar]
- 45.Ye B., Wu Y.-L. An efficient synthesis of qinghaosu and deoxoginghaosu from arteannuic acid. Chem. Commun. 1990:726–727. doi: 10.1039/c39900000726. [DOI] [Google Scholar]
- 46.Lansbury P.T., Nowak D.M. An efficient partial synthesis of (+)-artemisinin and (+)-deoxoartemisinin. Tetrahedron Lett. 1992;33:1029–1032. doi: 10.1016/S0040-4039(00)91851-3. [DOI] [Google Scholar]
- 47.Nowak D.M., Lansbury P.T. Synthesis of (+)-artemisinin and (+)-deoxoartemisinin from arteannuin b and arteannuic acid. Tetrahedron. 1998;54:319–336. doi: 10.1016/S0040-4020(97)10286-1. [DOI] [Google Scholar]
- 48.Zhu C., Cook S.P. A concise synthesis of (+)-artemisinin. J. Am. Chem. Soc. 2012;134:13577–13579. doi: 10.1021/ja3061479. [DOI] [PubMed] [Google Scholar]
- 49.Avery M.A., Jennings-White C., Chong W.K.M. The total synthesis of (+)-artemisinin and (+)-9-desmethyltemesinin. Tetrahedron Lett. 1987;28:4629–4632. doi: 10.1016/S0040-4039(00)96582-1. [DOI] [Google Scholar]
- 50.Avery M.A., Chong W.K.M., Jennings-White C. Stereoselective total synthesis of (+)-artemisinin, the antimalarial constituent of Artemisia annua L. J. Am. Chem. Soc. 1992;114:974–979. doi: 10.1021/ja00029a028. [DOI] [Google Scholar]
- 51.Hao H.-D., Li Y., Han W.-B., Wu Y. A hydrogen peroxide based access to qinghaosu (artemisinin) Org. Lett. 2011;13:4212–4215. doi: 10.1021/ol2015434. [DOI] [PubMed] [Google Scholar]
- 52.Roth R.J., Acton N. Isolation of arteannuic acid from Artemisia annua. Planta Med. 1987;53:501–502. doi: 10.1055/s-2006-962787. [DOI] [PubMed] [Google Scholar]
- 53.Paddon C.J., Keasling J.D. Semi-synthetic artemisinin: A model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014;12:355–367. doi: 10.1038/nrmicro3240. [DOI] [PubMed] [Google Scholar]
- 54.Tsuruta H., Paddon C.J., Eng D., Lenihan J.R., Horning T., Anthony L.C., Regentin R., Keasling J.D., Renninger N.S., Newman J.D. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. PLoS ONE. 2009;4:e4489. doi: 10.1371/journal.pone.0004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westfall P.J., Pitera D.J., Lenihan J.R., Eng D., Woolard F.X., Regentin R., Horning T., Tsuruta H., Melis D.J., Owens A., et al. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc. Natl. Acad. Sci. USA. 2012;109:E111–E118. doi: 10.1073/pnas.1110740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ro D.-K., Paradise E.M., Ouellet M., Fisher K.J., Newman K.L., Ndungu J.M., Ho K.A., Eachus R.A., Ham T.S., Kirby J., et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 57.Paddon C.J., Westfall P.J., Pitera D.J., Benjamin K., Fisher K., McPhee D., Leavell M.D., Tai A., Main A., Eng D., et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496:528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 58.Turconi J., Griolet F., Guevel R., Oddon G., Villa R., Geatti A., Hvala M., Rossen K., Göller R., Burgard A. Semisynthetic artemisinin, the chemical path to industrial production. Org. Process Res. Dev. 2014;18:417–422. doi: 10.1021/op4003196. [DOI] [Google Scholar]
- 59.Roth R.J., Acton N. A facile semisynthesis of the antimalarial drug qinghaosu. J. Chem. Ed. 1991;68:612. doi: 10.1021/ed068p612. [DOI] [Google Scholar]
- 60.Jung M., ElSohly H.N., Croom E.M., McPhail A.T., McPhail D.R. Practical conversion of artemisinic acid in desoxyartemisinin. J. Org. Chem. 1986;51:5417–5419. doi: 10.1021/jo00376a070. [DOI] [Google Scholar]
- 61.Feth M.P., Rossen K., Burgard A. Pilot plant pat approach for the diastereoselective diimide reduction of artemisinic acid. Org. Process Res. Dev. 2013;17:282–293. doi: 10.1021/op300347w. [DOI] [Google Scholar]
- 62.Pieber B., Glasnov T., Kappe C.O. Continuous flow reduction of artemisinic acid utilizing multi-injection strategies—Closing the gap towards a fully continuous synthesis of antimalarial drugs. Chem. A Eur. J. 2015;21:4368–4376. doi: 10.1002/chem.201406439. [DOI] [PubMed] [Google Scholar]
- 63.Castro B., Chaudret R., Ricci G., Kurz M., Ochsenbein P., Kretzschmar G., Kraft V., Rossen K., Eisenstein O. Nonclassical CH−π supramolecular interactions in artemisinic acid favor a single conformation, yielding high diastereoselectivity in the reduction with diazene. J. Org. Chem. 2014;79:5939–5947. doi: 10.1021/jo500233z. [DOI] [PubMed] [Google Scholar]
- 64.Hsing-Jang L., Wen-Lung Y., Sew Y.C. A total synthesis of the antimarial natural product (+)-qinghaosu. Tetrahedron Lett. 1993;34:4435–4438. doi: 10.1016/0040-4039(93)88052-K. [DOI] [Google Scholar]
- 65.Constantino M.G., Beltrame M., da Silva G.V.J., Zukerman-Schpector J. A novel asymmetric total synthesis of (+)-artemisinin. Synth. Commun. 1996;26:321–329. doi: 10.1080/00397919608003621. [DOI] [Google Scholar]
- 66.Yadav J.S., Thirupathaiah B., Srihari P. A concise stereoselective total synthesis of (+)-artemisinin. Tetrahedron. 2010;66:2005–2009. doi: 10.1016/j.tet.2010.01.051. [DOI] [Google Scholar]
- 67.Haynes R.K., Vonwiller S.C. Catalysed oxygenation of allylic hydroperoxides derived from qinghao (artemisinic) acid. Conversion of qinghao acid into dehydroginghaosu (artemisitene) and qinghaosu (artemisinin) Chem. Commun. 1990:451–453. doi: 10.1039/c39900000451. [DOI] [Google Scholar]
- 68.Vonwiller S.C., Warner J.A., Mann S.T., Haynes R.K. Copper(ii) trifluoromethanesulfonate-induced cleavage oxygenation of allylic hydroperoxides derived from qinghao acid in the synthesis of qinghaosu derivatives: Evidence for the intermediacy of enols. J. Am. Chem. Soc. 1995;117:11098–11105. doi: 10.1021/ja00150a009. [DOI] [Google Scholar]
- 69.Sy L.-K., Zhu N.-Y., Brown G.D. Syntheses of dihydroartemisinic acid and dihydro-epi-deoxyarteannuin b incorporating a stable isotope label at the 15-position for studies into the biosynthesis of artemisinin. Tetrahedron. 2001;57:8495–8510. doi: 10.1016/S0040-4020(01)00711-6. [DOI] [Google Scholar]
- 70.Sy L.-K., Brown G.D. The mechanism of the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron. 2002;58:897–908. doi: 10.1016/S0040-4020(01)01193-0. [DOI] [Google Scholar]
- 71.Sy L.-K., Brown G.D. The role of the 12-carboxylic acid group in the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron. 2002;58:909–923. doi: 10.1016/S0040-4020(01)01192-9. [DOI] [Google Scholar]
- 72.Brown G.D., Sy L.-K. Synthesis of labelled dihydroartemisinic acid. Tetrahedron. 2004;60:1125–1138. doi: 10.1016/j.tet.2003.11.069. [DOI] [Google Scholar]
- 73.Brown G.D., Sy L.-K. In vivo transformations of artemisinic acid in Artemisia annua plants. Tetrahedron. 2007;63:9548–9566. doi: 10.1016/j.tet.2007.06.062. [DOI] [Google Scholar]
- 74.Acton N., Roth R.J. On the conversion of dihydroartemisinic acid into artemisinin. J. Org. Chem. 1992;57:3610–3614. doi: 10.1021/jo00039a020. [DOI] [Google Scholar]
- 75.Kopetzki D., Lévesque F., Seeberger P.H. A continuous-flow process for the synthesis of artemisinin. Chem. Eur. J. 2013;19:5450–5456. doi: 10.1002/chem.201204558. [DOI] [PubMed] [Google Scholar]
- 76.Lévesque F., Seeberger P.H. Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem. Int. Ed. 2012;51:1706–1709. doi: 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]
- 77.Gilmore K., Kopetzki D., Lee J.W., Horvath Z., McQuade D.T., Seidel-Morgenstern A., Seeberger P.H. Continuous synthesis of artemisinin-derived medicines. Chem. Commun. 2014;50:12652–12655. doi: 10.1039/C4CC05098C. [DOI] [PubMed] [Google Scholar]
- 78.Chen H.-J., Han W.-B., Hao H.-D., Wu Y. A facile and scalable synthesis of qinghaosu (artemisinin) Tetrahedron. 2013;69:1112–1114. doi: 10.1016/j.tet.2012.11.056. [DOI] [Google Scholar]
- 79.Amara Z., BellamyJessica F.B., Horvath R., Miller S.J., Beeby A., Burgard A., Rossen K., Poliakoff M., George M.W. Applying green chemistry to the photochemical route to artemisinin. Nat. Chem. 2015;7:489–495. doi: 10.1038/nchem.2261. [DOI] [PubMed] [Google Scholar]
- 80.Gomes G.D.P., Vil’ V., Terent’ev A., Alabugin I.V. Stereoelectronic source of the anomalous stability of bis-peroxides. Chem. Sci. 2015;6:6783–6791. doi: 10.1039/C5SC02402A. [DOI] [PMC free article] [PubMed] [Google Scholar]